
Abstract
Adverse environmental and social conditions early in life have a strong impact on health. They are major risk factors for mental diseases in adulthood and, in some cases, their effects can be transmitted across generations. The consequences of detrimental stress conditions on brain metabolism across generations are not well known. Using high-field (14.1 T) magnetic resonance spectroscopy, we investigated the neurochemical profile of adult male mice exposed to traumatic stress in early postnatal life and of their offspring, and of undisturbed control mice. We found that, relative to controls, early life stress-exposed mice have metabolic alterations consistent with neuronal dysfunction, including reduced concentration of N-acetylaspartate, glutamate and γ-aminobutyrate, in the prefrontal cortex in basal conditions. Their offspring have normal neurochemical profiles in basal conditions. Remarkably, when challenged by an acute cold swim stress, the offspring has attenuated metabolic responses in the prefrontal cortex, hippocampus and striatum. In particular, the expected stress-induced reduction in the concentration of N-acetylaspartate, a putative marker of neuronal health, was prevented in the cortex and hippocampus. These findings suggest that paternal trauma can confer beneficial brain metabolism adaptations to acute stress in the offspring.
Keywords
Introduction
The brain is the key organ in appraising the environment and in triggering adequate physiological and behavioural responses to potential adverse and dangerous factors. Although stress responses allow adaptation to environmental threats, they can be maladaptive when excessively intense and/or persistent, becoming a risk factor for the development of psychopathology. Indeed, traumatic stress experienced during a sensitive period of brain development increases the susceptibility to develop psychosis, mood and anxiety disorders in adulthood.1–4 Further, the progeny of individuals exposed to stress early in life is often predisposed to psychopathology.5–8 Notwithstanding, there is also evidence for resilience in offspring of individuals exposed to trauma. 9 In mice, early-life stress can also have beneficial effects on the offspring, such as improved goal-directed behaviours, flexibility and adaptation to novel rules. 10
Regional concentrations of brain metabolites, the so-called neurochemical profiles, vary with the developmental stage, oscillate with local functional state, and are affected by acute injury or prolonged disease. 11 Neurochemical profiles measured non-invasively by 1H magnetic resonance spectroscopy (MRS) have been widely used to probe specific metabolic alterations in neurological disorders. 12 Here, we used state-of-the-art high resolution MRS to determine whether brain metabolism is altered by postnatal traumatic stress in adult mice and their offspring. We applied an established model of postnatal trauma based on unpredictable maternal separation combined with unpredictable maternal stress (MSUS). This paradigm causes depressive symptoms, antisocial behaviours, risk-taking and cognitive deficits, which are transmitted across several generations by males or females.13–15 Since trauma is known to impact limbic and reward circuits, 16 we examined the neurochemical profiles of mice exposed to MSUS in the dorsal hippocampus, prefrontal cortex and striatum.
Methods and materials
Animals
All experiments were conducted in C57BL/6JRj mice according to the Swiss animal welfare legislation under approval of the local ethics committee (Kanton Zürich Gesundheitsdirektion Veterinäramt) and are reported according to the ARRIVE guidelines. Mice were housed in the Zürich facility with controlled temperature (20–22℃) and humidity (50–56%) and free access to food and water, under a 12-h reverse light–dark cycle (light off at 08:00). One week before MRS experiments, mice were transferred to Lausanne and housed under similar conditions but with light off at 19:00.
The study involved control and MSUS-exposed mice (F1) and their offspring (F2). Sample size estimation was based on previous experiments.17,18 MRS experiments were performed on 3-month-old male mice (F1: 13 MSUS and 13 controls; F2: 14 MSUS and 15 controls). A second set of 7-month old F2 male mice also underwent brain MRS experiments (12 MSUS and 10 controls) in combination with an acute stress. One week after baseline MRS, mice from this second group were exposed to cold swim stress, and MRS experiments were immediately repeated after warming up for 10 min under a heat lamp.
MSUS paradigm
Male and female mice (F0; from Janvier SAS, Berthevin, France) were bred on one-to-one pairing, and gestating females were single-housed and either maintained in regular conditions and allowed to raise their pups undisturbed (controls), or subjected to MSUS from post-natal day 1 to 14. Female mice were randomly assigned to either experimental group. For MSUS, dams and litters were subjected to 3-h daily separation unpredictably, during which dams were randomly and unpredictably exposed to restraint stress for 20 min or forced swim stress for 5 min. 13 Control mice were left undisturbed except for weekly cage changing. Once weaned on post-natal day 21, pups (F1) were housed in groups of 4–5 of the same gender and treatment, but from different mothers to avoid litter effects. To obtain second-generation mice (F2), adult F1 control or MSUS males were mated to naïve adult females then removed after mating, so F1 male mice were not in contact with the gestating female or with their progeny.
Acute swim stress
Mice were single-housed one day before handling. Exposure to cold swim stress prior to MRS experiments was performed as previously described. 19 Briefly, mice were placed in a plastic cylinder (20 cm high, 16 cm diameter) filled with 18 ± 1℃ water up to 12 cm height for 6 min.
MRS
Timing (in min) of MRS acquisitions after cold swim stress.
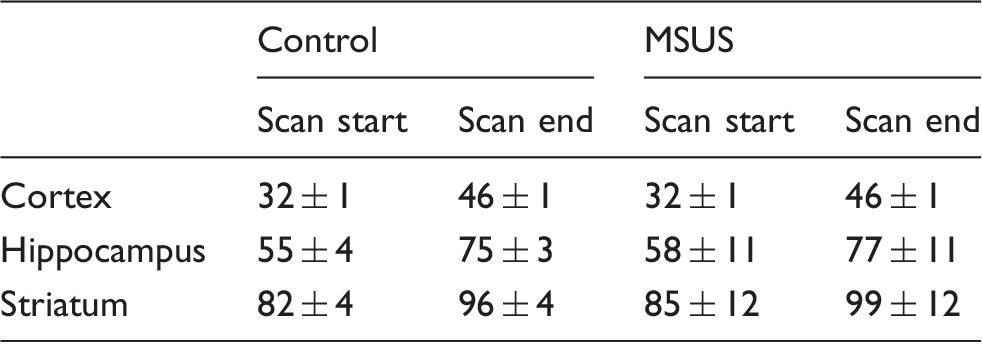
Data are mean ± SD of nine F2 controls and 11 MSUS F2 mice.
Metabolite concentrations were determined with LCModel (Stephen Provencher Inc., Oakville, Ontario, Canada), including a macromolecule (Mac) spectrum in the database and using the unsuppressed water signal measured from the same VOI as internal reference. 17 The following metabolites were included in the analysis: alanine (Ala), ascorbate (Asc), aspartate (Asp), creatine (Cr), γ-aminobutyrate (GABA), glutamine (Gln), glutamate (Glu), glutathione (GSH), glycine (Gly), glycerophosphorylcholine (GPC), glucose (Glc), lactate (Lac), myo-inositol (Ins), N-acetylaspartate (NAA), N-acetylaspartylglutamate (NAAG), phosphorylethanolamine (PE), phosphorylcholine (PCho), phosphocreatine (PCr), scyllo-inositol (Scyllo), taurine (Tau). The Cramér-Rao lower bound (CRLB) was provided by LCModel as a measure of the reliability of the quantification for each metabolite. In most measured spectra, scyllo-inositol was below the detection limit, and was excluded from subsequent data analyses. Remaining metabolites had CRLB below 30%.
Nine mice (out of 77) were excluded from the study because of abnormally high glutamine levels in all brain regions investigated, which suggests the occurrence of congenital portosystemic shunting: 18 1 F1 control, 2 F1 MSUS, 4 F2 control and 2 F2 MSUS mice.
Statistical analysis
Data are presented as mean ± SEM unless otherwise stated and were analysed using the analysis of variance (ANOVA) for all metabolites together (repeated measures), followed by independent Student’s t tests for post hoc comparisons. Significance was considered for P < 0.05. Effect of MSUS-exposure on baseline neurochemical profiles (for either F1 or F2 mice) was tested with independent ANOVAs for the hippocampus, cortex and striatum because their neurochemical profiles are distinct. 17 The effect of acute stress was tested in a single multivariate ANOVA together with the effects of brain region and MSUS-exposure, as well as their interactions. Statistical analyses were performed with IBM SPSS 21 (IBM Corp., Armonk, New York, USA).
Results
Localised MRS at 14.1 T in the hippocampus, anterior cortex and striatum in vivo resulted in spectra with signal-to-noise ratios of 21 ± 3, 21 ± 3 and 18 ± 2, and spectral line widths of 11 ± 3, 12 ± 3 and 11 ± 2 Hz, respectively (averaged for all acquired spectra, errors in SD of 101 scans/region; Figure 1). This excellent spectral quality allowed us to quantify a neurochemical profile composed of 19 metabolites in the three brain structures.
Representative 1H spectra acquired in vivo at 14.1 T from the mouse prefrontal cortex, dorsal hippocampus and striatum (a). For display, spectra were processed with a shifted Gaussian function (gf = 0.12, gfs = 0.02) prior to Fourier transformation. Peak assignment is as follows: 1, glucose; 2, lactate; 3, alanine; 4, phosphocreatine; 5, creatine; 6, glutamate; 7, glutamine; 8, GABA; 9, N-acetylaspartylglutamate; 10, aspartate; 11, glycine; 12, phosphorylethanolamine; 13, phosphrylcholine; 14, glycerylphosphorylcholine; 15, N-acetylaspartate; 16, glutathione; 17, ascorbate; 18, taurine; 19, myo-inositol. Consecutive mouse brain slices from fast-spin-echo images (6-mm thickness) are displayed below. Typical placement of VOIs in the cortex, striatum and hippocampus is represented in slices A–B, C–E and I–K, respectively. LCModel fit of the top spectrum (prefrontal cortex) is represented in (b). The fit is overlaid on the spectrum and the resulting residual is shown below. Mac: macromolecules; bk: background baseline; see text for metabolites.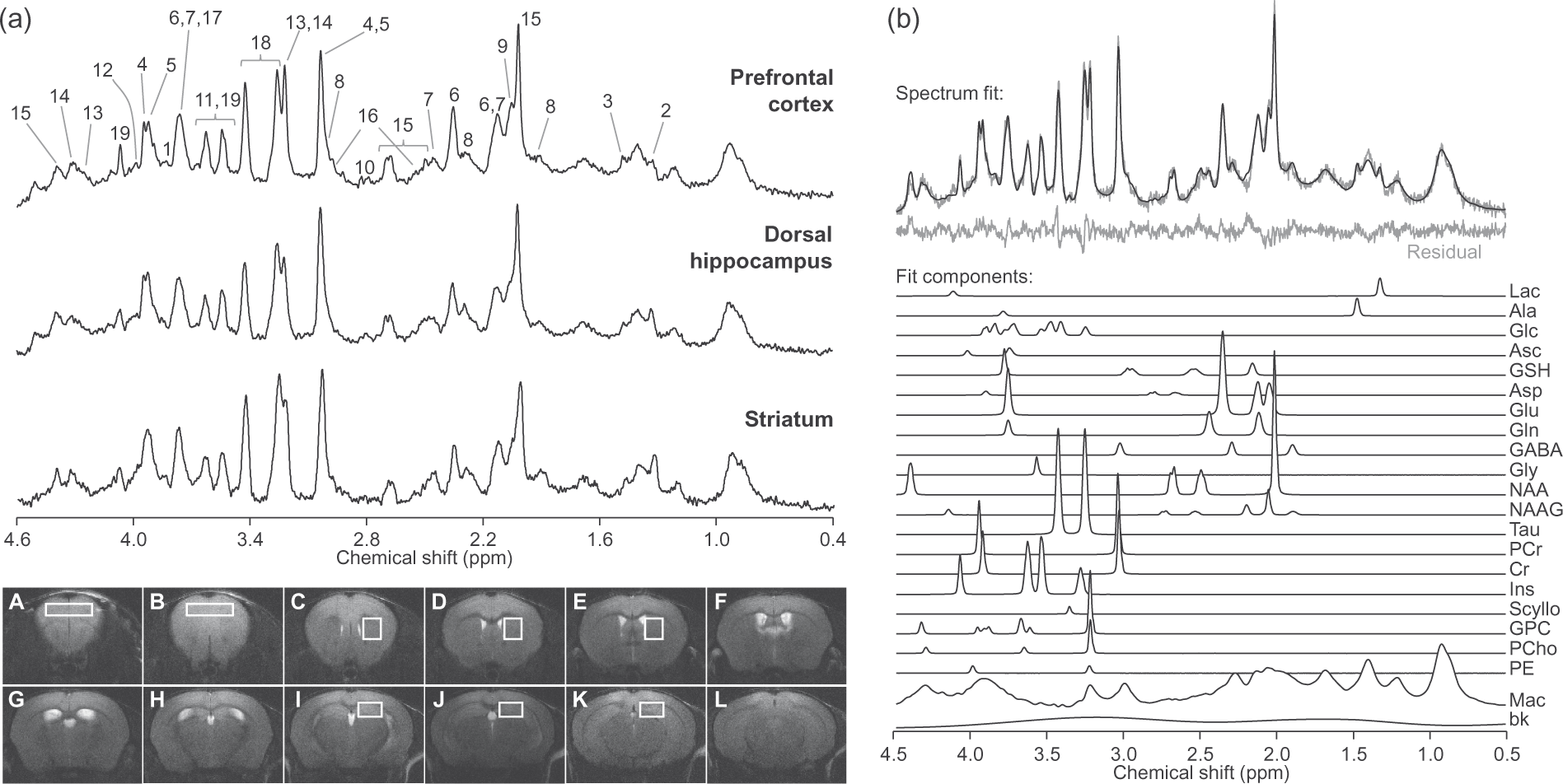
MSUS induced neurochemical alterations in the prefrontal cortex (F(1,22) = 9.574, P = 0.002) but not in the dorsal hippocampus (F(1,22) = 0.553, P = 0.457) or striatum (F(1,22) = 0.004, P = 0.951) of adult mice (F1; Figure 2). In particular, MSUS-exposed mice (F1) had reduced cortical levels of the putative neuronal marker NAA (−9.2%, P = 0.023), the neurotransmitters glutamate (−10.8%, P < 0.022) and GABA (−12.3%, P < 0.027) and the phospholipid precursor phosphorylcholine (−33.0%, P < 0.046) when compared with controls.
Neurochemical profiles in the hippocampus, prefrontal cortex and striatum of MSUS-exposed mice (F1, n = 11) and non-stressed controls (n = 12) mice. Data are mean ± SEM. *P < 0.05 for MSUS-exposed mice vs. controls. Ala: alanine; Asc: ascorbate; Asp: aspartate; Cr: creatine; GABA: γ-aminobutyrate; Glc: glucose; Gln: glutamine; Glu: glutamate; Gly: glycine; GPC: glycerophosphorylcholine; GSH: glutathione; Ins: myo-inositol; Lac: lactate; NAA: N-acetylaspartate; NAAG: N-acetylaspartylglutamate; PCho: phosphorylcholine; PCr: phosphocreatine; PE: phosphorylethanolamine; Tau: taurine.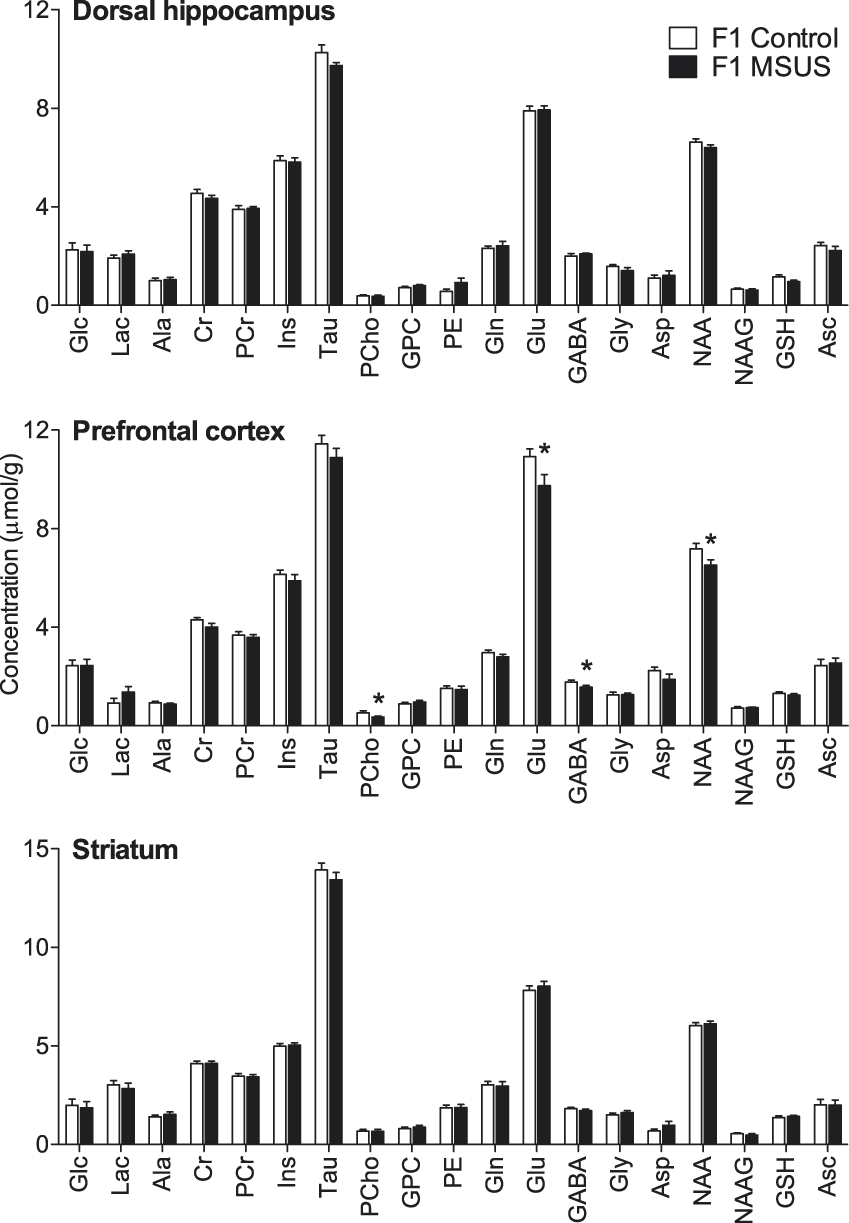
To investigate whether the observed effects are transmitted to the following generation, we bred MSUS-exposed males to naïve control females (non-stressed) to generate F2 progeny. MRS in the offspring of MSUS-exposed mice (F2) revealed no baseline alteration of metabolite concentrations in any of the three brain regions analysed when compared with controls: F(1,44) = 0.642, P = 0.423 for cortex; F(1,44) = 1.277, P = 0.259 for hippocampus; F(1,44) = 0.070, P = 0.781 for striatum (Figure 3).
Neurochemical profiles in the hippocampus, prefrontal cortex and striatum of the offspring (F2) of MSUS-exposed (n = 24) and control mice (n = 21). Data are mean ± SEM. Abbreviations are as in Figure 2.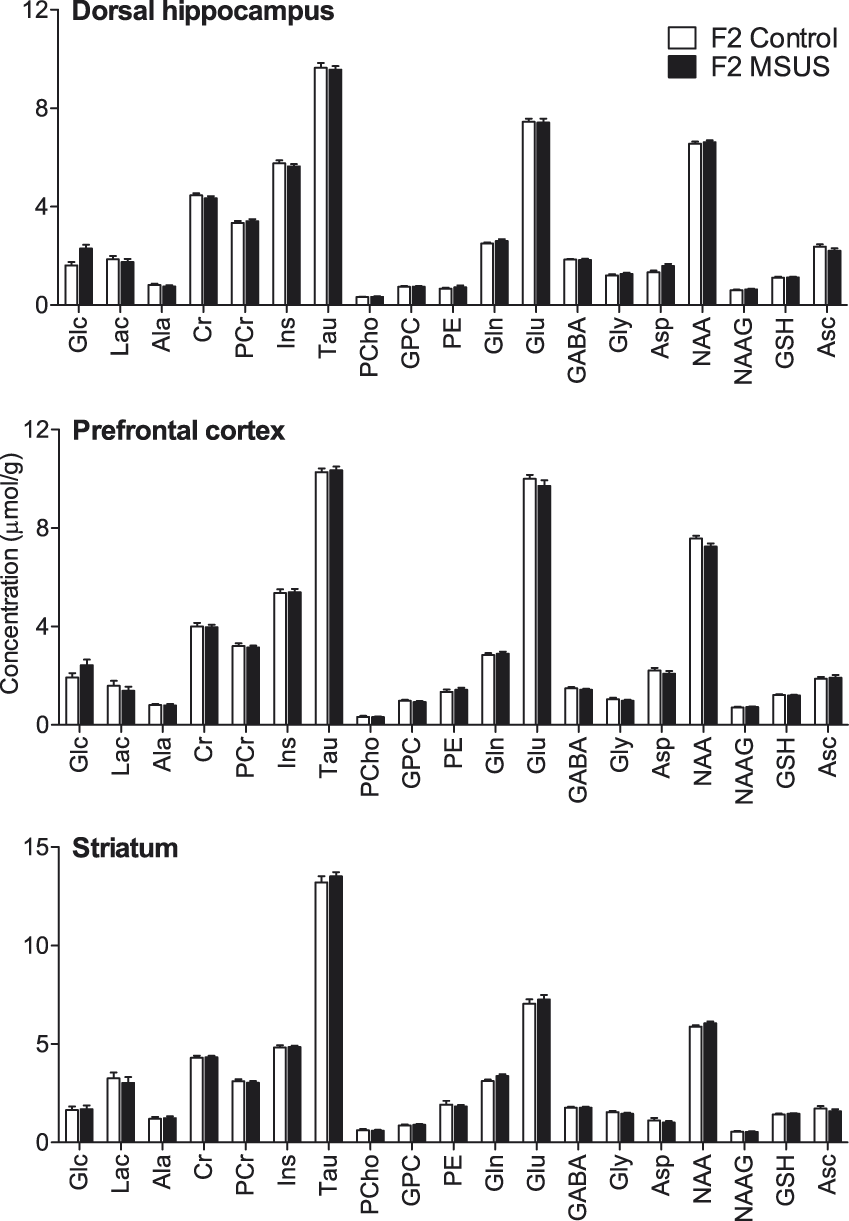
Since some systems may reveal their dysfunctions only when activated, we tested whether exposure to an acute stress affects brain metabolism in F2 MSUS offspring. Upon acute swim stress, we observed that the concentrations of several brain metabolites were modified in F2 MSUS mice compared with controls, and metabolic modifications were region-specific (Figure 4). Repeated-measures ANOVA with acute swim stress, brain region and paternal MSUS as fixed factors, as well as their interactions revealed the expected regional specificity of the neurochemical profile (F(36,182) = 101.0, P < 0.001), a strong effect of acute swim stress exposure (F(18,90) = 9.524, P < 0.001) that was region-specific (region-stress interaction: F(36,182) = 2.552, P < 0.001), and an effect of paternal MSUS (F(18,90) = 2.725, P = 0.001) that significantly interacted with the effect of acute stress exposure (stress-MSUS interaction: F(18,90) = 1.803, P = 0.032). Interactions of paternal MSUS exposure with brain region were not significant (interaction MSUS-region: F(36,182) = 0.816, P = 0.773; interaction MSUS-region-stress: F(36,182) = 1.166, P = 0.248).
Acute swim stress-induced variation of metabolite concentrations in the hippocampus, prefrontal cortex and striatum of the offspring (F2) of MSUS-exposed (n = 11) and control (n = 9) mice. Data are mean ± SEM. Multivariate ANOVA results: region F(36,182) = 101.0, P < 0.001; stress F(18,90) = 9.524, P < 0.001; MSUS F(18,90) = 2.725; P = 0.001; region*MSUS F(36,182) = 0.816, P = 0.773; stress*MSUS F(18,90) = 1.803; P = 0.032; region*stress F(36,182) = 2.552; P < 0.001; region*stress*MSUS F(36,182) = 1.166, P = 0.248. Post-hoc testing significant stress-induced modifications are depicted by †P < 0.05 and ‡P < 0.01 (baseline vs. post-stress); MSUS-exposure effect was tested on concentration differences between post-stress and baseline (*P < 0.05, **P < 0.01). Abbreviations are as in Figure 2.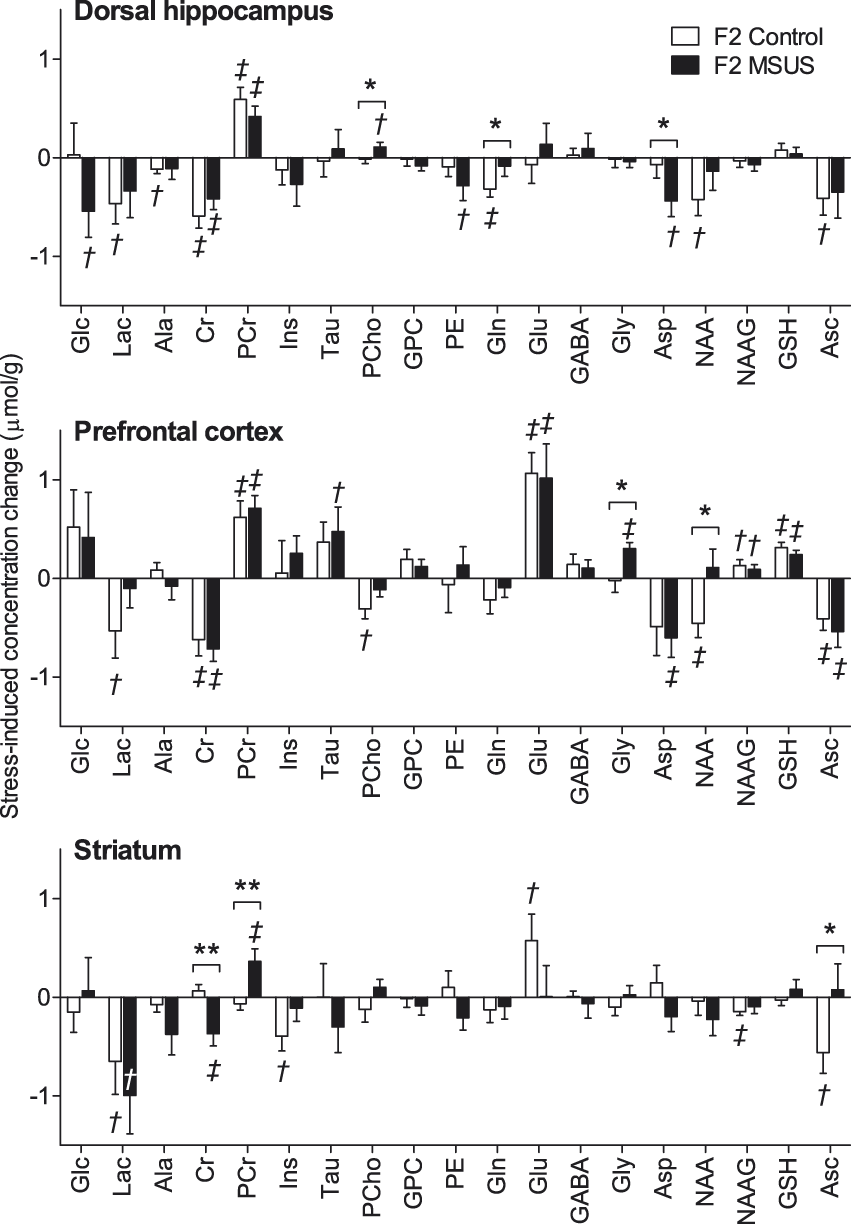
Modifications of metabolite concentrations caused by acute swim stress in F2 control animals were specifically in the dorsal hippocampus, reduced lactate (−30%, P = 0.028), alanine (−18%, P = 0.017), creatine (−14%, P < 0.001), glutamine (−13%, P = 0.003), NAA (−6%, P = 0.018) and ascorbate (−18%, P = 0.025), and increased phosphocreatine (+19%, P < 0.001); in the prefrontal cortex, reduced lactate (−37%, P = 0.047), creatine (−16%, P = 0.003), phosphorylcholine (−70%, P = 0.014), NAA (−6%, P = 0.008) and ascorbate (−23%, P = 0.005), and increased phosphocreatine (+18%, P = 0.003), glutamate (+11%, P < 0.001), NAAG (+18%, P = 0.030) and glutathione (+28%, P < 0.001); in the striatum, reduced lactate (−27%, P = 0.047), myo-inositol (−8%, P = 0.017), NAAG (−24%, P = 0.003) and ascorbate (−30%, P = 0.016), and increased glutamate (+9%, P = 0.035).
These neurochemical responses to acute swim stress were modified by paternal MSUS exposure. Namely, in the offspring of MSUS mice but not controls, acute swim stress exposure caused a 26% reduction in aspartate (P = 0.011 for pre- vs. post-stress; P = 0.047 for MSUS vs. control) and a 40% increase in phosphorylcholine (P = 0.027 for pre- vs. post-stress; P = 0.045 for MSUS vs. control) in the hippocampus. In contrast, it caused a 13% reduction of glutamine levels in the hippocampus of controls (P = 0.003 for pre- vs. post-stress), but not in the offspring of MSUS mice (P = 0.217 for pre- vs. post-stress; P = 0.046 for MSUS vs. control).
Interestingly, in prefrontal cortex, there was a 6% reduction in NAA levels (P = 0.008 for pre- vs. post-stress in controls) by acute swim stress in control offspring but not in the offspring of MSUS mice (P = 0.284 for pre- vs. post-stress; P = 0.014 for MSUS vs. control). In addition, a 31% increase in glycine concentration was induced in F2 MSUS mice (P = 0.011 for pre- vs. post-stress) but not in F2 controls (P = 0.433 for pre- vs. post-stress; P = 0.014 for MSUS vs. control).
In the hippocampus and prefrontal cortex of both control and MSUS mice, there was a reduction of creatine concentration caused by acute swim stress that was accompanied by an increase in phosphocreatine. In the particular case of the striatum, this effect occurred only in MSUS progeny, which had an 8% reduction in creatine (P = 0.008 for pre- vs. post-stress; P = 0.003 for MSUS vs. control) and 13% increase in phosphocreatine (P = 0.009 for pre- vs. post-stress; P = 0.004 for MSUS vs. control) concentrations. While ascorbate concentration in striatum was reduced by acute swim stress in the control offspring (−30%, P = 0.013 for pre- vs. post-stress), it was not modified in MSUS offspring (P = 0.389 for pre- vs. post-stress; P = 0.037 for MSUS vs. control).
Discussion
This study shows that traumatic stress in early postnatal life (MSUS) induces neurochemical alterations in the prefrontal cortex of adult mice and strikingly modifies the metabolic response to acute stress in their paternal line offspring.
In F1 adult mice, the alterations included reduced concentration of metabolites that are mainly synthesized in neurons, specifically the excitatory neurotransmitter glutamate, the inhibitory neurotransmitter GABA, and the putative neuronal marker NAA, suggesting neuronal dysfunction or neurodegeneration in the prefrontal cortex. 11 These results extend a previous report showing a reduction in NAA, glutamate and glutamine in the prefrontal cortex of rats exposed to maternal separation in the first 2 weeks of life. 22 Importantly, our observations in the prefrontal cortex parallel findings from MRS studies in the anterior cingulate cortex and other prefrontal areas of adult humans suffering from neurodevelopmental or psychiatric disorders.23–25 Particularly, meta-analysis of MRS studies of neurodevelopmental disorders including autism spectrum disorders, attention deficit hyperactivity disorder and obsessive compulsive disorder, found a general reduction of glutamate and GABA in areas of patients’ prefrontal cortex, relative to healthy individuals. 25 A reduction in NAA levels was generally observed in psychiatric disorders, most consistently in the anterior cingulate cortex, and often the decreased NAA was reversed with adequate therapy.24,26 Although MRS measurements of glutamate levels in schizophrenia patients are contradictory, there is a general consensus for an association between chronic schizophrenia and reduced glutamate in areas of the frontal cortex. 27 Likewise, the majority of MRS studies reported lower GABA concentrations in schizophrenia patients relative to healthy individuals, regardless of the brain area analysed and illness stage. 27 Also patients suffering from mood disorders display a functional impairment in the prefrontal cortex28,29 that is accompanied by reduced glutamate and/or glutamine MRS signals. 30
In addition to GABA, glutamate and NAA, also phosphorylcholine was reduced in the prefrontal cortex of early-life stress-exposed mice relative to controls. Phosphorylcholine is a water-soluble choline-containing compound involved in membrane lipid metabolism. 11 Namely, it is precursor of phosphatidylcholine and, in turn, of sphingomyelin, which is necessary for adequate myelination of axons. 31 In addition of integrating membranous myelin sheaths surrounding axons, sphingomyelin is implicated in immune responses. 32 Notably, the prefrontal cortex of mice exposed to post-natal maternal separation was shown to display a neuroinflammatory response with microgliosis. 33 However, baseline low-grade cortical inflammation in MSUS-exposed mice remains to be demonstrated. Overall, these data in adult mice exposed to MSUS integrate well with previous observations of prefrontal cortex integrity loss and a possible inflammatory state after early chronic life stress.
Neurochemical modifications elicited by MSUS exposure were observed only in prefrontal cortex in F1 mice. These results are also consistent with the observation that early-life stress reduces the number of parvalbumin-containing interneurons in the prefrontal cortex but not the hippocampus of the stress-exposed mice in adulthood.34,35 The absence of metabolic alterations in other brain regions contrasts with the fact that other phenotypes are observed across the brain, i.e. impaired synaptic plasticity in hippocampus 15 and altered serotoninergic signalling in different brain areas 36 in MSUS mice. This may suggest that the observed neurochemical modifications are not contributing to these phenotypes. Likewise baseline neurochemical profiles were not affected in any tested brain area in the F2 offspring. But because the F2 offspring does display many behavioural alterations including depressive-like symptoms, antisocial behaviours, impaired memory and altered risk assessment in adulthood,10,13,15,36 we explored the comprehensive neurochemical profile following an acute swim stress challenge. This indeed revealed broad modifications in the neurochemical profile in MSUS offspring.
Acute stress is known to prompt an increase in norepinephrine and glucocorticoids thereby rapidly modulating glutamatergic neurotransmission. It stimulates glutamate release in the hippocampus and prefrontal cortex, which results in learning facilitation. 37 In line with this, we observed a prominent increase of glutamate concentration in the prefrontal cortex of F2 MSUS mice after acute swim stress. In a study at low magnetic field (4.7 T), Kim et al. 38 reported similar variation in glutamate levels within the rat cortex and hippocampus immediately after 1 h immobilisation stress. Stimulation of glutamatergic neurotransmission is energetically demanding and requires increased glucose metabolism in neurons and astrocytes, namely to fuel glutamate synthesis, restore membrane potentials, clear synaptic glutamate and convert it into glutamine. 39 To match this glucose demand, stress simultaneously increases blood glucose levels and stimulates brain glucose utilisation, 37 which likely results in lactate production and release from the brain parenchyma. Indeed, stress is known to increase lactate release in the medial prefrontal cortex, hippocampus and striatum.40,41 After the stress event, the lactate in the tissue can be avidly consumed by neurons for energy production and production of neurotransmitter amino acids. 42 As expected in F2 animals, lactate levels after stress were generally lower than in the baseline scan.
Stress caused a general reduction of ascorbate (MRS-detected signal includes both oxidised and reduced forms) and a concomitant increase of glutathione levels in the prefrontal cortex. In line with this, administration of ascorbate to cultured neurons results in its rapid intracellular oxidation to dehydroascorbate and decreased glutathione levels. 43 Although the fate of ascorbate in the post-stress period is not evident, its reduction may result in stimulation of neuronal glycolysis. In fact, Cisternas et al. further showed that exposing cultured neurons to ascorbate inhibits glycolysis, stimulates glucose oxidation through the pentose phosphate pathway (probably to maintain redox homeostasis), and favours lactate utilisation. 43 In sum, the reduction of ascorbate levels is consistent with a stress-induced stimulation of brain metabolism, leading to a reduction of energy substrates such as lactate and alanine, and accumulation of phosphocreatine after stress.
The post-stress readjustment of brain energy metabolism to basal rates resulted in an excess of available energy reflected by higher phosphocreatine-to-creatine ratios in the prefrontal cortex and hippocampus. Delayed effects (in the range of hours) of the glucocorticoids released upon stress include regulation of gene expression, namely reduction of neuronal expression of genes involved in ATP synthesis, and of lactate dehydrogenase B that converts lactate into pyruvate for further mitochondrial oxidation. 37 Reducing ATP synthesis and lactate oxidation in the brain would later contribute to a normalisation of phosphocreatine, creatine and lactate concentrations. MRS in the striatum of acute stress-exposed F2 mice was performed 90 min after the acute swim stress challenge. Interestingly, levels of creatine and phosphocreatine in the striatum were modified after acute swim stress in F2 MSUS mice but not in controls. Possibly, the late effects of glucocorticoids on genes that regulate energy metabolism already took place at this time in controls but not yet in F2 MSUS mice. In agreement with this interpretation, we have identified MSUS-induced cerebral epigenetic changes involving histone post-translational modifications at the mineralocorticoid receptor gene, as well as decreased mineralocorticoid receptor expression in the offspring of MSUS-exposed mice. 10 In opposition, levels of ascorbate were reduced by acute swim stress in the striatum of controls but not of F2 MSUS mice, suggesting that the proposed stress-induced redox response that regulates neuronal metabolism (see above) is blunted in the striatum of F2 MSUS mice.
NAA was reduced after the acute swim stress episode in the dorsal hippocampus and the prefrontal cortex of control mice but not in the offspring of MSUS-exposed mice. Aside from an acute metabolic response to stress, NAA reductions may also result from impaired mitochondrial integrity in neurons and degeneration of neuronal processes. 11 A substantial retraction of dendritic branches was found in the medial prefrontal cortex after a single episode of forced swim, 44 and loss of excitatory synapses (PSD95-positive) may also occur in the hippocampus after acute stress. 45 NAA was also found to be specifically reduced in the anterior cingulate cortex and in the hippocampus of mice one day after exposure to fear conditioning, 46 and in the hippocampus of adult rats after repeated forced swim for 28 consecutive days. 47 Interestingly, the absence of a stress-induced decline in NAA levels in the prefrontal cortex and hippocampus in MSUS offspring alludes to an improved response to stress. This improvement suggests a beneficial adaptation in MSUS-exposed offspring, which may confer resistance to stress-induced neurodegeneration.
Glutamate levels in the prefrontal cortex of both MSUS-exposed offspring and controls were increased, consistent with the effects of acute swim stress. Curiously, this was accompanied by an increase in glycine in the offspring of MSUS-exposed mice, but not in controls. Glycine is as co-agonist of N-methyl-D-aspartate (NMDA) receptors, and tonic activation of the glycine modulatory site has an important role in the response to stress.48,49 Although stress can increase the potency of glycine at the NMDA receptor, 50 its involvement in inhibitory neurotransmission through glycine receptors within the prefrontal cortex is notably robust.51,52 Increased levels of glycine in the prefrontal cortex upon stress can thus counterbalance excessive excitatory glutamatergic transmission, positing a further mechanism of adaptive protection from deleterious effects of acute stress in MSUS-exposed offspring.
Hippocampal and cortical aspartate levels were reduced in F2 MSUS mice after acute swim stress, relative to basal concentrations (pre-stress scan). Conversely, such stress-induced modifications were not observed in non-stressed controls. Aspartate is synthesised from oxaloacetate, is essential to transport reducing equivalents into the mitochondrial matrix (though the malate-aspartate shuttle) and is also a precursor for NAA synthesis is neurons. 11 While NAA can be used by oligodendrocytes to produce myelin, aspartate released extracellularly damages myelinated axons through activation of NMDA receptors in oligodendrocytes. 53 Again, the reduction of aspartate levels upon stress in the prefrontal cortex and hippocampus of F2 MSUS mice appears to be linked to protective effects contributed by higher NAA. Moreover, post-stress levels of the sphingomyelin precursor phosphorylcholine 32 tended to be higher in F2 MSUS mice than controls, which may further result in protection of myelinated axons.
Acute swim stress induced a reduction of glutamine levels in the hippocampus, possibly reflecting impaired glutamatergic transmission and reduced glutamate clearance by astrocytes that are responsible for glutamine synthesis. In line with the beneficial effect of paternal MSUS exposure, 10 the offspring of MSUS-exposed mice was devoid of glutamine alterations after stress in the hippocampus.
In conclusion, we show that acute swim stress unmasks the heritable effects of early life stress on brain metabolism. Since acute stress blunted the reduction of a neuronal health proxy, NAA concentration, in the hippocampus and prefrontal cortex in MSUS offspring only, our study demonstrates a beneficial adaptation of the stress response following paternal early life stress.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Swiss National Science Foundation (SNSF), the University Zürich, the Swiss Federal Institute of Technology (ETHZ), Roche, and the ETHZ Foundation. JMND was supported by a SNSF Ambizione grant (#148250). KG was supported by a DocForte fellowship.
Acknowledgements
The authors are grateful to Martin Roszkowski for helping with logistics of mice transport, and to Prof. Rolf Gruetter for providing access to the animal house and MR scanners of the CIBM of the EPFL, UNIL, UNIGE, HUG, CHUV and the Leenaards and Jeantet Foundations.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
KG, IMM and JMND designed the study. KG and GvS performed MSUS experiments. AC and JMND performed MRS experiments. JMND analysed MRS data. All authors interpreted results and contributed to writing the final manuscript.