
Abstract
Mitochondria are dynamically active organelles, regulated through fission and fusion events to continuously redistribute them across axons, dendrites, and synapses of neurons to meet bioenergetics requirements and to control various functions, including cell proliferation, calcium buffering, neurotransmission, oxidative stress, and apoptosis. However, following acute or chronic injury to CNS, altered expression and function of proteins that mediate fission and fusion lead to mitochondrial dynamic imbalance. Particularly, if the fission is abnormally increased through pro-fission mediators such as Drp1, mitochondrial function will be impaired and mitochondria will become susceptible to insertion of proapototic proteins. This leads to the formation of mitochondrial transition pore, which eventually triggers apoptosis. Thus, mitochondrial dysfunction is a major promoter of neuronal death and secondary brain damage after an insult. This review discusses the implications of mitochondrial dynamic imbalance in neuronal death after acute and chronic CNS insults.
Introduction
Mitochondria are vital for cells to generate energy through oxidative phosphorylation and to regulate various physiological functions such as cell proliferation, apoptosis, neurotransmission, calcium (Ca2+) buffering, and oxidative stress.1–3 To meet the cellular bioenergetic requirements, mitochondria exist in a dynamic equilibrium between fission and fusion that also enables them to adopt to changes in the Ca2+ flux.4,5 Fission and fusion are controlled by a group of conserved GTPases of the dynamin family of proteins.5,6 While dynamin-related protein 1 (Drp1 or Dnm1l) and or fission 1 protein (Fis1) mediates fission, mitofusin 1 and 2 (Mfn1 and Mfn2) and optic atrophy 1 (Opa1) mediate fusion (Figure 1).
4
Drp1 assembles and oligomerizes on the mitochondrial surface to induce fission, whereas Fis1 promotes fission by binding and recruiting Drp1 to the mitochondrial surface.4,7 On the other hand, Mfn1 and Mfn2 regulate outer mitochondrial membrane (OMM) fusion, and Opa1 mediates inner mitochondrial membrane (IMM) fusion.
8
Mitochondrial dynamics and CNS insults. Under normal physiological conditions, mitochondria exist in a dynamic equilibrium by continuous fusion and fission events. Fission is regulated by dynamin-related protein 1 (Drp1), fission 1 protein (Fis1), and mitochondrial fission factor (Mff), whereas fusion is regulated by optic atrophy 1 (Opa1) and mitofusin (Mfn) 1 and 2. Fission leads to formation of small, rounded mitochondria, whereas fusion forms elongated, tubular interconnected mitochondrial networks. These dynamics are necessary to maintain the functionality and the location of mitochondria to meet the cell’s energy needs. CNS insults result in an imbalance of mitochondrial dynamics by shifting the balance toward fission and fragmentation leading to compromised mitochondrial membrane potential (mΨ), mitochondrial DNA (mtDNA) function, and oxidative phosphorylation.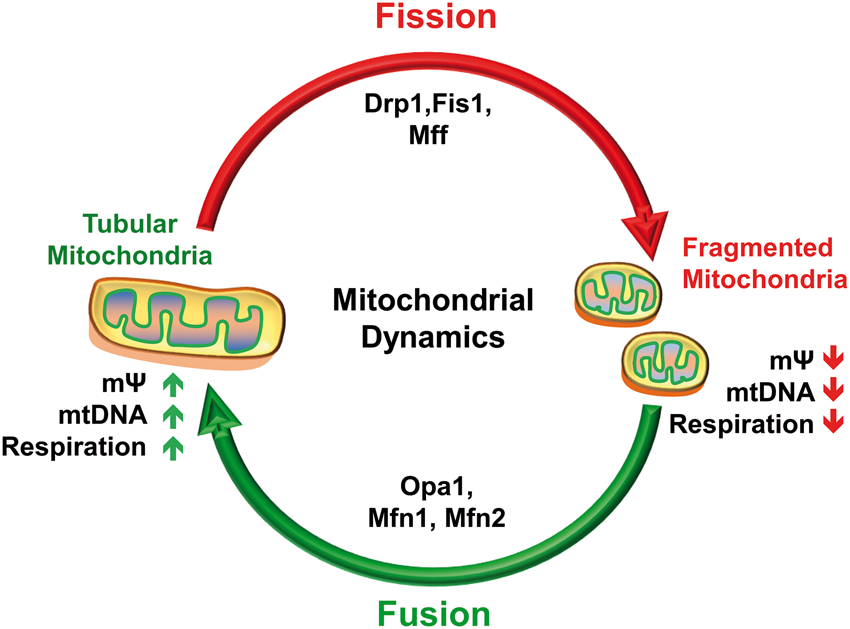
Fission is necessary to regulate the amount and the location of mitochondria, cell’s changing energetic needs and to dispose damaged mitochondria. 8 Fusion complements fission to achieve the exchange of mitochondrial DNA (mtDNA) in order to maintain mitochondrial functionality (Figure 1). 8 Unlike other cell types, neurons require continuous redistribution of mitochondria across long distances including dendrites, axons, and synapses to meet end-to-end energy requirements, Ca2+ buffering, and neurotransmission. This is achieved by dynamic regulation of mitochondria through fission and fusion events. Moreover, fission and or fusion determine the structural and functional status of mitochondria such that mitofusins or Opa1 maintain mtDNA content, membrane potential (mΨ), coenzyme Q levels, cristae shape and respiratory chain activity.9–13 These events are further supported by mitochondrial biogenesis that is co-ordinated between nuclear and mitochondrial genomes and involves replication of the mitochondrial genome followed by division of mitochondria. 14 Additionally, balance between synthesis of new mitochondria (biogenesis) and removal of aged and/or damaged mitochondria (mitophagy) regulates mitochondrial mass in the cell, whereas impaired mitophagy and or biogenesis could lead to neuropathologies. 15
Disturbances, defects, or imbalance in fission and fusion mechanisms contributes to the mitochondrial dysfunction that is linked to many acute and chronic neurological disorders including stroke, traumatic brain injury (TBI), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD).4,16–21 For instance, overexpression of Drp1 or Fis1 leads to increased fission and mitochondrial fragmentation, whereas overexpression of a dominant negative isoform of Drp1 (Drp1K38A, a GTP-binding defective mutant where an alanine is substituted with a lysine in the catalytic site) or knockdown of Fis1 prevents fission and promotes fusion leading to mitochondrial health.22–24 Likewise, mutations in fusion protein genes (Opa1 and Mfn2) lead to autosomal dominant optic atrophy and Charcot-Marie-Tooth disease type 2A.25,26 As regulating the dynamic balance between fission and fusion could restore the mitochondrial function and provide neuroprotection, 18 it is important to understand the functional significance of mitochondrial dynamics in neuronal death following pathological conditions. This review highlights the current understanding of mitochondrial fission–fusion after CNS insults.
Mitochondrial fission
Fission divides long tubular mitochondria into smaller, rounded mitochondria which is an essential physiologic function to recycle the healthy content and to remove the old, damaged content (Figure 1). However, abnormally increased fission results in mitochondrial fragmentation that contributes to neuronal death after acute and chronic CNS insults.4,17–19,21,27,28 Drp1, which is an ortholog of yeast Dnm1, is a major controller of fission in mammals.4,5,29,30 Fission is an essential process during development as Drp1 knockout mice show developmental abnormalities, particularly in the forebrain, and do not survive beyond post-embryonic day 12.5 31 . In the brain-specific Drp1 null mice, mitochondria show extensive networks, brain hypoplasia as a result of failed developmentally regulated apoptosis and die within 36 h after birth.31,32 Additionally, a rare lethal dominant-negative mutation has been reported within the middle domain of human Drp1 gene that substitutes the conserved alanine residue at position 395 with an aspartic acid residue, causes mitochondria to become unusually elongated, lose their respiratory function, and fail to distribute thereby leading to spatiotemporal deficits in ATP supply and Ca2+ signaling in high-energy demand area such as synapses.33,34 This leads to abnormalities including microcephaly, optic atrophy, hypoplasia, and persistent lactic acidemia. 33
Drp1 in humans is transcribed by a gene located on chromosome 12. 35 Drp1 protein is comprised of an N-terminal GTPase domain, a dynamin-like middle domain, a variable insert B domain, and a C-terminal GTPase-effector domain (GED). The interaction between the N-terminal GTPase domain and the C-terminal GED domain regulates the higher-order structure (dimer, tetramer and oligomer) of Drp1 which is essential for mitochondrial fission. 36 Drp1 activity is regulated by various post-translational modifications which occur mainly in the GED domain or within its close proximity. 37 Drp1 lacks lipid-interacting pleckstrin homology domain and thus its anchorage to the mitochondrial membrane is dependent on receptors that recruit Drp1 to mitochondria including Fis1, mitochondrial fission factor (Mff), and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51).7,38 While Fis1 promotes fission by recruiting Drp1 from cytosol to mitochondria,38,39 Mff recruits Drp1 to the OMM to facilitate fission.5,8,40 Mff down-regulation prevents mitochondrial recruitment of Drp1 resulting in inhibition of fission.5,8,40 Once recruited to mitochondria, Drp1 forms spiral tubules at the OMM and constrict the mitochondria using its intrinsic GTPase activity.4,29 Mdivi-1 is a BBB permeable inhibitor of Drp1 that when injected systemically achieves maximum concentration in the brain by 4 h, and has a half-life of about 12 h.41,18,20,30,41–47 Blocking Drp1 by Mdivi-1 could restore mitochondrial dynamics, improve mitochondrial function by preserving mitochondrial structural integrity, and mΨ and ATP levels. 41 Treatment with Mdivi was shown to significantly decrease the infarct volume, apoptotic neuronal death and neurological deficits following transient middle cerebral artery occlusion (MCAO) in adult mice and rats.18,41,43 Inhibition of Drp1-dependent mitochondrial fission was also shown to provide neuroptotection by activating cyclic adenosine monophosphate (cAMP), protein kinase A (PKA) and cAMP response element binding (CREB) pathway leading to increased adenosine levels and thus increased cerebral blood flow to the ischemic tissue. 41 Inhibition of Drp1 by Mdivi-1 was also shown to improve hippocampal-dependent learning and memory and decrease lesion volume in mice and rats following TBI.20,47 Taken together, these studies strongly suggest that Drp1 plays an important role in regulating neuronal death during CNS insults.
Post-translational modifications of Drp1 controls mitochondrial fission
Several post-translational modifications modulate Drp1 function.5,48 For example, phosphorylation of serine at various conserved sites (40, 44, 616, and 637) by various enzymes activates or inhibits Drp1-mediated mitochondrial fragmentation in neurons. Phosphorylation at serine 40 and serine 44 by glycogen synthase kinase 3β (GSK3β) increases the GTPase activity of Drp1 that is essential for mitochondrial fission. 49 Similarly, phosphorylation at serine 616 by extracellular signal-regulated kinase 2 (Erk2) or cyclin-dependent kinase 1, and dephosphorylation at serine 637 by calcineurin enable Drp1 to translocate from the cytosol to mitochondria to induce mitochondrial fragmentation.50,51 On the contrary, phosphorylation of Drp1 at serine 637 by PKA, and inhibition of calcineurin, or depletion of GSK3β blocks Drp1 translocation to mitochondria, suppresses the GTPase activity, prevents fission and delays apoptosis leading to neuroprotection and motor recovery after cerebral ischemia.4,52–57
Ubiquitination mediated by ubiquitin ligases including E3 (Parkin) and March5 also modulates Drp1 activity and the down-stream mitochondrial fragmentation.5,58 Parkin-induced ubiquitination of Drp1 leads to its degradation by proteasome 58 and deletion or mutation of Parkin inhibits Drp1 ubiquitination and degradation resulting in increased mitochondrial fragmentation. 58
SUMOylation, which is a ubiquitin-like post-translational modification, also modulates Drp1 activity and mitochondrial fragmentation. In mammals, SUMO is a family of small proteins comprised of four isoforms (SUMO1, 2, 3, and 4), which covalently conjugate to lysine residues in target proteins. 59 Drp1 has been shown to be SUMOylated by an ubiquitin-like conjugating enzyme 9 at multiple noncanonical sites in the B domain. 60 SUMO1 often colocalizes with Drp1 at fission sites in mitochondria undergoing fragmentation, and SUMO1 overexpression protects Drp1 from degradation leading to increased mitochondrial fragmentation in cells transfected with SUMO1.61,62 This suggests that SUMO1 conjugation to Drp1 is pro-apoptotic. Whereas, overexpression of deSUMOylating enzyme sentrin-specific protease 5 (SENP5) catalyzes the cleavage of SUMO1 from Drp1 and rescues SUMO1-Drp1-induced mitochondrial fragmentation, while silencing SENP5 induces mitochondrial fragmentation. 63 Unlike SUMO1, SUMO2/3 conjugation prevents mitochondrial translocation of Drp1 leading to decreased mitochondrial fragmentation, curtailed cytochrome C (cyto C) release, and cell death in the rat primary cortical neurons subjected to oxygen glucose deprivation (OGD). 64 SUMO2/3 conjugation is regulated by a deSUMOylating enzyme SENP3 and its expression was shown to be reduced after OGD. SENP3 unavailability thus prolongs Drp1 SUMOylation and this in turn suppresses Drp1-mediated cyto C release and caspase-mediated cell death during OGD. However, following reoxygenation, recovery of SENP3 deSUMOylates Drp1 to reverse this process, thereby allowing Drp1 mitochondrial localization and subsequent mitochondrial fragmentation and cyto C release. 64 Thus, both ubiquitination and SUMOylation modulate Drp1-mediated mitochondrial fragmentation, cyto C release, and cell death.
The GED domain of Drp1 is suggested to be important for stimulation of its GTPase activity, formation, and stability of higher-order complexes and efficient mitochondrial division. 36 Increased levels of nitric oxide (NO) lead to S-nitrosylation of cystine 644 residue of the GED domain that enhances Drp1 GTPase activity leading to excessive mitochondrial fragmentation and neuronal death in acute and chronic neurodegenerative diseases.28,65,66 In addition, NO could trigger Drp1 phosphorylation at serine 616 residue resulting in activation and recruitment of Drp1 to the mitochondria. 67 Disrupting nitrosylation by mutating the cysteine or with Drp1K38A could prevent mitochondrial fission.28,65
Mitochondrial fusion
Mitochondria fuse to form elongated tubular networks leading to recycling of the healthy mitochondrial content which plays vital roles in cell growth, maintenance of mΨ, and cellular respiration. 68 In addition, fusion allows mtDNA repair and redistribution of protein component and metabolites. Fusion is controlled by a number of proteins including Mfn1, Mfn2, and Opa1, and involves tethering and fusion of the OMM, docking, and fusion of the IMM, and mixing of intramitochondrial components including mtDNA (Figure 1).
OMM fusion
The outer membrane of a mitochondria can fuse together with the outer membrane of another mitochondria and this fusion is regulated by the GTPases Mfn1 and Mfn2.4,29,69 Mfn1 and Mfn2 are attached to the outer membrane in a way that their C-terminal coiled domains and N-terminal GTPase domain face outward from mitochondria and toward the cytosol.5,8 These mechanisms create an intracellular bridge that connects mitochondria with each other. 4 Deletion and/or inhibition of Mfn1 and Mfn2 results in failure of fusion leading to formation of very small mitochondrial fragments with reduced mass, and decreased transport of mitochondrial component proteins leading to neurodegeneration.19,70–73 Deletion of Mfn1 leads to no change in phenotype up to one year of age, whereas deletion of Mfn2 in a subset of dopaminergic neurons leads to hunched and hypoactive mice by 5 weeks of age. 73 Furthermore, loss of Mfn2 results in mitochondrial fragmentation and severe age-dependent motor deficits that precede the loss of dopaminergic terminals in the striatum. 73 These perturbations in mitochondrial dynamics is a risk factor for the neurodegeneration in PD. 73 On the contrary, overexpression of Mfn1 and Mfn2 results in suppression of mitochondrial fragmentation and cyto C release leading to reduced Bax activation and decreased apoptosis. 19
IMM fusion
The IMM fusion is mediated by the GTPase Opa1, which is autologous to yeast Mgm1. 29 As Mgm1 forms oligomers that are important for tethering and fusion of the inner membranes, mitochondria deficient in Mgm1 could only fuse their outer membranes. 69 In mammals, oligomerization of Opa1 restricts release of mitochondrial proteins including cyto C from the inter-membrane space.74–77 In contrast, loss of Opa1 could compromise the structural and functional integrity of IMM and could induce disorganization of cristae, loss of mΨ, cyto C release, mitochondrial fragmentation, and induction of apoptosis even without any other stimulus. 77 These effects can be seen in the dominant optic atrophy disease where mutation in Opa1 gene causes the degeneration of the retinal ganglion cells.26,76,78,79 At least 8 splice variants of Opa1 are formed in humans by differential splicing from 30 exons on chromosome 3. 80 Opa1 protein contains a GTPase domain, a middle domain, and a GED domain. The amino-terminal region preceding the GTPase domain of Opa1 carries a mitochondrial import sequence (MIS) and a transmembrane helix near MIS anchors Opa1 to the IMM.4,81 Various proteases including rhomboid protease presenilin-associated rhomboid-like, m-AAA protease paraplegin and i-AAA protease Yme1L process Opa1 to generate long (L-Opa1) and short (S-Opa1) isoforms. It has been shown that the long isoform together with one or more short isoforms, but not alone, mediates mitochondrial fusion.82,83 The loss of mΨ, but not the inhibition of oxidative phosphorylation or glycolysis destabilizes the long isoforms leading to processing of L-Opa1 to S-Opa1 and decreased mitochondrial fusion.83,84 Furthermore, processing of Opa1 is accelerated under reduced electrochemical potential across the IMM.29,69
Mitochondrial fragmentation and apoptosis
The fission-induced mitochondrial fragmentation by Drp1 plays an important role in release of various pro-apoptotic proteins including cyto C during intrinsic apoptosis (Figure 2).1,4,19,85,86 Bax, one of the proapoptotic Bcl-2 family proteins, translocates from cytosol to mitochondria and targets OMM during apoptosis.
87
Interestingly, Drp1 is required for translocation of Bax to mitochondria, whereas Drp1 mitochondrial translocation and oligomerization did not require Bax.
88
During apoptosis, Bax and Bak (proapoptotic Bcl-2 family of proteins) stabilize Drp1 in mitochondrial membrane by stimulating SUMO1 conjugation of Drp1.
62
Moreover, Drp1-dependent mitochondrial fragmentation sensitizes mitochondrial membrane to Bax insertion and activation during apoptosis, whereas Drp1K38A mutant or siRNA-mediated knockdown of Drp1 blocks Bax activation, insertion, and oligomerization in mitochondria.
19
Bax translocation from cytosol to mitochondria has been strongly linked with mitochondrial release of cyto C and caspase-9 after transient focal ischemia.
87
Drp1 is transcriptionally regulated by p53, which upregulates the p53 up-regulated modulator of apoptosis (PUMA) that induces the binding of Bax and Bak to the OMM.89,90 Hence, blocking p53 prevents mitochondrial fission and cellular apoptosis during oxidative stress in cells exposed to hydrogen peroxide.
89
Moreover, Drp1-mediated mitochondrial fragmentation promotes Bax insertion in the OMM and downregulation of Drp1 decreases this phenomenon resulting in a less-permeable membrane.
19
Furthermore, Bax/Bak-mediated SUMOylation of Drp1 was also linked to Drp1-mediated apoptosis and mitochondrial fission in cells treated with staurosporine (STS).5,29,30,62 A Drp1K38A inhibits cyto C release and apoptosis in cells treated with STS.
91
These studies define the relationship of apoptosis and mitochondrial fragmentation.
CNS insults promote mitochondrial fragmentation. Following a CNS insult, p53 activates dynamin-related protein 1 (Drp1) transcription. Drp1 is regulated by various post-translational modifications including phosphorylation by glycogen synthase kinase 3β (GSK3β), extracellular signal-regulated kinase 2 (Erk2), and calcineurin. These modifications induce translocation of Drp1 to the outer mitochondrial membrane (OMM) and promotes mitochondrial fragmentation in association with fission 1 protein (Fis1). Drp1-dependent fragmentation also sensitizes mitochondrial membrane to Bax/Bak insertion leading to the formation of membrane transition pore through which various pro-apoptotic proteins such as cytochrome C (cyto C) and apotosis inducing factor (AIF) are released. Once in the cytosol (cyto C) or nucleus (AIF), these proteins activate apoptotic pathways resulting in neuronal death. Drp1-dependent mitochondrial fragmentation and apoptosis can be prevented by pharmacological inhibitors such as Mdivi-1 or overexpression of proteins such as PTEN-induced putative kinase 1 (PINK1) or DJ1 to induce neuroprotection.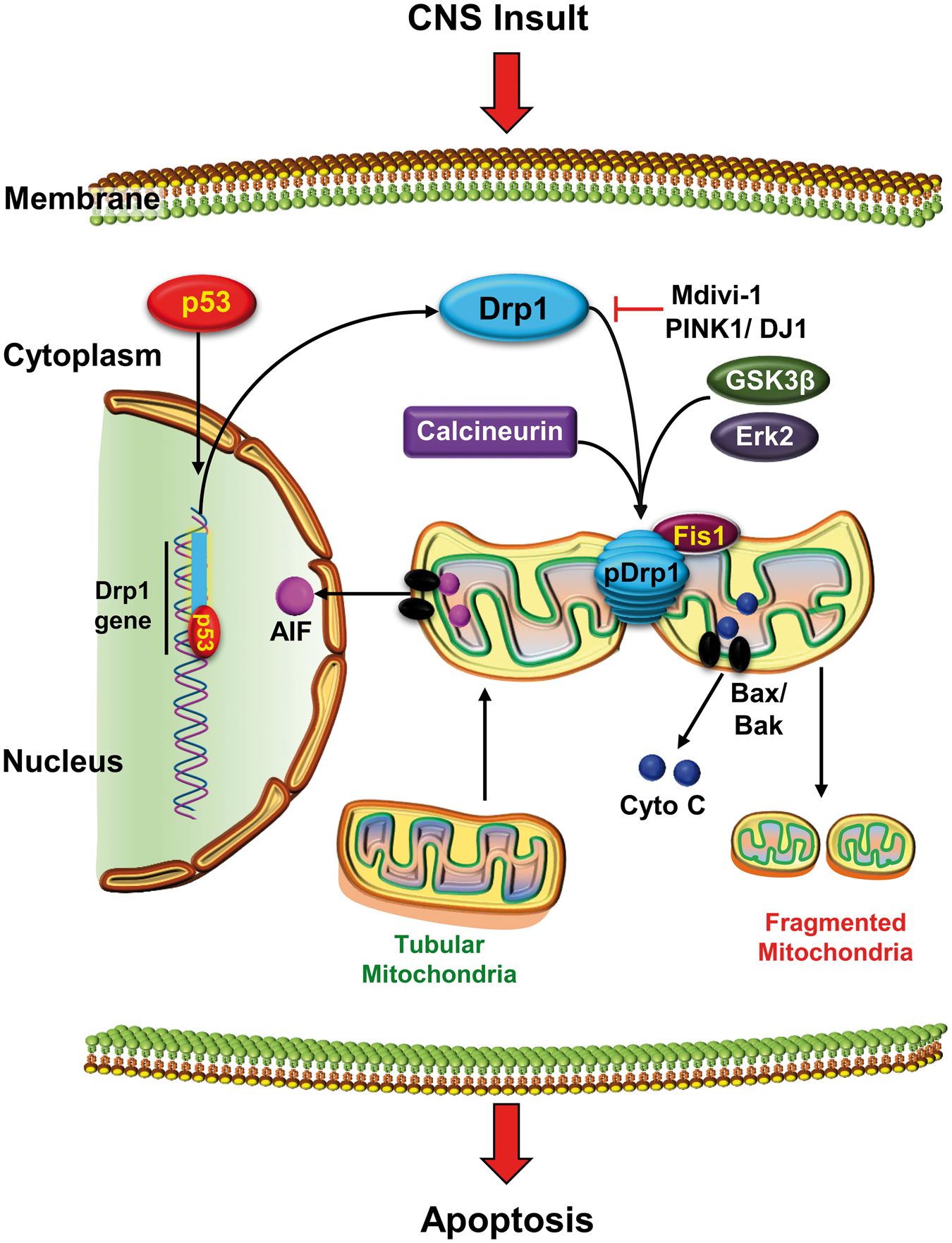
Mitochondrial fragmentation and mitophagy
Fission–fusion regulates cellular homeostasis by maintaining the quality of mitochondria. Mitophagy plays an important role in this quality control. Particularly mitophagy follows mitochondrial fragmention after CNS insults which results in lowered mΨ, 92 loss of Opa1 and Mfn1 and Mfn2, 93 and colocalization with autophagy promoter LC3.94,95 Interestingly, overexpression of Opa1, Drp1K38A, or silencing of Fis1 decreases mitophagy, suggesting that mitochondrial fragmentation is essential for mitophagy. 92 Conversely, elongated mitochondria resulting from fusion are spared from mitophagic degradation,96,97 as seen in starvation-induced autophagy where mitochondrial network remains intact. 97 Interestingly, these mitochondria possess more cristae, higher ATP synthase activity and hence will delay apoptosis, and thereby offer protective function.96,97
Mitochondrial fragmentation after stroke
Fission and fusion markers are altered after various CNS insults.

Note: The mitochondrial dynamic balance is tilted toward fission leading to mitochondrial fragmentation by differential regulation of fission (Drp1, Fis1, and Mff) and fusion (Mfn1, Mfn2, and Opa1) proteins. Mitochondrial fragmentation is further regulated by proteins including DJ1, PINK1, α-Syn, Tau, and Aβ. Drp1: dynamin-related protein 1; Fis1: fission 1 protein; Mff: mitochondrial fission factor; Mfn1, Mfn2: mitofusin 1 and 2; Opa1: optic atrophy 1; PINK1: PTEN-induced putative kinase 1; α-Syn: α-Synuclein; Aβ: amyloid β; AD: Alzheimer's disease; PD: Parkinson's disease; HD: Huntington's disease.
There are several proteins that influence Drp1 function including PTEN-induced putative kinase 1 (PINK1) and α-Synuclein (α-Syn). Of these, PINK1 has been shown to protect neurons from ischemic brain damage by regulating the translocation of Drp1 to the mitochondria following stroke, 6 whereas α-Syn shifts the equilibrium toward fission causing mitochondrial damage, 104 and hence studies showed that silencing α-Syn prevents Drp1 phosphorylation and decreases infarct volume leading to better functional recovery after transient focal ischemia. 105 Mitochondrial fragmentation might lead to neuronal damage by either Drp1-mediated translocation of Bax to mitochondria 88 or sensitization to Bax/Bak insertion into the OMM and activation of apoptosis.19,88,106–108 Drp1-dependent apoptotic mitochondrial fission has been suggested to occur prior to physical fragmentation of mitochondria.30,91 Fragmented mitochondria also produce reactive oxygen species that are toxic to neurons.30,109–112 Therefore, Drp1 not only increases apoptosis, but also induces oxidative stress after stroke.
Excitotoxicity is an early event after stroke that initiates neuronal death. Excitotoxicity has been shown to down-regulate Mfn2 (a protein of the mitochondrial fusion machinery) 113 and further increase Bax translocation to mitochondria leading to disruption of calcium homeostasis. Mitochondrial dysfunction and increased cell death resulted in Mfn2-deficient neurons. 113 On the contrary, overexpression of Mfn2 blocks mitochondrial fragmentation, and neuronal death during glutamate-induced excitotoxicity. 114 Interestingly, exercise preconditioning increases the expression of Opa1 which promotes fusion, and thus decreases brain edema and provides neuroprotection after cerebral ischemia. 115 Thus, mitochondrial fission increases and fusion decreases secondary brain damage after cerebral ischemia.
Mitochondrial fragmentation and neurodegenerative diseases
Mitochondrial dysfunction and oxidative stress are thought to be the early triggers of AD pathophysiology. 116 Studies also show that oxidative stress occurs much before amyloid-beta (Aβ) deposition, formation of plaques and neurofibrillary tangles, which are the hallmarks of AD. 117 Moreover, Aβ-mediated neurotoxicity was reported to be mediated in part through fragmented mitochondria. 49 In AD, mitochondrial fragmentation was shown to occur due to increased expression of Drp1 and Fis1, and decreased levels of Mfn1, Mfn2, and Opa1 (Table 1). 24 As discussed earlier, phosphorylation of Drp1 plays an important role in regulating the mitochondrial fragmentation.49,118 GSK3β, which phosphorylates Drp1 at serine 40 and serine 44, was shown to increase Drp1 GTPase activity and its mitochondrial distribution as well as induce mitochondrial fragmentation in a mouse model of AD. 49 Whereas, blocking GSK3β-induced Drp1 phosphorylation could restore mitochondrial length, lower caspase-3 activity, and improve memory performance in the transgenic APP/PS1 mouse model of AD. 49 Moreover, preventing Drp1 phoshprylation also reduces the levels of Aβ in APP/PS1 mice. 49 Interestingly, it was evidenced that Aβ and phosphorylated–tau (p-tau) strongly interact with Drp1 leading to increased neuronal mitochondrial fragmentation and impaired axonal mitochondria transport in AD.24,65,119 Although the exact mechanism of action of Aβ and Drp1 complex is not known, nitrosylatation of Drp1 by Aβ was thought to contribute to mitochondrial fragmentation. 65 Drp1 nitrosylation is dependent on the levels of NO formed by neuronal NO synthase (nNOS) and Aβ is known to increase the activity of nNOS. 120 Cerebral nitrosylated Drp1 levels were also shown to be increased in a transgenic AD mouse model (Tg2576) as well as in AD patients. 65 Similarly, p-tau was observed to interact abnormally with Drp1 in the brains from patients with severe AD. 119 GTPase activity of Drp1 also elevates at various stages (early, definite and severe) in AD patients as well as in AD mouse model. 119 Moreover, it was suggested that tau could regulate the transport of mitochondria and potentially impair synapses in AD. 121 All these studies indicate that reducing the levels of Drp1, Aβ and/or p-tau can protect brain after AD. 119 In HD, mutated Huntingtin protein (mtHtt) was shown to interact with Drp1 leading to impaired mitochondrial biogenesis and defective axonal transport and synaptic degeneration. 122 Hence, inhibition of Drp1 prevents mitochondrial fission and improves mitochondrial function/survival, and axonal transport in HD.122,123
Mitochondrial dysfunction due to impairment of mitochondrial complex I has been suggested to be a major contributor that intitates the onset of PD. 124 Mitochondrial dysfunction is mediated by many proteins associated with PD including Parkin, PINK1, DJ1, and α-Syn that modulate neuronal degeneration.125,126 Following mitochondrial depolarization, Drp1 and Parkin are co-recruited to mitochondria in proximity of PINK1 127 and Parkin prevents Drp1-induced mitochondrial fission.6,125 Hence, loss/silencing of Parkin and PINK1 lead to mitochondrial fragmentation and early onset of PD.128–131 Furthermore, downregulation of PINK1 leading to altered mitochondrial fusion–fission balance is known to sensitize dopaminergic cells to neurotoxins. 132 In animal models of PD, Drp1 inhibition was shown to attenuate neurotoxicity and restore striatal dopamine release. 133 DJ1 was also shown to have neuroprotective effects in mitochondrial pathology seen in PD 134 by controlling redox signaling and acting as a transcriptional regulator to induce antioxidative genes. 135 In mice, DJ1 deficiency results in nigrostriatal dopaminergic deficits, hypokinesia, sensitivity to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) toxicity, and oxidative stress, whereas its overexpression protects cell from oxidative stress and MPTP-induced nigral dopamine neuron loss.136–139 Interestingly, DJ1 has been shown to translocate to the mitochondria in such conditions and thereby plays an important role in regulating mitochondrial fragmentation and neuronal loss.139,140 Moreover, mitochondrial fragmentation induced by PD-associated DJ1 mutation could be prevented by either silencing Drp1 or overexpressing DJ1, which potentially indicates that DJ1 mutations may cause PD by impairing mitochondrial dynamics and function. 141
α-Syn is a protein known to be the hallmark of Lewy pathology in PD due to its misfolding and aggregation in perikarya, dendrites, and axons of dopaminergic neurons in the substantia nigra pars compacta of the CNS. 142 α-Syn is reported to localize with mitochondria due to its high affinity to mitochondrion-specific acidic phospholipids and cardiolipin. 143 Mitochondrial localization of α-Syn is directly linked to mitochondrial stress and oxidative stress. 144 Moreover, increased α-Syn is shown to promote mitochondrial fragmentation by shifting the dynamic equilibrium toward fission 145 leading to neuronal death. 146 A mutation in α-Syn (A53T) that causes familial PD has been shown to affect mitochondrial dynamics by reducing interconnectivity and increasing circularity of mitochondria in an age-dependent manner, thereby leading to neurodegeneration in an animal model of PD. 147 The above changes in mitochondria are also in part due to age-dependent reduction in the levels of Mfn1 and Mfn2 in A53T α-Syn mice. 147 Moreover, α-Syn has been shown to promote translocation of Drp1 to mitochondria, and Drp1 is required for α-Syn-induced mitochondrial fragmentation. 145 Taken together, all the evidence suggests that mitochondrial fragmentation is a major contributor in initiation and progression of neurodegenerative diseases.
Conclusion
Mitochondrial dynamic imbalance regulates the progression and outcome of neuronal dysfunction in many neurological disorders. This occurs when balance between fission and fusion is shifted toward fission partly due to Drp1 activation leading to mitochondrial fragmentation, energy failure, and activation of cell death pathways. Preventing mitochondrial dynamic imbalance could restore the equilibrium between fission and fusion leading to neuroprotection.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by NIH grants NS082957, NS083007, NS095192 and VA Merit Review Grant I01BX002985.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JB and SLM contributed equally to this work. RV coordinated the manuscript-writing and design.