
Abstract
The CO2/HCO3– buffer minimizes pH changes in response to acid–base loads, HCO3– provides substrate for Na+,HCO3–-cotransporters and Cl–/HCO3–-exchangers, and H+ and HCO3– modify vasomotor responses during acid–base disturbances. We show here that rat middle cerebral arteries express cytosolic, mitochondrial, extracellular, and secreted carbonic anhydrase isoforms that catalyze equilibration of the CO2/HCO3– buffer. Switching from CO2/HCO3–-free to CO2/HCO3–-containing extracellular solution results in initial intracellular acidification due to hydration of CO2 followed by gradual alkalinization due to cellular HCO3– uptake. Carbonic anhydrase inhibition decelerates the initial acidification and attenuates the associated transient vasoconstriction without affecting intracellular pH or artery tone at steady-state. Na+,HCO3–-cotransport and Na+/H+-exchange activity after NH4+-prepulse-induced intracellular acidification are unaffected by carbonic anhydrase inhibition. Extracellular surface pH transients induced by transmembrane NH3 flux are evident under CO2/HCO3–-free conditions but absent when the buffer capacity and apparent H+ mobility increase in the presence of CO2/HCO3– even after the inhibition of carbonic anhydrases. We conclude that (a) intracellular carbonic anhydrase activity accentuates pH transients and vasoconstriction in response to acute elevations of pCO2, (b) CO2/HCO3– minimizes extracellular surface pH transients without requiring carbonic anhydrase activity, and (c) carbonic anhydrases are not rate limiting for acid–base transport across cell membranes during recovery from intracellular acidification.
Introduction
Spontaneous hydration of CO2 is slow and may become rate limiting for H+ buffering and transport of acid–base equivalents in intra- and extracellular solutions and across cell membranes.1–3 Carbonic anhydrases catalyze the reaction CO2 + OH– ⇄ HCO3– and thereby accelerate equilibration of the CO2/HCO3– buffer system. Carbonic anhydrases expressed in erythrocytes facilitate the conversion of CO2 generated from tissue metabolism to HCO3– in blood and likewise liberation of CO2 in pulmonary capillaries with subsequent transfer to the alveoli. Carbonic anhydrases have also been proposed to play important roles in the cerebral vasculature, 4 where they potentially affect pCO2, pH and [HCO3–] inside cells and in the interstitial solution surrounding the vascular smooth muscle and endothelial cells. Acid–base disturbances modify cerebrovascular tone and contribute to local blood flow regulation in response to altered metabolic demand. Sustained decreases in extracellular pH (pHo) can inhibit voltage-gated Ca2+-channels 5 and via concomitant changes in intracellular pH (pHi) reduce the Ca2+-sensitivity of the vascular smooth muscle cell contractile machinery.6–9 However, abrupt intracellular acidification of vascular smooth muscle cells can also cause vasoconstriction probably due to competition between H+ and Ca2+ for buffer binding. 10 Additionally, decreases in extracellular [HCO3–] can enhance vasoconstriction via a receptor protein tyrosine phosphatase (RPTP)γ-dependent mechanism that requires intact endothelium-dependent vasorelaxant function. 7 The complex interplay between pHi and vasomotor tone has recently been reviewed in detail.11–13
H+ buffer systems minimize pH fluctuations. 11 The CO2/HCO3– buffer system is particularly important 13 because it (a) provides substrate for acid–base transporters that control pHi and local pHo,8,14,15 (b) can be sensed by cells to modify vascular tone, 7 and (c) acts as mobile buffer inside and outside of cells.16,17 Although carbonic anhydrase inhibitors have been shown to modify vascular tone, it is controversial whether these effects are due to altered acid–base regulation18,19 or pH-independent actions20,21 on elements of excitation-contraction coupling or the contractile machinery.
Under most physiological and pathophysiological conditions, cells of the vascular wall are prone to intracellular acidification due to the inside-negative membrane potential and net acid production from metabolism. 11 Elimination of intracellular acid from vascular smooth muscle cells in cerebral arteries occurs via NBCn1 (Slc4a7)-mediated Na+,HCO3–-cotransport and NHE1 (Slc9a1)-mediated Na+/H+-exchange.9,14 Maintaining normal steady-state pHi in the wall of arteries is important for vasoconstriction and -relaxation6,8,9,14,22 and for control of artery structure.3,6 Carbonic anhydrases have been proposed to take part in transport metabolons where anchoring of carbonic anhydrases to Na+,HCO3–-cotransporters,23,24 Cl–/HCO3–-exchangers,25–27 Na+/H+-exchangers 28 or monocarboxylate transporters 29 allows transfer of metabolites from one active site to the next and thereby increases transport function.30,31 In addition to enzymatically facilitating substrate delivery and removal at the inner and outer surfaces of acid–base transporters, transporter-anchored carbonic anhydrases may allosterically activate acid–base transporters 32 or serve as “proton-collecting antenna” that can direct acid–base equivalents to the active site of the transporters. 33 Some investigators have found, however, that carbonic anhydrases do not bind to Na+,HCO3–-cotransporters 34 or Cl–/HCO3–-exchangers 35 and that carbonic anhydrase activity does not facilitate acid–base transport activity.36,37
We performed the current study to explore the function of carbonic anhydrases in the cerebrovascular wall. Specifically, we tested the hypothesis that carbonic anhydrases are expressed in rat middle cerebral arteries, contribute to local acid–base homeostasis and facilitate acid–base transport across cell membranes, and thereby adjust cerebral artery tone during rapid disturbances in the chemical equilibrium between H+, CO2, and HCO3–. We find that (a) carbonic anhydrases contribute to intracellular CO2/HCO3– equilibration with consequences for cerebrovascular tone, (b) the CO2/HCO3– buffer system attenuates extracellular surface pH (pHs) transients without requiring carbonic anhydrase-mediated catalysis, and (c) net acid extrusion across the plasma membrane during intracellular acidification is not influenced by inhibition of carbonic anhydrase activity.
Materials and methods
Male Wistar Hannover rats were obtained from Taconic Biosciences (Denmark) or Janvier Labs (France), housed for approximately one week in the animal facility at Aarhus University under a 12-h light/12-h dark cycle with free access to food and water, and euthanized at 12-15 weeks of age by CO2 inhalation followed by decapitation. Middle cerebral arteries were isolated by micro-dissection and mounted in a custom-built wire myograph (CW121, DMT, Denmark) designed for use of out-of-equilibrium solutions 7 or in 4-channel wire myographs (620 M, DMT) for studies of tension development. The CW121 myograph was designed to allow mixing of two heated precursor solutions through a nylon mesh. The calculated transit time from the point of mixing to the vascular preparation mounted on 50-µm tungsten pins in a heated (37℃) stainless steel chamber was less than 100 ms. 7 Arteries were normalized to 90% of the internal diameter equivalent to a transmural pressure of 100 mmHg. 38 Tension development during changes in the CO2/HCO3– composition of the extracellular bath solution was investigated in arteries pre-constricted with the thromboxane analog U46619 to 50% of maximum active tension. Changing the bath solution caused a washing artifact superimposed on the effect of buffer change. To eliminate this artifact and show only the effect of adding CO2/HCO3–, the tension response was corrected by subtraction of tension development in time control experiments where the bath solution was washed from a CO2/HCO3–-free solution to the same CO2/HCO3–-free solution. Animal handling was approved by the Danish Animal Experiments Inspectorate and conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The reporting of animal experiments conforms with the ARRIVE guidelines.
Fluorescence-based pHi and pHs measurements
We measured pHi in middle cerebral arteries loaded with the pH-sensitive fluorophore 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF; Invitrogen, Denmark) by adding 10 µM of the acetoxymethyl (AM) ester form in 0.02% dimethyl sulfoxide (DMSO) to the myograph bath for 10–20 min at 37℃. Applying BCECF-AM to the myograph bath was previously found to load vascular smooth muscle cells in the wall of rat middle cerebral arteries with no detectable loading of endothelial cells. 39 We measured extracellular pHs in middle cerebral arteries incubated for 6 min at 37℃ with 12.5 µg/mL of fluorescein-conjugated wheat germ agglutinin (W834, Life Technologies, USA) that binds selectively to N-acetylglucosamine and N-acetylneuraminic acid residues in the glycocalyx. Based on a Photon Technology International DeltaScan imaging system (USA), BCECF- or W834-loaded arteries were excited alternately at 440 and 495 nm and emission light collected at 530 nm using photomultipliers. Background fluorescence accounted for a few percent of the signal from BCECF-loaded arteries and around 20% of the signal from arteries loaded with W834; in both cases, background fluorescence was measured before the arteries were loaded with fluorophore and subtracted from the measured emissions prior to calculation of the 495/440 ratio. W834 fluorescence signals were smoothed by Savitzky-Golay (least-squares polynomial degree 3, 31 samples; LabChart, AD Instruments, New Zealand) to minimize noise and corrected for linear baseline drift. Fluorescence ratios were calibrated to pH using the high-[K+] nigericin method. 40 Nigericin is an H+/K+-ionophore that equalizes gradients for H+ and K+, and by use of calibration buffer solutions with [K+] corresponding to the predicted intracellular level, pHi is expected to equal pHo, which is controlled to obtain corresponding values for pH and fluorescence ratio. The calibration solutions contained (in mM): 135 K+, 12 Na+, 1.6 Ca2+, 1.2 Mg2+, 138.2 Cl–, 2 H2PO4–, 1.2 SO42–, 5.5 glucose, 0.03 ethylenediamine tetraacetic acid (EDTA), 10 HEPES; adjusted to pH 6.5, 6.8, 7.2, and 7.55 at 37℃ when bubbled with CO2-free air (i.e. 21% O2/balance N2).
Intracellular acidification was induced with NH4+-prepulse technique 41 where 20 mM NH4Cl was added to the bath solution and washed out to a Na+-free solution after 15 min. We estimated intracellular buffering capacity from the change in pHi in response to washout of NH4Cl: 14 the acid load is determined as the intracellular NH4+ concentration at the end of the NH4+-prepulse and calculated based on the Henderson-Hasselbalch equation with the assumption that NH3 is in equilibrium across the cell membrane. Acid–base transport activities were compared at equivalent pHi values to take into account the activation profile for net acid extrusion as a function of pHi. 42 We observed no significant effect of acetazolamide compared to DMSO vehicle on intracellular buffering capacity when plotted as function of pHi (n=16, P=0.64, least-squares linear regression analysis): at pHi 6.91 (where the Na+-dependent net acid extrusion is evaluated) total buffering capacity was 39±10 mM in the presence of 100 µM acetazolamide and 38±6 mM in the presence of DMSO vehicle.
Standard equilibrated solutions
For experiments based solely on equilibrated solutions, the CO2/HCO3–-containing physiological saline solution contained (in mM): 140 Na+, 4 K+, 1.6 Ca2+, 1.2 Mg2+, 124.02 Cl–, 22 HCO3–, 1.2 SO42–, 1.18 H2PO4–, 5.5 glucose, 0.03 EDTA, 10 HEPES; pH was adjusted to 7.4 at 37℃ after vigorous bubbling with a gas mixture of 5% CO2/balance air. CO2/HCO3–-free solutions were obtained by replacing HCO3– with equimolar Cl– and aerated with CO2-free air. In Na+-free solutions, Na+ was substituted with equimolar N-methyl-D-glucammonium (NMDG+) except for NaHCO3 that was replaced by choline-HCO3. To minimize CO2/HCO3–-independent buffer capacity and mobility in experiments studying extracellular pHs transients, [HEPES] was reduced to 2 mM by substitution with NaCl.
Out-of-equilibrium CO2/HCO3– solutions
We used out-of-equilibrium CO2/HCO3– solutions to study the effect of adding CO2 under nominally HCO3–-free conditions at a constant extracellular pH (pHo) of 7.4. Two solutions—the first contained 10% CO2 and 220 µM HCO3– at pH 5.4, whereas the second adjusted to pH 7.55 contained 65 mM HEPES but no CO2 or HCO3–—were delivered by Pump 33 (Harvard Apparatus, UK) dual syringe pumps through CO2-impermeable Tygon® tubing and mixed at a rate of 7 mL/min shortly (<100 ms) before reaching the artery under investigation. Because of the relatively slow spontaneous equilibration of the reaction CO2 + H2O ⇄ H2CO3, the out-of-equilibrium condition of the instantaneously mixed solution degenerates only slowly and can be maintained for several hundred milliseconds. 43 Thus, with rapid continuous superfusion, arteries can be constantly bathed in solutions of virtually any desired CO2/HCO3–/pH composition. 7 The nominally HCO3–-free (containing ∼110 µM HCO3–) out-of-equilibrium solution with 5% CO2 at pH 7.4 was generated by one-to-one mixing of precursor solutions A and B with the following compositions (in mM): solution A, 157 Na+, 5.64 K, 3.2 Ca2+, 169.04 Cl–, 0.03 EDTA; aerated with 10% CO2/balance air and adjusted to pH 5.4 at 37℃. The calculated [HCO3–] in solution A is ∼220 µM and derives from hydration of CO2 followed by limited dissociation of the carbonic acid at the low pH. Solution B, 91 Na+, 2.4 Mg2+, 2.36 K+, 91 Cl–, 2.4 SO42–, 2.36 H2PO4–, 11 glucose, 0.03 EDTA, 65 HEPES; adjusted to pH 7.55 at 37℃ when aerated with CO2-free air. The equilibrated CO2/HCO3–-free solution used in experiments involving out-of-equilibrium solutions contained (in mM): 124 Na+, 4 K+, 1.6 Ca2+, 1.2 Mg2+, 130.02 Cl–, 1.2 SO42–, 1.18 H2PO4–, 5.5 glucose, 0.03 EDTA, 32.5 HEPES; adjusted to pH 7.4 at 37℃ when bubbled with nominally CO2-free air.
Carbonic anhydrase inhibitors
We used the membrane-permeable and -impermeable carbonic anhydrase inhibitors acetazolamide (ATZ, 100 µM) and 4-(aminomethyl)benzenesulfonamide (AMB, 30 µM), respectively, at concentrations that completely inhibit carbonic anhydrase activity.44,45 Acetazolamide and AMB were solubilized in DMSO and distilled water, respectively. In experiments based on cell lysates, 100 µM acetazolamide and 30 µM AMB were found to inhibit the rate of CO2-induced acidification equally (data not shown).
Reverse transcription and polymerase chain reaction
Primers for polymerase chain reactions based on cDNA reverse transcribed from total RNA isolated from rat middle cerebral and mesenteric arteries.

Note: All primer sequences are given from 5′ to 3′. CAI, CAII, CAIII, CAVII, and CAXIII have been localized to the cytosol; CAVα and CAVβ to mitochondria; CAIV, CAIX, CAXII, and CAXIV are membrane-associated extracellular carbonic anhydrases; and CAVI is secreted. We did not test the expression of the acatalytic isoforms CAVIII, CAX, and CAXI.
Experimental design and statistics
Data are given as mean±SEM. The n-values report number of rats except for the RT-PCR experiments where arteries from two rats were pooled for each analysis, i.e. for the RT-PCR experiments, the number of rats used was twice the n-value. Otherwise, each treatment group only included one unilateral middle cerebral artery preparation per rat. Effects of single interventions on arteries from the same rats were compared by paired two-tailed Student’s t-test. Distributions of single variables were compared to a hypothetical mean value using two-tailed one-sample t-tests. Effects of two variables on a third variable measured multiple times in arteries from the same rats were evaluated by repeated measures two-way ANOVA. Concentration–response relationships were analyzed by least-squares sigmoidal curve fits and the derived log(EC50)- and maximum-values were compared between groups using extra sum-of-squares F-tests. Buffering capacity estimates were plotted as function of pHi and analyzed by least-squares linear regression analyses. Probability (P) values below 0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism® 5.04 software. Experiments with inhibitors and vehicle controls, as well as matched time and intervention controls, were performed in parallel or alternatingly. The investigators were not blinded to the application of active drug vs. vehicle. A few arteries that did not respond to application of U46619 or did not load well with fluorophores were excluded from analyses.
Results
We investigate the expression of carbonic anhydrases and their functional role for regulation of pHi and extracellular pHs in the cerebrovascular wall based on rat middle cerebral arteries.
Multiple carbonic anhydrase isoforms are expressed in rat arteries
Transcripts for multiple carbonic anhydrase isoforms are expressed in the rat cerebrovascular wall including isoforms previously localized to the cytosol (CAII, CAIII, and CAVII), mitochondria (CAVβ), and extracellular space (CAIV, CAIX, CAXII, and CAXIV) or found to be secreted (CAVI) from cells (Figure 1 and Supplementary Table 1). Compared to cerebral arteries, rat mesenteric arteries are heavily influenced by sympathetic innervation and respond more modestly to acidosis. In mesenteric arteries, we identified transcripts for CAII, CAIII, CAIV, CAVβ, CAVI, and CAIX but—in contrast to middle cerebral arteries—no detectable levels of CAVII, CAXII, and CAXIV (Figure 1 and Supplementary Table 1).
Transcripts for multiple carbonic anhydrase isoforms are detected in rat middle cerebral and mesenteric arteries. We find expression of cytosolic (CAII, CAIII, and CAVII), mitochondrial (CAVβ), extracellular (CAIV, CAIX, CAXII, and CAXIV), and secreted (CAVI) carbonic anhydrase isoforms in rat middle cerebral arteries. RT + and − indicate experiments in which reverse transcriptase is added and omitted, respectively, to control for genomic amplification. H2O indicates reactions in which water is added instead of template to control for contamination. The positive control tissue for each reaction is listed in Table 1. Supplementary Table 1 summarizes the results of multiple independent experiments scored as “positive,” “weakly positive” or “negative.” Each experiment was performed on RNA pooled from two rats. Specificity of all bands is confirmed by sequencing with at least 99% identity to the mRNA sequences available from NCBI. The smaller amplicon size for CAVI in liver tissue indicates absence of a splice cassette present in the vascular wall.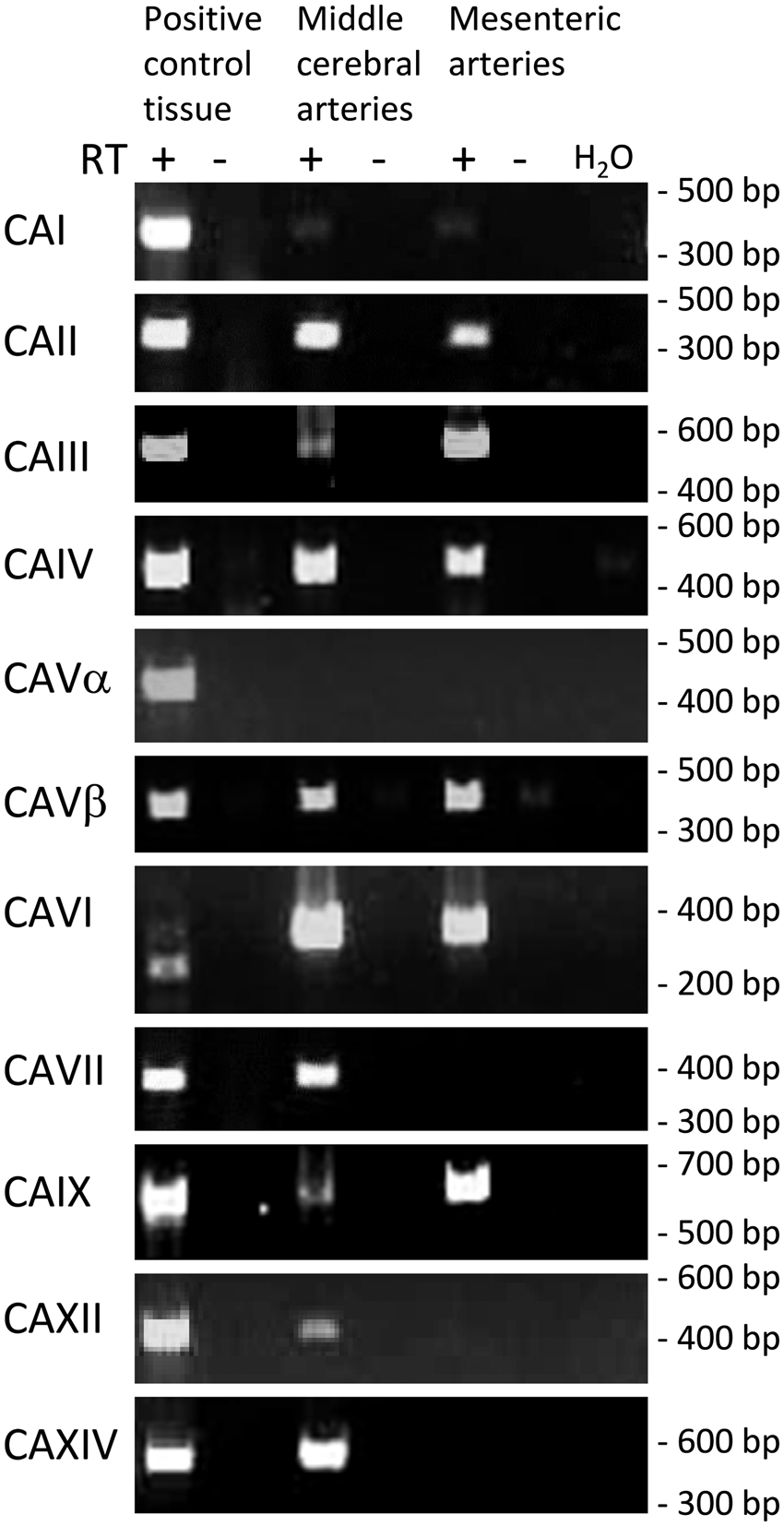
Carbonic anhydrase activity increases CO2/HCO3–-dependent pHi fluctuations
Steady-state pHi without CO2/HCO3– is unaffected by ATZ (ΔpHi=0.01±0.03 vs. DMSO; n=9, P=0.64, two-tailed one-sample t-test) and AMB (ΔpHi=0.01±0.15 vs. control; n=5, P=0.94, two-tailed one-sample t-test). Changing from CO2/HCO3–-free to CO2/HCO3–-containing bath solution at a fixed pHo of 7.4 under control experimental conditions (DMSO vehicle) causes a biphasic pHi response with initial acidification followed by partial, gradual pHi recovery (Figure 2(a)). In the presence of acetazolamide—that blocks intracellular as well as extracellular carbonic anhydrases—the rate of intracellular acidification is reduced (Figure 2(a) and (b)), whereas the final plateau pHi reached within 5 min after the application of CO2/HCO3– is similar in the presence of acetazolamide and DMSO vehicle (Figure 2(a); P=0.27, paired two-tailed Student’s t-test). Applying the cell-impermeable carbonic anhydrase inhibitor AMB—to block only extracellular carbonic anhydrases44—affects neither the rate of intracellular acidification nor the final plateau pHi after shift from CO2/HCO3–-free to CO2/HCO3–-containing bath solution (Figure 2(c) and (d)).
Inhibition of intracellular carbonic anhydrases decelerates acidification upon application of CO2/HCO3– without affecting steady-state pHi. Acetazolamide (ATZ, 100 µM) inhibits both intra- and extracellular carbonic anhydrase activity whereas 4-(aminomethyl)benzenesulfonamide (AMB, 30 µM) is cell impermeable and inhibits only extracellular carbonic anhydrase activity. (a and c) Average traces of pHi during changes from CO2/HCO3–-free to CO2/HCO3–-containing bath solution at pHo 7.4. Experiments were performed without superfusion. The absolute steady-state pHi under control conditions (DMSO vehicle) prior to the switch from CO2/HCO3–-free to CO2/HCO3–-containing solution was 7.31±0.05 (n=9). (b and d) Initial rates of intracellular acidification after addition of CO2/HCO3– (ATZ/DMSO: n=9; AMB/control: n=5). Data are compared by paired two-tailed Student’s t-test. *P<0.05 vs. DMSO vehicle. NS: not significantly different vs. control.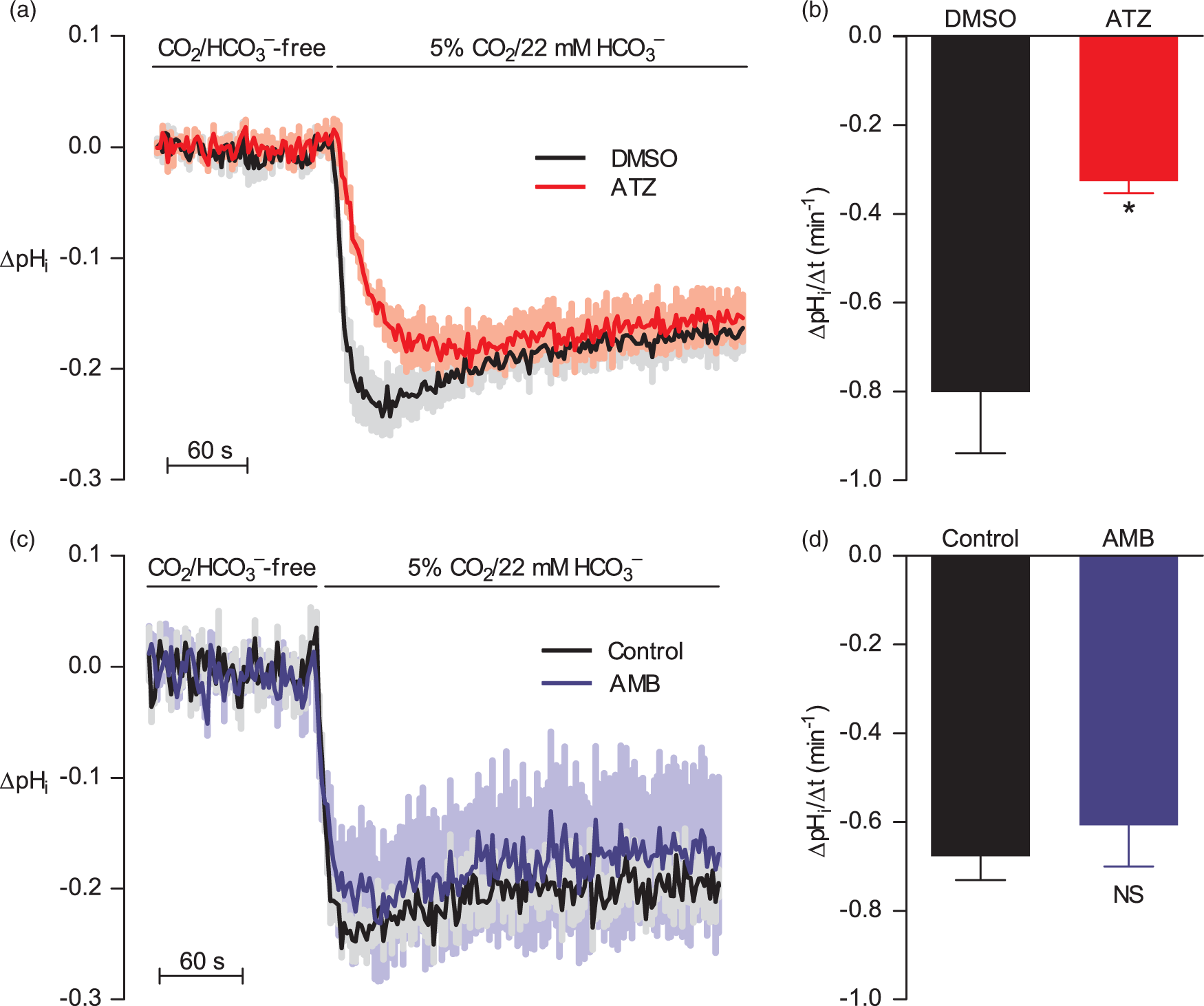
Under standard experimental conditions, steady-state pHi in vascular smooth muscle cells of rat middle cerebral arteries is 0.15±0.02 lower in the presence than absence of CO2/HCO3– (n=6, P<0.001, two-tailed one-sample t-test), which is in contrast to previous findings from mouse middle cerebral arteries where pHi is approximately 0.2 higher in the presence than absence of CO2/HCO3–. 14 The different pHi response on omission of CO2/HCO3– implies a greater activity of Cl–/HCO3– exchange relative to Na+,HCO3–-cotransport in rat compared to mouse middle cerebral arteries.
Carbonic anhydrase activity accentuates transient CO2/HCO3–-dependent tension development
We next investigate the effect of acetazolamide on U46619-induced vasoconstriction during (a) pHi-transients caused by changes in pCO2 and [HCO3–] (Figure 3(a)) and (b) steady-state pHi conditions in the presence and absence of CO2/HCO3– (Figure 3(b) and (c)).
Acetazolamide inhibits the transient contraction during CO2-induced intracellular acidification but has no effect on U46619-induced contractions under equilibrated acid–base conditions. (a) Effect of acetazolamide (ATZ)—compared to DMSO vehicle—on tension development during changes from CO2/HCO3–-free to CO2/HCO3–-containing bath solution (n=14). Tension is shown after subtraction of the response in time control experiments where the bath solution was washed from CO2/HCO3–-free solution to the same CO2/HCO3–-free solution. Tension development during the first 100 s after solution change is compared by repeated measures two-way ANOVA; the P-value reports significant interaction, i.e. tension development as function of time after change of buffer is significantly influenced by acetazolamide. Final plateau tension development (corresponding to the last 30 s of the traces) is not significantly different between experiments performed in the presence of acetazolamide or DMSO (P=0.96, repeated measures two-way ANOVA). (b and c) Effect of acetazolamide (ATZ)—compared to DMSO vehicle—on tension development during cumulative application of U46619 in the presence (b) or absence (c) of CO2/HCO3– (n=14). Data are fitted by least-squares regression analyses to sigmoidal curves and compared using extra sum-of-squares F-test. All experiments are performed without superfusion. ***P<0.001 vs. DMSO vehicle. NS: not significantly different vs. DMSO vehicle.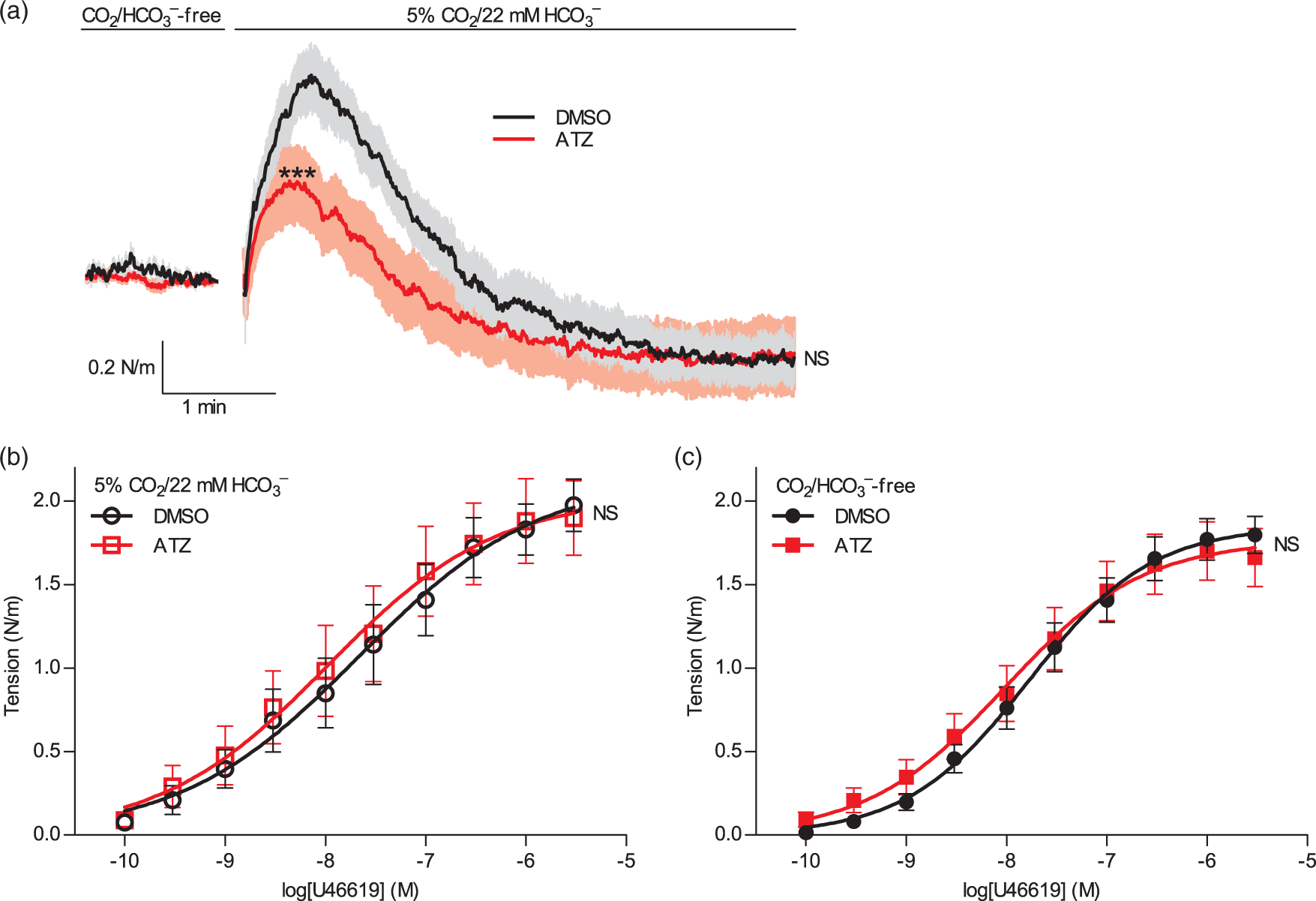
Arteries pre-constricted to 50% of maximal U46619-induced active tension respond biphasically to changes from CO2/HCO3–-free to CO2/HCO3–-containing bath solution at a fixed pHo of 7.4: first, arteries constrict, and then, they reach a stable plateau tension below the initial level of constriction (Figure 3(a)). The transient vasoconstriction caused by CO2/HCO3– is consistent with previous data from mesenteric arteries that acute hypercapnia-induced intracellular acidification at constant pHo results in vasoconstriction 51 likely due to competition between H+ and Ca2+ for buffer binding. 10 In congruence with the decelerated initial intracellular acidification after CO2/HCO3– application (Figure 2(a)), the transient vasoconstriction is attenuated in arteries pretreated with 100 µM acetazolamide (Figure 3(a)). The final steady-state tension is unaffected by 100 µM acetazolamide (Figure 3(a)) again reflecting the similar pHi levels under these conditions (Figure 2(a)). The lower steady-state tension in the presence than absence of CO2/HCO3– (Figure 3(a)) is consistent with the sustained lower pHi (Figure 2(a)), which can reduce vascular smooth muscle Ca2+ sensitivity,6,8,9 and the higher extracellular [HCO3–], which can be sensed and cause vasorelaxation in cerebral arteries via an RPTPγ-dependent mechanism. 7
Whether investigated in the presence (Figure 3(b)) or absence (Figure 3(c)) of CO2/HCO3–, arteries pre-incubated with 100 µM acetazolamide or exposed to equivalent amounts of DMSO vehicle for at least 10 min respond with similar tension development in response to cumulative application of U46619.
Together these findings support that acetazolamide—applied in concentrations providing full inhibition of carbonic anhydrase activity45—can affect vasomotor responses of rat middle cerebral arteries during transient phases where carbonic anhydrase-mediated catalysis is important for intracellular equilibration of the CO2/HCO3– buffer system.
Recovery from intracellular acidosis is unaffected by carbonic anhydrase inhibition
In order to investigate the role of carbonic anhydrases for transport of acid–base equivalents across the plasma membrane, we next induce intracellular acidification using the NH4+-prepulse technique (Figure 4(a) and (c)): addition of 20 mM NH4Cl to the bath solution causes abrupt intracellular alkalinization as NH3 enters into cells and takes up H+ from the intracellular environment. After 15 min recovery towards resting levels—due to NH4+ influx and activation of cellular net base extrusion—washout of NH4Cl to a Na+-free bath solution causes intracellular acidification as the imported H+ bound to NH4+ is liberated. Almost no pHi recovery is observed under Na+-free conditions (Figure 4) consistent with net acid extrusion mediated primarily via Na+,HCO3–-cotransport and Na+/H+-exchange.6,8,9,14 We find no effect of 100 µM acetazolamide on overall net acid extrusion activity (Figure 4(a) and (b)) or Na+,HCO3–-cotransport activity—measured as net acid extrusion in the presence of CO2/HCO3– and 100 µM of the Na+/H+-exchange inhibitor dimethylamiloride (DMA) (Figure 4(c) and (d))—during intracellular acidification. These findings also show that acetazolamide does not affect Na+/H+-exchange activity, i.e. the DMA-sensitive component of net acid extrusion (Figure 4).
Carbonic anhydrase inhibition does not affect net acid extrusion from vascular smooth muscle cells during intracellular acidification (a to d). Intracellular acidification is induced with NH4+-prepulse technique to study pHi recovery and net acid extrusion in the presence of 100 µM acetazolamide (ATZ) or equivalent DMSO vehicle. Experiments are performed in the presence of CO2/HCO3– with (c and d) or without (a and b) 100 µM Na+/H+-exchange inhibitor DMA added at the point of NH4Cl wash out and kept in the bath solution throughout the rest of the experiment. Panels a and c show average pHi curves. Panels b and d show rates of net acid extrusion. Na+-dependent net acid extrusion (n=7) is quantified at average pHi 6.92±0.10 (CO2/HCO3– + ATZ), 6.92±0.09 (CO2/HCO3– + DMSO vehicle), 6.82±0.03 (CO2/HCO3– + DMA + ATZ), and 6.88±0.06 (CO2/HCO3– + DMA + DMSO vehicle). Data are compared using repeated measures two-way ANOVA followed by Bonferroni post-tests. NS: not significantly different vs. DMSO vehicle.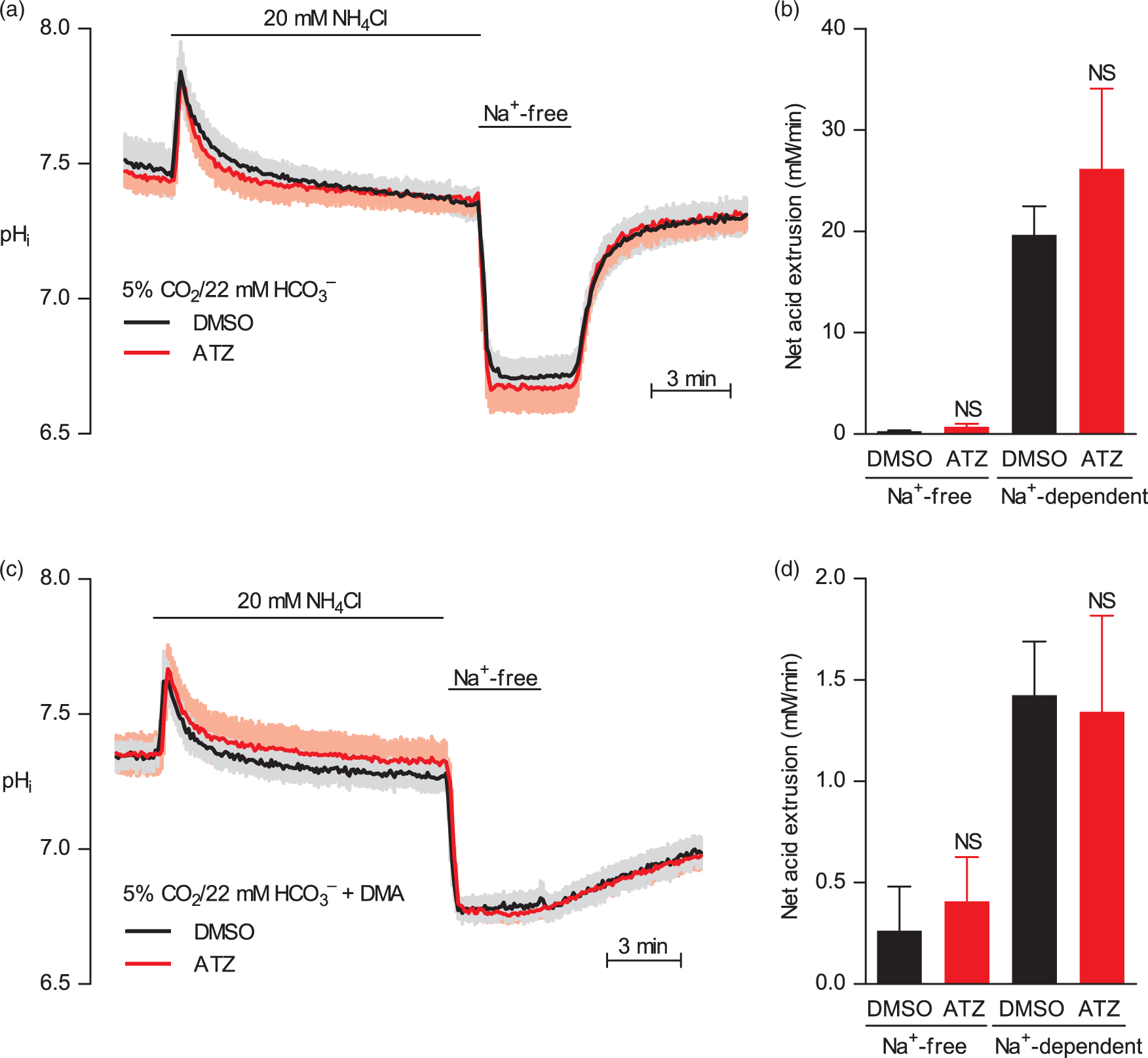
The relatively low contribution of Na+,HCO3–-cotransport activity (Figure 4(d)) to overall net acid extrusion activity (Figure 4(b)) in vascular smooth muscle cells of rat middle cerebral arteries is in contrast to mouse middle cerebral arteries—where Na+,HCO3–-cotransport and Na+/H+-exchange contributes approximately equally to net acid extrusion14—but consistent with the lower steady-state pHi in rat middle cerebral arteries in presence compared to absence of CO2/HCO3–.
Carbonic anhydrases accelerate intracellular CO2 hydration in rat middle cerebral arteries
We apply out-of-equilibrium CO2/HCO3– solutions to rat middle cerebral arteries in order to distinguish between individual effects of adding CO2 and HCO3–. We add 5% CO2 with only 110 µM HCO3– at a fixed pHo of 7.4 by exploiting the slow equilibration of the spontaneous reaction CO2 + H2O ⇄ H2CO3 in combination with a system of CO2-impermeable tubes that allow for rapid solution mixing immediately before the tissue preparation. Using this approach, [HCO3–] is maintained essentially similar to that of the immediately mixed solution.
7
As shown in Figure 5(a), adding CO2 under these nominally HCO3–-free conditions causes a monophasic acidification that is clearly different from that seen when 5% CO2 is added with the physiological level of 22 mM HCO3– (Figure 5(c)). When [HCO3–] is maintained in the micromolar range (Figure 5(a)), the pHi recovery from the CO2-induced acidification is virtually absent, and the rate of CO2 hydration can be studied in isolation. These experiments confirm that 100 µM acetazolamide decelerates intracellular CO2 hydration (Figure 5(a) and (b)) and support that the effect of acetazolamide on the pHi transient after application of CO2/HCO3– (Figures 5(c) and (d) and 2(a) and (b)) can be explained by decelerated CO2 hydration rather than accelerated HCO3– uptake.
Carbonic anhydrase inhibition decelerates intracellular hydration of CO2. Under continuous superfusion (7 mL/min) at pHo 7.4, CO2 is introduced either under out-of-equilibrium conditions (n=6–7) in the nominal absence (∼110 µM) of HCO3– (a and b) or under equilibrated conditions (n=13) with 22 mM HCO3– (c and d). Data are compared by two-tailed Student’s t-tests. ***P<0.001 vs. DMSO vehicle.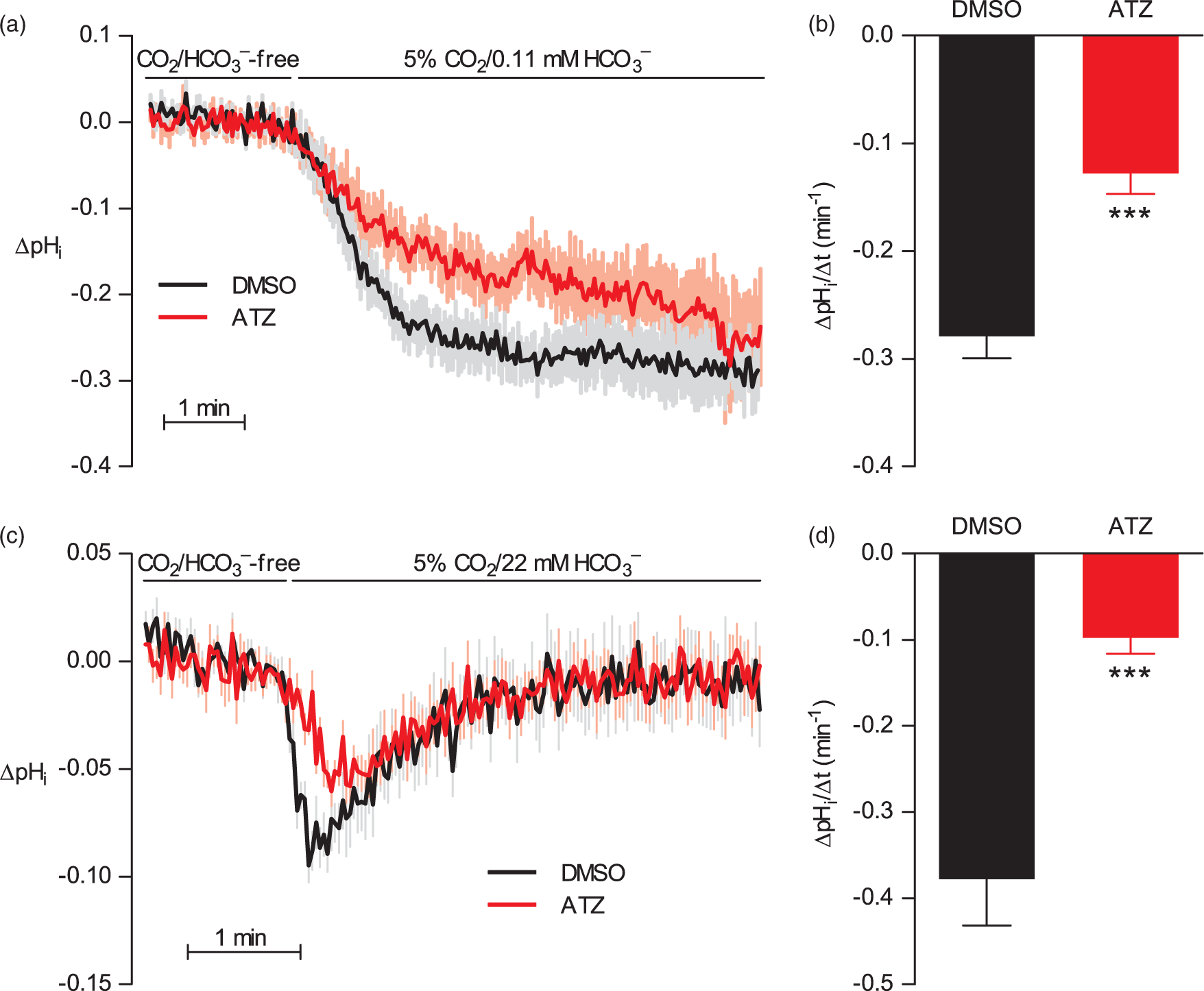
Interestingly, the high-flow superfusion (7 mL/min) necessary for maintaining out-of-equilibrium CO2/HCO3– conditions affects pHi regulation even when equilibrated solutions are applied: it is apparent that the rate of pHi recovery after initial CO2/HCO3–-induced intracellular acidification is enhanced during high-flow superfusion (Figure 5(c)) compared to standard no-flow (Figure 2(a)) experimental conditions. Also, whereas steady-state pHi under no-flow conditions is lower with than without CO2/HCO3– (Figure 2(a)), the final plateau pHi in the presence of CO2/HCO3– is similar to that observed in the absence of CO2/HCO3– when investigated during high-flow superfusion (Figure 5(c)). Altered acid–base transport activity in cells exposed to increased shear stress has previously been reported for cultured endothelial cells.52,53
CO2/HCO3– attenuates extracellular pHs fluctuations independently of carbonic anhydrases
Carbonic anhydrases have been suggested to affect extracellular pHs dynamics in cells with high acid–base transport rates across the plasma membrane.32,54 We investigate the consequences of carbonic anhydrase inhibition for extracellular pHs transients caused by addition of NH4Cl: on addition of NH4+ to the bath, NH3 generated from dissociation of NH4+ will traverse the plasma membrane. By taking up H+ from the cytosol, NH3 influx causes the abrupt increase in pHi (Figure 4(a) and (c)); and in parallel, the transport of NH3 into the cells causes a rightward shift in the NH4+ ⇄ NH3 + H+ equilibrium at the outer surface of the plasma membrane. The consequent release of H+ at the extracellular surface of cells in the cerebrovascular wall results—when buffer capacity and mobility are kept low in the absence of CO2/HCO3– and with only 2 mM HEPES—in a transient decrease in extracellular pHs (Figure 6(a)). This transient extracellular surface acidification is abolished in the presence of CO2/HCO3– and is not reestablished when arteries are pre-incubated with 100 µM acetazolamide (Figure 6(a) and (b)). These findings suggest that the spontaneous reactions HCO3– + H+ ⇄ H2CO3 ⇄ CO2 + H2O are sufficiently fast at facilitating buffering and dissipation of the liberated H+ that catalysis by carbonic anhydrases is dispensable for pHs control.
The CO2/HCO3– buffer eliminates extracellular pHs transients even after inhibition of carbonic anhydrase activity. (a) Average traces of extracellular pHs dynamics in response to addition of 20 mM NH4Cl. (b) Average rates (n=13–15) of initial surface acidification after addition of NH4Cl in the absence of CO2/HCO3– and in the presence of CO2/HCO3– with or without 100 µM acetazolamide (ATZ). Data are compared using one-way ANOVA followed by Bonferroni post-tests. *P<0.05, **P<0.01 vs. DMSO vehicle under CO2/HCO3–-free conditions. NS: not significantly different, as indicated.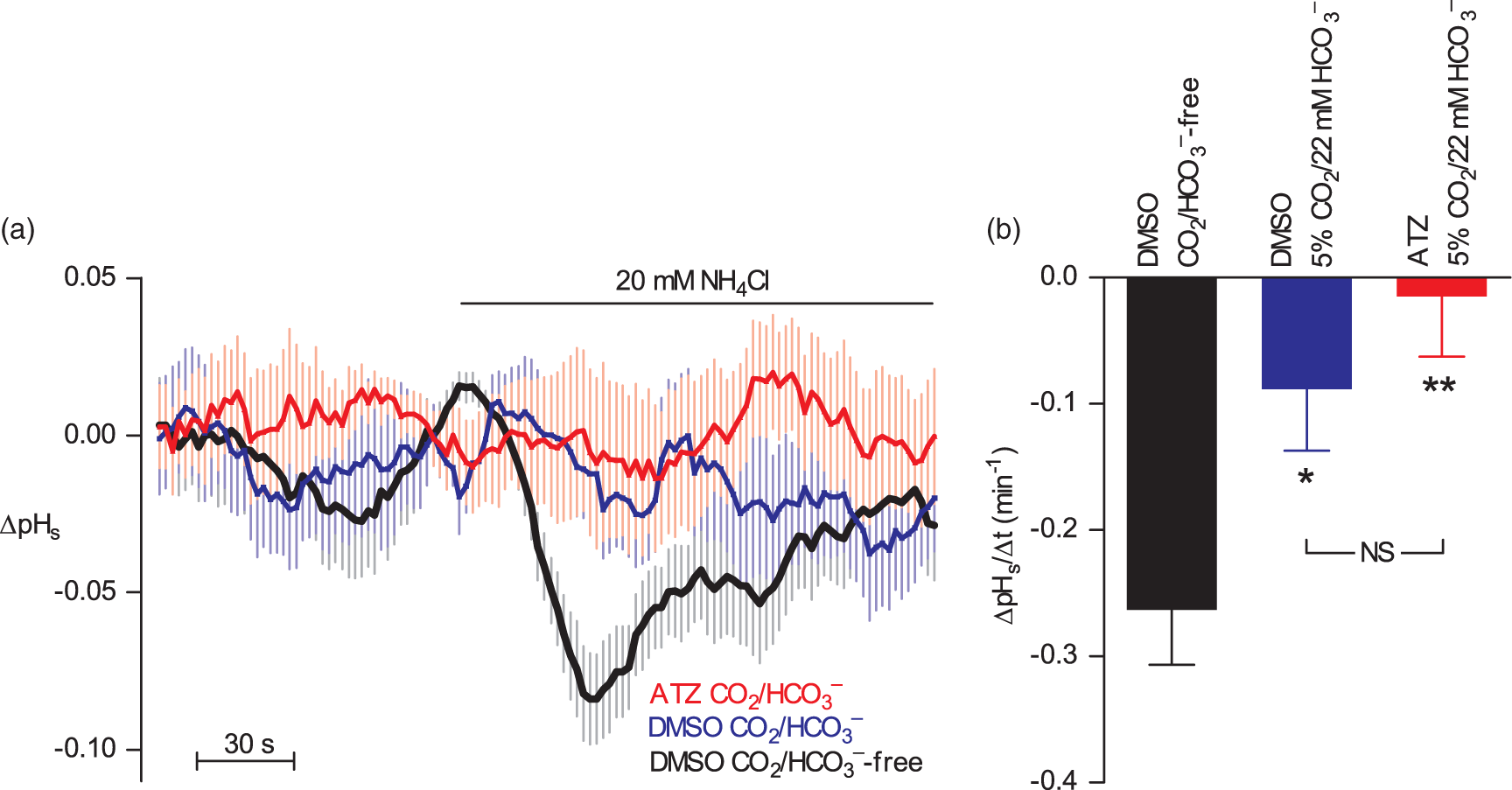
Discussion
We show here that carbonic anhydrases are expressed and functionally important in the cerebrovascular wall. Transcripts for multiple intra- and extracellular carbonic anhydrase isoforms are detectable in rat middle cerebral arteries; but functionally, carbonic anhydrases appear particularly important for intracellular CO2/HCO3– equilibration, which can affect acid–base regulation and cerebrovascular tone.
We investigate direct effects of carbonic anhydrase inhibitors in the cerebrovascular wall of isolated arteries. Noteworthy, under in vivo conditions, systemically administered carbonic anhydrase inhibitors will also decrease net urinary acid excretion, and the associated metabolic acidosis may cause cerebral vasodilation. Acetazolamide-induced vasodilation in vivo has been used to assess the cerebral perfusion reserve, although capacity for cerebral autoregulatory vasodilation has been demonstrated even after high-dose acetazolamide administration. 55 The mechanism by which carbonic anhydrases directly affect arterial tension development is controversial particularly with respect to its pH-dependency: some investigators observe the vasorelaxant effects of carbonic anhydrase inhibitors only at concentrations several-fold higher than the IC50 for carbonic anhydrase inhibition, 20 whereas others find good correlation between the effective concentrations causing carbonic anhydrase inhibition, pHi disturbances, and vasorelaxation. 19 In the current study, carbonic anhydrase inhibitors affect artery tension only during transient phases of CO2/HCO3– buffer equilibration (Figure 3(a)). When the CO2/HCO3– buffer system is given time to equilibrate, no apparent effect of acetazolamide is observed on pHi (Figure 2) or cerebrovascular tone (Figure 3(b) and (c)). Thus, our findings support that in rat middle cerebral arteries, the vasoactive effect of 100 µM acetazolamide is pH dependent. The reason that carbonic anhydrase inhibitors in some previous in vitro studies,19,20,56 yet not in the present study (Figures 2(a), 3 and 5(c)), decrease vasomotor tone and modify pHi at steady-state is not clear but could relate to differences in the concentration range (i.e. within or exceeding the range required to inhibit carbonic anhydrase activity), animal species (i.e. pig, rat, or guinea pig), and vascular bed (e.g. retinal, mesenteric or cerebral) investigated. Because the contraction of cerebral arteries has minimal effects on hypercapnia-induced changes in pHi, 57 we simplified the experimental setup by measuring pHi in the absence of vasoconstrictors. Contractile function was tested in arteries that were not loaded with fluorophores or exposed to excitation light in order to avoid artifacts that can result from organic solvents and prolonged light exposure. 9
Motile vascular smooth muscle cells are morphologically specialized with long narrow protrusions (i.e. filopodia), and carbonic anhydrases play a major role in minimizing pHi gradients in migrating vascular smooth muscle cells. 3 The long convoluted diffusion distances in filopodia—combined with local differences in metabolic acid production and transport of acid–base equivalents across the cell membrane—allow for the generation of spatial pHi gradients; and the amplitude of these pHi gradients can be modified by carbonic anhydrases that enhance the apparent H+ mobility.3,17 In the current study, we find that the effects of carbonic anhydrase-mediated catalysis on pH dynamics and tension development in contractile, spindle-shaped, vascular smooth muscle cells are more modest and restricted to transient phases of CO2/HCO3– buffer equilibration.
In middle cerebral arteries, we find expression of transcripts for carbonic anhydrase isoforms (CAIV, CAIX, CAXII, and CAXIV) with known extracellular localization (Figure 1). However, the potential functional role of extracellular carbonic anhydrase activity is not clear, because the inhibition of extracellular carbonic anhydrases with AMB does not affect the rate of cellular CO2 uptake and hydration (Figure 2(c) and (d)). Whereas CO2/HCO3– attenuates pH transients at the outer cell surface, this effect—which is most likely caused by the enhanced buffer capacity and mobility17—does not require carbonic anhydrase activity (Figure 6). It is noteworthy that the functional importance of extracellular carbonic anhydrases depends on the experimental conditions: whereas cell-impermeable carbonic anhydrase inhibitors have no effect in isolated cancer cells under rapid superfusion, they increase pH non-uniformity when the same cells are studied as multicellular preparations with unstirred extracellular spaces. 58 The cerebrovascular wall consists of multiple cell types in a complex three-dimensional tortuous space expected to amplify extracellular pHs transients and permit identification of carbonic anhydrase-mediated catalysis if it plays an important role. In congruence, although the application of NH4Cl in the presence of CO2/HCO3– does not produce detectable extracellular pHs transients even when acetazolamide is present, pHs transients are detectable when buffer capacity and mobility are reduced by omission of CO2/HCO3– (Figure 6).
It is controversial to what extent carbonic anhydrases facilitate acid–base transport across cell membranes. Based primarily on experiments with heterologous expression systems, carbonic anhydrases bound to the surface of acid–base transporters can facilitate transporter activity.23–29 In the current study, we find that carbonic anhydrase-mediated equilibration of the CO2/HCO3– buffer system is not rate limiting for Na+,HCO3–-cotransport or Na+/H+-exchange activity during intracellular acidification of vascular smooth muscle cells (Figure 4). A similar absence of carbonic anhydrase dependency has been reported for the electrogenic Na+,HCO3–-cotransporter NBCe1 (Slc4a4) expressed in Xenopus oocytes.36,37 Transport of acid–base equivalents across cell membranes is a multi-stage process involving transport to and from the membrane in addition to the transmembrane flux. Illustrating this complexity, carbonic anhydrases purportedly accelerate Na+,HCO3–-cotransport in isolated cardiomyocytes without affecting Na+/H+-exchange activity; and based on this finding, it was suggested that restricted access for intrinsic buffers limits delivery or removal of H+ equivalents to and from the cardiac Na+,HCO3–-cotransporter. 2 Thus, the local cell geometry may critically affect whether facilitated H+ transport by the CO2/HCO3– buffer system catalyzed by carbonic anhydrases is rate limiting for transmembrane acid–base transport.
Vasorelaxation in response to respiratory acidosis (i.e. a decrease in pH caused by elevated pCO2) is well-described 59 and can be explained by increased ryanodine receptor-dependent activation of large-conductance K+-channels 60 and an inhibitory effect of increased [H+] on voltage-gated Ca2+-channels. 5 In the current study, we unmask that increases in pCO2 at constant pHo elicit transient vasoconstriction (Figure 3(a)) that temporally coincides with the decrease in pHi (Figure 2(a)). Vasocontractile influences of (a) acute CO2-induced intracellular acidification and (b) low extracellular [HCO3–] via RPTPγ-dependent signalling 7 represent two “braking” mechanisms on vasorelaxation that come into play during respiratory and metabolic acidosis, respectively. Because carbonic anhydrases accelerate and amplify pHi decreases in response to elevated pCO2 (Figures 2(a) and 5), they may be important for limiting vasorelaxation and hence the decrease in pre-capillary resistance during respiratory acidosis and thereby reduce the risk of cerebral edema that could be the consequence of unopposed vasodilation.
Previous studies into the role of carbonic anhydrases for pH regulation in the vascular wall have focused primarily on their effects on steady-state pHi.19,56 We provide here additional investigations of pHi transients during CO2/HCO3– buffer equilibration, acid–base transport activities across cell membranes during intracellular acidification, and extracellular pHs transients in response to high transmembrane fluxes of acid–base equivalents. We also for the first time apply out-of-equilibrium technology—that can separate effects of adding CO2 from effects of adding HCO3– at constant pH—to studies of carbonic anhydrases in arteries (Figure 5). Because HCO3– plays multiple roles as part of a mobile buffer pair, as substrate for acid–base transporters, and as a sensed ion important for adaptation of vascular function during acid–base disturbances, facilitated equilibration of the CO2/HCO3– buffer system has potential to modify vascular biology. Indeed our findings show that multiple carbonic anhydrase isoforms are expressed in rat middle cerebral arteries where they accelerate intracellular CO2 hydration and amplify the associated transient vasoconstriction. In contrast, carbonic anhydrases have no effect on net acid extrusion across the plasma membrane during intracellular acidification and are dispensable for CO2/HCO3–-dependent attenuation of extracellular pHs fluctuations.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Lundbeck Foundation (grant no. R93-2011-8859) and the MEMBRANES research center at Aarhus University.
Authors’ contributions
EB conceived the project and wrote the manuscript. JR performed experiments. EB and JR designed experiments, analyzed and interpreted data, revised the manuscript, and approved the final version.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.