
Abstract
Alterations of the renin-angiotensin system have been implicated in the pathogenesis of Alzheimer's disease. We tested the efficacy of losartan (10 mg/kg/day for three months), a selective angiotensin II type 1 receptor antagonist, in alleviating cerebrovascular and cognitive deficits in double-transgenic mice (six months at endpoint) that overexpress a mutated form of the human amyloid precursor protein (APPSwe,Ind) and a constitutively active form of the transforming growth factor-β1, thereafter named A/T mice. Losartan rescued cerebrovascular reactivity, particularly the dilatory responses, but failed to attenuate astroglial activation and to normalize the neurovascular uncoupling response to sensory stimulation. The cognitive deficits of A/T mice were not restored by losartan nor were the increased brain levels of soluble and insoluble Aβ1–40 and Aβ1–42 peptides normalized. Our results are the first to demonstrate the capacity of losartan to improve cerebrovascular reactivity in an Alzheimer's disease mouse model of combined Aβ-induced vascular oxidative stress and transforming growth factor-β1-mediated vascular fibrosis. These data suggest that losartan may be promising for restoring cerebrovascular function in patients with vascular diseases at risk for vascular dementia or Alzheimer's disease. However, a combined therapy may be warranted for rescuing both vascular and cognitive deficits in a multifaceted pathology like Alzheimer's disease.
Keywords
Introduction
In addition to amyloid-beta (Aβ) plaques, neurofibrillary tangles, synaptic loss, and glial cell activation in Alzheimer's disease (AD), the vascular pathology has become a well-recognized hallmark of the disease. 1 The latter is defined functionally by chronic cerebral hypoperfusion, and anatomically by cerebral amyloid angiopathy (CAA) and thickening of the blood vessel wall due to accumulation of extracellular matrix proteins. 2 This fibrotic process has been partly imputed to transforming growth factor-β1 (TGF-β1), 3 a known pathological molecule in cardiovascular diseases including midlife hypertension, 4 a prominent risk factor for AD in late life. 5 Cross-talks in TGF-β1 signaling and that of angiotensin II (AngII) acting through angiotensin II type 1 receptors (AT1Rs) occur, and blockers of AT1Rs currently appear as the best option to block TGF-β1 pathological responses. 4 Interestingly, epidemiological and clinical studies have reported diverging results on the benefits of antihypertensive drugs targeting AT1Rs on the incidence of AD.6–8 Hence, additional investigations are needed to clarify the impact of antihypertensive therapy on AD pathogenesis.
Antihypertensive drugs such as AT1R antagonists selectively obstruct the actions of AngII on hypertension-linked AT1R sparing those on type 2 receptors (AT2R), with both receptor types being present in brain tissue. In AD mouse models, a range of AT1R antagonists showed efficacy in improving cognition with or without decreasing amyloidosis.9–13 However, these studies were conducted in Aβ1–40-infused mice or in transgenic mice overexpressing mutated forms of the human amyloid precursor protein (APP) models that exclusively recapitulate the Aβ pathology of AD. Studies have yet to investigate the possibility of reversing cerebrovascular and mnemonic impairments in mice that combine the amyloidogenic pathology with the vascular comorbidity pertaining to TGF-β1, 14 as seen in AD patients.3,15 It is therefore important that the reported benefits of selective AT1R antagonists be further assessed in a mouse model with higher relevance to the multifaceted human pathology.
Here, we examined the benefits of losartan (10 mg/kg/day, three months treatment starting at three months of age) in bitransgenic A/T mice concurrently overproducing Aβ and TGF-β1, 14 integrating the comorbid factor of cerebrovascular pathology to that of enhanced amyloidosis. 1 Specific AD landmarks were evaluated such as cerebrovascular reactivity, functional hyperemia, astrocyte activation, amyloidosis, and spatial learning and memory. Pial vessels and cerebral protein levels of specific markers were also evaluated. Our findings underscore the efficacy of losartan in improving cerebrovascular function, despite persisting high levels of Aβ and TGF-β1, astroglial activation, neurovascular uncoupling, and cognitive deficits. Together, our results bring to light the therapeutic value of targeting the RAS for functional improvement of a severely impaired brain vasculature as seen in AD and in patients with vascular diseases at risk for vascular dementia.
Methods
Animals
Bitransgenic A/T mice 14 co-overexpress a mutated form of the human APP (APPSwe,Ind) driven by the platelet-derived growth factor-β promoter (line J20) 16 and a constitutively active form of TGF-β1 driven by the glial fibrillary acidic protein (GFAP) promoter (line T64), 17 both on a C57BL/6J background. Identification of transgenes was done by touchdown PCR using tail-extracted DNA. 14 Three-month-old A/T mice and wild-type (WT) littermates (body weight, ∼30–50 g) were used for this study and were treated or not for a period of three months (six-months old at endpoint) with losartan at a dose previously shown to rescue spatial learning and memory in single transgenic APP J20 mice (L, 10 mg/kg/day, in drinking water). 13 All animals were randomly assigned and males and females were distributed equally in each group. Water and chow were available ad libitum. No significant difference in body weight between the groups was observed during the treatment period (weight gain in g: WT: 8.6 ± 1.2 WTL: 6.2 ± 1.5 A/T: 5.6 ± 1.5 A/TL: 4.4 ± 1.6; L referring to losartan), and mice did not display any apparent signs of side effects due to therapy. Experiments were approved by the Animal Ethics Committee of the Montreal Neurological Institute and complied with local and national regulations in accordance to the Canadian Council on Animal Care Institute, and the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
Morris water maze
Spatial learning and memory were tested in a modified version of the Morris water maze. 18 The paradigm consisted of eight days of two training sessions in a 1.4 m circular pool filled with opaque water (18 ± 1 ℃) located in a quiet room with distal visual cues, followed by a probe trial on day 9. Six-month-old mice (n = 8–12 per group) were first familiarized with the test by searching a visible platform (days 1–3, 3 trials/day, 60 s/trial), followed by a five-day training session, whereby mice have to learn the location of a hidden platform submerged ∼1 cm below the surface of the water (days 4–8, 3 trials/day, 90 s/trial). Platform location and distribution of visual cues were changed between the two training sessions. A 45 min inter-trial interval was respected. Spatial memory was evaluated during the probe trial (platform removed) on day 9 (60 s/trial, 1 trial). Escape latencies and probe trial parameters (percent time spent and distance traveled in the target quadrant where the platform used to be located, number of platform crossings above the previously located platform and swim speed) were recorded with the 2020 Plus tracking system and Water 2020 software (Ganz FC62D video camera; HVS Image, Buckingham, UK). 14 Swim speeds were the same for all groups (data not shown). Mice were kept warm with a heating lamp to avoid hypothermia. Subsequent experiments started two days later.
Cerebral blood flow
The evoked cerebral blood flow (CBF) response in the somatosensory cortex induced by whisker stimulation (neurovascular coupling or functional hyperemia) was measured with laser Doppler flowmetry (LDF, Transonic Systems Inc., Ithaca, NY). Mice (n = 4 mice/group) were anesthetized with ketamine (85 mg/kg, intramuscularly (i.m.); Bioniche, Belleville, ON, Canada) and xylazine (3 mg/kg, i.m.; Haver, Etobicoke, ON) and fixed in a stereotaxic frame. The LDF probe was placed on the bone of the left barrel cortex after thinning to translucency with a dental drill. An electric toothbrush was used to stimulate (8–10 Hz, 20 s) the whiskers on the right snout (for details, see Ongali et al. 14 ). Body temperature was kept stable at 37 ℃ with a heating pad. Four to five stimulations were performed for each mouse with ∼30 s interval, and the CBF responses were acquired and expressed as percent average increase relative to baseline. The experimenter was blind to the identity of the mouse and the entire procedure lasted about 20 min/mouse.
Mean arterial blood pressure
Mean arterial blood pressure (MAP) was measured in a subset of mice (n = 3–4/group) from each group except WT-treated mice, as we previously showed that losartan does not affect MAP in these mice. 13 Mice were anesthetized with isoflurane (5% in medical air during 2-min induction, 1.5–2%), and a small incision was performed under local analgesia (2% xylocaine) from the third superior part to the midline of the hindpaw, for insertion of a small catheter (SAI, #MAC-01) in the femoral artery. Mice were then placed in a restraining cylinder, anesthesia was switched off and MAP, heart rate, and body temperature were acquired for a 30-min period (Powerlab, ADinstruments, St-Laurent QC, Canada). Losartan did not alter these parameters and, particularly, MAP (in mmHg), WT: 104.8 ± 5.9, A/T: 108.8 ± 4.8, A/TL: 99.5 ± 2.3.
Tissue collection and preparation
Upon completion of in vivo experiments, mice (n = 4 mice/group) were sacrificed by cervical dislocation for functional reactivity of the posterior cerebral artery (PCA) (see below). The remaining vessels from the circle of Willis and their cortical branches in the pial membrane were isolated, frozen on dry ice, and stored at −80 ℃ together with the cortex and hippocampus from one hemibrain for subsequent use for ELISA and Western blot experiments. The other hemibrain was immersion-fixed overnight in 4% paraformaldehyde (PFA in 0.1 M phosphate buffer (PB), pH 7.4; 4 ℃), cryoprotected (30% sucrose in 0.1 M PB; 4 ℃) overnight, frozen in isopentane (−45 ℃) and stored (−80 ℃) until sectioning (25 µm-thick sections) on a freezing-microtome.
Vascular reactivity
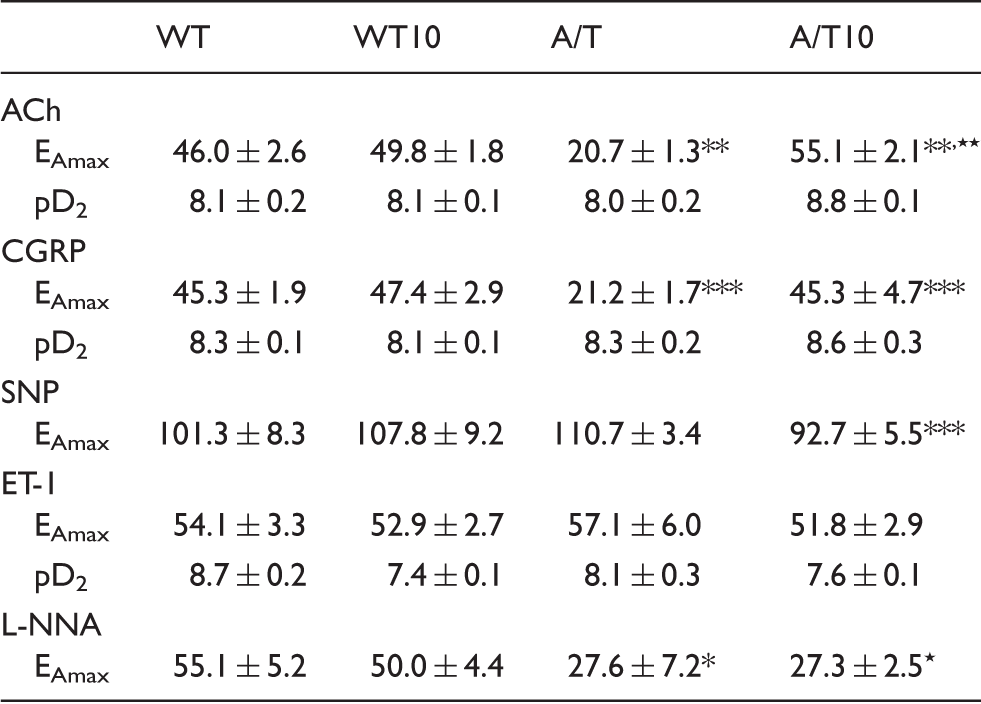
Vascular reactivity was measured in cannulated and pressurized PCA (60 mm Hg) segments (40–70 µm of intraluminal diameter) superfused with Krebs solution (37 ℃) using online videomicroscopy. 19 Dilations to acetylcholine (ACh; 10−10–10−5 M), calcitonin gene-related peptide (CGRP; 10−10−10−6 M), or to the NO donor sodium nitroprusside (SNP, 10−10–10−4 M) were measured on vessels slightly pre-constricted with serotonin (5-HT; 2 × 10−7 M). Contractions induced by endothelin-1 (ET-1; 10−10–10−6 M) and by inhibition of nitric oxide (NO) synthesis by superfusion of the NO synthase (NOS) inhibitor Nω-nitro-L-arginine (L-NNA; 10−5 M, for 35 min) were performed on vessels at basal tone. Changes in vessel diameter are presented in percent change from the basal or pre-constricted tone. Results were plotted as a function of agonist concentration or time for L-NNA superfusion. The maximal response (EAmax) and half maximal effective concentration (EC50 value or pD2 = −log EC50) were determined to compare agonist efficacy and potency, respectively.
ELISA measurement of Aβ species
Insoluble Aβ1–40 and Aβ1–42 levels were measured in cortex and hippocampus (two areas involved in cognition) obtained from one hemibrain using an enzyme-linked immunosorbent assay (ELISA; BioSource International, Camarillo, CA, USA), as described by the manufacturer (for details, Ongali et al. 14 ). Data are presented in micrograms per gram (µg/g) of protein in formic acid insoluble Aβ-fraction.
Western blot
Cortical and hippocampal protein (∼20 µg of ELISA supernatant, four mice/group) were loaded onto a 15% Tris/tricine SDS PAGE and transferred to nitrocellulose membranes for the detection of total levels of soluble Aβ species and full-length APP using a mouse anti-Aβ1-16 antibody (6E10, 1:1000, Covance, Emeryville, CA, USA). Hippocampal proteins (∼35 µg of ELISA supernatant) were also used for the detection of mouse TGF-β1 and AT4R using a rabbit anti-TGF-β1 (1:200; Cell Sciences, Canton, MA) or anti-AT4R (1:500; Chemicon, Temecula, CA), and AT1R and AT2R were detected using protein G-affinity purified monoclonal mouse antibodies (1:200 20 ). Cerebral blood vessel extracts were sonicated in Laemmli buffer (62.5 mM Tris-HCl, pH = 6.8; 2.35% sodium dodecyl sulfate (SDS); 100 mM DTT; 10% glycerol; 1 mM EDTA and 0.001% bromophenol blue) and protein concentration was measured by the method of Lowry. Proteins (∼20 µg) were also used for the detection of rabbit anti-AT4R (1:500; cat#AB9294, Chemicon, Temecula, CA) and mouse anti-AT1R and -AT2R (1:200 20 ). Membranes were subsequently incubated (1 h) with species-specific horseradish peroxidase-conjugated secondary antibodies (1:2000; Jackson ImmunoResearch, West Grove, PA, USA) in TBST blocking buffer (50 mM Tris-HCl, pH = 7.5; 150 mM NaCl; 0.1% Tween 20) containing 5% skim milk, and visualized with enhanced chemiluminescence (ECL Plus kit; Amersham, Baie d'Urfé, QC, Canada) using phosphorImager (Scanner STORM 860; GE Healthcare, Baie d'Urfé, QC, Canada). Band intensity was quantified by densitometry with Scion Image (Molecular Dynamics, Sunnyvale, CA, USA).
Histo- and immunohistochemical stainings
Thioflavin S (1%, 8 min) staining of mature, dense-core Aβ plaques was performed on thick sections (three sections per mouse, five mice per group), which were observed under a Leitz Aristoplan light microscope using epifluorescence and an FITC filter (Leica). Pictures were taken with a Nikon digital camera (Coolpix 4500), and were used for quantification using MetaMorph 6.1r3 (Universal Imaging). Thick sections were also immunostained with a rabbit anti-GFAP antibody, a marker of astrocyte activation (1:1000; DAKO), and visualized with a donkey anti-rabbit cyanin 2 (Cy2)-conjugated secondary antibody (1:400; Jackson ImmunoResearch, West Grove, PA, USA). The areas of interest (cingulate and somatosensory cortex, hippocampus) were manually outlined for percent area occupied by thioflavin S-positive staining.
Statistical analysis
Data are expressed as means ± SEM and were analyzed by two-way (genotype and treatment as variables) analysis of variance (ANOVA) or repeated measures ANOVA (for the cerebrovascular reactivity and Morris watermaze analysis), followed by Newman–Keuls post hoc multiple comparison test (Statistica Academic, Tulsa, OK, USA). Student's t-test was used to determine significance between two groups (GraphPad Prism 4, San Diego, CA, USA). A p value ≤ 0.05 was considered significant.
Results
Losartan improved cerebrovascular reactivity, but not neurovascular coupling
Isolated PCA of A/T mice displayed significantly reduced dilatory responses to ACh and CGRP relative to WT controls (Figure 1(a)), in line with previous data.
14
Receptor desensitization did not account for these alterations since agonist potencies at vascular receptors were comparable between WT and A/T mice (Table 1). Baseline NO synthesis or NO bioavailability was also significantly decreased compared to WT controls, as shown by the smaller diameter decrease during NOS inhibition with L-NNA superfusion (Figure 1(a), Table 1). In contrast, dilations induced by the NO donor SNP were not altered in A/T mice, indicative of preserved integrity of the smooth muscle, as also demonstrated by the intact contractile response to ET-1 (Figure 1(a), Table 1). Losartan fully normalized the ACh- and CGRP-mediated dilatory responses in A/T mice, but it failed to rescue baseline NO bioavailability, which is essential for maintaining resting tone (Figure 1(a), Table 1). Despite a small, albeit non-significant, rightward shift in the concentration-dependent contraction induced by ET-1 in both WT and A/T mice, the maximal response was unaltered by losartan (Figure 1(a), Table 1). Dilatations induced by SNP were comparable in all groups. Losartan did not affect vasomotor responses in WT mice (Figure 1(a), Table 1).
Losartan (L) normalized vasodilatory function, but not neurovascular coupling, in A/T mice. (a) The impaired cerebrovascular dilatations to ACh and CGRP in A/T mice (▾) were normalized by L (▿), but this was not accompanied by a recovery of baseline NO availability during NOS inhibition (L-NNA, 10−5 M). Dilatations induced by the NO donor SNP and contractile response to ET-1 remained unaltered in both treated- and untreated-A/T mice. Error bars represent SEM. n = 4 for each group. *p < 0.05, **p < 0.01, ***p < 0.001 when compared to WT, and ★p < 0.05, ★★p < 0.01, ★★★p < 0.001 when compared to treated-A/T mice using repeated measures ANOVA with genotype and treatment as the two variables, followed by Newman–Keuls post hoc test. (b) The impaired hyperemic response to whisker stimulation in A/T mice was not restored by L and remained reduced compared to the WT controls, as measured by LDF (n = 4 mice/group). Values represent the percent increase of CBF response relative to baseline. Error bars represent SEM. *p < 0.05 using two-way ANOVA followed by Newman–Keuls post hoc test. Effect of 10 mg/kg/day losartan on cerebrovascular responses of A/T mice. Note: Data are means ± SEM (n = 4 mice per group) and are expressed as the agonist maximal response (EAmax) or potency (pD2, −[logEC50]). EAmax is the percent maximal dilatation to ACh, CGRP, and SNP or the percent maximal diameter decrease to ET-1, 5-HT, or after 35 min inhibition with 10−5M L-NNA. *,⋆p < 0.05, **,⋆⋆p < 0.01, ***,⋆⋆⋆p < 0.001 when compared to untreated WT controls (*) or A/T mice (⋆) by two way ANOVA followed by Newman–Keuls post hoc multiple comparison test.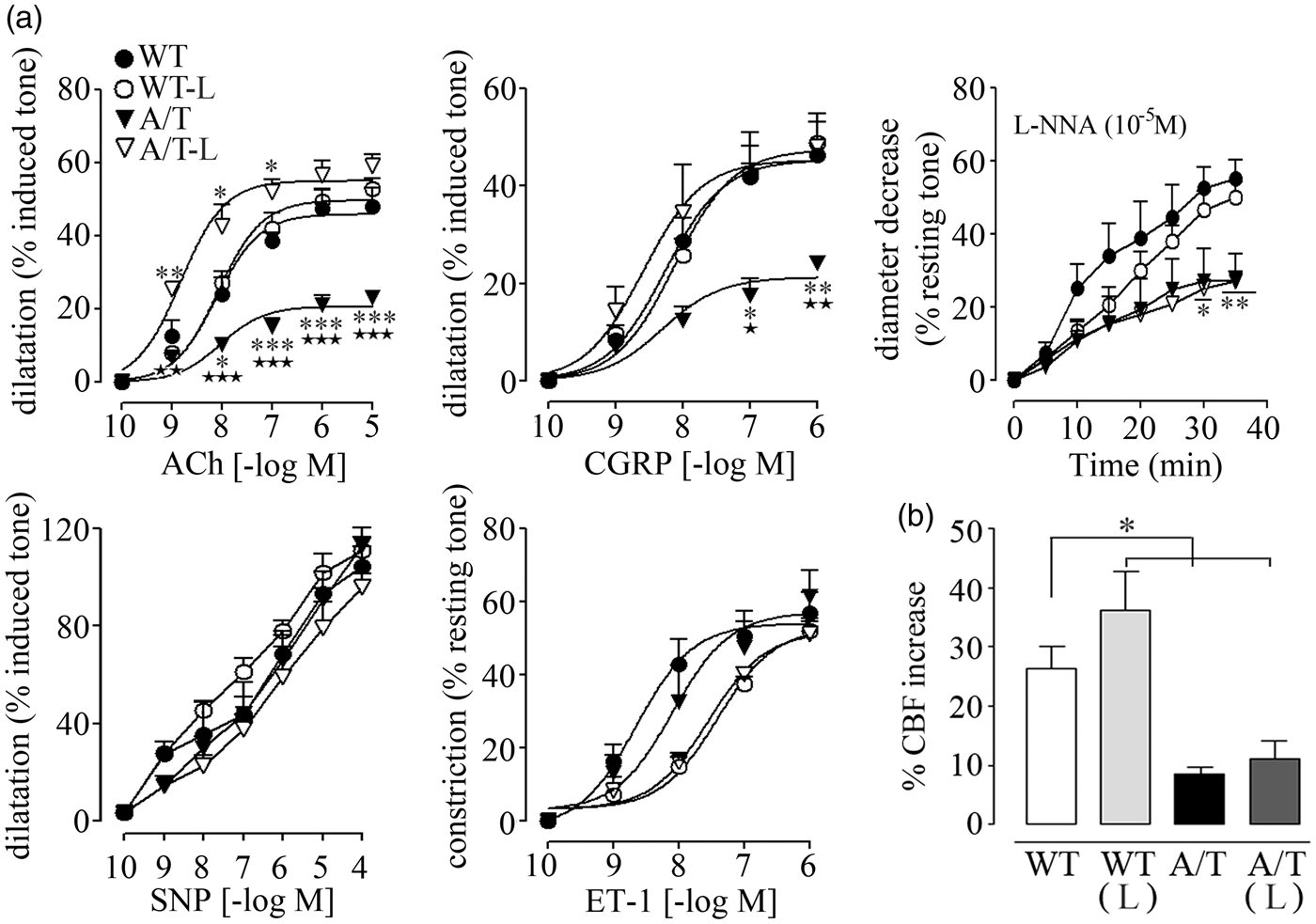
Upon neuronal activation, the evoked increase in CBF relies on concerted interactions between neurons, astrocytes, and vascular cells that compose the neurovascular unit. The latter is altered in AD 21 and in A/T mice. 14 Accordingly, the neurovascular coupling response to whisker stimulation was significantly reduced in A/T mice compared to WT controls (WT: 26.4 ± 3.7% vs. A/T: 8.6 ± 1.0%, p < 0.01, Figure 1(b)), and losartan failed to significantly improve this evoked response (11.0 ± 3.1%, p < 0.01; Figure 1(b)).
Unchanged vascular protein levels of Ang II receptors and oxidative stress marker SOD2
In pial vessels, Western blot analysis showed that chronic losartan treatment did not alter AT1R and AT2R protein levels (Figure 2), whereas AT4R were undetectable in brain vessels under our conditions. Similarly, the cerebrovascular levels of the antioxidant enzyme SOD2 were comparable among all groups (Figure 2).
Losartan (L) and protein levels in cerebral vessels from A/T mice. Protein levels of AT1R, AT2R, and SOD2 were not altered in pial vessels from A/T mice compared to WT controls, and L had no effect on any of these proteins in either A/T or WT mice, as measured by Western blot. Actin was used as a reference for loading. Error bars represent SEM (n = 4 mice/group).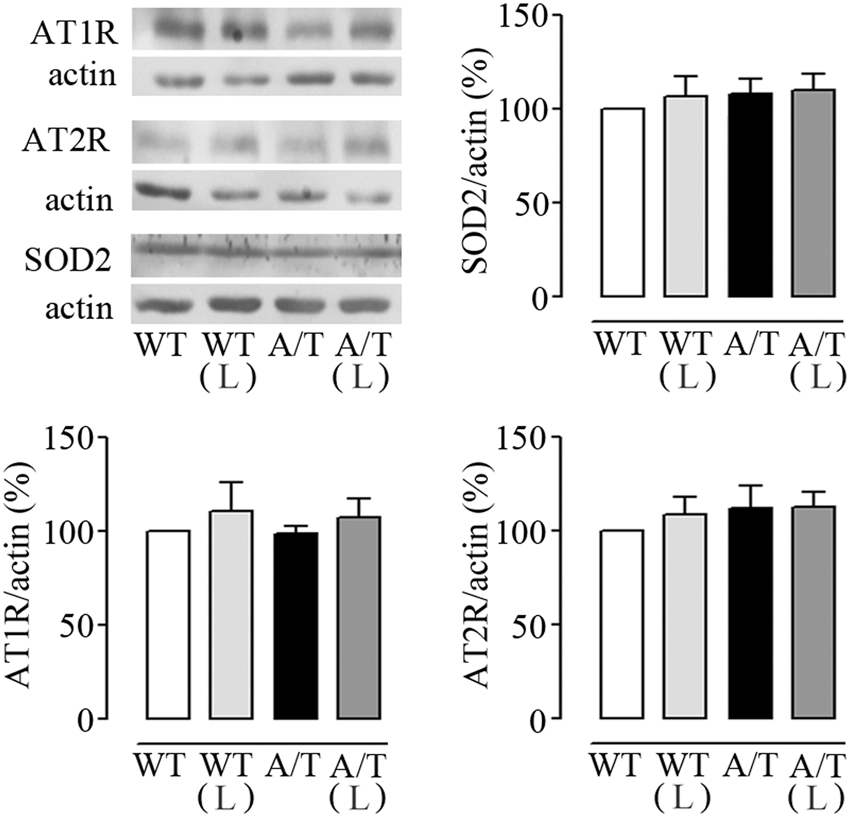
Losartan did not attenuate astroglial activation
As previously reported,
14
A/T mice displayed increased GFAP immunoreactivity in cortex and hippocampus compared to WT (p < 0.001), an indication of reactive astrocytes and an increased inflammatory response as seen in the brain of AD patients. Losartan exerted no reducing effect on this astroglial activation in either region (Figure 3).
Losartan (L) and astrogliosis in A/T mice. The percent area occupied by GFAP-immunopositive astrocytes in cortex (Co) and hippocampus (Hi) of A/T mice compared to WT littermates remained elevated despite L treatment. (n = 4 mice/group). ***p < 0.001 using two-way ANOVA followed by Newman–Keuls post hoc test.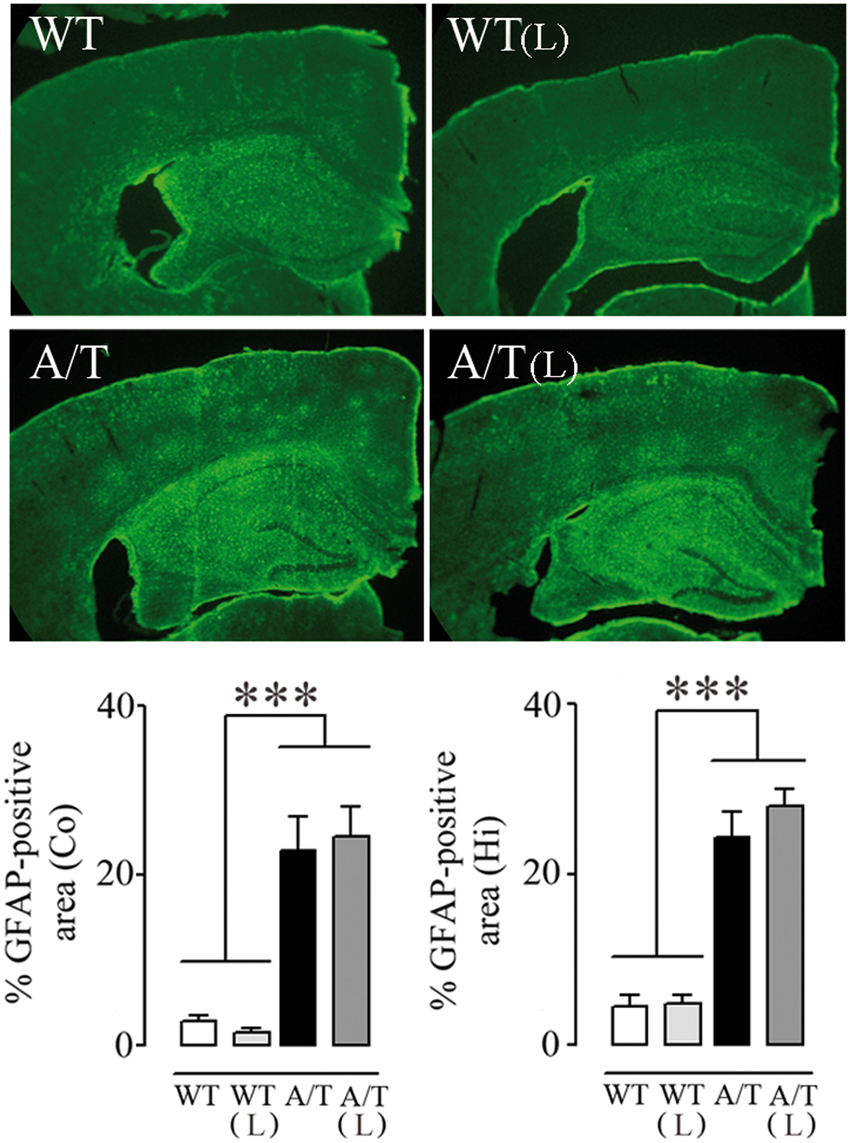
Losartan and amyloidosis
A/T mice displayed high levels of soluble and insoluble Aβ1–40 and Aβ1–42 species in cortex and hippocampus (Figure 4), and these remained unchanged after losartan therapy as measured by Western blot (Figure 4(a)) or ELISA (Figure 4(b)). Similarly, Aβ plaque load was not reduced by losartan as determined by thioflavin-S staining of dense-core Aβ plaques (Figure 4(c)). These results show that losartan did not alter the progression of the amyloidogenic process in A/T mice.
Losartan (L), amyloidosis, and TGF-β1 expression in A/T mice. (a) Western blot analysis with 6E10 antibody revealed no effect of L on soluble Aβ species in cortex and hippocampus of A/T mice. Actin was used as a reference for loading. (b) Insoluble Aβ1–40 and Aβ1–42 levels in L-treated-A/T mice remained unchanged as assayed in half-brain of cortex and hippocampus by ELISA. (c) The surface area occupied by Aβ plaque load was not changed among groups as determined by thioflavin-S staining of dense-core Aβ plaques in both cortex and hippocampus. Error bars represent SEM (n = 4 mice/group). *p < 0.05, **p < 0.01, ***p < 0.001 for comparison to A/T mice using two-way ANOVA followed by Newman–Keuls post hoc test.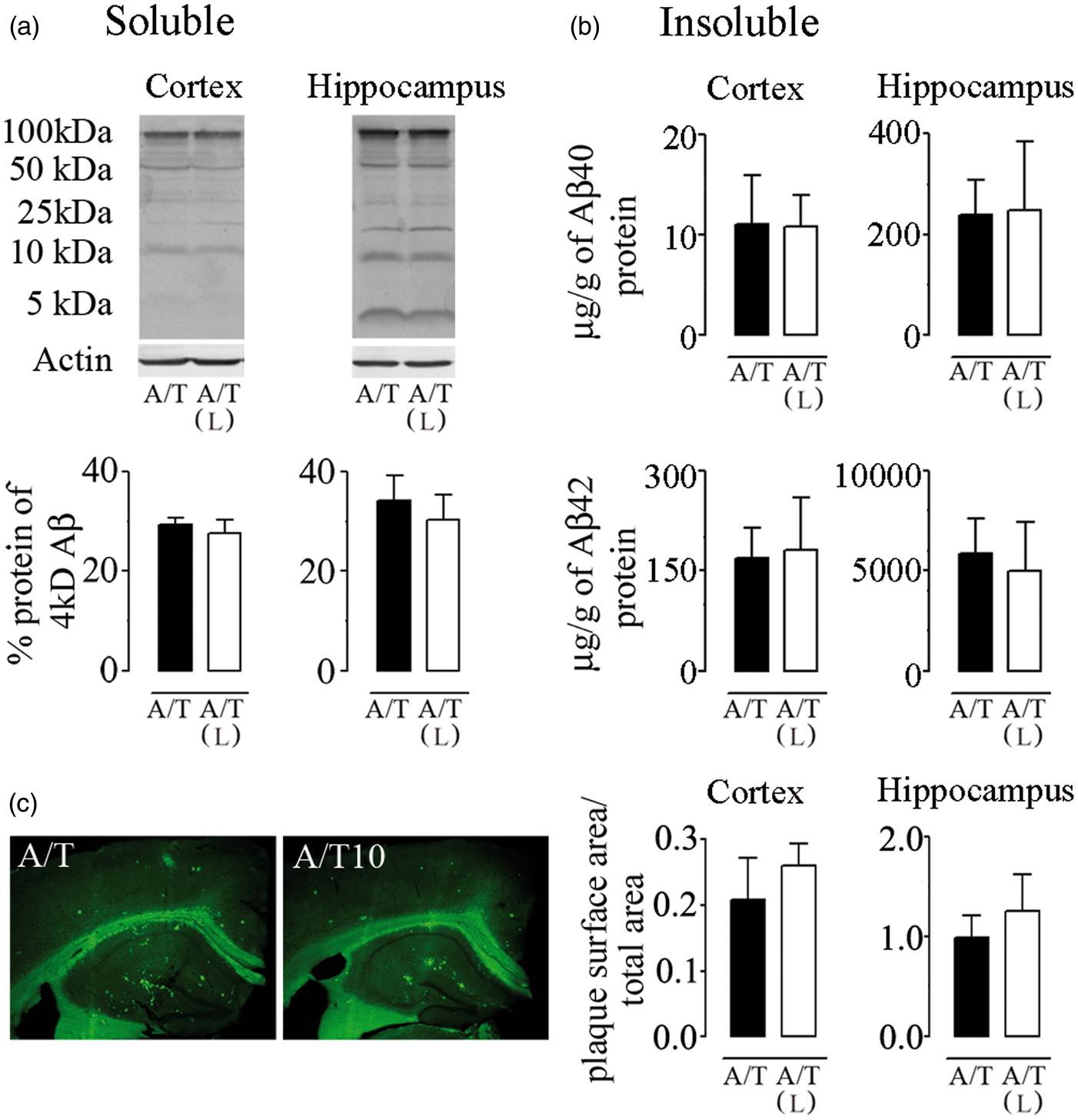
Losartan and spatial learning and memory
A/T needed slightly, albeit significantly, more time than WT mice to locate the visible platform (days 1–3) in the Morris water maze despite comparable time to find the platform on the first day of hidden platform testing (day 4), suggesting no major visual and motor disabilities, or lack of motivation. However, A/T mice were severely impaired in finding the hidden platform in subsequent days, as depicted by their elevated escape latencies compared to age-matched WT controls (significant on day 8, Figure 5(a)). A/T mice also displayed memory deficits in the probe trial as illustrated by the significantly decreased percent time spent and distance traveled in the target quadrant, and by the few platform crossings over the previously located platform (Figure 5(b)). These findings are in line with our previous studies.
14
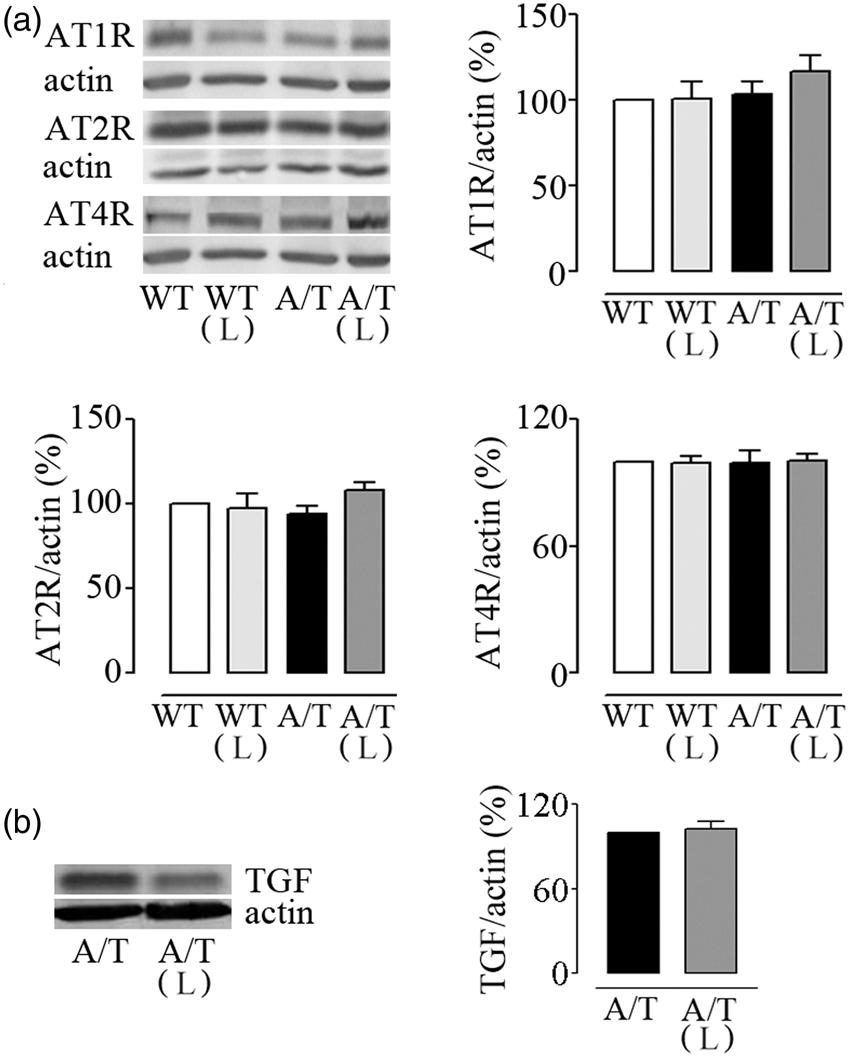
Losartan did not exert any beneficial effects on spatial learning measured with the escape latencies (Figure 5(a)), and it failed to significantly improve spatial memory, as depicted by the percent time spent and distance travelled along with the persistent decreased platform crossings (Figure 5(b)). Hippocampal AT1R, AT2R, and AT4R protein levels were similar between groups, as evaluated by Western blot (Figure 6(a)). In addition, losartan had no effect on hippocampal protein levels of TGF-β1 (Figure 6(b)).
Losartan (L) on spatial learning and memory in A/T mice. (a) Visible platform session (days 1–3) demonstrated slightly albeit significantly longer latency only on day 3 in A/T mice compared to WT controls. A/T mice (▾) displayed impaired learning during hidden platform testing compared to aged-matched wild-type (WT) littermates (•). L did not improve the learning deficit in A/T-treated mice (▿) nor change that of the WT-treated mice (○). (b) L did not exert any effect on spatial memory relative to the untreated A/T mice as evidenced by the percent time spent and distance travelled in the target quadrant, as well as fewer crossings over the previously located platform during the probe trial. Error bars represent SEM (n = 8–12 mice/group). *p < 0.05, **p < 0.01, ***p < 0.001 using repeated measures ANOVA with genotype and treatment as the two variables, followed by Newman–Keuls post hoc test. Losartan (L) and hippocampal protein levels in A/T mice. (a) Hippocampal protein levels of AT1R, AT2R, and AT4R were not altered in A/T mice compared to WT controls. In addition, L had no effect on AT1R proteins in either A/T or WT mice. (b) L did not counter the overexpression of TGF-β1 in treated A/T mice. Actin was used as a reference for loading (n = 4 mice/group). Error bars represent SEM.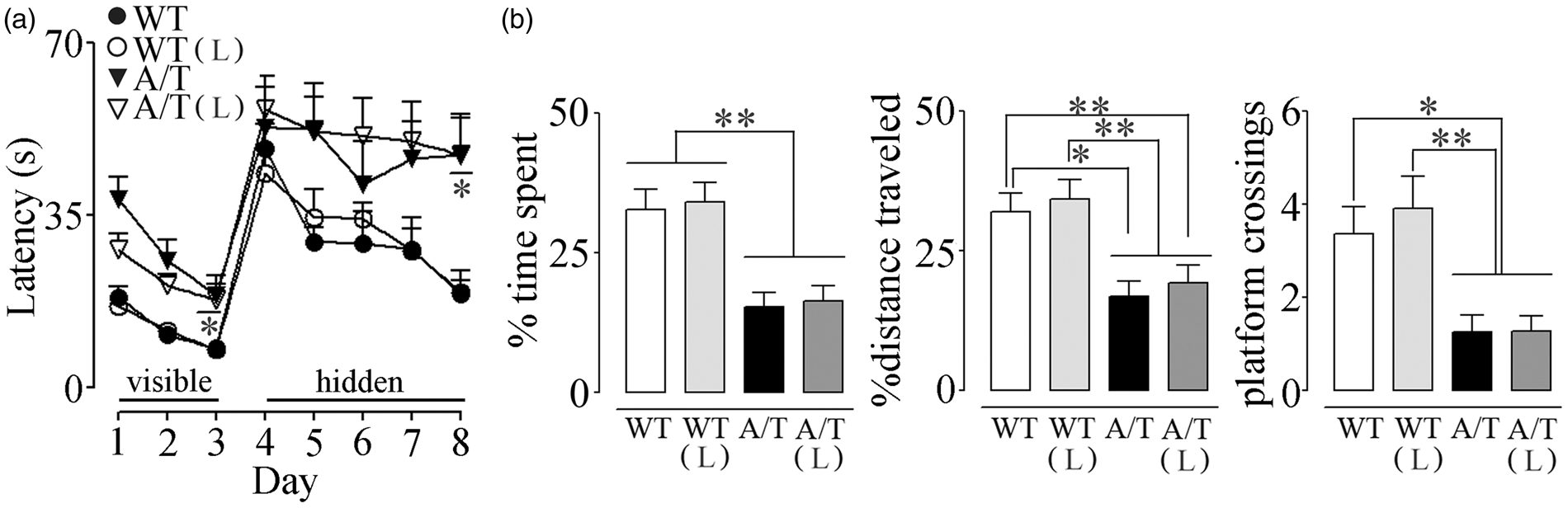
Discussion
The present study highlights the unique capacity of losartan to improve dilatory function in cerebral arteries from A/T mice with combined Aβ and TGF-β1 pathology, despite a lack of recovery of baseline NO production or bioavailability in the vessel wall. Losartan-related benefits occurred despite neurovascular uncoupling, astroglial activation, and a persistent amyloid pathology. Moreover, cerebrovascular improvements were not accompanied by recovery of spatial learning and memory in A/T mice. Compared to the benefits of losartan on both cerebrovascular and cognitive deficits in APP mice, 13 these findings draw attention to the potential limitations of losartan therapy in AD patients with a pathology not limited to Aβ but also encompassing TGF-β1-mediated inflammation and cerebrovascular fibrosis.
Losartan and cerebrovascular function
A most fascinating finding from our study was that losartan restored endothelium-dependent dilatations to ACh and CGRP in A/T mice. This recovery was achieved despite a persistent deficit in baseline NO production or bioavailability, a response previously found to be rescued by losartan 22 or valsartan 23 in hypertensive animal models and, recently, in singly transgenic APP mice. 13 The fully recovered ACh and CGRP-mediated dilations may suggest normalization of receptor-mediated NO-dependent and -independent responses. Alternatively, other dilatory pathways may have benefited from losartan therapy. Indeed, ACh-induced cerebrovascular dilatation occurs not only through m5 muscarinic ACh receptor-mediated activation of NO release, 24 but also via hydrogen peroxide (H2O2) derived from endothelial NOS activity 25 or endothelial TRPV4 channel activation. 26 Similarly, CGRP-induced cerebral dilatations are primarily mediated by smooth muscle KATP 27 and Ca2+-activated K+ (KCa2+) channels, 28 which function can be improved by losartan in conditions of Aβ pathology, 13 diabetes type 2, 29 renal failure, 30 or following AngII inhibition. 31
Aβ-induced cerebrovascular dysfunction has been ascribed to increased reactive oxygen species, particularly
The neurovascular coupling response to sensory stimulation in A/T mice remained impaired after losartan therapy, which contrasted with its rescuing effects in APP mice. 13 This evoked hemodynamic response by activated thalamocortical (glutamatergic) afferents requires astroglial signaling to blood vessels. 39 Hence, losartan's failure to reduce astrogliosis in A/T mice despite the reported anti-inflammatory effects of AT1R including telmisartan in Aβ1-40-infused mice 12 or adult C57BL/6J male mice subjected to bilateral common carotid artery stenosis 40 may point to dysfunction at this level of the tripartite neurogliovascular unit. These findings further underscore the inability of losartan to fully counteract the synergistic pathological effects of combined high levels of TGF-β1and Aβ.
Losartan and cognitive function
Studies with losartan have yielded conflicting results related to its ability to confer protection against the onset of cognitive dysfunction. In our study, losartan failed to restore spatial learning and memory in adult A/T mice, in line with a recent study in aged eprosartan-treated triple transgenic AD mice. 41 However, neuroprotective effects on cognition have been found in Aβ1–40-injected mice 10 and in adult and aged J20 APP mice. 13 Similar benefits were observed with other AT1R antagonists such as olmesartan 11 and telmisartan 12 in Aβ1–40-injected mice, and with valsartan 9 in Tg2576 APP mice. None of these mouse models encompassed the comorbid TGF-β1-mediated cerebrovascular fibrosis seen in AD.3,19 Our study is the first to present limitations of AT1R antagonism in improving cognitive function in a mouse model recapitulating the Aβ- and TGF-β1-related pathology, and this despite initiating therapy in the early stages into the disease process.
Chronic upregulation of TGF-β1 or its signaling, a key extracellular matrix regulator, has been documented in the brain and the cerebral vasculature of AD patients,15,42 patients with ischemic stroke, 43 hypertension and diabetes who are at increased risk for AD, 38 and in patients with hereditary small vessel diseases (SVD) with discrete regions of infarction 44 and a decline in cognitive performance. 45 While some studies suggest a neuroprotective role of a transient increase in TGF-β1, 46 chronic TGF-β1 elevations, akin to our mouse model and reminiscent of the AD or SVD pathology, have been reported to have negative outcomes. 47 In fact, a study highlighted the distinction between temporarily limited and prolonged TGF-β1 augmentation using a tetracycline-regulated gene expression system on a transgenic mouse model with inducible neuron-specific expression of TGF-β1. 48 Neuroprotective effects were observed after short-term expression of TGF-β1 but the consequences of a chronic upregulation were unfavorable. The harmful contribution of long-term TGF-β1 in mice with an underlying Aβ pathology is further supported by the lack of beneficial effects of simvastatin in A/T mice, 49 whereas the same treatment fully rescued both cognitive and cerebrovascular function in J20 APP mice. 50 Reductions in brain glucose utilization, 51 baseline brain perfusion, 52 and neurovascular coupling 53 are other consequences of chronically increased brain TGF-β1 levels that suggest alterations at multiple levels of the neurogliovascular functional unit. These alterations, when combined to those induced by high levels of Aβ through the mutated human APP, add in complexity and severity of the pathology and likely better recapitulate human AD. In this respect, the failure of losartan in rescuing cognitive performance in A/T mice concurs with the limited capacity of therapeutic strategies to interrupt AD progression or reverse its deleterious effects in patients.
Losartan-dependent attenuation of TGF-β1 increases has been involved in preventing or reversing disease progression in animal models of chronic renal insufficiency, 54 cardiomyopathy, 55 and Duchenne muscular dystrophy. 56 In our study, Western blot analysis showed that TGF-β1 overexpression persisted in the brain of losartan-treated-A/T mice. However, it is unlikely that high brain levels of TGF-β1 underlie the cognitive failure of A/T mice since learning and memory are largely preserved in singly transgenic TGF mice even at a very advanced stage of the disease (∼20 months).57,58
Other mechanisms credited for the benefits of AT1R blockade on learning and memory are the concomitant increases in memory-enhancing AT2R 59 or AT4R 13 that bind with high affinity the AngII active metabolite Ang IV60,61. Both receptor subtypes are distributed in cognitive-processing areas including the hippocampus,59,61 and their role in memory processes has been confirmed in cognitively impaired AT2R-KO 59 and AT4R-KO 62 mice. Similarly, in mice with scopolamine-induced amnesia, selective AT4R activation facilitated hippocampal synaptogenesis and spatial memory. 63 However, in the present study, AT2R and AT4R protein levels were comparable in all groups, including in losartan-treated and -untreated A/T mice, precluding conclusion on the role of these receptors in the cognitive deficits in A/T mice.
Losartan and amyloidosis
Brain levels of soluble Aβ oligomers – reportedly the most detrimental to neuronal and synaptic function 16 – and insoluble Aβ species remained unchanged with losartan therapy in A/T mice, in agreement with studies using APP transgenic mice 13 and triple transgenic mice. 41 However, other studies showed significant reductions in soluble or deposited Aβ in Aβ1–40-injected,10,12 APP/PS1, 64 and Tg2576 9 mice following administration of sartans. These findings underscore the possibility of obtaining cognitive restoration independently of amyloid reduction, as previously documented.10,13,32,50 Hence, the failure of losartan to rescue memory in A/T mice may be unrelated to its inability to counter the amyloid pathology. Additionally, studies showing cognitively unaffected elderly individuals having equivalent Aβ plaque densities than AD patients further support the notion that Aβ plaques are not good correlates of dementia. 65 Further studies would be needed to decipher any potential role of AT1R antagonists like losartan on hyperphosphorylated tau,66,67 a biomarker of AD not found in A/T mice.
Conclusion
The efficacy of losartan against cerebrovascular dysfunction of adult A/T mice highlights the possible involvement of the brain RAS in the vascular aspects of AD pathology. Nevertheless, its failure to normalize the cognitive deficits, the functional hemodynamic response, and the increased astroglial activation may suggest that a multi-therapeutic approach may be more effective or, alternatively, that a higher efficacy would require earlier therapy. Overall, our findings in A/T mice with losartan would be supportive of the benefits reported herein for losartan in ameliorating the vascular aspect of AD or in vascular dementia and SVD, where brain vasculature alterations are the key contributors to the pathology.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants (to EH) from the Canadian Institutes of Health Research (CIHR grant MOP-84275 and MOP-126001) and the Heart and Stoke Foundation of Québec, as well as a Jeanne Timmins Costello Fellowship (PP).
Acknowledgements
We thank Lennart Mucke (Gladstone Institute of Neurological Disease and Department of Neurology, UCSF, CA) and the J. David Gladstone Institutes for the hAPPSwe,Ind and TGF-β1 transgenic mouse breeders.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
PP contributed to the design of the study, performed experiments, analysis, prepared figures and wrote the manuscript.
XT helped in experiments, blinding of the experimental groups, data analysis, and in reviewing the manuscript.
HI provided AT1R and AT2R antibodies and protocols, and reviewed the manuscript.
EH designed the study, reviewed experiments, analysis and figures, and wrote the manuscript.