
Abstract
In this study, we investigated the role of vitamin D3 (VitD3) on endogenous osteopontin (OPN), a neuroprotective glycoprotein, after subarachnoid hemorrhage (SAH). The endovascular perforation SAH model in Sprague-Dawley rats was used to study the effect of intranasal VitD3 (30 ng/kg) before (Pre-SAH + VitD3) and after (Post-SAH + VitD3) subarachnoid hemorrhage. Vitamin D3 (30, 60, 120 ng/kg/day) increased more than one fold endogenous OPN expression in astrocytes and endothelial cells of rat brain. Vitamin D3 significantly decreased brain edema and Evans blue extravasation. In addition, neurobehavioral scores were significantly higher in Pre-SAH + VitD3, but partly higher in Post-SAH + VitD3, group compared with SAH group. These protective effects of vitamin D3 were completely attenuated by intracerebroventricular injection of transcription inhibitor Actinomycin D and significantly inhibited by small interfering ribonucleic acid (siRNA) for vitamin D receptor and OPN in Pre-SAH + VitD3 rats. OPN expression was significantly higher in Pre-SAH + VitD3 rats, specifically A and C, but not B, isomers were upregulated in the astrocytes, leading to CD44 splicing, and P-gp glycosylation in brain endothelial cells. The results show that intranasal vitamin D3 attenuates blood–brain barrier (BBB) disruption through endogenous upregulation of OPN and subsequent CD44 and P-gp glycosylation signals in brain endothelial cells. Furthermore, this study identifies a novel strategy for the cost-effective management of subarachnoid hemorrhage.
Introduction
SAH accounts for up to 5–7% of all stroke cases 1 and has a high rate of mortality. Approximately 15% of SAH patients die before hospital admission and the 30-day mortality is 40–50%. 2 In the past, cerebral vasospasm was the primary focus of therapeutics for SAH, yet mortality and clinical outcomes after SAH remain considerably high,3,4 thus new concepts in the treatment of SAH patients are needed. 5 In recent years, efforts have been directed at elucidating the mechanisms of early brain injury after SAH. 6
OPN is a multifunctional extracellular and intracellular glycoprotein with known protective effects for ischemic injuries involving the brain and other organs.7–9 OPN consists of about 300 amino acids and can be post-translationally modified and has three isoforms that are differentially expressed in various tissues and subcellular locations. 7 Our previous studies have showed that OPN has neuroprotective effects for SAH in rats.10,11 Cerebral OPN is primarily expressed by astrocytes and endothelial cells after SAH. 12 Interestingly, some studies have suggested that OPN is upregulated by vitamin D in vitro and vivo.13–15
Vitamin D is a neuroprotective hormone which activates its vitamin D receptor (VDR), a nuclear transcription receptor, leading to activation transcription number of downstream proteins, including OPN. Increasing evidence from large epidemiological and basic science studies, suggests that vitamin D deficiency is a risk factor for neurological and cerebrovascular diseases, as well as stroke.16–18 Previous studies have also indicated that poor vitamin D levels are associated with both an increased risk of strokes and adverse outcomes for post-stroke patients. 19 To date, evidence for the use of vitamin D for stroke treatment is lacking and few studies have been performed in vivo.20,21 The molecular mechanisms of vitamin D signaling lead to a number of genomic and non-genomic pathways.22,23 However, the neuroprotective effects of vitamin D still have not been elucidated after SAH and might involve the beneficial effects of OPN. In the current study using a translational approach, we aimed to identify the role of intranasal administration of vitamin D on endogenous OPN expression for treatment of early brain injury after SAH in rats.
Materials and methods
This report is conducted according to the NIH guidelines for the use of animals and ARRIVE guidelines.
All experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee of Loma Linda University, are in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines, and comply with the NIH Guidelines for the Use of Animals in Neuroscience Research.
Animals and treatment
Adult male Sprague-Dawley rats (n = 279, 300–350 g; Harlan Indianapolis, IN) were housed in a light-and-temperature-controlled environment and fed ad libitum. Naïve rats were given 30, 60, and 120 ng/kg VitD3 (calcitriol, 71820, Cayman Chemical, MI, USA) by intranasal route to study the effect of VitD3 on endogenous expression of OPN in the brain (Supplementary Figure 1(a), n = 8 per group). Next, animals were divided randomly into the following groups: Sham operated plus vehicle (5% ethanol) (Sham), subarachnoid hemorrhage plus vehicle (SAH), SAH plus 30 ng/kg of VitD3 pretreatment (24 h before surgery, Pre-SAH + VitD3), and SAH plus 30 ng/kg of VitD3 post-treatment (1 h after surgery, Post-SAH + VitD3) (Supplementary Figure 1(b); n = 28 per group). Finally, to examine the molecular mechanism, 50 animals were divided randomly into the following groups: Pre-SAH + VitD3 + Vehicle (n = 5), Pre-SAH + VitD3 + Actinomycin (n = 5), Pre-SAH + VitD3 + Control siRNA (n = 11), Pre-SAH + VitD3 + VDR siRNA (n = 6), Pre-SAH + VitD3 + OPN siRNA (n = 11) (Supplementary Figure 1(c)). Of the 279 total animals, 187 rats were subjected to SAH, with 23 of 134 (16.5%) animals died within 24 h and 10 of 43 (23.3%) animals died between 24 and 72 h after SAH (Supplementary Figure 4(a)). All animals were sacrificed at either 24 h or 72 h after surgery.
Surgery procedures
The endovascular perforation model of SAH was performed as previously described.24,25 Briefly, rats were anesthetized and kept on a ventilator during surgery with 2–3% isoflurane in air (isoflurane was reduced to 1.5% at the moment of puncture). A sharpened 4–0 nylon suture was inserted into the left internal carotid artery through the external carotid artery and common carotid bifurcation. The 3 cm suture was advanced until resistance was felt at the bifurcation of the anterior and middle cerebral arteries. The suture was then further advanced to puncture the vessel and then immediately withdrawn after artery perforation. During sham operations, the suture was inserted into the left carotid artery; however, no perforation was performed. After removal of the suture, the skin incision was sutured, and rats were individually housed in heated cages until recovery.
Neurological score evaluation
At 24 h and 72 h after surgery, neurological scores were evaluated in a blinded fashion, using a modification of the Garcia scoring system as previously described. 26 This composite sensorimotor assessment evaluated the rodent’s spontaneous activity, spontaneous movement of all limbs, vibrissae touch, forelimbs outstretching, and climbing wall of cage. Each subtest was scored from 0 (minimum) to 3 (maximum). Animals were also evaluated for walking distances on a wooden beam for 1 min and wire hanging ability for 30 s, each scored from 0–4.
SAH grading
The severity of SAH was blindly evaluated by the SAH grading system immediately after euthanasia as previously described. 24 Briefly, after removing the brain from the skull, a photograph of the base of the brain was divided into six parts that were scored (0–3) according the amount of subarachnoid blood present, and a total score was calculated as the sum of each sections score. Operated animals received a total score ranging from 0 (no SAH) to 18 (most severe SAH), and SAH rats with a score of 7 or less were excluded from this study (Supplemental Table 1).
Brain water content analysis
Brains were collected at 24 h and 72 h after surgery and separated into left hemisphere, right hemisphere, cerebellum, and brain stem as previously described. 12 Each part was weighed immediately after removal (wet weight) and then dried at 100℃ for 72 h (dry weight). The percentage of brain water content was calculated as [(wet weight − dry weight)/wet weight] × 100%.
BBB disruption detection
At 24 h after surgery, Evans blue dye (2%; 5 mL/kg) was injected into the right femoral vein, over a period of 2 min, allowing the dye to circulate for a total of 60 min as previously described. 12 Under deep isoflurane anesthesia, rats were subjected to transcardial perfusion with 120 mL phosphate-buffered saline (PBS) through the upper part of the body, and brains were removed. The brains were then divided into left hemisphere, right hemisphere, cerebellum, and brain stem. Brain specimens were weighed, homogenized in PBS (1 mL PBS for each 300 mg of tissue), and then centrifuged at 14,000 r/min for 30 min; 0.5 mL of the supernatant was added to an equal volume of trichloroacetic acid (T0699, Sigma). After overnight incubation at 4℃ and centrifugation at 14,000 r/min at 4℃ for 30 min, 0.8 mL of the supernatant was used for spectrophotometric quantification of extravasated Evans blue dye at 610 nm.
Intracerebroventricular administration
Intracerebroventricular (i.c.v) drug administration was performed as previously described. 27 Briefly, rats were placed in a stereotaxic apparatus under 2.5% isoflurane anesthesia. The needle of a 10 μL Hamilton syringe (Microliter 701; Hamilton Company, Reno, NV) was inserted through a burr hole into the right lateral ventricles at the following coordinates relative to bregma: 0.92 mm posterior, 1.5 mm lateral, and 3.3 mm below the horizontal plane of the skull. A solution of Actinomycin D was prepared in dimethyl sulfoxide (DMSO) and diluted at 6.8 µg/μL in sterile 0.9% NaCl before administration; 10 μL of the Actinomycin D solution (68 µg) was injected 24 h before SAH induction surgery at a rate of 2 μL/min. Vehicle control animal received 10 μL of 0.9% NaCl including equal amount DMSO. Ten μL of the VDR siRNA (SR508804, OriGene Technologies, Inc., MD, USA), OPN siRNA Sigma-Aldrich, MO, USA), 13 and a scrambled, negative control siRNA were prepared at 10 μM final concentration each in RNAse free resuspension buffer by company instruction and was injected 48 h before SAH induction surgery at a rate of 2 μL/min.
Western blot
Western blot analyses were done as previously described.10–12 Briefly, equal amounts of sample protein (50 µg) were loaded onto an SDS-PAGE gel. After electrophoresis and transfer of the samples to a nitrocellulose membrane, the membrane was blocked and incubated with the primary antibody overnight at 4℃ with the following primary antibodies: VDR (1:500, sc-13133), OPN (1:1000, sc-21742), β-Actin (1:5000, I-19) (Santa Cruz Biotechnology Inc., Texas, USA), and P-glucoprotein (P-gp) antibody (1:2000, #13978, Cell signaling, Boston, MA, USA); CD44 antibody (1:2000, ab119348, Abcam, MA, USA). Following that, the membranes were incubated at room temperature for 1 h with the appropriate secondary antibodies (1:5000, Santa Cruz Biotechnology Inc.). Immunoblots were then probed with an ECL Amersham Western blotting detection reagents (Amersham Biosciences UK Ltd, PA, USA). Blot bands were quantified by densitometry using image J software (Image J 1.4, NIH, USA). β-Actin was used as loading control.
Immunofluorescence staining
Deeply anesthetized rats were transcardially perfused with 60 mL of PBS followed by 60 mL of 10% paraformaldehyde through the upper part of the body. The brains were removed, and then fixed for 24 h in 10% paraformaldehyde followed by 30% sucrose for three days, and then embedded into OCT compound (Scigen Scientific Gardena, CA, USA) and frozen at −80° until use. The tissues were cut into 10 µm thick slices at the level of bregma on a cryostat (LM3050S; Leica Microsystems, Bannockburn, III, Germany) and mounted on poly-L-lysine-coated glass slides. Double-fluorescence labeling was performed as described previously. 28 Briefly, anti-CD34 (1:100, 250591, Abbiotec, San Diego, CA), anti-CD31 (1:100, ab119339, abcam, MA, USA) anti-GFAP (1:500, ab33922, ab10062), anti-NeuN (1:500, ab104224), anti-P-gp (1:500, #13978, Cell signaling, Boston, MA, USA), and anti-OPN (1:100, sc-21742, Santa Cruz) antibodies were applied and incubated overnight at 4℃, followed by incubation with secondary antibodies 1 h at room temperature at a dilution of 1:500. The slides were viewed and pictures taken in LAS X software with a fluorescence microscope Leica DMi8 (Leica Microsystems, Germany).
Quantitative real-time polymerase chain reaction
Total ribonucleic acid (RNA) was extracted using the RNeasy kit (Qiagen) from 30 mg of brain tissue at the level of bregma. cDNA synthesis was carried out using GoScript Reverse Transcriptase (Promega, Madison, USA); 1 µg of total RNA was used for synthesis of cDNA. OPN splice variants were amplified with a specific forward primer 5-GTT TGG CAG CTC AGA GGA GA-3 and reverse primer 5-CCG AGT TCA CAG AAT CCT CG-3 which can produce OPN three isomers as shown in Figure 5(b). VDR and housekeeping gene β-Actin were amplified with forward primers as shown in Supplementary Table 2. Quantitative real-time polymerase chain reaction (qRT-PCR) reactions were conducted using the SYBR Green detection reagent in Bio-Rad iQ5 system (Hercules, California). Conditions for OPN isoforms qRT-PCR amplification were 50℃ for 2 min, 94℃ for 5 min followed by 40 cycles of 94℃ for 30 s, 58℃ (54℃ for VDR; 56℃ for β-Actin) for 30 s, and 72℃ for 90 s; and 72℃ for 15 min and finally a melting curve analysis (60–90℃ with a heating rate of 0.2 C and continuous fluorescence measurement) as described previously. 29
Statistical analysis
Data analysis was performed by using IBM SPSS Statistics software version 21 (IBM, Armonk, New York, USA). Data are shown as mean ± SD and analyzed by one-way ANOVA followed by LSD post hoc analysis for comparison among all groups. Independent Samples T-Test was used to compare two groups. A P value of < 0.05 was considered statistically significant.
Results
Intranasal administration of VitD3 increases endogenous OPN expression in the brain
Western blot analysis showed that a single intranasal administration of VitD3 (30, 60, and 120 ng/kg/day) significantly increased endogenous OPN expression in both hemispheres after 24 h (Figure 1(a)). In the left hemisphere of the brain, the expression of OPN was remained significantly higher after 48 and 72 h treatment of 30 ng/kg VitD3 (Figure 1(b)). After the treatments, body weight was significantly decreased in the 30 and 60 ng/kg treatment groups compared with naïve animals at 48 h, but body weight recovered in the 30 ng/kg treatment group by 72 h. OPN expression did not change significantly for any studied dose and time in the kidneys (Supplementary Figure 2). OPN expression was increased in the astrocytes and endothelial cells after intranasal treatment of 30 ng/kg/day VitD3 (Figure 1(c) and (d) and Supplementary Figure 3).
Intranasal administration of vitamin D3 (VitD3) upregulates endogenous osteopontin (OPN). (a) Representative Western blots of VitD3-induced dose-dependent (30, 60, 120 ng/kg/day) expression of OPN (upper panel) and quantitative analysis (lower panel) 24 h after intranasal administration in brain hemispheres of naïve rats (n = 5 per group). (b) Representative Western blots of time-dependent expression of OPN (upper panel) and quantitative analysis (lower panel) after intranasal administration of 30 ng/kg/h VitD3 in left hemisphere of naïve rats (n = 5 per group). *P < 0.05 vs. naïve rats. (c) Immunohistochemical staining of OPN (red) and astrocytes (GFAP, green) (n = 3 per group). Nuclei are stained with DAPI (blue). Arrows indicate colocalization of OPN and GFAP. Top panel indicates the location of immunohistochemical staining (small black box). Bar = 50 µm. (d) Immunohistochemical staining of OPN (red) and endothelial cells (CD34, green) (n = 3 per group). Nuclei are stained with DAPI (blue). Arrows indicate colocalization of OPN and CD34. Top panel indicates the location of immunohistochemical staining (small black box). Bar = 50 µm.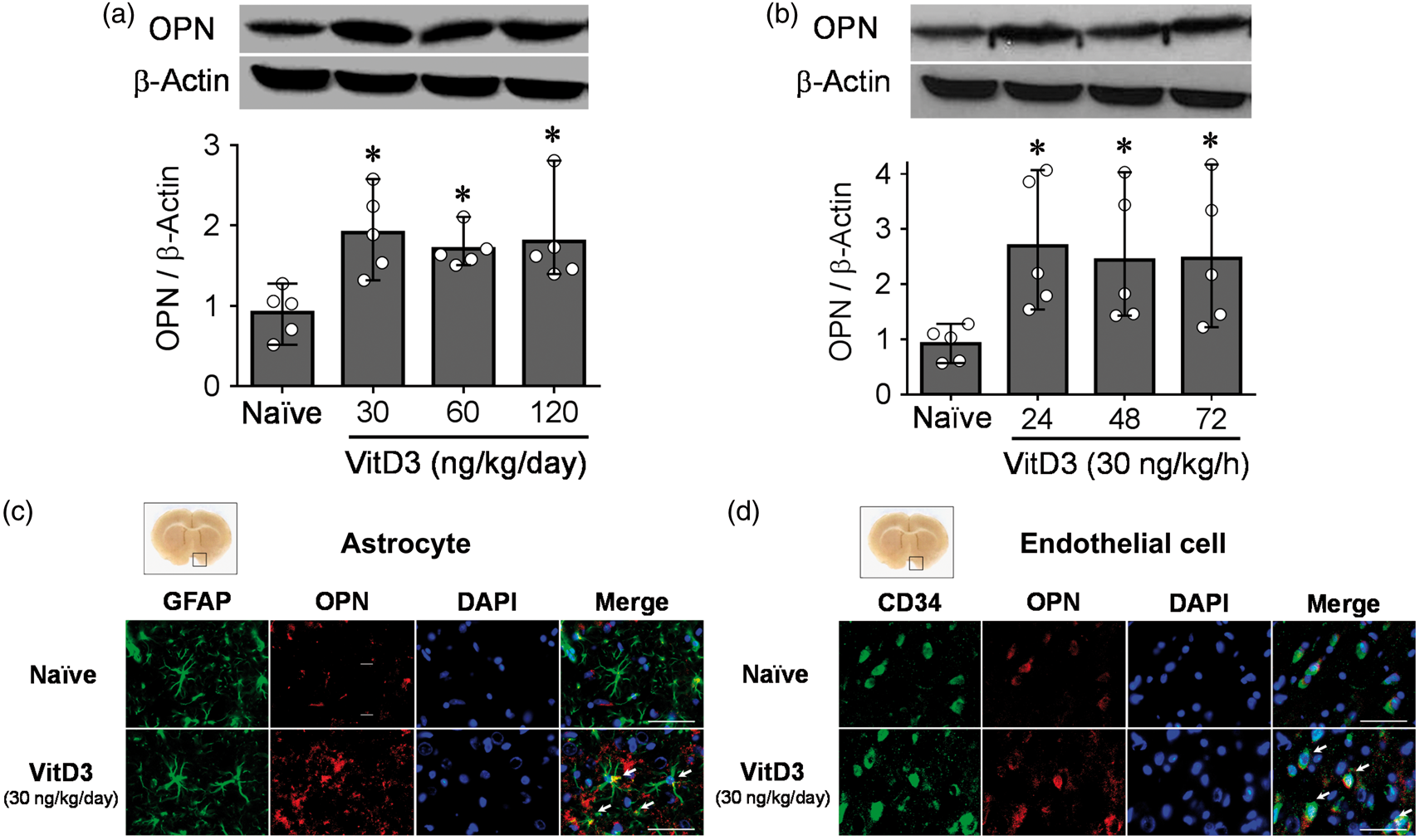
Mortality and SAH severity
After SAH, blood clots were around the basal cistern and the SAH grades were decreased gradually as 72 h passed. There was a significant difference in SAH grade between sham and SAH animals, but no difference was observed between all SAH groups (Supplementary Figure 4(a) and (b)) at either 24 or 72 h post-ictus. Body weight loss was significantly higher in the SAH groups compared with the sham group for 24 h, but body weight was significantly recovered in the Pre-SAH + VitD3 group at 72 h (Supplementary Figure 4(c)).
Intranasal administration of VitD3 prevents early brain injury after SAH
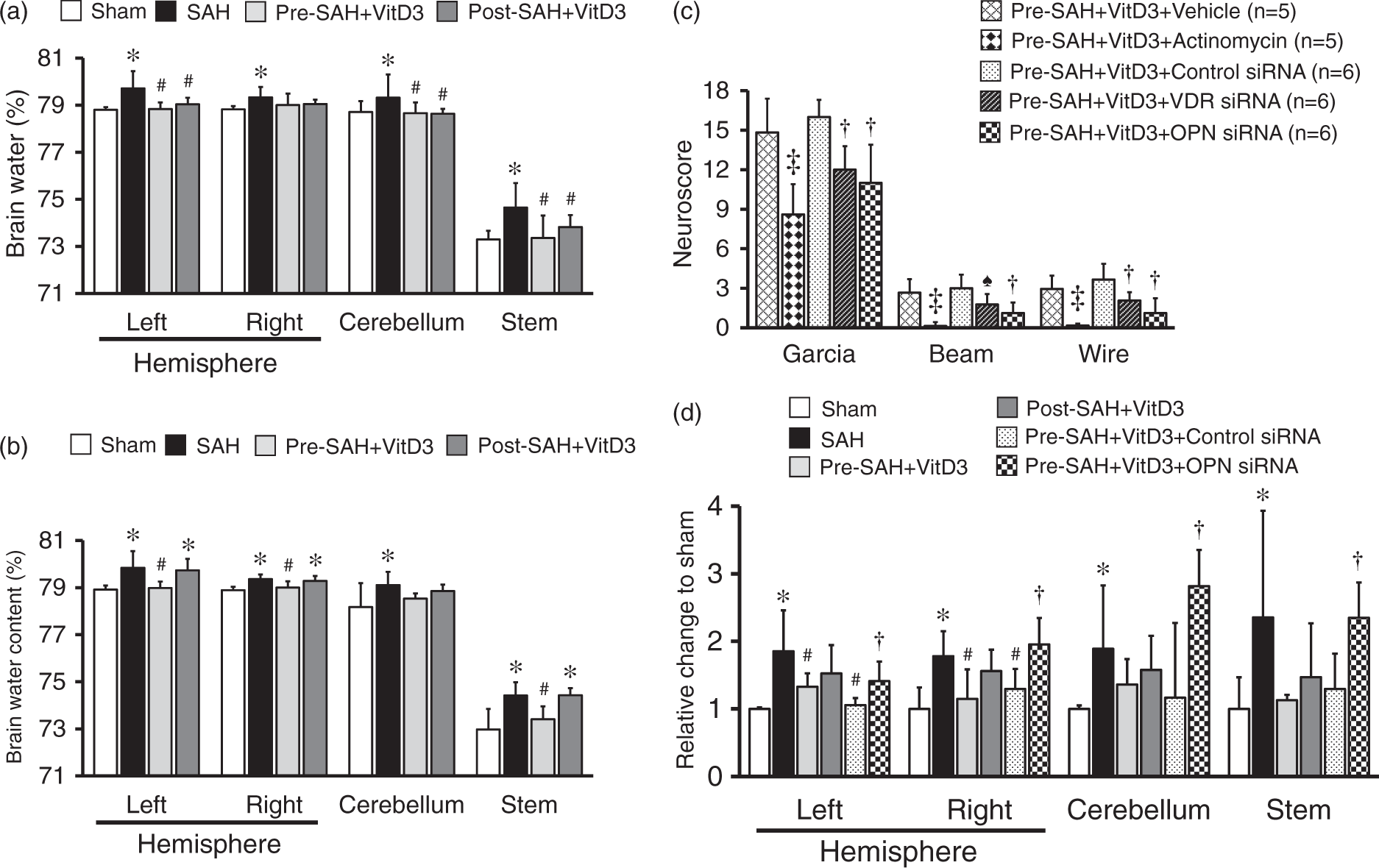
Pre-SAH intranasal administration of 30 ng/kg VitD3 significantly increased neurological score of neurological function at 24 (Figure 2(a)) and 72 (Figure 2(b)) h post-SAH compared with SAH given vehicle only. At 24 h after SAH, VitD3 treatment significantly attenuated brain edema in the brain stem, cerebellum, and left and right cerebral hemispheres (Figure 3(a)), with the protective effects of Pre-SAH + VitD3 lasting up to 72 h post-injury (Figure 3(b)). The preventive effects of Pre-SAH VitD3 were abolished by Actinomycin and siRNAs for VDR and OPN (Figure 3(c)). VitD3 also prevented blood brain disruption (measured by Evans blue extravasation) at 24 h, which was abolished by OPN siRNA (Figure 3(d)).
Intranasal administration of vitamin D3 (VitD3) improves neurological deficits after subarachnoid hemorrhage (SAH). Pre- and Post-treatment of VitD3 (30 ng/kg) significantly increased the modified Garcia (left), beam balance (middle), and wire hanging test (right) scores 24 h in Pre-SAH + VD3 and Post-SAH + VD3 groups (a, n = 10 per group) and 72 h in Pre-SAH + VD3 group (b, n = 6 per group). Pre-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 24 h before SAH; Post-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 1 h after SAH. *P < 0.05 vs. sham. #P < 0.05 vs. SAH. Role of vitamin D3 (VitD3), its receptor (VDR), and osteopontin (OPN) in neurological function and BBB disruption. (a) Pre- and Post-treatment of VitD3 (30 ng/kg) decreased brain water content at 24 (n = 8 per group) and (b) decreased brain water content in Pre-SAH + VD3 group at 72 (n = 6 per group) h after SAH. Pre-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 24 h before SAH; Post-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 1 h after SAH. *P < 0.05 vs. sham. #P < 0.05 vs. SAH. (c) Neurobehavioral tests. siRNA, small interfering ribonucleic acid; ‡P < 0.01 vs. Pre-SAH + VitD3 + Vehicle; †P < 0.05 vs. Pre-SAH + VitD3 + Control siRNA; n = 5-6 per group; ♠P = 0.054 vs. Pre-SAH + VitD3 + Control siRNA. (d) Evans blue extravasation. n = 5 per group.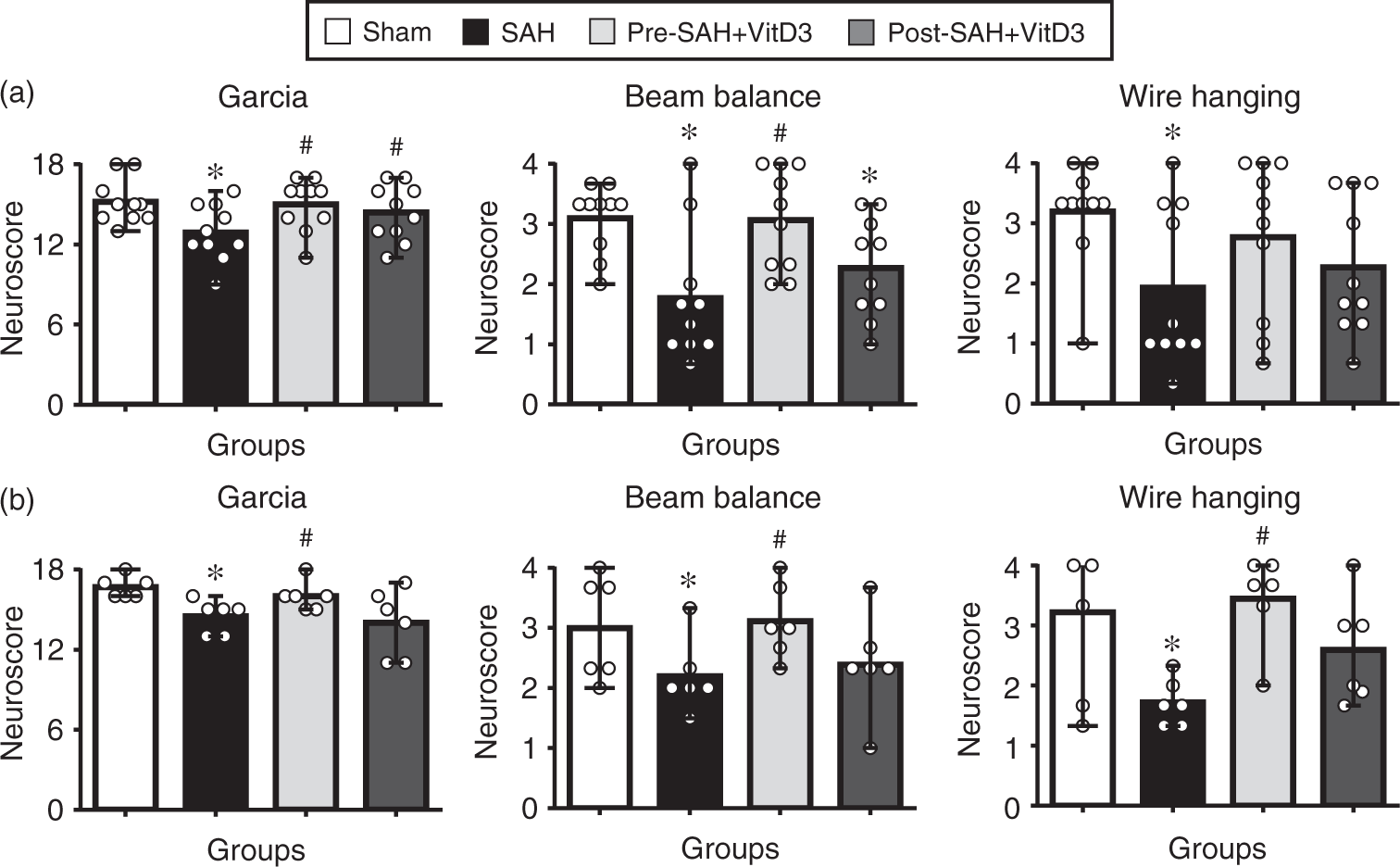
VitD3 upregulates endogenous OPN isomers through VDR dependent mechanism in astrocytes
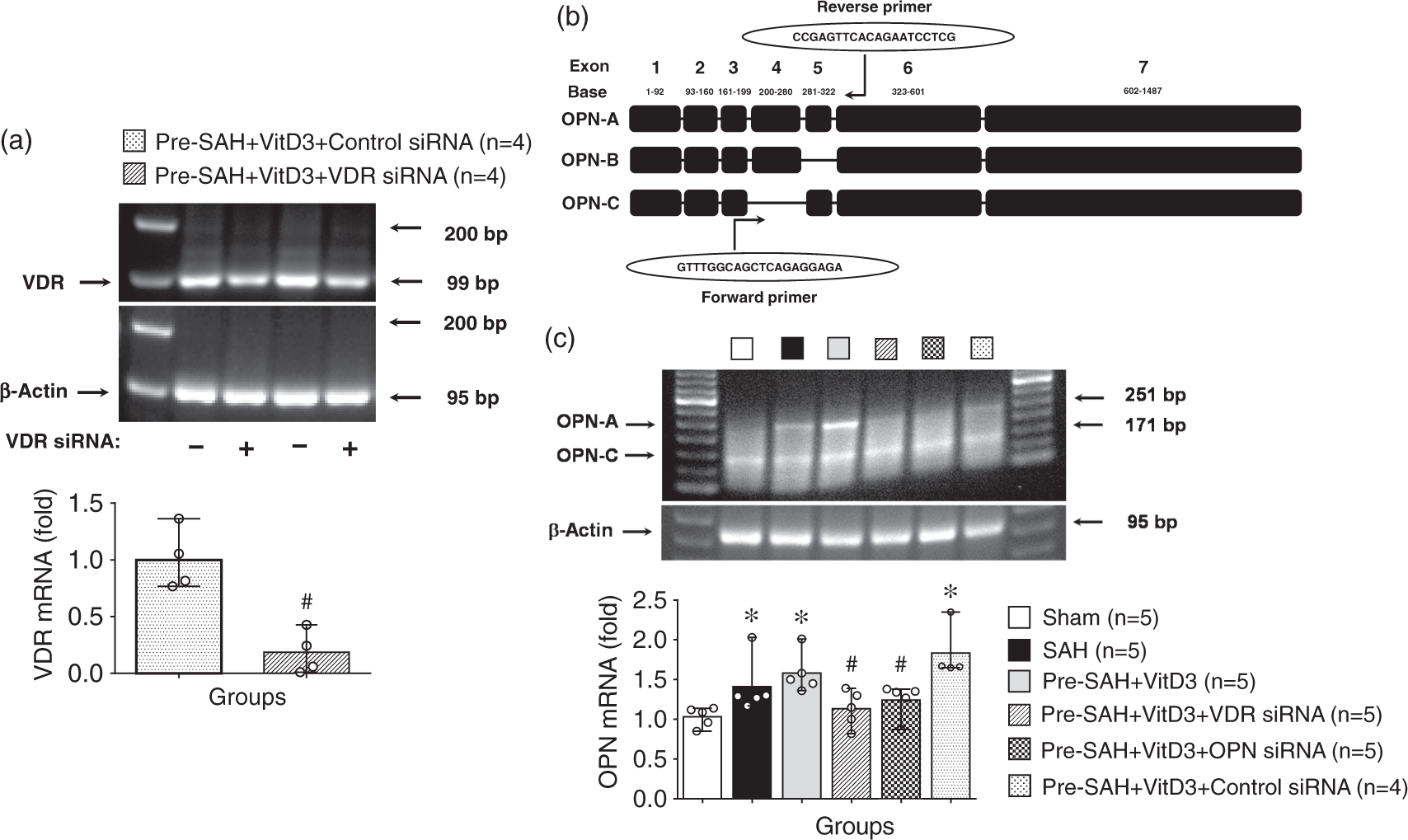
After SAH induction, either VDR or OPN expression was significantly higher than that sham group. Moreover, Pre-SAH intranasal administration of VitD3 significantly increased expression of VDR as well as endogenous OPN in the brain (Figure 4(a) and (b)). Expression of OPN was significantly higher in Pre-SAH + VitD3 rats compared with that of SAH rats (Figure 4(b)), and it was higher in astrocytes compared with that of SAH and Post-SAH + VitD3 rats (Figure 4(c), Supplementary Figure 5). Intracerebroventricular injection of VDR siRNA significantly inhibited VDR expression in the left hemisphere of Pre-SAH + VitD3 + VDR siRNA rats compared with Pre-SAH + VitD3 + Control siRNA rats (Figure 5(a)). Using qRT-PCR (Figure 5(b)), pretreatment of VitD3 significantly upregulated the endogenous OPN-A and -C isomers compared with that of the sham rats, but VDR siRNA, and OPN siRNA were reversed these effects compared with that Pre-SAH + VitD3 + Control siRNA rats (Figure 5(c)).
Role of vitamin D3 (VitD3) in upregulation of endogenous osteopontin (OPN) expression after subarachnoid hemorrhage (SAH). (a) Pre- and Post-treatment of VitD3 increased VDR brain expression. Pre-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 24 h before SAH; Post-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 1 h after SAH. *P < 0.05 and †P = 0.057 vs. sham; n = 5 per group. (b) Pre- and Post-treatment of VitD3 increased OPN cerebral expression. *P < 0.05 and **P < 0.01 vs. sham; #P < 0.05 vs. SAH. n = 5–6 per group. (c) Immunohistochemical staining of OPN (green) and astrocytes (GFAP, red). Nuclei are stained with DAPI (blue). Top panel indicates the location of immunohistochemical staining (small black box). Arrows indicate colocalization of OPN and GFAP. n = 3 per group. Bar = 50 µm. Vitamin D3 (VitD3) upregulates endogenous osteopontin (OPN) -A and -C isomers via a VDR-dependent pathway after subarachnoid hemorrhage (SAH). (a) Effect of VDR siRNA on the VDR mRNA expression. Pre-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 24 h before SAH; siRNA, small interfering ribonucleic acid. (b) Structure of OPN isomers and qRT-PCR universal primer design. (c) VitD3 upregulates endogenous OPN-A and -C isomers. *P < 0.05 vs. sham; #P < 0.05 vs. Pre-SAH + VitD3 + Control siRNA; n = 4–5 per group.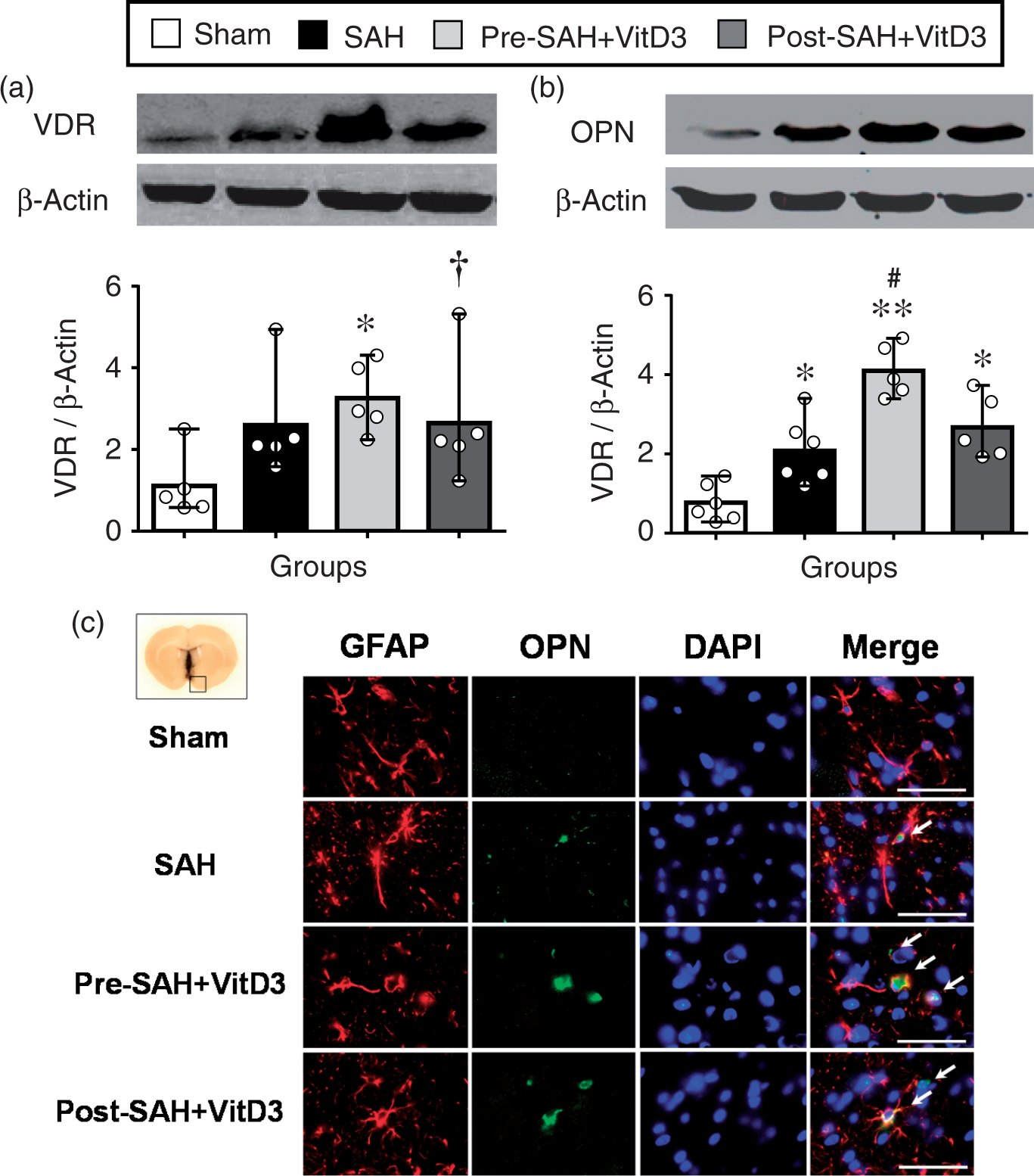
Endogenous OPN regulates CD44 splicing and P-gp glycosylation in endothelial cells
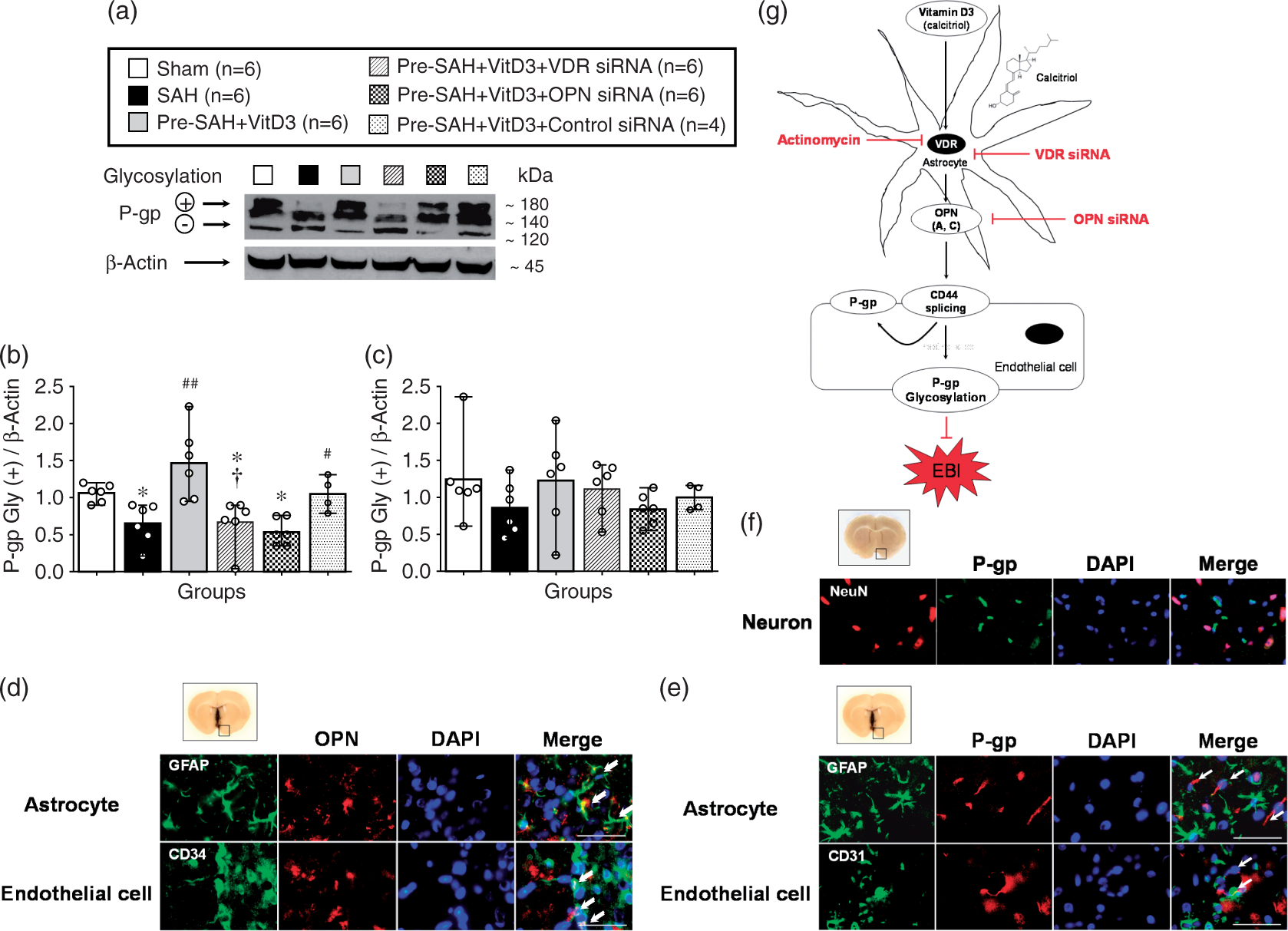
Expression of full length CD44 (85 kDa) was significantly higher in all SAH-induced groups compared with the sham group, but it was lower in Pre-SAH + VitD3 group compared with that of the SAH group, which was significantly abolished in the Pre-SAH + VitD3 + VDR siRNA group compared with the Pre-SAH + VitD3 + Control siRNA group (Figure 6(a) and (b)). CD44 splice variant (55 kDa) was higher after SAH compared to that of the sham group, and it was significantly higher in the Pre-SAH + VitD3 + VDR siRNA and Pre-SAH + VitD3 + OPN siRNA groups (Figure 6(a) and (c)). The ectodomain of CD44 (25 kDa) was significantly higher in the SAH group compared with sham group and was lower in the Pre-SAH + VitD3 group, such that is was not different from that of the sham group, but this effect was diminished by VDR and OPN siRNA interventions (Figure 6(a) and (d)). After SAH induction, P-gp glycosylation (180 kDa) was significantly lower in the SAH group compared with the sham group, and Pre-SAH treatment of VitD3 significantly increased P-gp glycosylation. However, the increase in P-gp glycosylation was reversed by VDR siRNA and OPN siRNA (Figure 7(a) to (c)). In the Pre-SAH + VitD3 group, expression OPN was observed in both astrocytes and endothelial cells and P-gp was predominantly expressed in the endothelial cells of the brain micro vessels (Figure 7(d) to (e) and Supplementary Figure 6), but not in the neuron (Figure 7(f) and Supplementary Figure 7).
Vitamin D3 (VitD3) regulates CD44 splicing after subarachnoid hemorrhage (SAH). Representative Western blots of CD44 (a) and quantitative analysis of full length CD44 (b), splice variant (c), and ectodomain (d) of CD44 after 24 h. Pre-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 24 h before SAH; siRNA, small interfering ribonucleic acid; *P < 0.05, **P < 0.01, and ***P < 0.001 vs. sham; #P < 0.05 vs. SAH; †P < 0.05, and ††P < 0.01 vs. Pre-SAH + VitD3 + Control siRNA; n = 6 per group. Vitamin D3 (VitD3) induces P-gp glycosylation after subarachnoid hemorrhage (SAH). Representative Western blots of P-glycoprotein (P-gp) (a) and quantitative analysis of mature/fully glycosylated P-gp (b) and immature P-gp (c) 24 h after SAH. Pre-SAH + VitD3, intranasal treatment 30 ng/kg/day VitD3 24 hours before SAH; siRNA, small interfering ribonucleic acid; *P < 0.05 vs. sham; #P < 0.05 and ##P < 0.001 vs. SAH; †P < 0.05 vs. Pre-SAH + VitD3 + Control siRNA. Colocalization of OPN (red) with astrocytes (GFAP, green) and endothelial cells (CD34, green) (d), and colocalization of P-gp (red) with endothelial cells (CD34, green), but not astrocytes (GFAP, green) (e) and neurons NeuN, red) (f). Nuclei are stained with DAPI (blue). Top panel indicates the location of immunohistochemical staining (small black box). Arrows indicate colocalization. n = 3 per group. Bar = 50 µm. (g) Proposed mechanism of VitD3 neuroprotection via endogenous OPN/CD44/P-gp glycosylation after SAH. EBI, early brain injury.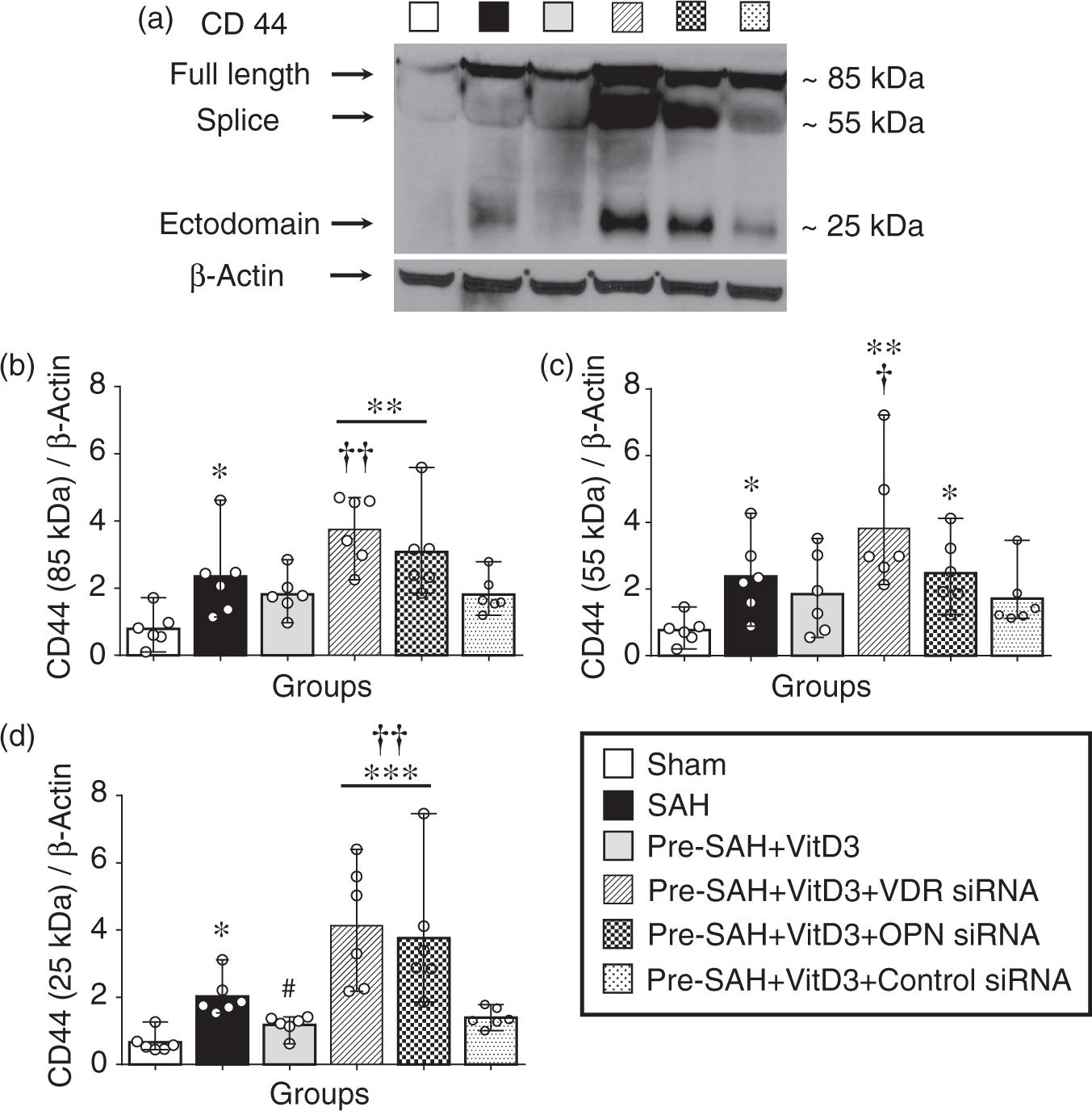
Discussion
The novel finding of the present study was that intranasal administration of VitD3 upregulates expression of endogenous brain OPN in astrocytes and it protects against BBB disruption by CD44 splicing and P-gp glycosylation in the endothelial cells of the BBB after SAH in rats. Intranasal administration of VitD3 (calcitriol), the most active form of vitamin D, improved neurological impairment and brain edema, and was associated with upregulation of endogenous OPN – A and – C isomers within 72 h after SAH. Blockage of VDR and OPN exacerbated neurologic impairment and BBB disruption was associated with a decrease of mature/fully glycosylated P-gp in brain endothelial cells of the rat (Figure 7(g)).
Vitamin D is an important key molecule that has a long history in the treatment of vitamin D deficiency. The VDR is ubiquitously expressed in all tissues 23 and VDR dependence has been shown in a number of genomic and non-genomic pathways,22,23 and number of these molecular mechanism involved to neuro protective effects of vitamin D. 30 Stroke is a significant cause of mortality in patients with vitamin D deficiency and prolonged vitamin D therapy could reduce risk and mortality of stroke.16,19,31 However, limited studies have described the precise molecular mechanism of vitamin D treatment after stroke. Studies showed that long-term vitamin D therapy is protective against ischemic stroke and does not increase the risk of intracerebral infarct. The underlying molecular mechanism of vitamin D may involve GDNF, NR3A/CREB pathways in neurons,20,21but the molecular mechanism of vitamin D has never been studied after SAH. In the present study, for the first time we demonstrated that VitD3 prevents EBI administered intranasally before SAH in rats and that the molecular mechanism of VitD3 involved OPN – A and – C isomer upregulation.
OPN is a neuroprotective glycoprotein8,9 and its expression can be upregulated by vitamin D.13–15 Our previous study showed that endogenous OPN expression increased after SAH induction and that exogenous OPN treatment has protective effects against vasospasm and BBB disruption after SAH through integrin receptor-dependent activation of MKP-1 and MAPK phosphatase in astrocytes and endothelial cells.10,12 Additionally, studies have shown that OPN protects against brain ischemic injury via Akt/p42/p44/MAPK phosphorylation and GDNF upregulation.8,21 Recently, we also showed that intranasal administration of OPN protects against neuronal apoptosis after SAH through FAK/PI3K/Akt-induced inhibition of caspase-3 cleavage. 11 The present study results showed that intranasal administration of VitD3 upregulates the A and C isomers of OPN in the rat brain and triggers the CD44/P-gp glycosylation pathway for protection against BBB disruption after SAH.
CD44, one of OPN’s receptors, is a widely distributed cell transmembrane adhesion molecule with diverse biological processes which is dependent outside–inside or inside–outside regulation of ectodomain and intracellular domain (ICD) cleavage in the cell membrane.32,33 Cell to cell molecular signaling of CD44 is initiated by substrate-selective cleavage regulation either within or outside of the cell. 34 Sequential proteolytic cleavage of CD44 is regulated by multiple signaling pathways, for example, the activation of metalloprotease and PKC, the influx of extracellular Ca2+, members of the Rho family of small GTPases, and the Ras oncoprotein. 35 In this study, we found that expression of the full length, splice, and the ectodomain of CD44 were both significantly increased after SAH. VitD3 treatment prevented the increase in the ectodomain, such that it was indistinguishable from sham levels. These results indicate that CD44 cleavage and its downstream signaling is dependent on VitD3-induced upregulation of endogenous OPN. Other studies have also shown that OPN/CD44 signaling plays a more active role in cell signaling and that the CD44 receptor interacts with P-gp to promote cell migration and invasion.36,37
P-gp, ATP-binding cassette, is an efflux transporter at the BBB 38 which is highly expressed in brain endothelial cells. 39 Functional activity of P-gp is dependent on glycosylation (180 kDa) of this transporter (mature P-gp)40,41 and the inactive, immature P-gp is unglycosylated or only partially glycosylated (120 and 140 kDa). 42 This seems to hold true for BBB preservation after SAH. Most studies have studied the drug efflux function of P-gp in tumor models, but there have been limited studies on the maturity of P-gp during brain diseases.43,44 Vitamin D has been shown to protect BBB disruption through upregulation of tight junction proteins after ischemic stroke. 45 A recent study showed that deficiency one of P-gp exacerbated BBB damage after stroke. 46 Durk et al.39,47 showed that vitamin D treatment upregulates P-gp expression and function in the capillary endothelial cells of rat and human brains, but the mechanism of P-gp activation by vitamin D was not investigated. In this study, the active mature P-gp (180 kDa) was significantly decreased after SAH and VitD3 treatment was able to restore mature P-gp back to baseline levels through OPN/CD44 signaling in endothelial cells. While numerous studies have identified a plethora of deleterious molecules which are increased after stroke, discovering and targeting molecules which are capable of reducing accumulation or promoting clearance of these harmful molecules seem to be of great importance. Since the primary role of P-gp is to remove foreign and harmful molecules from the cell, understanding the role of P-gp in clearance of critical pathological molecules is important for effective treatment of stroke. To this end, the current results shed new light on the role of vitamin D in promoting glycosylation of P-gp through OPN and CD44, and provide a functional link between activation of P-gp and promoting BBB stability after SAH.
Geographical and ethnical causes of vitamin D deficiency in the world are well documented 48 and vitamin D deficiency is strongly correlated with incidence of cancer, diabetes, and cardiovascular diseases,16–18 as well as increased stroke incidence and worse outcome. 19 Moreover, SAH is highly prevalent in the non-white population, 49 and SAH incidence, as well as worse prognosis is remarkably increased during the winter when vitamin D synthesis is reduced due to less ultra violet radiation, the stimulator of vitamin D synthesis. 50 Based on this and other studies, and the incidence of SAH, vitamin D and its regulatory mechanism of P-gp suggest that vitamin D is critical to reducing SAH sequela and reducing the occurrence of SAH. The intracellular signaling of the ICD of CD44 in P-gp maturation and specific role of the OPN – A and – C isomers in the brain after stroke remain to be elucidated in further studies.
For the first time, the beneficial effect of VitD3 was shown to be involved in increased endogenous OPN expression causing reduced levels of the CD44 ectodomain and subsequent maturation of P-gp in endothelial cells, thereby attenuating BBB disruption and EBI after SAH. Previous studies have supported OPN treatment as therapeutically beneficial for treatment of stroke patients. However, clinical translation of recombinant OPN as a stroke treatment may be hindered by the large cost (about $1500 per day/human). Interestingly, a number of studies have confirmed that vitamin D upregulates endogenous OPN expression in various tissues using in vitro and in vivo models.13–15 The current results also demonstrated that intranasal administration of VitD3 leads to increased expression of endogenous OPN, especially the A isomer, in the brain. As described our previous study, the intranasal route is an important method for OPN therapy after SAH, 11 and currently vitamin D is available to use by the oral route. Further studies are needed to clarify the difference between oral and intranasal treatment of vitamin D, and the fact that Vitamin D penetrates the BBB. In addition, there is a need to confirm clinical prevalence of this targeted treatment on the central nervous system. Moreover, pre-treatment of vitamin D was more protective compared with post-treatment after SAH. In order to explain this discrepancy, more studies are required on vitamin D deficiency followed by more complex neurobehavioral tests. Thus, intranasal administration with vitamin D would be a cost-effective way (about $0.15 per day/human) to upregulate OPN in SAH patients.
In conclusion, we demonstrated that intranasal administration of VitD3 attenuated EBI after SAH through a VDR/OPN/CD44/P-gp glycosylation-dependent mechanism in astrocytes and endothelial cells after SAH in rats, and have identified one of the novel regulatory mechanisms of vitamin D in the brain. Intranasal administration of vitamin D might be a potentially new therapeutic strategy for stroke patients.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partially supported by grants NS081740, NS082184 from the National Institutes of Health to J.H.Z.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
The conception and design were made by Budbazar Enkhjargal, John H Zhang, and Jiping Tang. Drafting the article was done by Budbazar Enkhjargal, Yasushi Sakai, and Devin McBride. Budbazar Enkhjargal, Anatol Manaenko, and Cesar Reis performed most experiments. Critically revising the article was made by all authors. Reviewed submitted version of manuscript was made by all authors. John H Zhang approved the final version of the manuscript on behalf of all the authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.