
Abstract
Blood–brain barrier (BBB) P-glycoprotein activity is rapidly reduced by vascular endothelial growth factor (VEGF) acting via Src and by tumor necrosis factor-α acting via protein kinase C (PKC)β1. To probe underlying mechanism(s), we developed an
Introduction
The blood–brain barrier (BBB) protects the central nervous system from xenobiotics in part by expression of multispecific efflux pumps including P-glycoprotein in the luminal membrane of brain capillary endothelial cells. Diminished function/expression of P-glycoprotein increases vulnerability to neurotoxicants. However, P-glycoprotein also limits brain accumulation of therapeutic drugs used to treat brain tumors, epilepsy, and HIV encephalitis; thus, reducing P-glycoprotein activity may be one way to overcome central nervous system drug resistance (Miller et al, 2008).
We have identified two distinct signaling pathways in brain capillaries that mediate acute, reversible downregulation of BBB P-glycoprotein transport activity, without changing P-glycoprotein expression. One is a ‘pro-inflammatory’ pathway involving tumor necrosis factor-α signaling through endothelin-1, nitric oxide, and protein kinase C (PKC)βI (Rigor et al, 2010). The other is a ‘proangiogenic’ pathway involving vascular endothelial growth factor (VEGF) signaling though Src kinase (Hawkins et al, 2010). Both pathways are active in isolated rat brain capillaries and in intact rats
We describe here a proteinase K (PK)-based protection assay designed to assess changes in P-glycoprotein exposure to capillary lumens
Materials and methods
All animal procedures were approved by the Animal Care and Use Committee of the National Institute of Environmental Health Sciences and conform to the guidelines and standards of the National Institutes of Health. Male Sprague-Dawley rats were obtained from Charles River (Raleigh, NC, USA) and maintained under standard conditions. Mouse anti-P-glycoprotein (C219) was purchased from Signet (Dedham, MA, USA). Mouse anti-Mrp2 was purchased from Axxora (San Diego, CA, USA). Mouse anti-Na,K-ATPase α1 was purchased from Millipore (Billerica, MA, USA). Mouse anti-β-actin, VEGF, and proteinase K were purchased from Sigma (St Louis, MO, USA). 12-deoxyphorbol-13-phenylacetate-20-acetate (dPPA) was purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA). Specific activity of PK was confirmed by a kinetic fluorimetric activity assay (Anaspec, San Jose, CA, USA).
Proteinase K Infusion, Capillary Isolation, and Immunoblot
Rats were anesthetized with ketamine cocktail and heparinized. The left common carotid artery was exposed to the bifurcation of the internal and external carotid arteries. The external carotid artery was ligated with a 3-0 silk suture, and the common carotid artery was cannulated with PE10 tubing connected to a perfusion circuit. Phosphate-buffered saline was infused by a peristaltic pump at 3.0 mL/min. The left jugular vein was cut to allow for drainage and the left cardiac ventricle was cut to stop the heart. On thorough flushing of the brain (∼1 min), a PK solution (0–60 U/mL in phosphate-buffered saline) was introduced into the circuit and perfused for 5 minutes, followed by flushing with 5 mmol/L phenylmethanesulphonyl fluoride in phosphate-buffered saline for another 5 minutes to arrest proteolysis. The brain was removed and placed into ice-cold phosphate-buffered saline. After removal of the meninges and choroid plexuses, the brain (or hemisphere ipsilateral to intracerebroventricular injection, see below) was homogenized and capillaries were isolated by density centrifugation and purification on a glass bead column as previously described (Hawkins et al, 2010).
Capillaries were snap frozen in liquid nitrogen, resuspended in lysis buffer (CelLytic MT, Sigma, St Louis, MO, USA) containing 0.1% sodium dodecyl sulfate, sonicated, and protein was extracted from the supernatants for electrophoresis and Western blot per standard protocols as previously described (Hawkins et al, 2010).
Pretreatment with Vascular Endothelial Growth Factor and 12-Deoxyphorbol-13-Phenylacetate-20-Acetate
We recently reported that both intracerebroventricular injection of VEGF and carotid infusion of the PKCβ activating phorbol ester dPPA increase brain uptake of the P-glycoprotein substrate verapamil (Hawkins et al, 2010; Rigor et al, 2010). To test whether internalization of P-glycoprotein underlies the loss of functional activity observed, we performed these treatments as previously described (Hawkins et al, 2010; Rigor et al, 2010) before infusion of PK. Briefly, rats were placed in a stereotactic frame for injection of either VEGF (500 ng in 2 μL artificial cerebrospinal fluid) or artificial cerebrospinal fluid alone into the lateral ventricle 30 minutes before PK infusion. Alternatively, rats were cannulated as described above and infused with either dPPA in mammalian Ringer or Ringer alone for 20 minutes before infusion of PK.
Results and Discussion
Regulation of P-glycoprotein at the BBB is complex, with multiple signaling pathways modifying protein expression and transport activity (Miller et al, 2008). Recent reports show that rapid loss of P-glycoprotein activity in response to tumor necrosis factor-α/PKCβI or VEGF/Src
In preliminary experiments, we infused PK into the rat brain vasculature at various concentrations and exposure times, with the goal of finding conditions under which P-glycoprotein would be cleaved but intracellular proteins would not. Infusing PK for 5 minutes caused a concentration-dependent loss of P-glycoprotein immunoreactivity without loss of β-actin (Figures 1A and 1B). Exposure times > 10 minutes and PK concentrations > 60 U/mL resulted in cleavage of β-actin, indicating proteolysis within the brain capillary endothelial cells. Immunoreactivity for a second luminal transport protein, multidrug resistance-related protein 2 (Mrp2) was also reduced by PK (Figures 1A and 1B). As with actin, immunoreactivity for Na,K-ATPase, a transporter expressed primarily at the abluminal membrane, was unchanged by PK, suggesting that PK-mediated proteolysis was limited to the luminal membrane of the capillary endothelium (Figures 1A and 1B). The lack of actin proteolysis observed was not due to an inability of PK to cleave actin, as permeabalizing the vasculature with Triton X-100 in the presence of PK (60 U/mL) decreased actin immunoreactivity at 44 kDa and induced the appearance of a lower molecular weight band, presumably a degradation product that retained the antigenic epitope (Figure 1C). Thus,
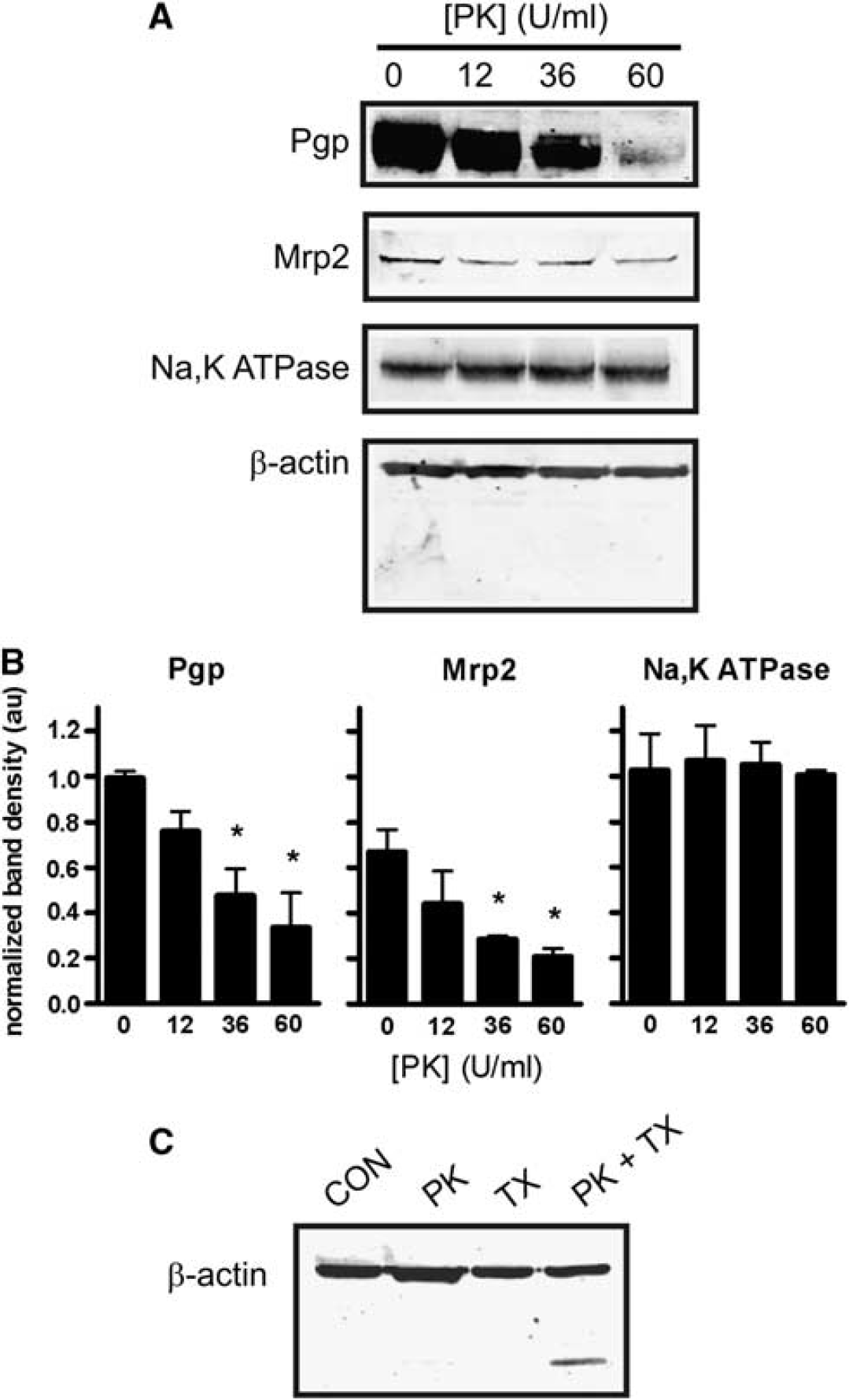
Proteinase K (PK) cleaves luminal membrane proteins
To function as an efflux transporter at the BBB, P-glycoprotein must be situated at the luminal surface of capillary endothelium. The extent to which P-glycoprotein localization is polarized in the BBB endothelium is controversial. In studies using immunofluorescence or electron microscopy, localization is reported as primarily luminal (Miller et al, 2000; Virgintino et al, 2002), and both luminal and abluminal (Bendayan et al, 2006). Roberts et al (2008) recently used
Intracerebroventrivular injection of VEGF at a dose previously shown to significantly increase net transport of P-glycoprotein substrates morphine and verapamil into the brain (Hawkins et al, 2010) protected P-glycoprotein from cleavage by PK. This was indicated by significantly increased immunoreactivity for P-glycoprotein after perfusion with PK in brain capillary lysates from VEGF-injected rats compared with those isolated from controls injected with artificial cerebrospinal fluid (Figure 2A). In a separate group of animals not perfused with PK, VEGF injection did not change total protein expression of P-glycoprotein (data not shown). The VEGF injection also did not protect Mrp2 from cleavage by PK (Figure 2B). This is an important negative control, as we previously showed that VEGF had no effect on Mrp2-mediated transport of Texas Red in isolated brain capillaries (Hawkins et al, 2010). In contrast to VEGF, carotid infusion of dPPA, also shown to increase transport of verapamil into brain (Rigor et al, 2010), did not alter P-glycoprotein immunoreactivity after infusion of PK (Figure 2C).
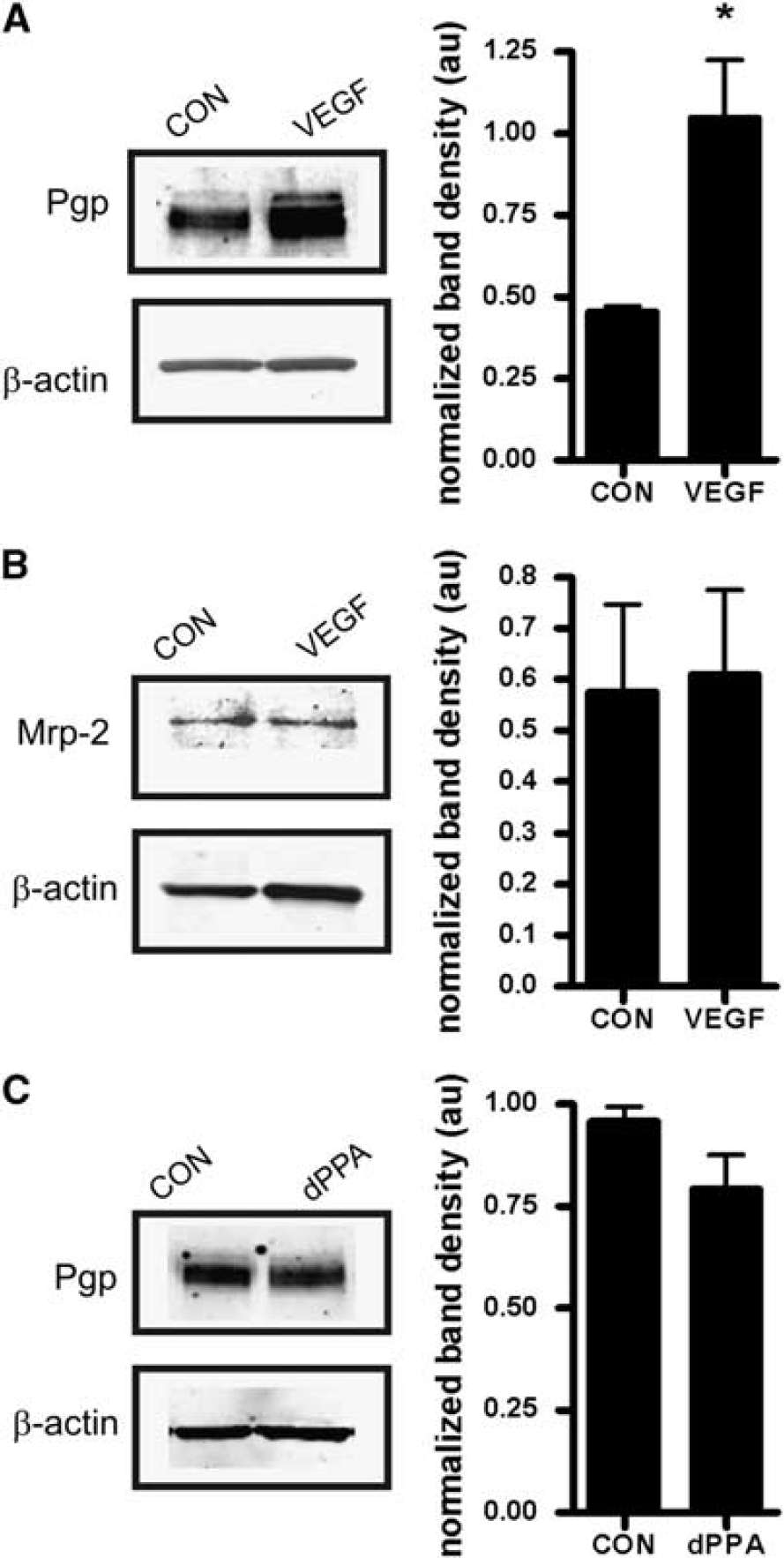
Intracerebroventricular injection of vascular endothelial growth factor (VEGF) internalizes P-glycoprotein at the blood–brain barrier (BBB). (
The results of the proteolysis protection assay are consistent with mechanistically distinct signals mediating rapid loss of P-glycoprotein activity by VEGF and dPPA. With VEGF, the increase in P-glycoprotein immunoreactivity could indicate movement of the transporter to a compartment not accessible to luminal PK, either through internalization or induction of a conformational change that protects from PK proteolysis. Our previous work indicated that Src-dependent phosphorylation of caveolin-1 accompanies rapid inhibition of P-glycoprotein in response to VEGF treatment, suggesting a role for caveolin-dependent internalization (Fagerholm et al, 2009; Hawkins et al, 2010). With dPPA, the lack of change in P-glycoprotein immunoreactivity indicates that transport activity was reduced while the transporter was still exposed to luminal PK. We have not observed phosphorylation of caveolin-1 in response to treatment with dPPA (RR Rigor, unpublished data), suggesting that a different phosphorylation-dependent pathway targets P-glycoprotein in response to tumor necrosis factor-α/PKCβI signaling.
On the basis of selective site-directed mutagenesis of P-glycoprotein, putative PKC phosphorylation sites on the P-glycoprotein molecule are not important for PKC control of transport activity (Germann et al, 1996; Goodfellow et al, 1996). Thus, P-glycoprotein inhibition by PKCβI likely involves phosphorylation of an ancillary protein. This could be achieved by recruitment of PKCβI and other binding partners to the adapter protein RBCK1, which is abundant in brain capillaries (Rigor et al, 2010).
P-glycoprotein at the BBB is a major obstacle for delivery of drugs to the brain, especially in diseases with enhanced central nervous system drug resistance. Previous attempts to inhibit P-glycoprotein pharmacologically have caused significant toxicity in combination with chemotherapeutic compounds (Friedenberg et al, 2006; Hubensack et al, 2008). Targeting signal transduction offers an alternative strategy for inhibiting P-glycoprotein selectively at the BBB. Although it is not known whether the same regulatory mechanisms control P-glycoprotein activity at the BBB as in peripheral organs responsible for drug clearance and metabolism (i.e., kidney or liver), a mechanistic understanding of P-glycoprotein regulation is critical to developing this approach to suppress central nervous system drug resistance. Our current work suggests rapid inhibition of P-glycoprotein activity by Src kinase or PKCβI occurs via distinct mechanisms, involving either putative endocytosis of the transporter or modulation within the membrane via accessory proteins, respectively. Targeting one of these processes may prove useful for clinical treatment of drug resistance, depending on desired magnitude and/or duration of P-glycoprotein inhibition.
In summary, we developed a novel
Footnotes
The authors declare no conflict of interest.