
Abstract
Wnt signaling is a conserved pathway involved in expansion of neural progenitors and lineage specification during development. However, the role of Wnt signaling in the post-stroke brain has not been well-elucidated. We hypothesized that Wnt-3a would play an important role for neurogenesis and brain repair. Adult male mice were subjected to a focal ischemic stroke targeting the sensorimotor cortex. Mice that received Wnt-3a (2 µg/kg/day, 1 h after stroke and once a day for the next 2 days, intranasal delivery) had reduced infarct volume compared to stroke controls. Wnt-3a intranasal treatment of seven days upregulated the expression of brain-derived growth factor (BDNF), increased the proliferation and migration of neuroblasts from the subventricular zone (SVZ), resulting in increased numbers of newly formed neurons and endothelial cells in the peri-infarct zone. Both the molecular and cellular effects of Wnt-3a were blocked by the Wnt specific inhibitors XAV-939 or Dkk-1. In functional assays, Wnt-3a treatment enhanced the local cerebral blood flow (LCBF) in the peri-infarct, as well as improved sensorimotor functions in a battery of behavioral tests. Together, our data demonstrates that the Wnt-3a signaling can act as a dual neuroprotective and regenerative factor for the treatment of ischemic stroke.
Introduction
Stroke is a leading cause of mortality both in the US and worldwide. Ischemic stroke, which accounts for 75% of all strokes, is defined as the loss of blood flow to the brain due to blockage of vessels, causing severe damage to the local brain tissue. Patients who survive the initial ischemic attack often suffer from associated complications, such as hemiparesis, dementia, and depression. 1 Early management of neurological complications in patients with acute stroke is critically relevant in preventing further brain damage. 2 There remains a large unmet treatment gap for stroke intervention. The current intravenous thrombolysis treatment is limited to 5% or less of patients because of the requirement for treatment within the appropriate time window. 3 Furthermore, neuroprotective drugs that were developed in preclinical studies have thus far failed clinical translation. 4 As an alternative therapeutic approach, neurorestorative or regenerative therapy in the subacute and chronic phases following stroke has provided promising therapeutic outcomes for functional recovery after stroke.5,6 In this investigation, we explored the benefits of activating the canonical Wnt pathway, which is a central mediator of neurogenesis during development and in adults.
Wnt signaling has been shown to regulate cell proliferation in the central nervous system (CNS) during development7–9 and following injury. 8 Furthermore, Wnt is involved in the asymmetric division of stem cells, by both maintaining stem cell pluripotency10,11 and mediating cell fate specification and differentiation of neural stem cells both in vitro and in vivo.12–14 Many recent reports have also highlighted Wnt signaling as a potential target in the treatment of cancer,15,16 in stem cell differentiation to blood vessels, 17 cardiac tissues 18 and myelinating oligodendrocytes. 19 To date, 19 Wnt proteins 20 have been identified. Of the Wnt proteins, Wnt-3a serves as a particularly promising therapeutic candidate, because previous studies have established its involvement in hippocampal and cortical neurogenesis,11,21 as well as its requirement in neural stem cell (NSC) differentiation. Furthermore, Wnt-3a overexpression by lentiviral injection has been shown to provide regenerative and functional benefits following stroke. 22 However, while this provided the basis for the therapeutic relevance of Wnt-3a, the neuroprotective effect of Wnt-3a after ischemic stroke has not been thoroughly examined, and the translational potential using a clinically relevant drug treatment paradigm has not been explored.
In the present investigation, we investigated a translationally feasible form of Wnt-3a – a recombinant Wnt-3a that can be administered noninvasively via the intranasal route. We proposed that Wnt-3a signaling enhances neurogenesis in the subventricular zone (SVZ) and subgranular zone (SGZ) promote neurovascular repair in the ischemic brain. The hypothesis was tested in the mouse model of focal ischemic stroke targeting the primary somatosensory (S1 barrel) cortex. Our results suggest that recombinant Wnt-3a is a promising therapeutic candidate that can promote tissue repair and functional recovery after ischemic stroke.
Materials and methods
Animals and experimental groups
Experimental design and sample sizes for in vivo experiments.

Note: Sample sizes for different groups were chosen based on preliminary studies that were performed with a small group of animals per group (n = 2 for Westerns and immunohistochemistry, and n = 3 for behavior). Using the difference in means (estimate of effect size) determined with the preliminary studies and the sample sizes used, priori power analysis was performed (two tailed, α error probability = 0.05, power (1−β error probability) = 0.8) using G × Power (version 3.1.9.2, Universitat Düsseldorf, Germany). Detailed sample sizes for each experimental group are specified in figure legends.
Plasmid construction and viral infection
To express genes with an mCherry tag using a single plasmid, pEGIP was modified by restriction enzyme digestion and ligation to replace the GFP-IRES-Puro sequence with the BamHI-EcoRI-IRES-mCherry-WPRE (EIMW) sequence. pEGIP was a gift from Linzhao Cheng (Addgene plasmid # 26777). 24 For expression of Wnt-3a, the mouse DNA sequence of Wnt-3a was amplified from pAd-Wnt-3a and cloned into FUIMW through BamHI and EcoRI cutting sites. pAd-Wnt-3a was a gift from Tong-Chuan He (Addgene plasmid # 12518). 25 The primers used for cloning are listed as followed. Forward primer: 5′-GGGGGATCCA TGGCTCCTCT CGGATACCTC-3′. Reverse primer: 5′-GGGGAATTCC TACTTGCAGG TGTGCACGTC ATA-3′. EIMW-Wnt-3a lentivirus was produced and purified as described. 26 EIMW-Wnt-3a lentivirus was added into primary neuronal culture at seven days in vitro. At four days post-infection, cells were subjected to oxygen-glucose deprivation (OGD) treatment.
Primary neuronal cultures and OGD
Cortical neurons were isolated from embryonic day 14 (E14) mouse pups by dissection of the cerebral cortex as previously described. 27 Cells were maintained in a neurobasal media with B-27 serum-free culture supplement and L-glutamine (Invitrogen, Carlsbad, CA) until time of experiments. Cortical neurons of days in vitro (DIV) 7–11 days were incubated with the IRES-Wnt-3a lentivirus or Wnt-3a protein, both with and without inhibitors XAV-939 or Dkk-1 for 24 h. In lentiviral overexpression groups, the inhibitor was added three days after viral infection. The proteins were then isolated for Western blotting analysis. Cells were washed twice with ice-cold PBS and lysed in a medium containing 50 mM Tris–HCl (pH 8.8), 150 mM NaCl, 2 mM EDTA, 1% SDS, 2 mM sodium orthovanadate, 1% NP-40, 1% sodium deoxycholate, 20 µg/ml leupeptin, 20 µg/ml aprotinin, and 1 mM phenylmethanesulfonyl fluoride (PMSF). Cells were scraped from the dish, vortexed, and spun at 14,000g for 25 min to remove cell debris. Supernatants were then used for determination of protein concentration.
To perform the OGD insult, cells were cultured as mixed neuronal and glial population for 12–13 days. In the OGD group, media was exchanged for a physiological buffer solution lacking glucose (120 mM NaCl, 25 mM Tris-HCl, 5.4 mM KCl, 1.8 mM CaCl2, pH to 7.4 with NaOH). Cells were then incubated in a calibrated hypoxia chamber perfused with 5% CO2 and balanced nitrogen for a final ambient oxygen level of 0.2% for 3 h. Oxygen level was established, maintained, and monitored by the ProOx 360 sensor (Biospherix, NY). Wnt-3a (10 ng/250 µL) and/or XAV-939 (10 μM) were added into OGD medium. After 3 h, cells were returned to the normal incubator and the existing OGD media was completely changed out into normal oxygenated complete neuronal culture media with a series of half-media changes.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) Assay
The MTT assay was carried out to assess for mitochondrial and cell injury using a preassembled kit (Sigma-Aldrich, St. Louis, MO) as previously described; 28 24 h following the end of OGD exposure, the primary cortical neurons in 250 µL proliferation medium were plated into 48-well plates. At the predetermined time, 25 µL MTT reagent was added into each well, and plates were incubated for 2–4 h at 37℃ in the dark. Plates were read on FL600 Microplate Fluorescence Reader (BIO TEK, Winooski, VT) using a 570-nm wavelength filter.
Focal ischemic stroke model of the mouse
The sensorimotor cortex ischemic stroke was induced based on previous reports of the barrel cortex stroke, with modified artery occlusion procedures.29,30 Briefly, anesthesia was induced using 3.5% isoflurane followed by the maintenance dose of 1.5% isoflurane. The right middle cerebral artery (MCA) was permanently ligated using a 10–0 suture (Surgical Specialties Co., Reading, PA), accompanied by a bilateral common carotid artery (CCA) ligation for 7 min. This modified ischemic procedure was suitable and sufficient for the induction of focal ischemia in the mouse brain, resulting in specific infarct formation in the right sensorimotor cortex (see Figure 1). Animal body temperature was monitored during the surgery and recovery periods using a rectal probe and maintained at +37℃ by a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, US). Animals were kept in a ventilated humidity-controlled incubator (Thermocare, Incline Village, NV, USA). All animals were given 1 dose of meloxicam (oral, 1 mg/kg) prior to surgery and 1 daily dose of meloxicam (1 mg/kg) for three days post-surgery. Furthermore, animals were monitored for 60 min following the surgery to ensure adequate full recovery from anesthesia, as well as daily surveillance post-stroke for illnesses and locomotor activity. Among all mice in this study, there was approximately 5% rate of mortality necessitating exclusions following stroke. Mice were sacrificed by decapitation at 0, 6, or 12 h, or on days 1, 3, 14, 21, or 28 post-ischemia. The brains were immediately removed and preserved in OCT compound at −80℃ until further processing.
Wnt-3a treatment was neuroprotective after stroke. (a) Representative Western blots of cell lysates following infection with IRES-mCherry control vector or IRES-Wnt-3a-mCherry vector with or without XAV-939 cotreatment. (b) Quantification of Western blot analyses for Wnt-3a and β-catenin. (c) and (d) Representative images of in vitro primary cortical neurons subjected to 16-h reperfusion following 3-h OGD treatment among different groups. The cell density is higher in (d) due to the Wnt-3a protective effect against OGD. Cell density before OGD was similar between the two groups (data not shown). (e) Quantification of cell viability via MTT assay (24 h after OGD) demonstrated increased neuronal cell viability with Wnt-3a overexpression. Viability data are presented as box and whisker plot (min to max). All data are represented as mean ± SD; *p < 0.05 compared to mCherry; #p < 0.05 compared to IRES-Wnt-3a. (f) Application of recombinant Wnt-3a into media resulted in greater neuronal cell viability following OGD and reperfusion, and inhibition of Wnt signaling with XAV-939 reversed the neuroprotective effects. All data are represented as mean ± SD; *p < 0.05 compared to OGD; #p < 0.05 compared to OGD + Wnt-3a. (g) and (i) Confirmation for in vivo dosages of intranasal Wnt-3a and XAV-939 administration at 3d after stroke. (g) Representative Western blots for peri-infarct tissue lysates. (h) Quantification of protein levels of Wnt-3a and β-catenin. N = 4 for saline and Wnt3 + XAV-939 groups, and 5 for Wnt-3a group. (i) and (j) Representative TTC staining images and quantification of infarct volume at 72 h after stroke. TTC data are represented as mean ± SD; *p < 0.05 compared to saline; #p < 0.05 compared to Wnt-3a. N = 12 for saline group, 16 for Wnt-3a group, and 16 for Wnt-3a + XAV-939 group.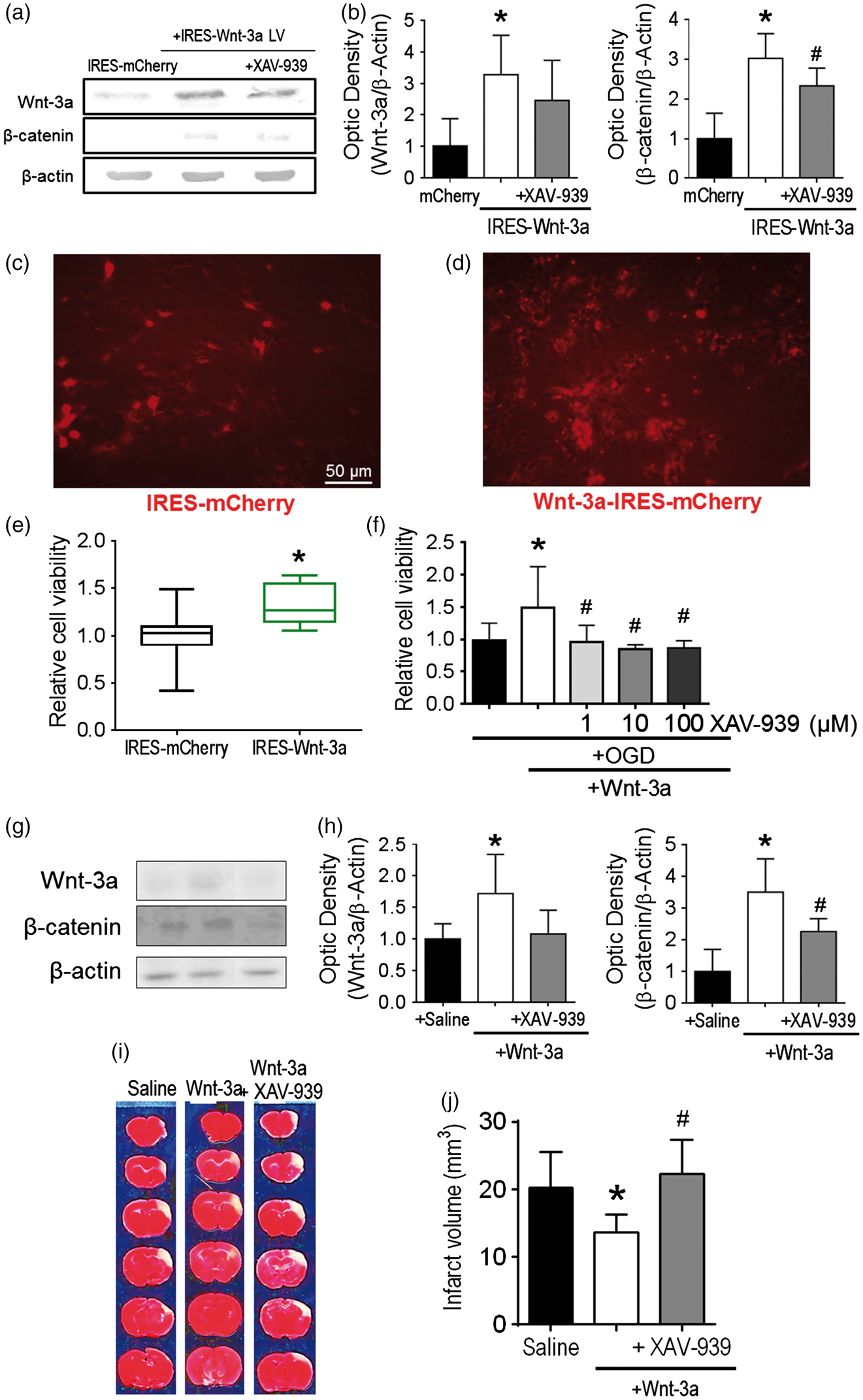
Dosage selection for Wnt-3 and XAV-939/Dkk-1 in vivo applications
To calculate the dosing required for both Wnt-3a and XAV-939 administration, we relied on in vitro findings and prior studies. For Wnt-3a dosing, the in vitro cell death assay determined that the effective concentration for driving a neuroprotective effect is 1 ng/ 25 µL (Figure 1(d)). We performed calculations under the assumption that the maximum average volume of an adult C57BL/6 male brain is approximately 460 mm3, which is the equivalent of 460 μl. 31 Thus, from the established concentration of 1 ng/25 μl, we calculated the total effective dose to be 18.4 ng. Furthermore, intranasal administration allows for a direct passage into the brain, allowing for greater distribution into the brain parenchyma. 32 To account for potential exhalation of the drug and fractions that may get remain trapped within the nostrils, we used a 3× higher dosage than the calculated, which ultimately resulted in 50 ng of Wnt-3a injected per animal, or 2 µg/kg for a 25 g mouse. For XAV-939 dosage, prior studies have used 1 mg/mouse (approximately 40 mg/kg) when delivering XAV-939 via i.p. injections. 33 However, intranasal delivery allows for 6 × greater cortical uptake of the drug than by i.p. injections. 32 Thus, we used a dosage (200 µg/animal, or 8 mg/kg for a 25 g mouse) that was slightly greater than the expected effective dosage (1/6 of 1000 µg = 166 µg/animal/day). Dkk-1 dosing was calculated in a similar manner. Briefly, prior studies established that the effective concentration for in vitro applications of Dkk-1 is 50 ng/ml.34,35 To apply this to our in vivo model, we performed the following calculations: 50 ng/mL ×0.46 mL = 23 ng per day for each animal. We used a 3.5× greater dose of 80 ng for each animal, or approximately 3 µg/kg/day.
Wnt-3a and XAV-939/Dkk-1 intranasal administration
Wnt-3a (50 ng/25 μl or 2 µg/kg) was administered using intranasal delivery in 0.9% NaCl with 0.1% bovine serum albumin. The dosage was given each day starting at 1 h after stroke and continuing for a total of three or seven days. For each day, 5 μl was delivered to each alternating nostril every 2 min until the full amount (25 μl) was delivered. Animals were restrained by the scruffing maneuver, in which the index finger and thumb were pinched together at the base of the neck to limit rotational range of motion. The Wnt-3a inhibitors XAV-939 (R&D Systems; 8 mg/kg) and Dickkopf-1 (Sigma, St. Louis, MO, USA; Dkk-1, 3 µg/kg) were dissolved in saline and delivered daily via intranasal administration. In the Wnt inhibition groups, the inhibitor was delivered immediately following Wnt-3a administration.
TTC staining of infarct volume measurement
Staining 2,3,5-triphenyltetrazolium chloride (TTC) was performed as previously described. 36 Briefly, at three days post-stroke, animals in different groups were sacrificed and TTC staining was used to reveal damaged/dead brain tissue. Brains were removed and placed in a brain matrix and then sliced into 1-mm thick coronal sections. Slices were incubated in 2% TTC (Sigma) at 37℃ for 5 min, followed by storage in 10% buffered formalin for 24 h. Digital images of the caudal aspect of each slice were obtained by a flatbed scanner. Infarct, ipsilateral hemisphere, and contralateral hemisphere areas were measured using ImageJ software (NIH, Bethesda, MD, USA). Infarct volume was calculated using the indirect method. 37
5-bromo-2′-deoxyuridine injections
For labeling of proliferating cells, 5-bromo-2′-deoxyuridine (BrdU) (Sigma) was administered to all animals at a dosage of 50 mg/kg body weight by intraperitoneal injection. Injections began on day 3 after stroke and continued once daily until sacrifice at either 14 or 21 days after stroke, depending on the nature of the experiment (see Table 1 for more info).
Immunohistochemistry
Frozen brain sections were sliced into coronal sections (10 µm thick) using a cryostat (Leica Microsystems, Buffalo Grove, IL). The sections were completely air dried and fixed with 10% buffered formalin (Fisher Scientific, Pittsburgh, PA, USA). Brain sections were then submerged in an ethanol/ acetic acid solution (2:1) for 10 min, washed three times with 1× PBS solution, and incubated with 0.2% Triton X-100 for 45 min. Slides were then blocked with 1% fish gelatin (diluted in PBS; Sigma, St. Louis, MO, USA) for 1 h at room temperature. Incubation with mouse anti-NeuN (1:400; Millipore, Billerica, MA), rabbit anti-Col-IV, and goat anti-DCX (1:50, Santa Cruz Biotechnology, Dallas, TX, USA) primary antibodies were diluted in PBS at 4℃ overnight. For double-staining of slides with BrdU, brain sections were post-fixed with formalin in 10% buffered formalin and washed in PBS. Slides were then treated with methanol (−20℃) and allowed to air dry. After rewetting in PBS, sections were treated with 0.1 mL borate buffer at pH 8.4. Slides were incubated with rat anti-BrdU (1:400; ABD Serotec, Raleigh, NC) overnight at 4℃. Primary antibodies were washed with PBS and replaced with secondary antibodies, Alexa Fluor®488 goat anti-mouse (1:300; Life Technologies, Grand Island, NY) and Cy3-conjugated donkey anti-rat (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature before rinsing again with PBS. After a final PBS wash, slides were mounted with Vectashield fluorescent mounting medium (Vector Laboratory, Burlingame, CA), and coverslipped for microscopy and image analysis. Fluorescent images (10×, 20×, and 40×) were acquired using the Olympus BX61 upright epifluorescent microscope (Olympus, Center Valley, PA, USA). High magnification confocal images (60×, 100×) were acquired using the Olympus FV1000 inverted confocal IX81 microscope.
Stereological cell counting
Stereology was carried out as previous described.38,39 For systematic random sampling in design-based stereological counting, every 10th (90 µm apart) brain section across the entire region of interest were counted. For multi-stage random sampling, six fields per brain section were randomly chosen under 10, 20×, or 40× magnification of an epifluorescent microscope or in confocal images. Neurogenesis in the ischemic border region was evaluated by counting the number of NeuN+/BrdU+ colabeled cells at 21 days post-stroke. Angiogenesis in the peri-infarct zone was evaluated by counting the number of Col-IV+/BrdU+ colabeled cells at 21 days post-stroke. For hippocampal neurogenesis and angiogenesis, NeuN+/BrdU+ and GLUT1+/BrdU+ colabeled cells were counted, respectively, in the subgranular zone (SGZ) of the dentate gyrus ipsilateral to the injury at 21 days post-stroke. For neuroblast migration, counting of DCX+ and DCX+/BrdU+ colabeled cells in white matter between the SVZ and ischemic cortex was performed at 14 days post-stroke. Counting was performed in matched coronal sections verified using the well-established mouse brain atlas. 40 Counting assays were performed under blind conditions.
Local cerebral blood flow measurement
Laser Doppler scanner imaging of cortical cerebral blood flow above the territory of the right MCA was conducted as previously described. 41 Briefly, animals were anesthetized and an incision was made to expose the skull above the territory of the right MCA. The laser was centered over the right coronal suture. A 3 × 3 mm square area around the initial spot was scanned by the Periscan Laser Doppler perfusion imaging system and analyzed by the LDPI Win 2 software (Perimed AB, Stolkholm Sweden). This technique is based on the principle that photons from the laser interact and are Doppler shifted only by moving red blood cells. Tissue perfusion is calculated by the LDPI program as the mean and amplitude of the Doppler shift. These parameters translate to average velocity and the concentration of the moving blood cells. 42 During the laser Doppler study, we measured blood flow in the same location for each animal at time points immediately before stroke, during the MCA and CCA occlusions, and then 7 and 14 days after stroke. Blood flow values are presented as percent of baseline (before stroke) flow for each animal.
Western blot analysis
The peri-infarct region was homogenized on ice using the Mammalian Protein Extraction Reagent (M-PER) Lysis buffer (Pierce, Rockford, IL) with phosphatase and proteinase inhibitors. Tissue samples were centrifuged at 12,000 r/min for 10 min at 4℃, and supernatant was collected. Isolated protein samples were stored in −80℃ until used. Protein concentration was determined by Bicinchoninic Acid Assay (BCA; Sigma Aldrich, St. Louis, MO). Proteins samples (40 ug/30 μl) were loaded onto a 4%–20% polyacrylamide gel before being separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in a Hoefer Mini-Gel system (Amersham Bioscience, Piscataway, NJ). Afterwards, the proteins were transferred in a Hoefer Transfer Tank onto a polyvinylidene fluoride (PVDF) membrane (BioRad, Hercules, CA). The membrane was blocked using 5% BSA diluted with TBS containing 0.05% Tween 20 (TBST) at room temperature for 1 h and incubated overnight at 4℃ with primary antibodies for β-catenin (1:1000, R&D Systems, USA), BDNF (1:500, Santa Cruz Biotechnology, Santa Cruz, CA) and Wnt-3a (1:1000, R&D Systems). β-actin (Sigma, St. Louis, MO) was used as a protein loading control. After washing with TBST, membranes were incubated with AP-conjugated or GRP-conjugated secondary antibodies (GE Healthcare, Piscataway, NJ) for 2 h at room temperature. After final washing with TBST, the signals were detected with either bromochloroidolylphosphate/nitroblue tetrazolium (BCIP/NBP) solution (Sigma) or film. Signal intensity was quantified by ImageJ (NIH) and normalized to the β-actin intensity.
Adhesive-removal test
The adhesive-removal test is a sensitive method to monitor the severity and restoration of the sensorimotor deficits after cerebral focal ischemia in mice. A small quarter-circle adhesive was fixed on alternating forepaws of the mouse. The time it took for the mouse to contact and to remove the adhesive was recorded. Animals were trained for three days prior to stroke induction to adequate performance of the task. Animals that demonstrated inability to perform the task expediently were excluded from future experiments. We then carried out the adhesive removal task 1 day prior to stroke to establish baseline behavior, and then at 3, 7, 14, 21 and 28 days after stroke. The task was carried out by a blinded investigator. The mean times (seconds, averaged from three to four trials) required to contact and to remove stimuli from each paw was recorded. All testing trials were conducted during the daytime.
Corner test
The corner test was carried out as previously described with slight modifications. 30 It is a well-established test used to detect integrated sensorimotor function involving both stimulation of the vibrissae (sensory input) and rearing (motor output). We designed a modified star-shaped chamber comprised of ten cardboard pieces with dimensions of 30×20×0.3 cm, with each corner forming a 30° angle. At 28 days post-stroke, each mouse was placed into the center area of the star and allowed to freely roam for 10 min, during which animal activity was recorded using an overhead camera. Afterwards, a blinded investigator watched back the replay of the animal movements, and observed the instances in which the animal crawled into the corner and reared up, and then documented whether the animal turned left or right. 15 turns were quantified per trial, and final data analysis was carried out using the right/total turn ratio.
Statistical analysis
GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used for statistical analysis and graphic presentation. Data was analyzed using either Student’s t-test or one-way ANOVA with Bonferroni’s correction for multiple comparisons. Data are reported in Mean ± SD. Significant difference is determined as P < 0.05
Results
Protective effect of Wnt-3a in cortical neuronal cultures and after ischemic stroke
Previous reports suggested that Wnt-3a signaling may be neuroprotective in CNS pathological models, such as Huntington’s disease, Alzheimer’s disease, traumatic brain injury, and striatal stroke.22,43–46 To evaluate whether Wnt-3a can also confer neuroprotection after focal ischemic stroke, cortical neuronal cultures overexpressing either Wnt-3a (tagged with mCherry) or mCherry-only controls were created by lentiviral infection. Wnt-3a overexpression, as well as the effective dosage of XAV-939 (10 µM) was confirmed by Western blotting (Figure 1(a) and (b)). The transgenic neurons were subjected to oxygen glucose deprivation (OGD) for 3 h. At one day after OGD, there were ostensibly greater numbers of surviving neurons in the Wnt-3a overexpression group, as compared to the mCherry control group (Figure 1(c) and (d)). Furthermore, cell viability was assessed using MTT assay. In Wnt-3a over-expression neuronal cultures, cell viability was significantly increased compared to vehicle controls (Figure 1(e)). This effect was corroborated using the recombinant Wnt-3a protein paradigm, both in primary cortical neuron cultures and in our mouse model of stroke. Neurons receiving Wnt-3a (1 ng/25 µL per well in 48-well plates) following OGD demonstrated greater resilience against cell death, while administration of Wnt-3a inhibitor XAV-939 (1, 10, 100 µM) abolished these neuroprotective effects (Figure 1(f)). This dosage assay was also used to determine the optimal concentration of XAV-939 for Wnt inhibition by MTT assay.
Young adult C57BL/6 mice were subjected to a focal ischemic insult targeting the right somatosensory cortex, including the barrel fields, which receives sensory input for whisker deflections. 29 Three days after the focal ischemia, we performed a Western blot to confirm Wnt-3a and XAV-939 activity, as well as TTC staining. Western blot data validated that Wnt-3a injections resulted in significantly greater levels of Wnt-3a and β-catenin, and these elevations were abolished by XAV-939 administration (Figure 1(g) and (h)). TTC staining demonstrated a significant reproducible infarct developed in the somatosensory cortex (Figure 1(i) and (j)). To determine whether the Wnt signaling pathway had an impact on infarct formation, stroke mice were given Wnt-3a treatment (2 µg/kg/day × 3 days, intranasal) starting at 1 h after stroke and repeated once a day for a total of three days. The intranasal route is an established noninvasive delivery for drugs, 47 molecules, 48 and cells48–50 to the brain because perforations within the olfactory epithelium allow the reagents to cross the blood brain barrier. As observed using TTC assay, the infarct volume at three days was significantly reduced by Wnt-3a treatment. On the other hand, co-application of the specific Wnt-3a inhibitor XAV-939 (8 mg/kg/day × 3 days) completely blocked the neuroprotection (Figure 1(g) and (h)).
Changes in Wnt/β-catenin signaling and its impact on downstream BDNF regulation after ischemic stroke
After the confirmation of an important role for Wnt-3a in ameliorating brain ischemic damage, we next wanted to reveal how the endogenous Wnt-3a pathway was altered in the post-stroke brain. Western blotting showed that the protein expression of Wnt-3a and its intracellular mediator β-catenin were not significantly changed in the peri-infact region (Figure 2(a) and (c)). However, in the SVZ region, both Wnt-3a and β-catenin were significantly decreased 72 h after stroke (Figure 2(b) and (d)). Because the Wnt/β-catenin pathway is an integral component in regulating endogenous neurogenesis, we explored whether restoration of the Wnt-3a levels via a pharmacological approach could drive native brain tissue repair.
Wnt-3a supplementation restored Wnt signaling and neurotrophic expression following stroke. (a–d) Endogenous Wnt-3a and β-catenin levels were determined in wildtype animals following stroke by Western blot and densitometry analysis. (a) and (b) Representative Western blot analysis of tissue from the subventricular zone (SVZ, A) and the peri-infarct (b). (c) and (d) In the SVZ (d), β-catenin and Wnt-3a are significantly downregulated during the subacute phase after stroke. No significant differences were observed in the peri-infarct (c). *p < 0.05 compared to hour 0; **p < 0.01 compared to hour 0; ***p < 0.001 compared to hour 0. N = 5 for each group for each timepoint. (e–h). Animals were subjected to focal ischemic stroke and then were given intranasal injections of saline, Wnt-3a, or Wnt-3a + Dkk1. Then, Western blot and densitometry analysis were performed to detect the expression of Wnt-3a, β-catenin and BDNF in peri-infarct regions at 14 days after stroke. Wnt-3a supplementation increased levels of Wnt-3a, its intracellular signaling mediator β-catenin, and the neurotrophin BDNF. All data represented as mean ± SD; *p < 0.05 compared to saline; #p < 0.05 compared to Wnt-3a. N = 7 for saline group, seven for Wnt-3a group, and eight for Wnt-3a + Dkk1 group.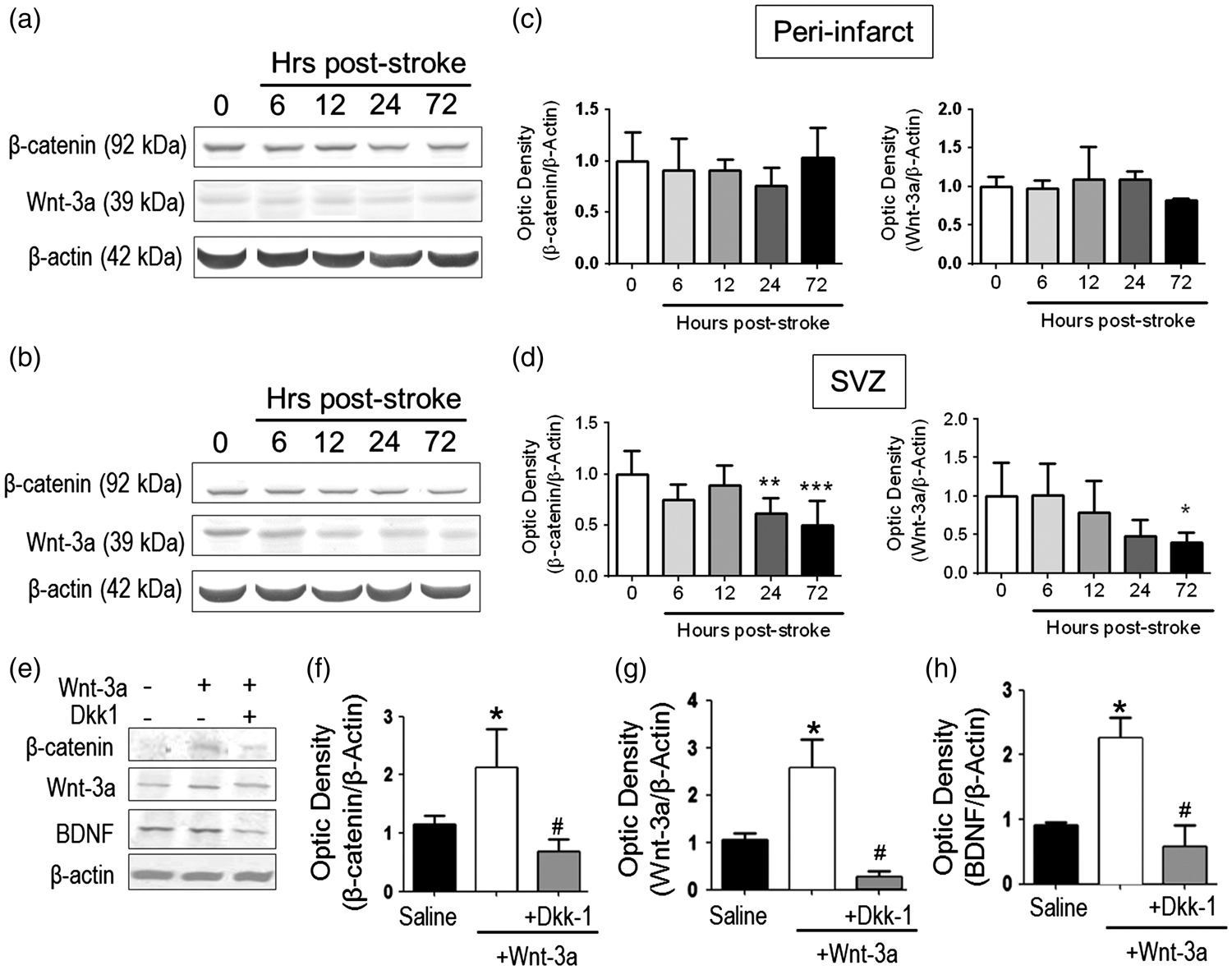
To compensate for the downregulation of Wnt-3a signaling, we tested the non-invasive intranasal administration of recombinant Wnt-3a (2 µg/kg) starting at one day after stroke and continuing for seven days. In order to confirm the successful delivery of Wnt-3a into the ischemic region, Western blotting was used to measure the Wnt-3a level and its downstream signal β-catenin. At 14 days after stroke, mice that had received Wnt-3a treatment showed an approximate two-fold increase in levels of Wnt-3a and β-catenin in the peri-infarct region, as compared to those of saline controls (Figure 2(e) to (g)). To confirm that this effect was Wnt-3a-dependent, we utilized the Wnt-3a inhibitor Dkk-1. Co-administration of Wnt-3a inhibitor Dkk-1 (3 µg/kg/day, intranasal) completely blocked the increased expression of Wnt-3a, as well as the expression of β-catenin (Figure 2(e) to (g)).
Although most of the genetic targets are mitogens, we hypothesized that Wnt-3a may affect neurotrophic activity as well, given the important of neurotrophins on neuronal viability. Specifically, we focused on brain-derived neurotrophic factor (BDNF) and discovered that Wnt-3a administration significantly upregulated BDNF expression (Figure 2(e) and (h)). As similar to the previous findings, Dkk-1 abolished the increase in BDNF levels, suggesting that the Wnt pathway is directly involved in mediating BDNF upregulation (Figure 2(e) and (h)).
Effect of Wnt-3a inhibition on neurogenesis in the ischemic cortex
In addition to the neuroprotective effect, previous data and our observation of Wnt-3a’s role in enhancing BDNF levels both indicate that Wnt-3a harbors regenerative potential within the post-stroke brain. However, we already found that Wnt-3a levels are pathologically attenuated in the SVZ following stroke (Figure 2(c) and (d)). To elucidate the effects of the reduction in endogenous Wnt/β-catenin activity in the CNS, the Wnt inhibitor, XAV-939 (8 mg/kg/day, intranasal administration), was given to the mice daily for seven days following stroke, immediately following Wnt-3a delivery. BrdU (50 mg/kg, i.p.) was injected daily starting from three days after stroke until the day before sacrifice to label newly formed cells. Through immunohistochemical assessments, attenuation of Wnt-3a signaling with XAV-939 reduced numbers of newly formed neurons (BrdU+/NeuN+ colocalized cells) in the peri-infarct region (Figure 3(a) to (d)). Colocalization of BrdU and NeuN fluorescence was confirmed with high magnification Z-stack images (Figure 3(c)).
The role of the Wnt signaling pathway in neurogenesis after stroke. (a–d) Immunohistochemical examinations of newly formed neurons in the peri-infarct region. (a) Left panel: the region of interest for immunofluorescence analysis following stroke is defined as the peri-infarct region surrounding the core region (as indicated by the asterisk). Right panel: cartoon of the coronal section depicting the peri-infarct region (green boxes) that was acquired and quantified (pink: ischemic core). (b) Representative immunofluorescence images for NeuN+ (green), BrdU+ (red), and NeuN+/BrdU+ co-labeled cells (white arrows) among different treatment groups. (c) High-magnification (60×) confocal three-dimensional image confirming colocalization of BrdU and NeuN fluorescence. (d) Quantification of neurogenesis by NeuN+/BrdU+ co-labeled cells in the peri-infarct following administration of either saline (negative control) or the Wnt-signaling inhibitor, XAV-939. All data represented as mean ± SD; *p < 0.05 compared to sham; #p < 0.05 compared to saline. N = 5 for sham group, 10 for saline group, and 4 for XAV-939 group. (e–i) Immunostaining inspection of neuroblasts migration from the SVZ to the ischemic cortex at 14 days after stroke. (e) Representative images of clusters of newly divided migratory neuroblasts (marked by co-labeling of BrdU, red, and DCX, green) emerging from the ipsilateral SVZ. (f) Higher magnification image to demonstrate colocalization of BrdU and DCX fluorescence. (g) Cartoon of the coronal section depicting the SVZ region of interest (green box) that was acquired and quantified. (h) High-magnification confocal three-dimensional image confirming colocalization of BrdU and DCX fluorescence. (i) Wnt-3a injection increased the number of migrating neuroblasts from the SVZ toward the peri-infarct, as determined by analysis of co-labeling of DCX and BrdU. Inhibition of the Wnt pathway with XAV-939 abolished the pro-regenerative effects. All data represented as mean ± SD; *p < 0.05 compared to saline; #p < 0.05 compared to Wnt-3a. N = 6 for saline group, 13 for Wnt-3a group, and 8 for Wnt-3a + XAV-939 group.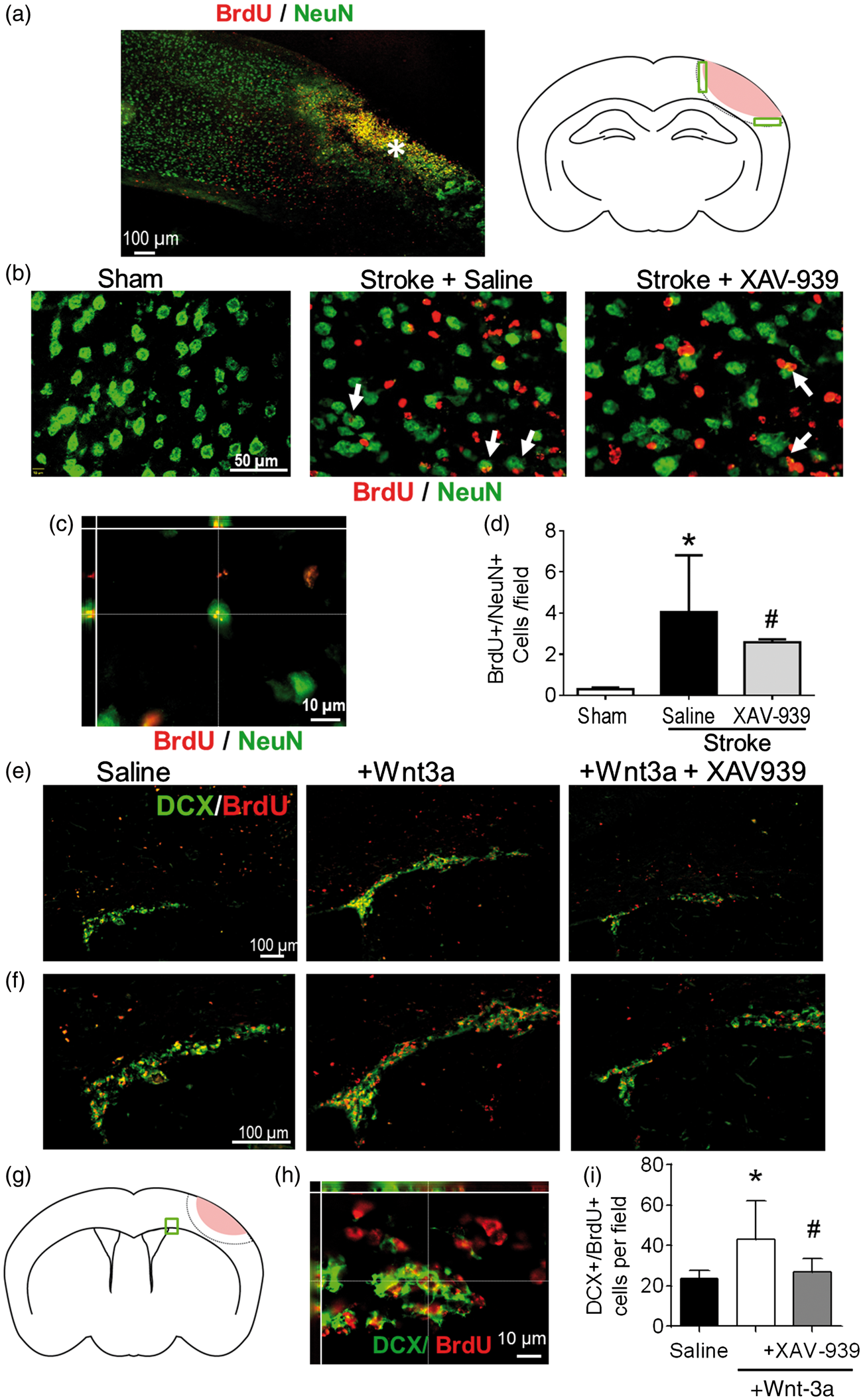
Wnt-3a enhanced migration of neural stem cells from the SVZ after focal ischemia
Given the maladaptive reduction of Wnt-3a in the SVZ following stroke, we investigated whether compensatory Wnt-3a supplementation could augment neurogenesis, through Wnt’s effects on regenerative activity and/or cell migration from the SVZ to the ischemic cortex. To this effect, Wnt-3a (2 µg/kg/day) was intranasally delivered each day starting from one day after stroke and continuing for seven days. Furthermore, to trace the proliferation of native cells, BrdU (50 mg/kg) was injected daily starting from three days after stroke. Pluripotent radial glial cells (RGCs) reside in the regions flanking the lateral ventricles, where they differentiate into doublecortin+ (DCX+) neuroblasts that migrate in clusters towards areas of injury. Within the proximity of the SVZ, Wnt-3a treatment (as compared to saline control) resulted in a more robust clustering pattern, suggesting greater proliferation and differentiation of NSCs in the SVZ niche (Figure 3(e) and (f)). At 14 days after stroke, Wnt-3a treatment significantly elevated numbers of DCX+/BrdU+ colocalized cells from the SVZ towards the peri-infarct (Figure 3(f) and (i)). The enhanced migration then evolved into greater levels of reparative activity around the injury, which was examined at 21 days after stroke. At this later timepoint, BrdU+ cells in the ipsilateral cortex were abundant and distributed throughout the peri-infarct (Figure 4(a)). High magnification confocal images were acquired to confirm colocalization of BrdU and NeuN (Figure 4(b)). Compared to the stroke-saline control group, Wnt-3a significantly increased the number of BrdU+/NeuN+ colabeled cells in the peri-infarct at 21 days after stroke. Infusion of XAV-939 reversed the increase in NeuN+/BrdU+ cells induced by Wnt-3a (Figure 4(a) and (c)), suggesting that supplementation of Wnt-3a activity does indeed contribute directly to mediating neural regeneration.
Wnt-3a signaling enhanced neurogenesis in the peri-infarct following ischemic stroke. (a) Representative immunofluorescence images for NeuN+ (green), BrdU+ (red), and NeuN+/BrdU+ co-labeled cells (white arrows) among different treatment groups. (b) Left panel: cartoon of the coronal section depicting the peri-infarct region (green boxes) that were acquired and quantified. Right panel: high magnification (100×) confocal image to confirm colocalization of BrdU and NeuN. (c) Quantification of neurogenesis by NeuN+/BrdU+ co-labeled cells in the peri-infarct following stroke induction and administration of either saline (negative control), Wnt-3a, or Wnt-3a+XAV-939. All data represented as mean ± SD; *p < 0.05 compared to saline; #p < 0.05 compared to Wnt-3a. N = 10 for saline group and Wnt-3a groups, and 4 for Wnt-3a + XAV-939 group.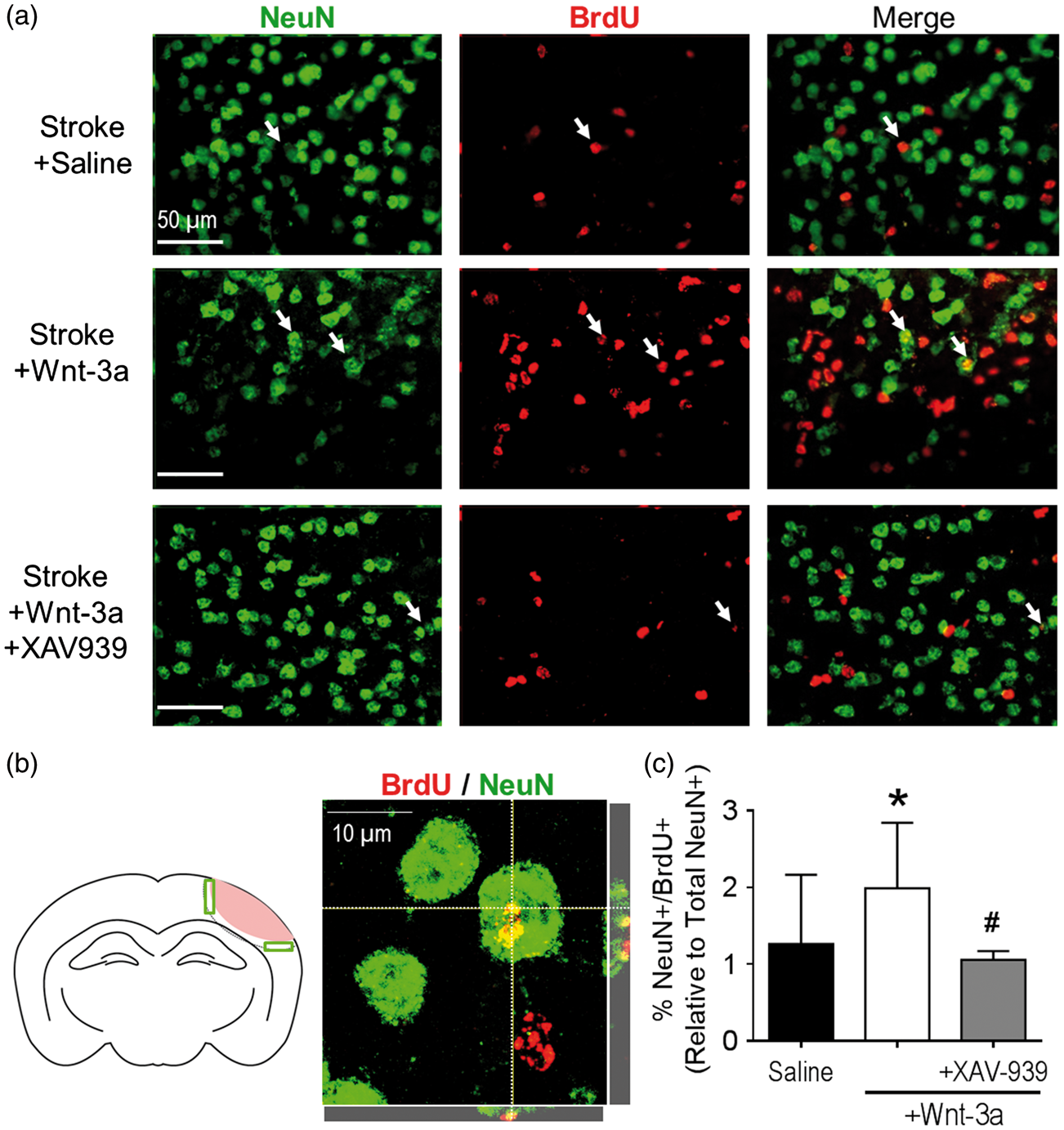
Wnt-3a promoted hippocampal neurogenesis and angiogenesis after ischemic stroke
We next investigated the effects of Wnt-3a on overall plasticity in the brain by examining a region that was distant from the injury and peri-injury zone. To this end, we evaluated the magnitude of neurogenesis within the ipsilateral hippocampus, specifically the SGZ of the dentate gyrus. We observed significantly greater levels of total BrdU+ cells, NeuN+/BrdU+, and GLUT1+/BrdU+ colabeled cells in the Wnt-3a treatment group, as compared to the Saline group, suggesting greater levels of proliferation of the stem cell pool in the SGZ, greater neurogenesis, and greater angiogenesis, respectively (Figure 5(a) to (d)). This effect was abolished in the Wnt-3a + XAV-939 group, suggesting that the effect was specific to the canonical Wnt pathway (Figure 5(a) and (b)).
Wnt-3a activation promotes hippocampal neurogenesis in the SGZ after cortical stroke. (a) Representative immunofluorescence images for NeuN+ (green), BrdU+ (red), and GLUT1 (blue). (b) Quantification of neurogenesis by NeuN+/BrdU+ co-labeled cells and angiogenesis by GLUT1+/BrdU+ co-labeled cells in the SGZ following stroke induction and administration of either saline (negative control), Wnt-3a, or Wnt-3a+XAV-939. N = 7 for all groups. (c) 40x images demonstrating BrdU + non-colocalized cell (arrowhead), colocalization of NeuN and BrdU (dashed arrow), and colocalization of GLUT1 + and BrdU (solid arrow). (d) Top panel: cartoon of the coronal section depicting the SGZ region of interest (green box) that was acquired and quantified. Bottom panel: representative high magnification (60×) confocal image to confirm colocalization of BrdU and NeuN. All data represented as mean ± SD; *p < 0.05 compared to saline; #p < 0.05 compared to Wnt-3a + XAV-939. N = 5 for saline and Wnt-3a groups, and 4 for Wnt-3a + XAV-939 group.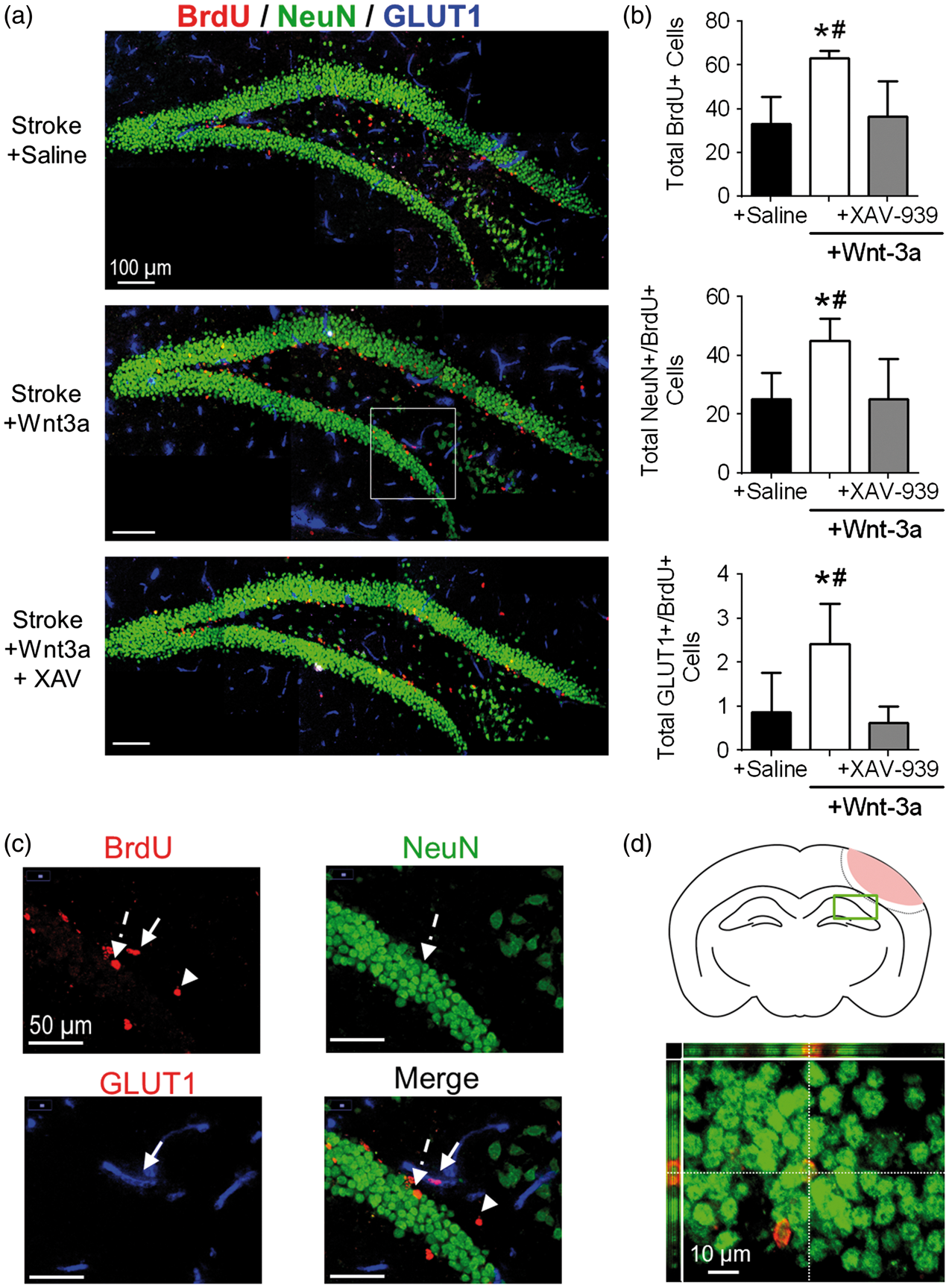
Wnt-3a enhanced angiogenesis and blood flow in the peri-infarct region after stroke
Given the observation that Wnt-3a treatment increased newly formed GLUT1-positive cells and the close coupling between the regeneration of neurons and endothelial cells,
51
we examined the effect of Wnt-3a on post-stroke angiogenesis in the peri-infarct region. Immunohistochemical analysis demonstrated that Wnt-3a administration increased Collagen IV (ColIV) density and ColIV+/BrdU+ colocalized cells in the peri-infarct region at 21 days after stroke (Figure 6(a) and (b)). Furthermore, local cerebral blood flow (LCBF) was measured using Doppler laser scanner at 7 and 14 days after stroke. Wnt-3a significantly restored the levels of LCBF in the peri-infarct to pre-stroke levels (Figure 6(c) and (d)). The Wnt-3a inhibitor XAV-939 attenuated the flow recovery (Figure 6(c) and (d)), corroborating the direct effect that Wnt-3a activity contributes towards angiogenesis and restoration of blood flow.
Wnt-3a enhanced angiogenesis in the peri-infarct and improved functional recovery after stroke. Angiogenesis was assessed 14 days after stroke in the peri-infarct region using immunohistochemical and LCBF measurements. (a) Representative immunofluorescence image for Col-IV (green), BrdU (red) and NeuN (blue). Right panel demonstrates a high magnification (100×) confocal image to confirm Col-IV+/BrdU+ colocalization. (b) Quantification for BrdU+/ColIV+ colabeled cells. All data represented as mean ± SD; *p < 0.05 compared to saline. N = 7 for each group. (c) Local cerebral blood flow (LCBF) was measured in ipsilateral hemispheres of vehicle (saline)-treated stroke control, Wnt-3a and Wnt-3a plus XAV-939 treated stroke mice using laser Doppler imaging. (d) Quantified data for LCBF measurement, represented as a percentage normalized to the baseline (before stroke). All data represented as mean ± SD; * p < 0.05 compared to saline (for the respective timepoint); #p < 0.05 compared to Wnt-3a group (for the respective timepoint). N = 6 for each group for each timepoint. (e) and (f) Mice receiving Wnt-3a treatment performed better on the adhesive-removal test following stroke, with a decrease in both latency to contact (e) and removal (f). The data represented as box and whisker plots (min to max). (g) In the corner test, mice receiving Wnt-3a treatment demonstrated a significant decrease in left/right turn ratio (closer to the value of 1) after stroke, suggesting restoration of normal turning behavior. The data are represented as scatterplots with mean ± SD; *p < 0.05 compared to sham; #p < 0.05 compared to saline. N = 15 for sham group, 12 for saline group, and 16 for Wnt-3a group.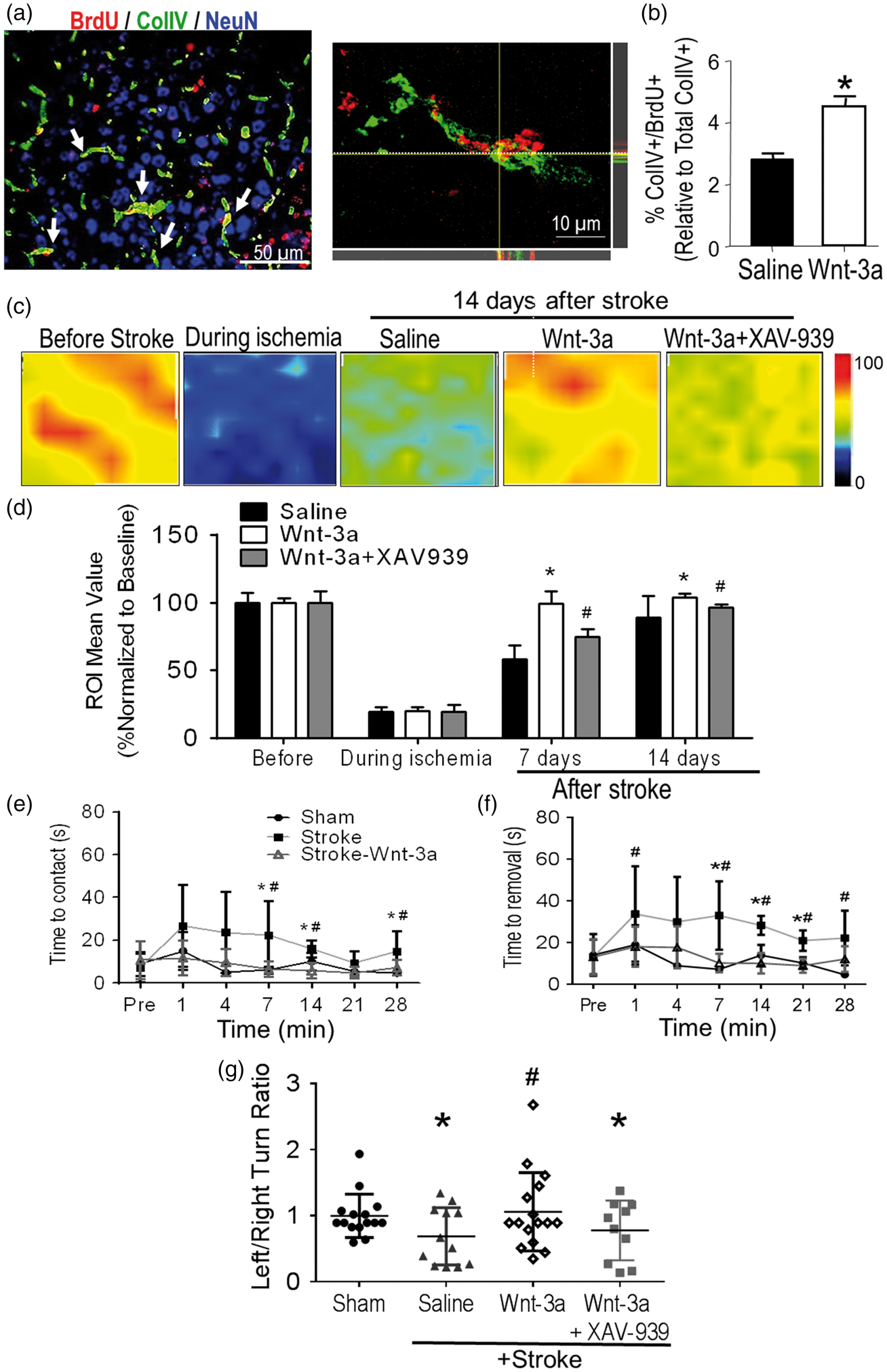
Wnt-3a intranasal delivery attenuated neurological deficits after stroke
The adhesive-removal test and corner test were performed at varying timepoints to assess sensorimotor impairment and functional recovery following stroke (Supp. Figure 1). In the adhesive removal test, stroke mice took much longer time to sense and remove the adhesive irritant attached to the contralateral forepaw, which corresponded to the ischemic cortex (Figure 6(e) and (f)). For stroke animals that received Wnt-3a, both the latency to contact and the latency to removal of the adhesive were reduced, indicating improved sensorimotor function (Figure 6(e) and (f)). No deficits were observed in the ipsilateral forepaw, the phenotype corresponding to the uninjured barrel cortex (Supp. Figure 2). At 28 days, we also observed a significant restoration of the left turn/right turn ratio in the corner test within the Wnt-3a treated group, as compared to the saline-stroke control group (Figure 6(g)).
Discussion
The present investigation explored both the neuroprotective and the regenerative potential of canonical Wnt signaling in the post-stroke adult brain. Wnt-3a is an endogenous pro-regenerative protein found in the CNS.11,52,53 The Wnt proteins are crucial in regulating the symmetric and asymmetric division of multiple cell types, including the self-renewal of hematopoietic stem cells 54 and the differentiation of neural progenitor cells (NPCs) to neurons and astrocytes.12,53,55 Although Wnt dysregulation results in many distinct pathological processes, including hepatic fibrosis, heart failure, and tumor formation,56–58 the therapeutic effects of Wnt-3a have not been thoroughly examined, especially in the context of ischemic stroke. In order to elucidate underlying neuroregenerative changes following ischemia, we investigated the peri-infarct zone and the NSC niche zones. In the peri-infarct zone and the SVZ, we probed the levels of the upstream Wnt-3a ligand, as well as its downstream secondary mediator, β-catenin. From these initial studies, we demonstrated the pathological decline in Wnt activity in the SVZ following stroke, which provided the rationale behind pharmacological supplementation of Wnt-3a. Next, we showed that administration of exogenous Wnt-3a significantly increases proliferation of NSCs in the SVZ and promotes migration of new neuroblasts to the ischemic cortex and subsequent neuronal differentiation. Finally, key to the ultimate goal of therapeutic translation, we demonstrated that Wnt-3a treatment efficaciously enhanced functional recovery following stroke. This was an important finding that suggested improved underlying brain repair. Previous studies demonstrated that neurogenesis in the adult brain probably contributes towards not only brain repair, but global plasticity as well. 59 This was corroborated by our investigation of hippocampal neurogenesis, which instantiated the distal plasticity that is upregulated as a result from cortical injury. Nonetheless, the greater numbers of new neurons observed in the peri-infarct region, as well as the improved functional outcomes suggest that upregulation of the Wnt signaling pathway and the associated endogenous neurogenesis, especially in the SVZ, could contribute to brain repair.
After focal cerebral ischemia, SVZ-derived NPCs are induced to proliferate and asymmetrically divide into migratory neuroblasts. They migrate toward the injured region of the ischemic brain and differentiate into mature neurons, serving as the primary source of endogenous neurogenesis.60,61Wnt-3a enhances NSC division and differentiation in the spinal cord,10,62 as well as NSC proliferation and division in the SVZ.53,63 Furthermore, to probe the mechanisms of Wnt-related effects, we utilized two inhibitors of the Wnt pathway, Dikkopf-1 (Dkk-1) and XAV-939. Dkk-1 selectively blocks Wnt signaling by inhibiting the LDL-receptor-related protein 5/6 (LRP5/6) interactions. Dkk-1 contributes to NSC death and blocks Wnt-3a-induced neuronal differentiation.63,64 XAV-939 negatively regulates the Wnt pathway by inhibiting tankyrases and subsequent stabilization of Axin.
In this investigation, we focused on Wnt-3a because of its established roles in neural development during embryogenesis,63,65 as well as in adult neurogenesis.13,53,62,66 Knock-out of Wnt-3a shows embryonic lethality during organogenesis. 65 Activation of Wnt-3a/β-catenin signaling directly regulates neurogenesis events, which includes a set of transcriptional changes leading to enhanced division and differentiation of DCX+ neuroblasts out of the SVZ.66–68 Furthermore, Wnt-3a is neuroprotective and may protect neurons during neurodegenerative processes, such as Alzheimer’s disease and Huntington’s disease.43,44,69
Our data demonstrated that Wnt-3a is down-regulated in the SVZ following a focal ischemic insult, which may hinder the brain’s capacity for endogenous neurogenesis. This was an interesting finding given the robust neurogenesis that is normally observed after stroke. 70 A possible explanation for this finding may be that there are multiple mediators involved in neurogenesis, including upregulation of other factors that activate NSC division, such as epidermal growth factor (EGF) and fibroblast growth factor-2 (FGF2). 71 Indeed, infusion of these two factors results in enhanced neuronal proliferation following stroke. 72 Another possibility may be the PTEN/PI3K/Akt (Phosphatase and tensin homologue/phosphoinositide 3-kinase/protein kinase B (PKB, AKA Akt)) pathway. Disinhibition of PI3K/Akt through degradation of PTEN following stroke contributes to greater levels of neurogenesis, 73 and PTEN blockade has been pursued as a possible stroke therapy. 74 Our data, on the other hand, uncovers a novel viable target after stroke, given the observed pro-regenerative properties of Wnt.
Wnt-3a is a canonical Wnt protein, whose activity is mediated by upregulating β-catenin, which is then able to turn on a set of genes, including BDNF 75 and Krùppel-like factor 8 (KLF8). 76 The role of Wnt/β-catenin signaling in neurogenesis has been demonstrated in the neural lineage commitment of embryonic stem (ES) cells.14,77 Firstly, our data confirmed that intranasal Wnt-3a administration can activate Wnt/β-catenin signaling through Western blot interrogation of β-catenin. We also interrogated the levels of BDNF, a neurotrophic factors whose expression has been tied to Wnt activity. 78 Our findings demonstrate that Wnt-3a can significantly upregulate protein levels of both β-catenin and BDNF in the peri-infarct region at 14 days after stroke. BDNF has been shown to promote neurite outgrowth in vitro and to protect against neural cell death from ischemia after middle cerebral artery occlusion (MCAO). 79 Furthermore, BDNF is integral for normal neural development and repair because it induces the growth and differentiation of new neurons and synapses and helps to support the survival of existing neurons.80,81 Our data demonstrate that the increased BDNF production is consistent with enhanced neurorestorative processes, including greater neurogenesis and angiogenesis, after focal brain ischemia.
We further detected the impact of Wnt signaling on neurogenesis in the peri-infarct during the chronic phase after stroke. During this period, neurogenesis is a key primary step in remodeling damaged neural circuits and eventually restoring lost function. The redirected migration of neuroblasts to the injured region is a normal adaptive response observed following stroke in various regions of the brain. 82 Previously, it was shown that lentiviral overexpression of Wnt-3a in the mouse SVZ leads to an increase in the number of newly divided neurons and improved functional recovery after stroke. 22 Similarly, our data provide consistent observations. We demonstrated that intranasal delivery of recombinant Wnt-3a in the post-stroke brain also resulted in significant enhancement of Wnt-3a activity. As compared to intracranial delivery of lentivirus, our method provides a superior translational option by two factors: (1) non-invasive delivery via the intranasal route, and (2) a pharmacological agent that will not have the potential immunogenicity or other adverse events associated with lentiviral infection, such as off-target genomic integration. 83 Furthermore, we expounded upon additional therapeutic effects of Wnt-3a, including the enhanced angiogenesis associated with Wnt-3a activity, as well as its restoration of vascular function through laser Doppler imaging. Finally, we demonstrated that intranasal Wnt-3a administration is capable of enhancing hippocampal neurogenesis and angiogenesis as well.
The barrel cortex receives sensory input originating from contralateral whisker deflections. 84 In the focal ischemic stroke model, damage to the barrel cortex results in impaired sensory perception, a manifestation that can be evaluated with the corner test. 30 Through this assay that is sensitive for whisker sensation, we demonstrated that Wnt-3a promoted whisker-dependent functional recovery at 28 days after stroke.
In ischemic stroke patients, the number of new vessels surrounding injured tissue correlates to longer survival. 85 Microvascular proliferation and remodeling occur after cerebral ischemia in the rat barrel cortex, which increases blood flow and stimulates collateral growth, linking increased angiogenesis to improved performance in neurological and behavioral tests. 86 Our data suggest that the enhancement of Wnt-3a signaling promoted both the survival and regeneration of blood vessels in the peri-infarct, which corresponded to more robust blood flow during the chronic phase after stroke. Thus, Wnt-3a activity is able to enhance restoration of both neurons and the associated vasculature, which is a crucial component to ensuring differentiation and survival of the newly generated neurons. 87 However, while we demonstrated elevation of neurogenesis and angiogenesis following Wnt-3a administration, it is inconclusive whether Wnt-3a directly contributed to enhanced angiogenesis, or whether it was a consequence of the greater neurogenesis and the pairing of the neurovascular unit. Additionally, further studies are required to determine whether the greater numbers of newly formed neurons from Wnt-3a therapy resulted in greater connectivity and integration into the existing neuronal circuit and establishment of functional synaptic activity. There may be other factors at play, such as enhanced synaptic plasticity as a result of Wnt stimulation of neurogenesis, which could result in upregulation of growth factors. Further analyses will be required to disentangle these different cellular components. Finally, as we noted, Wnt also promotes neuroprotection, which preserves the original neurons and neural connections and contributes towards the reduction of behavioral deficits, as we also observed through the time course of adhesive removal test.
The present investigation has determined the critical decline in Wnt/β-catenin signaling in SVZ that is the major regenerative niche after cortical injury in the adult brain. This decline supports the supplementation of Wnt-3a after stroke by exogenous Wnt-3a administration. We demonstrate that intranasal delivery of recombinant Wnt-3a protein after stroke contributed to robust neuroprotective and regenerative activities, as well as improvements in functional outcomes. Although future studies are needed to characterize the long-term benefits and effects of Wnt therapy, we propose that treatment using Wnt-3a can be considered as a viable neuroprotective and regenerative therapy for ischemic stroke.
Conclusion
Overall, the present investigation produced compelling preclinical evidence for recombinant Wnt-3a as a therapeutic option following brain injury. One of the primary advantages of this method is that it can be delivered noninvasively. This allows for greater feasibility in its translational application. These initial findings provide a promise for future investigations of Wnt-3a as a therapeutic potential in different stroke models.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH grants NS075338 (LW), NS085568 (LW), NS091585 (LW), an American Heart Association (AHA) Established Investigator Award (LW), an AHA Postdoctoral Fellowship POST25710112 (ZZW), and an NIH Predoctoral Fellowship T32 007480-15 (JZ), National Natural Science Foundation of China 81500989 (YZ).
Acknowledgements
We would like to thank Michka G Sharpe and Dr. Wenyuan Cao for their technical help.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author’s contributions
Wei ZZ performed experiments, data analysis and prepared manuscript; Zhang JY performed experiments, data collection and analysis, revised manuscript and figures; Taylor TM participated in concept development, performed experiments, data collection and analysis, Gu X performed stroke surgery, data collection and analysis; Zhao Y participated in data analysis and final revision of the paper; Wei L oversaw concept development, data analysis, manuscript revision and financial support.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.