
Abstract
The critical role of the vasculature and its repair in neurological disease states is beginning to emerge particularly for stroke, dementia, epilepsy, Parkinson’s disease, tumors and others. However, little attention has been focused on how the cerebral vasculature responds following traumatic brain injury (TBI). TBI often results in significant injury to the vasculature in the brain with subsequent cerebral hypoperfusion, ischemia, hypoxia, hemorrhage, blood–brain barrier disruption and edema. The sequalae that follow TBI result in neurological dysfunction across a host of physiological and psychological domains. Given the importance of restoring vascular function after injury, emerging research has focused on understanding the vascular response after TBI and the key cellular and molecular components of vascular repair. A more complete understanding of vascular repair mechanisms are needed and could lead to development of new vasculogenic therapies, not only for TBI but potentially vascular-related brain injuries. In this review, we delineate the vascular effects of TBI, its temporal response to injury and putative biomarkers for arterial and venous repair in TBI. We highlight several molecular pathways that may play a significant role in vascular repair after brain injury.
Introduction
Over the last decade, traumatic brain injury (TBI) research has focused almost exclusively on neuroprotective strategies that reduce neuronal cell loss. 1 Unforutantely, this approach has failed to develop any meaningful therapeutics for clinical treatment. One less explored potential target has been the brain vasculature and its impact on TBI outcomes.2–4 A major consequence of TBI is direct damage to the cerebral vasculature. Hemorrhage, edema, blood flow abnormalities, and blood–brain barrier (BBB) disruption are immediate and early events that are commonly observed in patients and in animal models of TBI. These early events are then later followed by hypoperfusion, altered delivery of metabolic substrates, and hypoxic and ischemic tissue damage. The vasculature comprises several key cellular elements termed the expanded neurovascular unit (eNVU) and includes neurons, astrocytes, endothelial cells, pericytes, and smooth muscle cells. The eNVU elements work together to regulate cellular metabolism and activity through local blood flow. 5 After TBI, secondary injury mechanisms result in the dysregulation of the eNVU leading to subsequent cellular degeneration in severe injuries to altered function in more mild injuries.4,6
Emerging research now suggests that the injured vasculature attempts to undergo repair.7–9 Angiogenesis and vasculogenesis are the two primary processes initiated after TBI. The multiple components of the eNVU are actively involved in this response and are regulated by a myriad of cellular and molecular injury-induced signaling factors and processes. A greater understanding of how these signaling pathways coordinate vascular repair will provide insight into new potential drug targets and open up unexplored avenues for therapeutic intervention. Elucidating these pathways will also have broad implications for a host of other brain injuries that impact the vasculature.
The initial portion of this review will describe the cerebrovascular dysfunctions that occur after TBI. The second half of this review will focus on the vascular response to TBI and important cellular and molecular elements that may be active in the vascular repair process.
Traumatic brain injury
TBI is the most common acquired brain injury worldwide with millions of individuals affected across a broad age spectrum. 10 In the United States, the incidence of TBI is 1.7 million, resulting in 52,000 deaths annually. 11 The number of trauma-related deaths is higher in European countries. 12 Along with causing significant mortality, the short and long-term consequences associated with TBI have become more apparent and include cognitive deficits (loss of memory, loss of reasoning) and behavioral abnormalities (anxiety, depression, poor social interactions). 13 Indeed, it is estimated that 43% of TBI survivors develop subsequent chronic disabilities for which there are no clinically effective treatments to assist in functional recovery after TBI. 14
TBI is a heterogeneous condition with regard to etiology. In general, TBI has been defined as damage to the brain caused by an external impact, such as penetration by an object or blast wave. 15 The primary causes of TBI are sports-related injuries, blast exposures, automobile accidents, and falls. In sports, concussions (or repeated TBIs) have gained increased public awareness and are known to increase the risk of dementia and can lead to chronic traumatic encephalopathy and other cognitive disorders in retired athletes. 16 Blast TBIs are the result of blast pressure waves from explosive devices and are frequently observed in soldiers from Iraq and Afghanistan. 17 Clinical studies usually classify brain injuries based on severity (mild to severe). However, in studies using animal models, the resulting injury can be sub-classified based on the form of injury (open or closed head injury), type of impact (impact or blast), location within the brain (focal or diffuse), and symptoms (sub-concussive or post-concussive).
The severity of the injury is initially scored on the Glasgow Coma Scale, a tool used to evaluate a patient’s motor, verbal, and eye opening response. Advances in medical imaging, such as computed tomography (CT) and magnetic resonance imaging (MRI), are used extensively to confirm the existence and severity of injury. TBI is grouped into three severities: mild, moderate, or severe. Mild TBI (mTBI) is the most common and results in no overt changes within the brain. Skull fractures, hemorrhage, and altered brain structure(s) are rarely observed by CT scan and standard MRI following mTBI. 18 mTBI often leaves the patient with temporary symptoms; however, clinical studies along with research in animal models show progressive long-term neuropsychological disruptions.19,20 In contrast, moderate and severe TBIs result in visible neuroimaging changes within the brain, including hemorrhage and edema. 21 Most survivors are left with permanent cognitive and physical impairments. 21 Severe TBIs require immediate medical attention and often exhibit involvement in multiple brain regions. The long-term pathological consequences in these mild to severe TBI cases appears to be a disruption of the white matter tracts and systemic inflammation. 22
Neuropathology of TBI from the initial primary injury is followed by a secondary injury cascade. Primary injury is the mechanical damage that occurs at the time of injury and results in shearing of neurons, glia, and blood vessels. Primary injury is untreatable and only preventable by limiting exposure to the inciting event and taking safety precautions. Secondary injury, on the other hand, occurs over time and initiates a variety of signaling cascades that alter cell function and/or cause cell death. Some common events include oxidative stress, excitotoxicity, mitochondrial dysfunction, and inflammation. 23 These secondary events can last hours to years after the injury and can contribute to functional deficits and long-lasting disabilities. 24 Thus, preventing or mitigating the effects of secondary injury is potentially critical to improving the lives of TBI victims.
Vascular consequences of TBI
Extensive research has been conducted to identify potential therapeutic targets for TBI, but none have successfully been translated to the clinic. One major consequence of TBI that has not been extensively investigated is damage to the cerebral vasculature. The disrupted vasculature participates in the pathogenesis of TBI and can lead to secondary injury in the form of hemorrhage, edema, changes in blood flow, and BBB disruption. A better understanding of how these vascular effects after TBI result in chronic disabilities could lead to the development of improved therapeutics. Here, we will review different types of cerebrovascular complications that occur after TBI and their impact on brain pathology.
Hemorrhage
Hemorrhage occurs in 46% of all TBIs and is increasingly prevalent in moderate and severe injuries. 25 An insult to the brain results in bleeding that can gradually progress over the first 24–48 h. In some cases, the leaking blood collects outside the vessel forming a hematoma. Three distinct types of hemorrhage are reported after TBI and are based on their location in the brain: epidural hematomas (between the dura mater and skull), subdural hematomas (between dura and arachnoid mater), and subarachnoid hemorrhage (between the arachnoid and pia mater). Of the three types, post-traumatic subarachnoid hemorrhage is the most severe and is associated with increased mortality and poor outcomes. 26 The effects of hemorrhage result in increased intracranial hypertension, vasospasms, and compression of structures in the brain. 27 However, this depends on the amount and degree of bleeding into the parenchyma. While most hemorrhage resolves on its own, microbleeds can still be present causing deposition of hemosiderin that is detectable by MRI. 28 Accumulation of ferritin/hemosiderin is known to be toxic to brain cells and can elicit an inflammatory response. 29 Interestingly, preclinical studies demonstrated that delayed microbleeds following TBI are associated with white matter damage.30,31
Edema
Cerebral edema, the excess accumulation of fluid within the brain, accounts for 50% of deaths in severe head injuries. 32 Pediatric patients are at greater risk of developing edema than adults. 33 After TBI, cerebral edema forms at the lesion and incorporates into the surrounding tissue. Two distinct types have been observed, cytotoxic and vasogenic edema. Cytotoxic edema results in water accumulation in cells and is caused by dysregulation of the sodium and potassium pumps on the cell membrane. 34 The retention of water and ions can adversely affect cell function and even lead to apoptosis. The BBB remains intact in cytotoxic edema. 34 Vasogenic edema results in water accumulation in the extracellular space and is caused by the disruption of the BBB. 34 Vasogenic edema is associated with elevated intracranial pressure, tissue swelling, changes in blood flow, and compression of brain structures. 35 Although it has been accepted that cytotoxic edema occurs immediately after injury and leads to vasogenic edema, recent studies have expanded on this concept and proposed two transitional forms of edema that occur after stroke.6,34 In this model, anoxic and ionic edema results in dysregulation of the components of eNVU which leads to increased vascular permeability and development of vasogenic edema. 34
Changes in cerebral blood flow
Altered cerebral blood flow (CBF) is one of the most common outcomes of TBI. Both regional and global changes in blood flow are observed but depend on the size, location, and type of tissue injury (i.e. contusion, hematoma, or diffuse edema). 36 A majority of TBI patients have significant reductions in CBF within the first 12 h after injury,36,37 but this is dependent on the severity of the injury. It has been reported that CBF values can reach ischemic levels falling to 18–20 ml/100 ml/min after severe TBI (normal values in humans are ∼45–50 ml/100 ml/min).36,38 After this initial period, many patients experience hyperemia (increase in CBF values compared to normal tissue) while others have shown low or normal CBF values.39,40 Interestingly, at later times after injury, there is an apparent uncoupling between blood flow and tissue metabolism, with long-term outcomes being vasospasm and hypoperfusion. 39 These decrements in CBF can result in ischemic brain injury, which is the leading cause of death after severe TBI. 39 Although CBF typically normalizes within several weeks, therapeutic procedures to enhance blood flow acutely are needed and could have significant clinical benefits for TBI patients. 41
Vasospasms
Vasospasms, or the abnormal constriction of blood vessels, can adversely affect functional recovery after TBI. The reported incidence of vasospasms after head injury ranges from 19 to 68%. 42 While most vasospasms are short-lived, severe episodes can last longer but typically resolve by 14 days in closed head injuries. 43 Blast TBI victims have been reported to have a higher frequency of developing vasospasms than other forms of head injuries and can last up to 30 days after injury. 44 The cause of vasospasms after TBI remains undetermined, but others have suggested that it triggered by hypertension and subarachnoid hemorrhage. 45 A study by Dore-Duffy indicated that pericytes release endothelin-1, a potent vasoconstrictor molecule, near the blood vessels after TBI. 46 Vasospasms can reduce cerebral perfusion to regions of the brain, which could lead to ischemic injury and infarction if untreated. 45 Clinicians undertake extensive efforts to manage vasospasms early in order to avoid long-term injuries.
BBB disruption
BBB disruption following TBI has been well-established. The most common technique to assess BBB disruption in humans is by cerebrospinal fluid/serum albumin quotient, which measures albumin in cerebral spinal fluid in reference to blood serum. Ho et al. 47 in a retrospective cohort study of patients with severe non-penetrating TBI found that 44% of patients experience BBB disruption which was associated with poor long-term outcome. BBB disruption typically returns to normal by several weeks, but in some patients, it has been reported to last months and years after mild injuries. 48 The BBB regulates the movement of substrates between the blood and central nervous system (CNS) and is critical in maintaining cellular homeostasis. This selective barrier is composed of tightly connected endothelial cells together with astrocytes, neurons, and pericytes (forming the eNVU). The endothelial tight junction proteins that seal the BBB become disrupted after TBI, either by the contusion and/or increased intracranial pressure. 49 Neutrophils also exacerbate BBB disruption by secretion of proteases and cytokines. 50 The disruption of BBB leads to the release of harmful molecules into the local injury microenvironment such as iron, reactive oxygen species, and blood toxins. 51 Such molecules can augment inflammation and increase the risk of edema and hypertension. 47
Coagulopathy
Altered coagulation profiles after TBI have been reported in numerous studies.52–54 Harhangi et al. 52 in a systematic review of 82 studies confirmed that coagulopathies were present in TBI patients with a prevalence of 33% and that there was a significant correlation between mortality and poor outcomes. One key finding of most studies is that the severity of the TBI is related to the degree of coagulopathy which is in turn related to outcomes and mortality. Further, the time course of the coagulation abnormalities is also dependent upon the severity of injury. 55 The coagulation cascade appears to be dramatically altered after moderate and severe TBIs, initiated in large part by the release of tissue factor (TF) into the circulation leading to depletion of coagulation factors (extrinsic pathway), decreased platelets, altered BBB and leading to increases in circulating microparticles.56–58 Hyperfibrinolysis, with increases in d-Dimer, has also been proposed to be a marker of outcomes via a potential cascade, wherein TF can increase activation of the extrinsic pathway. 59 Alternatively, low platelet levels may also contribute to increased hemorrhagic evolution particularly early after hospitalization. 60 The hemostasis mechanisms that can be impacted by TBI are clearly varied but ultimately lead to inflammation, hyperfibrinolysis, endothelial dysfunction, and platelet abnormalities. Thus, it is clear that early coagulation mechanisms play a significant role in TBI outcomes but more research is warranted on improved biomarkers and therapeutic interventions.61,62
Chronic inflammation
While the inflammatory response is initiated minutes after injury and protects against pathogens, eliminates necrotic cells, and promotes tissue repair, it has become apparent that chronic inflammation is associated with CNS damage.63,64 Microglia cells, the resident macrophages in the brain, have drawn much attention. Microglia are rapidly activated in response to injury and work to limit the extent of tissue damage. 63 However, persistent microglial activation is a key contributor to secondary brain injury. Several studies have shown that microglia can become activated for weeks to years after brain injury.65–67 Assessment of postmortem tissue of TBI patients who died months after the injury revealed increased activation of microglia in the white matter tracts. 67 Microglia can become over-active in response to danger-associated molecular pattern factors that are released by damaged cells. 68 As a result, over-active microglia produce high levels of inflammatory cytokines and cytotoxic factors that cause further neurodegeneration. 69 Additionally, the sustained release of these factors disrupts the BBB, resulting in the transit of inflammatory factors and peripheral immune cells into the brain thereby further increasing the inflammatory response. 51
Cerebral vasculature injury: Repair OR pathology?
While the effects of vascular injury in acquired neurological injury, such as stroke, have been extensively evaluated, our understanding of mechanisms underlying repair is poorly understood. Several studies have suggested that the injured vasculature appears to initiate repair in the course of two to three weeks.9,70 Using a novel vessel painting technique to document the acute changes to the cerebral blood vessels after TBI, we observed dramatic alterations in vascular parameters not only at the injury site but also in the contralateral brain in a rodent model of TBI.
71
Extending these early studies, we observed that moderate TBI which results in gross vascular injury is subsequently followed by repair over the course of two weeks (unpublished results, Figure 1). Smaller vessels, such as capillaries appear to recover over a similar time frame. Morgan et al.
7
using a rodent acceleration impact model observed premature capillary structures at two days post injury. Lateral fluid percussion injury resulted in an early loss of capillaries which slowly increased and reached baseline levels by 14 days.
72
This study also revealed widespread hypoperfusion throughout the injured brain, with CBF values showing no correlation with vessel density at 14 days. Interestingly despite restoration of capillary structure, hypoperfusion was still present, suggesting that structural repair does not implicitly result in improved function. Similarly, Jia et al.
73
using an optical micro-angiography technique found that a penetrating brain injury results in a sudden loss of blood flow at the lesion site and is gradually reestablished by two to three weeks.
73
Therefore, there is evidence that indicates that vascular impairments are immediate but repair, albeit delayed, does occur after TBI.
TBI results in vascular injury with subsequent repair. (a) We induced a moderate controlled cortical impact (CCI) in the right hemisphere of adult rats (n = 5-6/group). Animals underwent vessel painting to stain the cerebral vasculature at 1 and 14 days after injury. Our vessel painting technique is performed by injecting Dil (lipophilic 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine Perchlorate) solution into the left ventricle followed by cardiac perfusion and fixation. Whole brain axial images were obtained using wide-field fluorescent microscopy (representative images are shown). Moderate CCI elicits a loss of vasculature that extends beyond the injury site. Over the ensuing 14 days, there is increased vascular density at the injury site. Injury site (star). (b) We performed a classical vascular analysis to quantify the features of labeled vessels in the injured and uninjured hemisphere. Vascular analysis of the injured hemisphere revealed a reduction in vessel density at 1 day compared to the uninjured hemisphere (t-test, **p < 0.01). Vessel density was restored and returned to baseline levels by 14 days (t-test, p = 0.82). All error bars are presented as standard error of mean.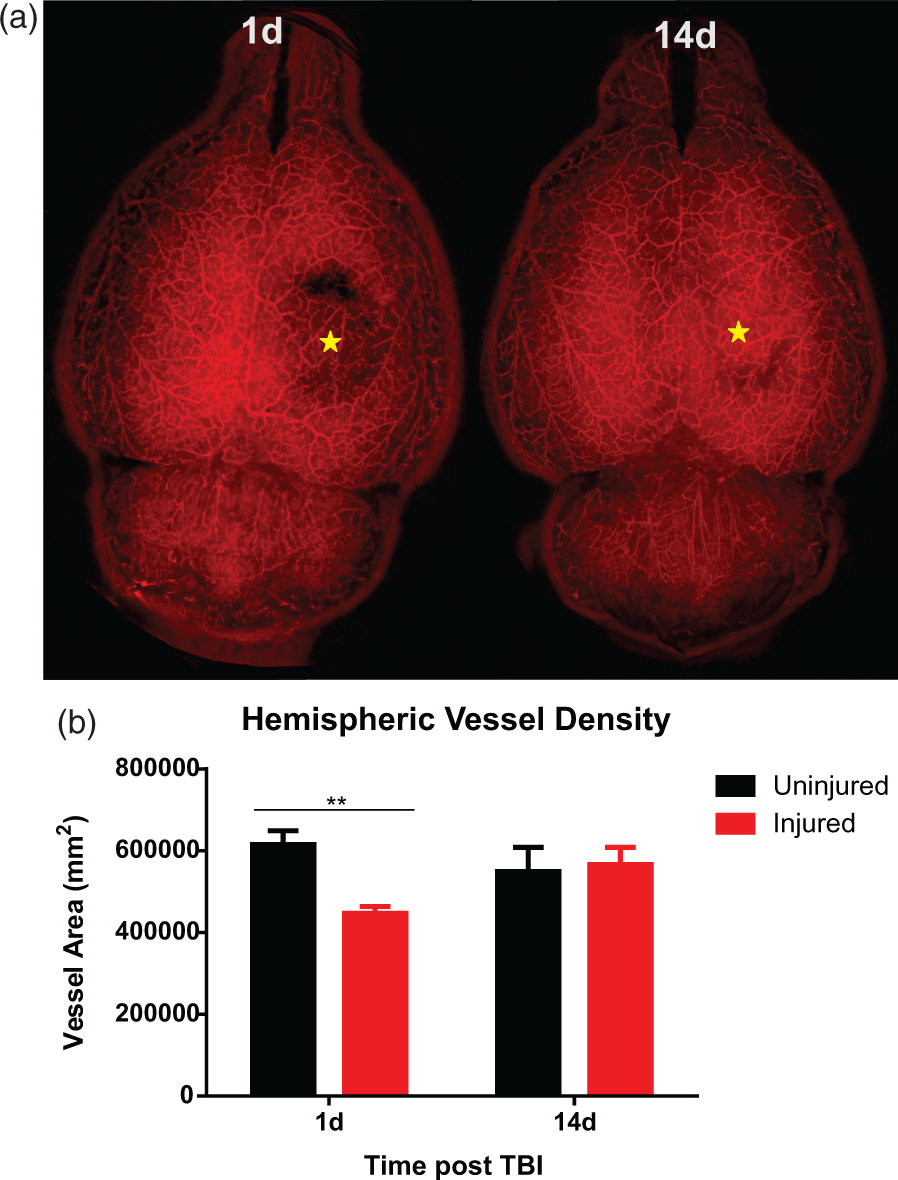
It is clear that the repair process is influenced by the severity of the injury. Park et al. 9 explored vascular loss and recovery using a fluid percussion injury model. Their study utilized two injury severities, a moderate (2 atm) and a severe (3 atm) injury and evaluated the vasculature at 1 and 14 days post injury. Both severities resulted in significant vascular injury at one day post injury compared with sham animals. Only moderately injured animals showed improvements by 14 days after injury, where microvascular density in the injured cortex recovered to near-sham levels. The repaired vasculature of severely injured animals was abnormal, with an increased number of irregular and torturous vessels. These animals also had a significant reduction in microvascular density. Their study highlights that vessel repair can be abnormal and may contribute to the pathological outcomes after brain injuries. It remains to be seen how vascular repair is affected following mTBI.
Other studies have noted significant alterations to the newly formed blood vessels. While morphological changes to the cerebral vessels are apparent early after TBI,74,75 it appears that these deficits do not fully recover and can persist for weeks and months after the initial injury. Electron microscopy showed aberrations to the vessel walls, ridged elastic lamina, swelled and apoptotic endothelial cells, and degraded extracellular matrix.76–78 Both large and small vessels appear to be affected. In a fluid percussion injury model, injured rats had fragmented capillaries and reduced capillary diameter at 30 days post injury. 70 Gama Sosa et al. 79 utilized a blast model of mTBI and observed a unique vascular pathology that led to chronic changes in the vessels six months after injury. These malformed vessels likely increase the risk of delayed hemorrhage, hypoperfusion, and other secondary effects.31,80 Also, an outcome of poor vascular repair when coupled with cerebrovascular dysfunction can lead to further exacerbation of tissue injury. Thus, damaged and abnormal repair of the vasculature may be an important contributing factor to poor outcomes often observed after TBI in humans and in rodent models.
New vessel formation following injury
The formation of new blood vessels (or neovascularization) after acquired brain injury has increasingly gained interest. It has been well-documented that after TBI, two primary mechanisms, angiogenesis and vasculogenesis occur (Figure 2). While both angiogenesis and vasculogenesis are active during embryonic development and lead to the formation of a primitive vascular network, they also play a role in promoting vascular repair in the adult after injury. These complex but distinct processes play an important role in repairing the injured vasculature after TBI.
New vessel formation after TBI. Brain trauma leads to damage to the cerebral vasculature with subsequent repair via angiogenic and vasculogenic processes. Mature endothelial cells, in particular endothelial tip and stalk cells, are involved in various steps of angiogenesis. Tip cells (blue oval shape) are found at the end of the vessel sprout and they use their filipodia to guide the sprout toward the angiogenic growth factor source, while stalk cells (blue trapezoid shape) are found inside the vessel lumen and they proliferate to extend the sprout. Circulating endothelial progenitor cells (light blue rectangular shape) play a key role in postnatal vasculogenesis but also contribute to angiogenesis. Endothelial progenitor cells are recruited from the bone marrow that then incorporate into the damaged vessels and differentiate into endothelial cells. Endothelial progenitor cells also are important for the release of angiogenic factors, which regulate angiogenesis. Other cell types support or augment angiogenesis and vasculogenesis. Astrocytes (red), pericytes (green), and perivascular macrophages (light purple globular shape) cooperate with the newly formed vessel and promote vessel stabilization. Neuronal cells (yellow) are associated with the blood vessels and release angiogenic factors that may guide the vessel sprout toward the injury site. Macrophages (dark purple star shape) have been shown to adhere and ligate ruptured vessels in the brain. Additionally, a majority of these cells release angiogenic growth factors into the hypoxic tissue.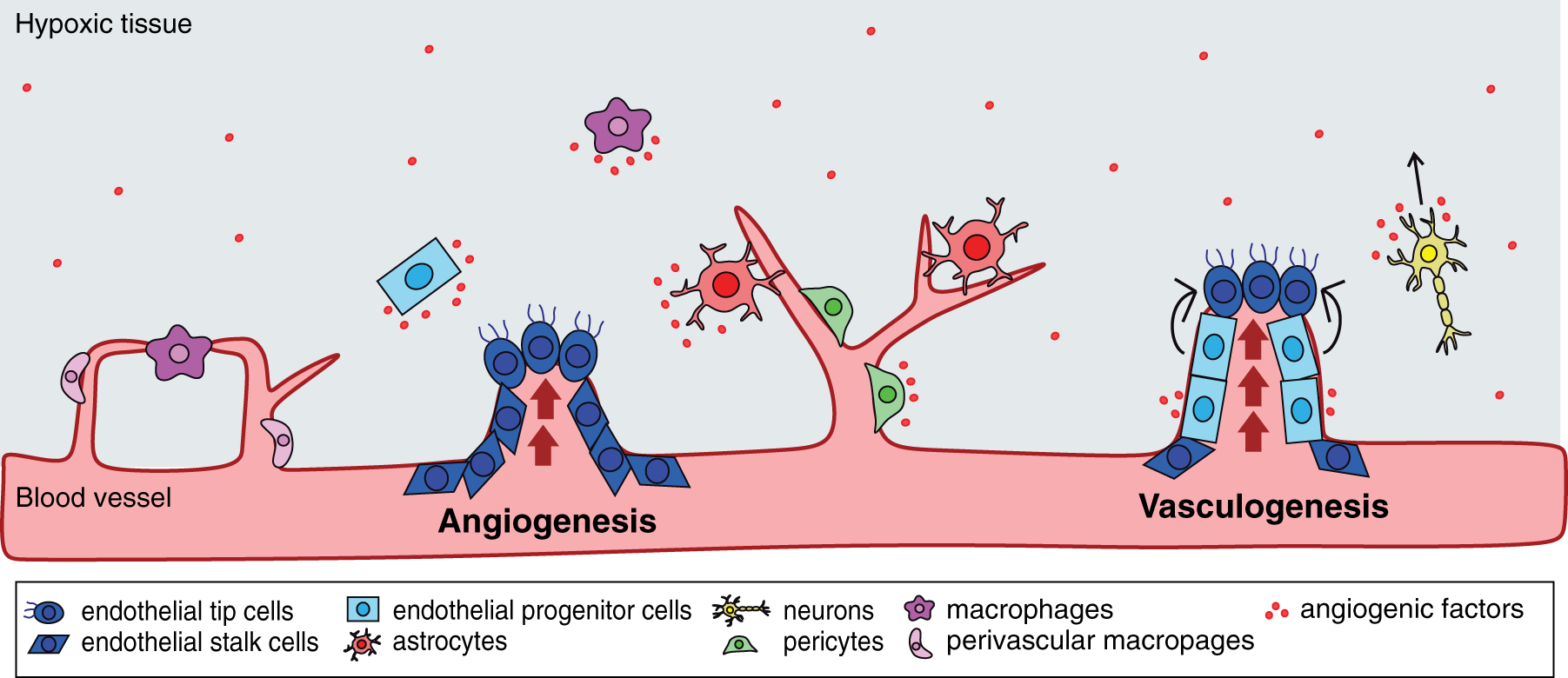
Angiogenesis
Angiogenesis is the formation of new blood vessels from an existing vasculature. New vessels form by sprouting from the main vessels (sprouting angiogenesis) or splitting in two daughter vessels (intussuscepted angiogenesis). Sprouting angiogenesis is commonly observed after injury and is mediated by resident endothelial cells (ECs), which line the luminal surface of the blood vessels forming the endothelium. 81 As previously mentioned, new vessel sprouts are detected at a relatively short time after injury with many studies reporting increased capillary density after TBI,7,9,72,82 where new capillaries appear to migrate from the peri-lesional tissue into the injury site (unpublished results).
Tissue hypoxia, or low tissue oxygenation, is one of the primary triggers of angiogenesis. Clinical studies have reported the presence of hypoxia after TBI, which is typically caused by cerebral hypoperfusion and ensuing ischemia.37,83 Experimental TBI animal models revealed an up-regulation of hypoxia-driven molecules in the injured brain. One key reporter molecule is hypoxia-inducible factor-1 (HIF-1a),9,84 which has been associated with vascular repair in stroke and other disease models. Low oxygen inhibits the degradation of HIF-1α where it accumulates and binds to hypoxia-inducible factor-1β. This transcription factor promotes expression of a number of angiogenic growth factors. 85 These factors constitute a class of signaling molecules that are involved in vascular repair. For example, vascular endothelial growth factor (VEGF) exhibits robust expression in the latter stages of experimental and clinical TBI.86,87 Other factors are also up-regulated and contribute to vascular repair (a more thorough discussion of angiogenic factors will be described later in this review).
Sprouting angiogenesis is an important repair process that requires the coordinated interactions of ECs. The endothelium is activated in response to brain trauma. 88 The hypoxic tissue causes release of angiogenic growth factors into the nearby tissue. 85 These factors signal to the nearby vessels to detach the pericytes and degrade the local extracellular matrix. 85 ECs form vessel sprouts and then migrate and proliferate in the direction of the hypoxic tissue. 89 To facilitate this process, ECs specialize into distinct phenotypes known as endothelial tip and stalk cells: tip cells migrate and lead the vessel sprout, while neighboring stalk cells proliferate and form the vessel lumen. 81 Initially, an immature and unstable vascular structure is formed. Following recruitment of the pericytes, this immature vessel is remodeled to form a stable, functioning vessel. 85
Vasculogenesis
Vasculogenesis is the de novo formation of new blood vessels. The term vasculogenesis has been used in various injury models and refers to a subset of bone marrow-derived cells with vasculogenic potential. Increasing evidence suggests that endothelial progenitor cells (EPCs) play an important role in postnatal vasculogenesis. 90 These CD34+ cells originate from the bone marrow and circulate through the vasculature in response to injury. Many TBI studies have detected increased numbers of EPCs in the blood as well as blood vessels at the injury site.91–93
The initiator(s) of vasculogenesis has been a subject of debate, but many speculate that it is triggered in a hypoxic-dependent fashion. A prime example where this is known to occur is in tumor biology, in which many tumors create a hypoxic environment that leads to vascularization. In hypoxic tumors, HIF-1α mobilizes EPCs to induce vascular growth. 94 Stromal-derived factor-1α (SDF-1α), a chemo-attractant factor that is regulated by HIF-1α, helps recruit EPCs to the tumor site. 95 Similar to tumors, tissue hypoxia is evident at the TBI lesion site and leads to the expression of angiogenic growth factors including SDF-1α. 85 These factors enter the blood circulation and signal to the bone marrow to mobilize EPCs. Serum VEGF and angiopoietin-1 (Ang-1) levels show a significant correlation with circulating EPCs in patients with severe TBIs suggesting that these growth factors may mediate recruitment of EPCs into the blood stream. 91 Thus, hypoxia is likely the driving force for vasculogenesis after moderate and severe TBI.
Unlike angiogenesis, vasculogenesis is initiated in the bone marrow. EPCs remain adhered to the marrow stromal cells under physiological conditions. After injury, EPCs become activated where they are released from the stromal cells and enter the blood stream. 94 In TBI patients, EPC levels decrease at one to two days post injury and rapidly increase and peak by five to seven days after injury.91,92,96 Within the blood, EPCs mature and acquire similar phenotypes as ECs. 97 EPCs are known to home to sites of vascular injury. 94 Guo et al. 93 detected CD34+ EPCs around the lesion at one day after TBI which peaked at seven days post injury. The levels of EPCs correlated with angiogenesis after TBI. The function of EPCs has been much debated, but many studies suggest that EPCs play a dual structural and instructive role in vasculogenesis. EPCs can incorporate into the injured vessels and differentiate into new ECs.7,93 Additionally, others have shown that EPCs act as supporting cells that release trophic factors to the injured vessels.98,99 These secreted factors induce angiogenesis by promoting proliferation and migration of ECs. 99
Supportive role of the eNVU
Active cell types in vascular repair.
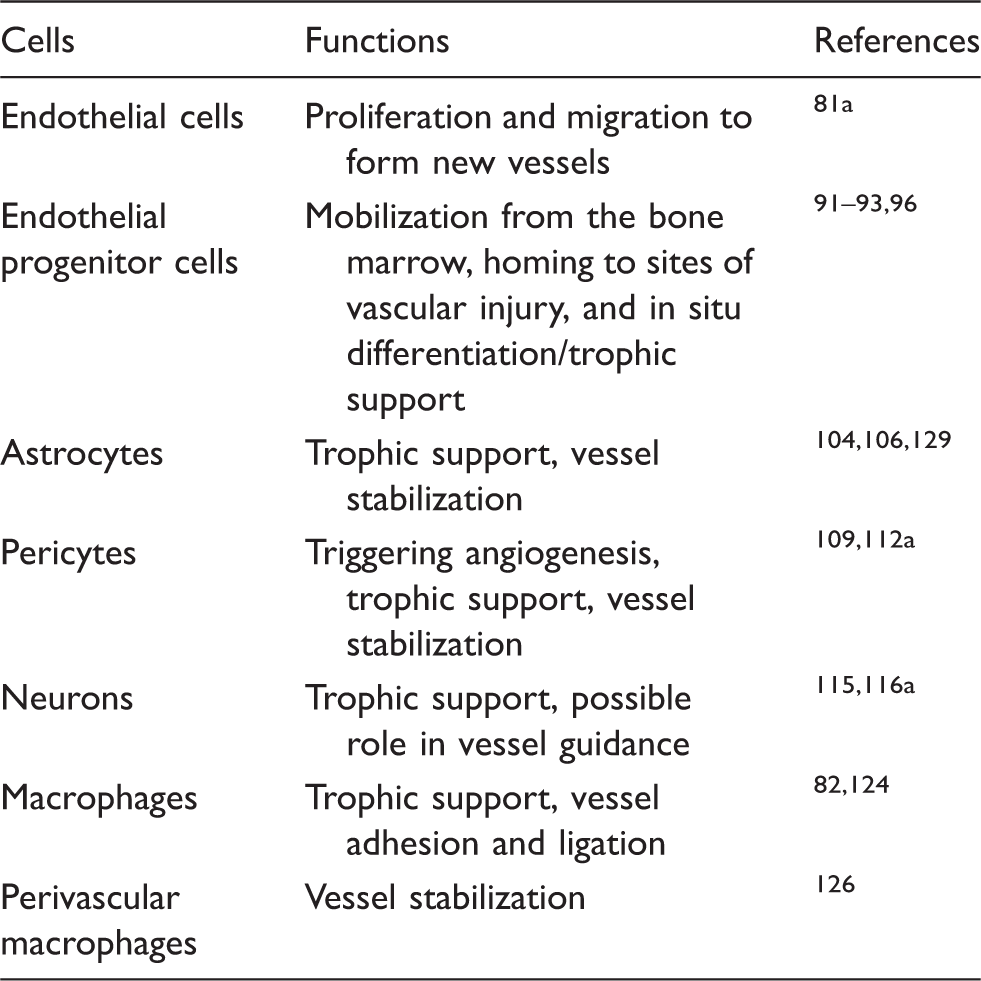
Papers covers general aspects of vascular repair.
Astrocytes
Astrocyte activation after injury has been well described and are known to accumulate around a lesion, in a process known as astrogliosis. These cells perform numerous functions, some of which are related to vascular repair. Reactive astrocytes are known to secrete trophic factors that promote angiogenesis and BBB repair. Astrocytes are an important source of angiogenic growth factors after TBI, including VEGF and SDF-1α 82,100 High mobility group box 1 (HMGB-1), a danger-associated molecular pattern molecule, has been suggested to have beneficial effects in the chronic stages after brain injury. HMGB-1 mediates crosstalk between astrocytes and ECs and EPCs after injury and can lead to neurovascular remodeling.101–103 Additionally, astrocytes can produce tenascin, thrombospondin, and other proteins found in the extracellular matrix. 104
Astrocytes also participate in glial scarring. Although much of the focus has been placed on its inhibition related to axonal regeneration after injury, 105 glial scarring may play a beneficial role in vascular repair. During scarring, reactive astrocytes have close interactions with peri-lesional vessels. 106 These intimate interactions with blood vessels are evident at seven to sixty days post injury, which coincides with the formation of the new vasculature. Astrocytes are integral components of the BBB and are essential for normal BBB function. 107 Thus, these astrocyte-vessel interactions may function in stabilizing the blood vessels and restoring blood flow to the injured tissue.
Pericytes
Accumulating evidence suggests that pericytes are important players in vascular repair. Pericytes, or mural cells, cover the surface of the micro-vessels where they reside within the basement membrane in direct contact with the endothelium. 108 The pericyte population declines in the first few days after controlled cortical impact (CCI). 109 Additionally, a majority of pericytes are observed leaving the blood vessels after weight drop impact. Their detachment from the vessels facilitates a critical step for angiogenesis, 46 as pericytes are required to destabilize and detach from the vessel wall which allows ECs to form vessel sprouts. A number of angiogenic factors are responsible for their detachment from the vessels. 110
At later stages of injury, pericytes become reactive and dramatically increase around the lesion, in a process known as pericytosis. When active, pericytes secrete angiogenic growth factors that mediate EC activity and vascular integrity. 108 Ang-1 is one such factor and has been shown to induce expression of the tight junction protein occludin. 111 Along with providing trophic support, active pericytes make close interactions with newly formed vessels. 112 Pericyte recruitment is critical for the latter stages of angiogenesis and contributes to stabilizing the new vessel. 112
Neurons
While neurons are essential for functional recovery after injury, there is some evidence that they may also participate in vascular repair. Following brain injury, increased neurogenesis is observed in the sub-ventricular zone, 113 in which new neurons are localized to the peri-lesional vessels. 114 Administration of neural progenitor cells after TBI results in a transient increase in angiogenesis adjacent to the lesion border. 115 Neural stem and progenitor cells are known to secrete trophic factors into the injury microenvironment, including a number of angiogenic growth factors. 116 Interestingly, however, one study showed that neurons limit angiogenesis by binding and sequestering angiogenic factors in the retina. 117 Although this work was specific to retinal neurons, it is plausible that cortical neurons act in a similar manner and may guide angiogenic branching toward the injury site. Thus, this raises the possibility that neuronal cells may be important in regulating post-injury angiogenesis.
Macrophages
Macrophages, a monocyte-derived cell that is part of the inflammatory response, have been implicated in vascular repair. Activated macrophages are grouped into two forms: classically activated, anti-angiogenic macrophages (M1) or alternatively activated, pro-angiogenic macrophages (M2 including the M2a and M2c subsets). 118 However, primary human M1 macrophages have been shown to secrete pro-angiogenic molecules in vitro. 119 After CCI, macrophages are broadly activated adopting both an M1 and M2 phenotype,118,120 but appear to be a shift to M1 phenotype at later time points. 121 Macrophages can express angiogenic growth factors and matrix remodeling proteases after TBI, including VEGF, FGF, and matrix metalloproteinase-9 (MMP-9).82,122 In fact, they contribute significantly to late increases in VEGF levels. 82 Additionally, macrophages may mediate vascular pruning through secretion of pro-apoptotic factors. 123 Along with providing trophic support, these cells physically interact with the sprouting vessels. Liu et al. 124 demonstrated that macrophages promote repair of cerebrovascular ruptures in a zebrafish model. Macrophages were seen adhering to the ends of the ECs and ligating the ruptured vessels. Also, during development, macrophages mediate fusion between endothelial tip cells leading to vessel growth and anastomoses. 125
Perivascular macrophages
Unlike peripheral macrophages that infiltrate the brain parenchyma, perivascular macrophages reside in the perivascular space and are closely associated with vessel walls. 126 After weight drop injury, there was a significant increase in the number of CD163+ perivascular macrophages between two and four days post injury, which was mainly localized to the lesion site. 127 The extent to which perivascular macrophages mediate vascular repair after TBI is presently unknown, but are likely to have similar functions as peripheral macrophages. Perivascular M2 macrophages have been implicated in tumor angiogenesis and depletion of these cells attenuates vascularization and tumor growth. 128 Additionally, perivascular macrophages are critical in maintaining vascular integrity under physiological conditions. 126 M2 macrophages, but not M1 macrophages, have a significant impact on barrier integrity which is likely due to strong interactions with the endothelium. 126 Thus, perivascular macrophages may function in stabilizing the new vessels after trauma.
Injury-induced signaling factors involved in vascular repair
Role of extracellular factors in modulating vascular response.

VEGF: vascular endothelial growth factor; FGF2: fibroblast growth factor 2; SDF-1α: stromal-derived factor-1α; MMΠ−9: matrix metalloproteinase-9; ANG-1: angiopoietin-1; EPO: erythropoietin; vWF: von Willebrand factor
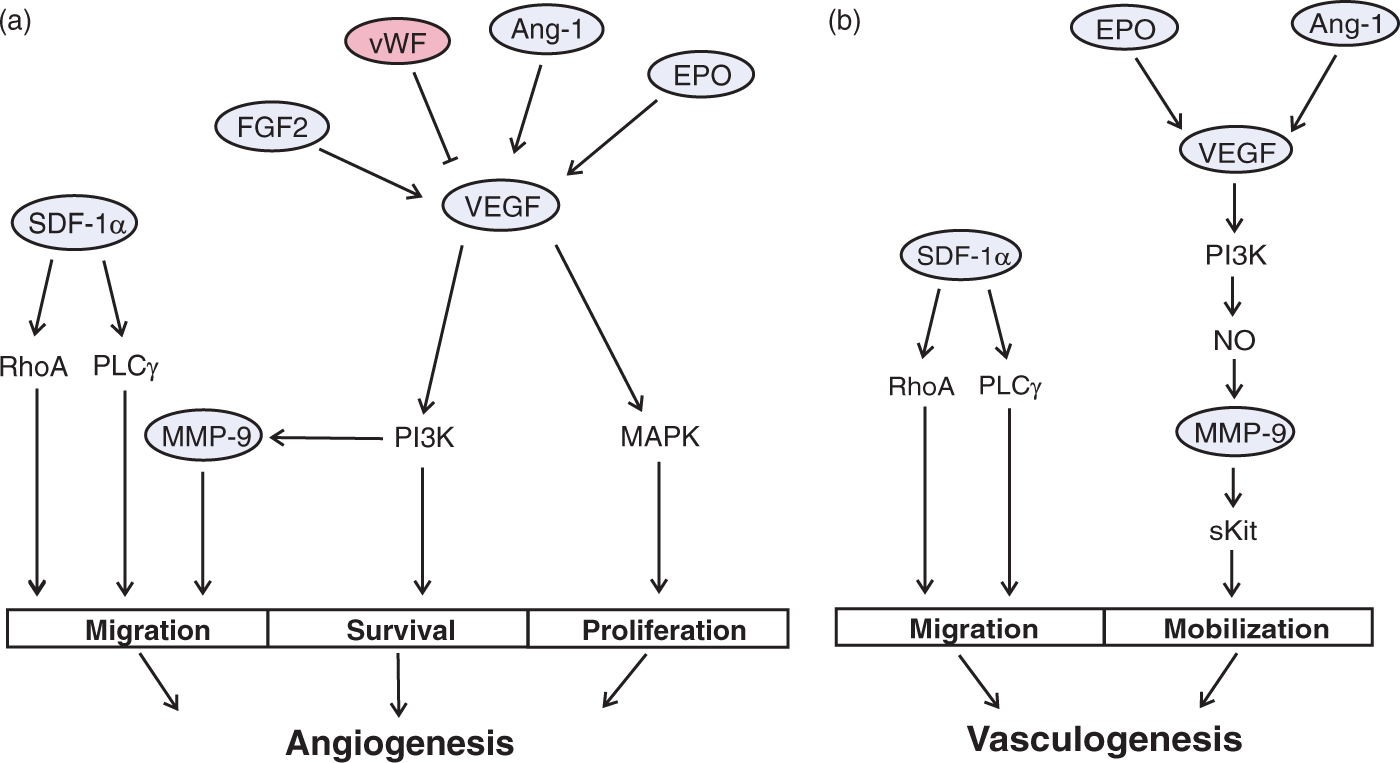
Possible interactions between signaling molecules. (a) The interplay between these angiogenic factors is critical in understanding the molecular mechanism(s) of vascular repair. After trauma, active cells release angiogenic factors into the injured tissue. VEGF is the key inducer of angiogenesis, which activates PI3K and MAPK pathway leading to EC survival and proliferation. FGF2, Ang-1, and EPO are also up-regulated at distinct times after TBI and these factors signal through VEGF signaling, either by increasing expression of VEGF and/or initiating down-stream PI3K and MAPK pathways. Along with pro-angiogenic factors, anti-angiogenic factors, including vWF, have been shown to inhibit angiogenesis. Migratory processes are controlled by SDF-1α, which activates down-stream PLCγ and RhoA pathway. MMP-9, a target of the PI3K pathway, is pivotal for EC migration. (b) Angiogenic factors are released into the blood stream which can activate cells from bone marrow. Serum VEGF, EPO, and Ang-1 are elevated after TBI and have been associated with postnatal vasculogenesis. Similar to angiogenesis, vasculogenesis is mediated by the VEGF signaling pathway. VEGF causes activation of the PI3K pathway and leads to the up-regulation of NO and MMP-9. MMP-9 converts the Kit ligand to its active, soluble form (sKit). sKit is responsible for mobilizing EPCs from the bone marrow. Activated EPCs then migrate to sites of injury in a process known as chemotaxis. SDF-1α activates down-stream PLCγ and RhoA pathway to induce EPC migration.
VEGF
VEGF is a key regulator of angiogenesis and modulating its levels has a significant effect on vessel formation. 86 Krum et al. 87 utilized a specific neutralizing antibody to inhibit endogenous VEGF and tested its effect on tissue repair after brain infusion injury. Neutralizing VEGF levels resulted in a significant reduction in vessel proliferation and an increase in vessel degeneration. Administration of VEGF has emerged as a potential treatment for vascular injuries. Several studies have shown that VEGF treatment enhances angiogenesis in TBI,130,131 ischemia,132,133 and stroke. 134 VEGF signaling is mediated through Flt-1 (VEGF-R1) and Flk-1 (VEGF-R2) that are expressed on ECs. 135 Flk-1 is the predominate receptor and is associated with angiogenesis. 135 Binding of VEGF to Flk-1 (and neurophilin-1 co-receptor) induces down-stream signaling pathways such as mitogen-activated protein kinase (MAPK) and phosphoinositide 3′ kinase (PI3K). 136 Together these pathways promote EC proliferation, migration, and survival, which are indispensable for angiogenesis. 136 These effects are prevalent in the chronic stages of TBI, where VEGF levels are increased around the injury site. 82
VEGF has been shown to increase EPC expression and induce vessel formation after hind limb and myocardial ischemia as well as physiological conditions, confirming its role in postnatal vasculogenesis. 90 As we have stated earlier in this review, much of the work on VEGF-mediated vasculogenesis was performed in tumors and its role in TBI is limited to correlative studies in patients.91,137 VEGF regulates EPC activity in a similar manner as ECs. In EPCs, VEGF binds to Flk-1 and leads to their mobilization and release from the bone marrow and induces differentiation. 94 The effects are mediated by the PI3K pathway which leads to expression of nitric oxide (NO; a potent vasodilator) and MMP-9. 138
Fibroblast growth factor 2
Similar to VEGF, fibroblast growth factor 2 (FGF2) is a potent mitogen and stimulator of angiogenesis. Given its angiogenic effects, FGF2 administration has been utilized in several brain injury models. FGF2 treatment after closed head injury has been shown to increase vessel density compared with non-treated animals. 131 However, animals treated with VEGF alone showed higher vessel growth than FGF2. ECs express both FGF receptors (FGFR-1 and FGFR-2) and are sensitive to the FGF family. Binding of FGF2 to its receptors leads to activation of proliferative, migratory, and survival processes. 139 MAPK, PI3K, and Phospholipase C gamma (PLC γ) pathways have been linked with these important angiogenic processes. 139 Alternatively, Kano et al. 140 have proposed a mechanism involving FGF2 induction of VEGF with FGF2 signaling acting in conjunction with VEGF signaling to promote angiogenesis.
FGF2 is critical in maintaining vascular integrity. Wang et al. 141 showed that FGF2 treatment after CCI preserved BBB integrity and improved neurological function. The authors found that FGF2 activates the PI3K pathway and leads to up-regulation of tight junction proteins (claudin-5 and occludin) and inhibits RhoA, an antagonist of BBB formation. Rac-1, a down-stream effector of the PI3K, is responsible for FGF-2 protective effects of the BBB. 141 Similar findings were observed in a model of intracerebral hemorrhage, wherein FGF2 treatment reduced BBB damage, cerebral edema, and improved outcomes. 142
Stromal cell-derived factor 1 alpha
Stromal cell-derived factor 1 alpha (SDF-1α) is an important chemokine for various cell types. Recent studies found that SDF-1α attracts CD34-expressing cells from the bone marrow, which include EPCs and ECs. Inhibition of endogenous SDF-1α after TBI reduces the number of CD34+ cells, which coincides with decreased vessel density around the injury site. 143 Further evidence suggests that exogenous SDF-1α has angiogenic benefits. Administration of SDF-1α after fluid percussion and weight drop injury enhances vessel formation, reduces BBB damage, and improves neurological outcomes.143,144 The most extensively studied SDF-1α receptor is CXCR4, which is expressed on EPCs, ECs, and smooth muscle cells. 145 Following injury, neural stem/progenitor cells and other damaged cells release SDF-1α into the injury microenvironment. ECs are known to up-regulate SDF-1α expression when exposed to hypoxia. 145 SDF-1α binding to CXCR4 activates PLCγ and Rho pathways to promote a chemotactic response. 146 As a result, CD34-expressing cells are activated and migrate toward the SDF-1α gradient. 147 Along with recruiting cells, SDF-1α has been shown to directly regulate EC and EPC function via the PI3K and MAPK pathway. 146
MMP-9
MMP-9 is a protease enzyme that is necessary for initiating angiogenesis. 148 This enzyme is expressed by ECs and functions by degrading the extracellular matrix, which provides for sufficient space for EC migration and the formation of new vessel lumens. 149 Some matrix proteins that are degraded by MMP-9 include collagen, laminin, thrombospondin, and fibronectin. 150 Additionally, it is critical in the processing and activation of growth factors thereby affecting the levels of pro- and anti-angiogenic factors in the tissue. 150 For example, MMP-9 can cleave and release activated VEGF in tumors cells. 150
MMP-9 also mediates vasculogenesis by affecting EPC activity. In animal models of TBI, MMP-9 levels increase and localize to EPCs and cortical vessels around the lesion. 151 Serum MMP-9 levels correlate with increases in EPC levels in clinical TBI. 137 Huang et al. 152 showed that MMP-9-deficient mice have reduced vascularization after ischemic injury resulting from reduced EPC expression and stem cell activating cytokines such as Kit ligand. These cytokines promote activation of stem cells into the bone marrow and have been implicated in hematopoiesis. EPCs are normally quiescent and remain attached to the stromal cells of bone marrow mediated by membrane bound Kit ligand (inactive form). 153 After injury, bone marrow cells activate the PI3K pathway to increase production of NO which in turn up-regulates MMP-9. 154 MMP-9 converts the inactive Kit ligand to its active soluble form, sKit. 153 Consequently, sKitL disrupts EPC adhesion to the bone marrow allowing for their mobilization and release into the blood circulation.
Ang-1
Ang-1 promotes vessel stabilization in the latter stages of vascular repair. Because of its angiogenic effects, delivery of exogenous Ang-1 may have significant benefits if administered early. Pharmaceutical compounds, such as simvastatin, can increase Ang-1 expression and are known to improve vascular function in stroke models. 155 Under physiological conditions, cerebral vessels express high levels of Ang-1. After cryogenic injury, there is a marked decrease in Ang-1 in the acute stages. 156 This is accompanied by increases in Ang-2, which is an Ang-1 antagonist and is associated with BBB breakdown. At two to six days post injury, Ang-1 levels slowly increase which correlates with angiogenesis. 156 Ang-1 is primarily produced by pericytes and directly interacts with ECs. 157 Both Ang-1 and Ang-2 signal through the Tie-2 receptor, with Ang-1 acting as an agonist and Ang-2 as an antagonist. 157 The balance between Ang-1 and Ang-2 determines its signaling. Ang-1/Tie-2 signaling promotes EC proliferation, migration, and survival, in part through activation of the VEGF pathway. 157
Ang-1 has also been linked to vasculogenesis. Serum Ang-1 and EPCs showed similar expression profiles in patients with severe TBIs, suggesting a possible role in mobilizing EPCs from the bone marrow. 91 The mechanism is not completely understood, but many suggest that Ang-1 stimulates vasculogenesis through the VEGF pathway. Activation of the PI3K pathway is critical and leads to the up-regulation of NO, MMP-9, and sKit. 153
Erythropoietin
Erythropoietin (EPO), a well-recognized hematopoietic cytokine, can act as a growth factor in different tissues. 158 Although EPO controls erythropoiesis (production of red blood cells), emerging evidence suggests that EPO contributes to angiogenesis. EPO treatment has shown promising results in animal models of TBI. Delayed EPO treatment after CCI increases vascularization around the injury site and improves vascular function in a dose-dependent manner. 159 EPO treatment after CCI resulted in enhanced CBF induced by L-arginase administration, likely by stimulating production of NO. 160 Although EPO is mostly produced in the kidney, increased expression of EPO and its receptor (EPO-R) are evident in ECs, neurons, and astrocytes after injury. 161 EPO expression is regulated by HIF-1α. 143 In ECs, EPO/EPO-R signaling stimulates VEGF expression which in turn activates VEGF signaling. 162 Neural progenitor cells are also affected and may interact with ECs to promote angiogenesis. 163 In addition, EPO can activate the PI3K pathway independent of VEGF. 162
The angiogenic effects of EPO are partly the result of vasculogenesis. Wang et al. 164 studied the effects of recombinant human EPO treatment in a rat fluid percussion injury model. Human EPO treated animals showed significantly higher EPC levels after TBI, with increased numbers of CD31+ and CD34+ cells found around the lesion. Treated animals also showed improved spatial learning and memory. Similarly, EPO has been shown to increase EPC expression and enhance angiogenesis after myocardial infarction and stroke.165,166 EPO is thought to stimulate vasculogenesis through VEGF signaling and the down-stream PI3K pathway, increasing the expression of NO and MMP-9. 162
von Willebrand factor
von Willebrand factor (vWF) is a glycoprotein that is increased in response to TBI, with elevated serum levels an indicator of poor outcomes. 167 vWF is well-recognized for its role in blood coagulation by promoting platelet adhesion to sites of damaged vessels and aggregation to form the hemostatic plug.168–170 It is also known to complex with coagulation Factor VIII. This protein is normally present in its inactive form when bound to vWF, but is activated and released following blood vessel damage. 169 Upon release, Factor VIII interacts with other coagulation factors to promote blood clotting. After vascular injury, the endothelium rapidly secretes vWF into the plasma and basement membrane. 168 vWF binds to collagen near sites of vessel damage and acts as a bridging molecule that interacts with circulating platelets. vWF interaction with the platelet receptor (GPIb-IX) leads to platelet activation and integrin-dependent adhesion to the vessel wall. 168
Unlike most growth factors, vWF has been shown to inhibit angiogenesis. Loss of vWF results in increased vessel formation in vitro and in vivo. 171 vWF modulates VEGF-R2 dependent angiogenesis by either interacting with integrin αvβ3 on the cell surface and/or affecting storage of Ang-2 within Weibel-Palade bodies. 172 Thus, vWF likely controls premature vessel formation by countering angiogenic factors. Additionally, staining for vWF is routinely used to detect for activated vessels that are subjected to high levels of pro-angiogenic factors. 173
WNT/β-catenin cascade: A novel pathway for vascular repair
Given the need to develop new therapies, it is important to identify other signaling mechanisms that may be activated after TBI. One such potential mechanism is the Wnt/β-catenin (or canonical) cascade, which has been a major focus of research for CNS injuries and diseases. 174 Numerous studies reported activation of Wnt/β-catenin signaling after TBI.175–178 White et al. 175 showed that Wnt/β-catenin signaling is enhanced in glial cells after CCI, which coincides with astrogliosis. Other studies have revealed that Wnt/β-catenin signaling regulates expression of key survival factors that affect neurogenesis.176,178 Thus, it is clear that Wnt/β-catenin signaling and its down-stream elements participate in regenerative processes after brain injury.
There is strong evidence in support of the Wnt/β-catenin pathway in vascular repair. This pathway plays a major role during embryonic vascular development and controls many aspects of vascular repair processes including angiogenesis,
179
BBB formation,
180
vascular remodeling, sprouting,
181
and vascular stability.
179
During development, Wnt/β-catenin signaling works in conjunction with Notch signaling leading to the formation of nascent blood vessels.
182
Notch activation results in the expression of arterial genes leading to arterial differentiation, whereas Notch inhibition results in the expression of venous genes leading to venous differentiation.
182
Along with vascular development, Wnt/β-catenin signaling has angiogenic functions in the adult. Many angiogenic factors are regulated by Wnt/β-catenin signaling in cancer, including VEGF, MMP-9, and FGF2.
183
Based on its importance during development and post injury, we strongly advocate further research of the Wnt/β-catenin pathway and its effects on vascular repair is warranted (Figure 4).
Wnt/β-catenin signaling in TBI. The Wnt/β-catenin pathway is tightly regulated by soluble Wnt factors. In the presence of these factors, β-catenin escapes degradation where it accumulates and promotes transcription of Wnt target genes. The role of Wnt/β-catenin signaling after TBI is not completely understood, but many suggest that it is involved in regenerative processes such as astrogliosis and neurogenesis. It is unclear at the present times what role Wnt/β-catenin signaling plays in vascular repair, but there is data in support for it. During vascular development, Wnt/β-catenin signaling is required for the development of CNS blood vessels and works in conjunction with Notch signaling in arterial-venous specification. Additionally, many angiogenic growth factors (VEGF), metalloproteases (MMP-9), and tight junction proteins (Claudin 3 and ZO-1) are both directly and indirectly modulated by the Wnt signaling cascade. Thus, Wnt/β-catenin pathway and its associated molecules may be involved in repairing the injured vasculature after TBI.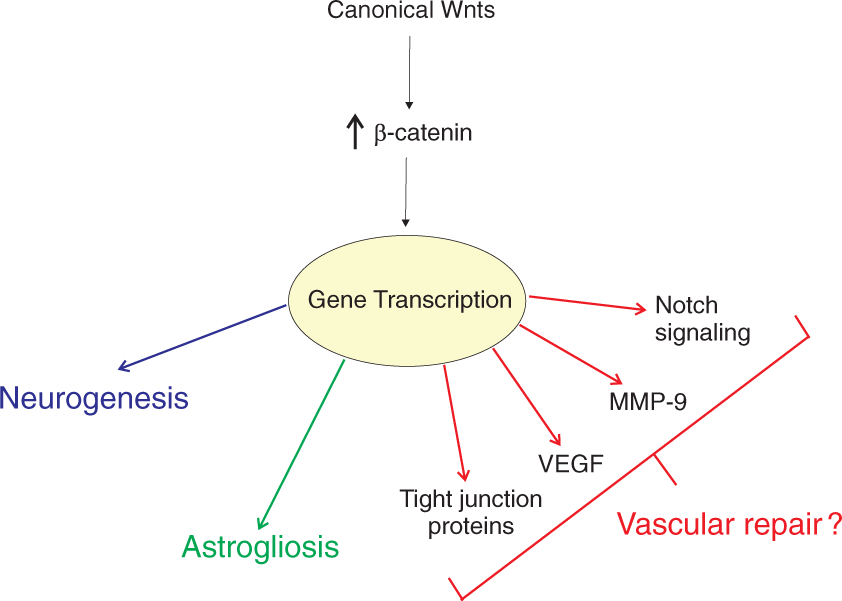
Distinct response in stroke
Unlike TBI, the cerebral vasculature responds differently after stroke. Ischemic stroke is the result of impeded blood flow due to an occlusion of a vessel. 174 The resulting infarct region is divided into the ischemic core and penumbra, tissue surrounding the core. 184 While the core experiences the greatest degree of tissue injury, the penumbra experiences limited blood flow allowing cells to survive. 185 Acutely after stroke, reduced blood flow activates signaling cascades that lead to vessel collateralization, or arteriogenesis. 186 This process is responsible for bypassing occluded vessels and increasing blood flow to the ischemic tissue. At one day after stroke, increased numbers of anastomosing microvessels are evident in the penumbra. 187 Arteriogenesis is initiated by fluid shear stress and leads to the activation of the endothelium (unlike angiogenesis that is hypoxia-driven). 185 Immune cells, such as monocytes and macrophages, are recruited where they release angiogenic growth factors and cytokines that promote EC and smooth muscle cell migration and proliferation and ultimately leading to the growth of new collateral arteries. 185 While arteriogenesis plays a key role in maintaining blood flow after ischemic stroke, there is no evidence of such a mechanism occurring after TBI.
Angiogenesis is thought to be initiated in the chronic phases of stroke. Post-mortem tissue of patients with ischemic stroke has revealed activated endothelial cells and increased microvessel density in the penumbra. 188 However, age plays a significant factor in post-ischemic angiogenesis. 189 Experimental studies using MRI suggest that the late phases of increased blood flow are the result of angiogenesis. 186 Thus, it appears that TBI and stroke both undergo a similar angiogenic process at these late time points.
Therapeutic perspective
Treatment strategies that target the cerebral vasculature are desperately needed for TBI and other vascular-related brain injuries, such as ischemic stroke, subarachnoid hemorrhage, intracerebral hemorrhage, to name a few. In the last decade, TBI research has primarily focused on the delivery of angiogenic proteins. This approach enhances vascular function in animal models of TBI, but is hampered by various complications that limit its effectiveness for translation to the clinic. Common issues include loss of protein activity, degradation, off-target effects, and ineffective concentrations at the injured tissue. Recently, a greater focus has been placed on delivery devices that increase efficacy and minimize side effects. Degradable hydrogels are used in research and can be designed to deliver multiple angiogenic factors in a controlled and efficient manner, but these are only effective for superficial TBI lesions. Furthermore, these devices can be expanded to administer drugs, living cells, and other bioactive agents. For more information, readers are encouraged to read recent reviews.89,190,191
A majority of the signaling factors discussed above can have detrimental effects after injury. VEGF is a prime example of how one factor can have dual functions. Although VEGF induces angiogenesis, it also enhances vessel permeability in the injured tissue. 192 Studies have demonstrated that VEGF treatment can lead to increased cerebral edema, but this is dependent on the timing of the treatment.134,193 Delayed administration has been shown to be the most efficacious in terms of vascularization.70,130 Furthermore, co-application of VEGF and other angiogenic factors may help in reducing deleterious effects. 194 Another consideration is the quality of the newly formed vessels. VEGF-induced vessels are inherently unstable and leaky as they develop, which can be attributed to a failure to express specific BBB proteins. 195 Thus, VEGF and other candidate proteins must be carefully selected when used for any future therapeutics.
Cell-based therapies have emerged as a possible treatment option for TBI. Recently, a great deal of attention has been placed on adult mesenchymal stem cells (MSCs). These cells are isolated from the bone marrow, have low antigenicity, and express high levels of growth factors and cytokines. 196 Administration of MSCs after experimental TBI ameliorates functional deficits and improves neurological outcome.197–199 In concert with neurogenesis, MSCs enhance angiogenesis and vasculogenesis.200–202. Interestingly, Zhang et al. 203 revealed that delivery of MSC-derived exosomes after CCI increases the number of nascent ECs and neurons in the injured tissue thereby improving outcomes. Exosomes are micro-vesicles that have RNA and protein as cargo and could be an intriguing cell-free treatment for TBI. Based on these promising preclinical studies, clinical trials are underway to test the safety and efficacy of autologous MSC treatment in TBI patients. 204
Conclusion
TBI is a major clinical problem that is associated with long-term morbidity among survivors. While the underlying mechanism(s) that lead to functional decline after TBI remains poorly understood, emerging research suggests that the damaged cerebral vasculature participates in the development of chronic disabilities. Ameliorating vascular dysfunction remains a key priority in the clinic, but therapeutic options are limited. Moving forward, it is critical to understand the mechanisms in which these cellular and molecular components may affect vascular repair. The eNVU has been understudied in its role in vascular repair and thus requires more attention. Targeting the PI3K and MAPK pathways may be effective therapeutic options but other upstream molecular targets need to be investigated to enhance vascular repair. A greater understanding of the vascular response to TBI and molecular signals that mediate vascular repair can lead to the development of new vasculogenic therapies. Most importantly, improvements in vascular function would limit secondary injuries leading to improved outcomes for patients with TBI.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by an NIH Program Project grant from National Institute of Neurological Disorders and Stroke (1P01NS082184, Project 3).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.