
Abstract
Inhibition of angiotensin II AT1 receptors protects against stroke, reducing the cerebral blood flow decrease in the periphery of the ischemic lesion. To clarify the mechanism, spontaneously hypertensive rats (SHR) and normotensive control Wistar Kyoto (WKY) rats were pretreated with the AT1 receptor antagonist candesartan (0.3 mg · kg−1 · d−1) for 28 days, a treatment identical to that which protected SHR from brain ischemia, and the authors studied middle cerebral artery (MCA) and common carotid morphology, endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) messenger RNA (mRNA), and protein expression in cerebral microvessels, principal arteries of the Willis polygon, and common carotid artery. The MCA and common carotid artery of SHR exhibited inward eutrophic remodeling, with decreased lumen diameter and increased media thickness when compared with WKY rats. In addition, there was decreased eNOS and increased iNOS protein and mRNA in common carotid artery, circle of Willis, and brain microvessels of SHR when compared with WKY rats. Both remodeling and alterations in eNOS and iNOS expression in SHR were completely reversed by long-term AT1 receptor inhibition. The hemodynamic, morphologic, and biochemical alterations in hypertension associated with increased vulnerability to brain ischemia are fully reversed by AT1 receptor blockade, indicating that AT1 receptor activation is crucial for the maintenance of the pathologic alterations in cerebrovascular circulation during hypertension, and that their blockade may be of therapeutic advantage.
Keywords
The brain maintains a constant blood flow by cerebrovascular autoregulation, compensatory hemodynamic responses in principal, larger conductance vessels, resistant arteries, and microvessels as a consequence of changes in perfusion pressure. Temporary alterations in systemic blood pressure are counterbalanced by cerebrovascular dilation or constriction (Nishimura et al., 2000b). In chronic hypertension, cerebral blood vessels increase vasoconstriction and develop morphologic changes characterized by inward eutrophic remodeling with increased wall thickening, reduced lumen diameter, and increased media–lumen ratio (Baumbach and Heistad, 1992; Ito et al., 2002) a process similar to that of peripheral principal and resistance arteries (Intengan and Schiffrin, 2001). The remodeling of larger cerebral arteries attenuates the hypertension-induced pressure increase in cerebral microvessels, a protective effect at the cost of decreased arterial compliance and a shift of the autoregulatory curve to the right as a result of decreased dilatory capacity (Nishimura et al., 2000b). During vascular occlusion, cerebral arteries have diminished capacity to dilate and compensate for decreased blood flow, and this results in increased vulnerability to ischemia and more frequent and severe strokes (Fujii et al., 1992).
Angiotensin II (Ang II), a vasoconstrictor hormone and neuromodulator, participates in the pathogenesis of hypertension through AT1 receptor stimulation not only in peripheral organs but also in the brain (Nishimura et al., 2000b). AT1 receptor stimulation results in cerebral vasoconstriction (Näveri et al., 1994) and is one important factor contributing to the shift in cerebrovascular autoregulation in the direction of higher blood pressures (Nishimura et al., 2000b). In peripheral and brain vessels, Ang II stimulates, through AT1 receptors present in endothelium and macrophages (Ito et al., 2001), smooth vascular cell growth, collagen formation, and fibrosis, leading to pathologic remodeling (Ledingham and Laverty, 2000) and increases expression of adhesion molecules, microvessel permeability, and inflammation (Ito et al., 2001). AT1 receptor blockade with a peripheral and brain receptor antagonist (Nishimura et al., 2000a) normalizes cerebrovascular autoregulation, improving arterial compliance and reducing the loss in blood flow after middle cerebral artery (MCA) occlusion in spontaneously hypertensive rats (SHR) (Nishimura et al., 2000b; Ito et al., 2002). The reduction in cerebral ischemia parallels the decrease in the size of the brain infarct and tissue swelling (Nishimura et al., 2000b; Ito et al., 2002). Reduction in blood pressure is not sufficient for protection against ischemia, as evidenced by the failure of other medications of similar antihypertensive efficacy to reduce the size of the ischemic infarct (Nishimura et al., 2000b). It appears that blockade of the Ang II system, either by angiotensin-converting enzyme (ACE) inhibitors or AT1 receptor antagonism, is necessary to reduce brain vulnerability to ischemia in hypertension (Saavedra et al., 2001).
Nitric oxide (NO) participates in the peripheral vascular alterations in hypertension, and the Ang II and NO systems are intimately associated (Briones et al., 2002). The literature results, however, are contradictory with respect to the relative role of nitric oxide synthase (NOS) isoenzymes in arterial relaxation, remodeling, and inflammation (Barsotti et al., 2001; Zhuo et al., 2002). To clarify the mechanism of the protective effect of AT1 antagonists against brain ischemia, we studied MCA and common carotid artery morphometry and endothelial NOS (eNOS) and inducible NOS (iNOS) protein and messenger RNA (mRNA) in common carotid artery, principal arteries forming the Willis polygon, and cerebral microvessels of SHR and their normotensive WKY controls treated with the AT1 antagonist candesartan. We chose a long-term treatment with a dose of candesartan that had previously been shown to protect from experimental ischemia in SHR (Ito et al., 2002).
MATERIALS AND METHODS
Materials
Osmotic minipumps (2004 Alzet osmotic minipumps, mean pumping rate 0.28 μL/h, mean fill volume 236 μL) were from Durect Corporation, Cupertino, CA, U.S.A.; TRIzol reagent, polymerase chain reaction (PCR) supermix, and SuperScript II first-Strand Synthesis System from Invitrogen Life Technologies, Carlsbad, CA, U.S.A.; glycogen and protease inhibitor cocktail (Complete Mini), from Roche Molecular Biochemicals (Mannheim, Germany); RNeasy minikit from Qiagen GmbH (Hilden, Germany); l-γ-glutamyl-p-nitroanilide from Sigma, St. Louis, MO, U.S.A.; the Bradford protein assay from BioRad (Hercules, CA, U.S.A.); anti-eNOS and anti-iNOS antibodies from Transduction Laboratories, Lexington, KY, U.S.A.; immunoglobulin G and enhanced chemiluminescence (ECL) immunoblotting detection systems from Amersham Life Science Inc., Arlington Heights, IL, U.S.A. The Zeiss LSM 5 Image Browser was from Carl Zeiss GmbH (Jena, Germany). The image processing system for protein quantification was from Scion Corporation, Frederick, MD, U.S.A. Candesartan was a gift from Dr. Peter Morsing, Astra Zeneca R&D, Mölndal, Sweden.
Animals
Adult, 12-week-old male SHR and WKY normotensive control animals, weighing 300 to 500 g, purchased from Taconic Farms, Germantown, NY, U.S.A., were housed under standard laboratory conditions, with room temperature at 25°C and 12 hours of light each day for 1 week before the experiments. The National Institute of Mental Health Animal Care and Use Committee approved all procedures.
With the animals under pentobarbital anesthesia (50 mg/kg, intraperitoneally), we implanted osmotic minipumps subcutaneously 1 week after their arrival, to deliver candesartan (0.3 mg·kg−1·d−1) or vehicle (0.1N Na2CO3) at constant infusion rate for 28 days to groups of 10 animals, and measured systolic blood pressure in conscious rats by the tail-cuff method on treatment days 0 and 28. Reported blood pressure measurements are the mean of four determinations in each of the individual animals studied, and were performed between 9:00 a.m. and 12:00 m.
First experiment
Effect of pretreatment with an AT1 antagonist on brain ischemia in spontaneously hypertensive rats subjected to middle cerebral artery occlusion
Permanent occlusion of distal middle cerebral artery. To confirm the protective effect of pretreatment with AT1 antagonists in brain ischemia, we studied two groups of SHR, treated with vehicle or candesartan, as described previously. We used a modified method (Tamura et al., 1981; Ito et al., 2002) to electrocoagulate and transect the left MCA 2 mm proximal to the inferior cerebral vein, distal to the origin of the lenticulostriatal arteries, on day 28 of treatment. Animals were anesthetized with 3.0% halothane and maintained with 1.3% halothane in 70% nitrous oxide and 30% oxygen under spontaneous respiration.
Measurement of volume of injury
We determined the infarct volume after 24 hours of ischemia with the 2,3,5-triphenyltetrazolium chloride (TTC) method (Nishimura et al., 2000b; Bederson et al., 1986) with image scanning and computerized microdensitometry after correction for brain swelling (Swanson et al., 1990). Tissue swelling was measured by subtracting the volume of the nonaffected hemisphere from the volume of the affected hemisphere divided by the volume of the nonaffected hemisphere (Nishimura et al., 2000b).
Second experiment
Effect of an AT1 receptor antagonist in cerebrovascular and carotid morphology in hypertensive and normotensive rats not subjected to ischemia
Morphometric analysis. To determine differences in arterial morphology in hypertension, and the effect of AT1 antagonists on arterial morphology of hypertensive and normotensive rats, we studied groups of nonischemic SHR and WKY rats, treated with vehicle or candesartan, as described herein. After 4 weeks of treatment, we anesthetized SHR and WKY rats by intraperitoneal injection of 50 mg/kg of pentobarbital and perfused them through the heart.
To study carotid artery morphometry, we dissected the common carotid arteries of SHR and WKY rats from the site proximal to their origin to their bifurcation after perfusion with 250 mL of physiologic saline, dissected a section of the arteries at a site proximal to their origin, and immersed the tissue in 2-methylbutane chilled with dry ice at −40°C and storage at −80°C. We sliced the tissues in 6-μm-thick sections in a cryostat at −20°C, mounted the sections on glass slides, fixed the sections in cold acetone for 20 minutes, and stained them with hematoxylin and eosin. We studied only artery sections with a round circumference in three consecutive 6-μm-thick sections for each animal, measured individually.
To study MCA morphometry, different groups of SHR and WKY rats were anesthetized as already described and were perfused with 250 mL of a solution of 0.4% glucose, 0.8% sucrose, and 0.8% NaCl4, followed by 4% paraformaldehyde in phosphate-buffered saline with 1% boric acid (pH 7.4). After perfusion, we removed the brain and immersed it in 30% sucrose at 4°C for 3 days followed by immersion in 2-methylbutane chilled with dry ice at −40°C and storage at −80°C. We sliced the brains in 20-μm-thick horizontal sections at −20°C, followed by staining with hematoxylin and eosin. The MCA was located at the level just above the inferior horn of the lateral ventricles (horizontal sections corresponding to H4.2 (plate 84 of Kruger et al., 1995). Artery sections were measured in the right and left hemisphere of three consecutive 20-μm-thick sections, and the results were pooled for each animal. Groups of SHR consisted of five animals, measured individually.
We measured the external, internal circumferences, and media thickness with an image analysis system at ×100 magnification, and calculated the lumen diameter and the cross-sectional area as internal circumference/π, and ([external circumference]2 – [internal circumference]2) × π, respectively.
Third experiment
Effect of an AT1 receptor antagonist in eNOS and iNOS messenger RNA expression in brain microvessels and common carotid artery, and eNOS and iNOS protein expression in brain microvessels, Willis polygon, and common carotid artery of hypertensive and normotensive rats not subjected to ischemia
Isolation of brain microvessels. To determine differences in eNOS and iNOS protein and mRNA expression in hypertension, and the effect of AT1 antagonists on NOS isoenzymes in hypertensive and normotensive rats, we studied groups of nonischemic SHR and WKY rats, treated with vehicle or candesartan, as described herein. Different groups of SHR and WKY rats were used for determination of NOS mRNA or protein.
To study brain microvessels, we treated additional groups of SHR and WKY rats with vehicle or candesartan, as already described. At the end of the 28-day treatment, the animals were anesthetized and perfused through the heart with saline as described herein. Brains were removed, rinsed in cold isotonic sucrose buffer (0.32 mol/L sucrose, 3 mmol/L HEPES, pH 7.4), cleared of pia mater and choroid plexus, and cut in pieces of approximately 1 mm3 (Tontsch and Bauer, 1989). We homogenized the brain with a Dounce homogenizer with tightly fitting pestle in 3 vol ice-cold sucrose buffer, followed by centrifugation at 4°C for 10 minutes at 1,000g. We discarded the supernatant, removed the dense white layer of myelin in the upper part of the pellet, and resuspended the pellet again in 3 vol of cold sucrose buffer on ice, followed by homogenization and centrifugation at 4°C for 10 minutes at 1,000g. We resuspended the sediment in sucrose buffer, centrifuged twice for 30 seconds at 100g, pooled the supernatants, and washed them twice with sucrose buffer and once with phosphate-buffered saline + 0.1% bovine serum albumin at 200g, 2 minutes each step. We resuspended the final pellet in 1 mL of phosphate-buffered saline + 0.1% bovine serum albumin, separated aliquots to study enzymatic activity and morphology, centrifuged the pellet at 14,000g and stored the precipitate at −70°C.
We followed the purification process by morphologic evaluations on dried smears fixed by 10% formaldehyde and stained with toluidine blue (Tontsch and Bauer, 1989; Orlowski et al., 1974), and we examined γ-glutamyl transpeptidase activity, a marker enzyme localized to brain small vessels, with 2.5 μmol/L l-γ-glutamyl-p-nitroanilide as substrate (Orlowski and Meister, 1963). We incubated tissue samples for 120 minutes at 34°C with 20 mmol/L glycylglycine, 10 mmol/L MgCl2, 100 mmol/L Tris, pH 9.0, and 2.5 μmol/L l-γ-glutamyl-p-nitroanilide as a substrate, in a total volume 1.0 mL, stopped the reaction with 500 μL of acetic acid to a final concentration of 1.0N, and 300 μL of methanol. After centrifugation for 15 minutes at 400g, we measured the absorbance of the supernatant at 415 nm. The activity of γ-glutamyl transpeptidase was 9 to 10 times higher in microvessels than in whole brain, with no significant differences between the two strains (unpaired t-test, P > 0.05, N = 16), consistent with the relative enrichment reported for highly purified brain microvessels (Arnerić et al., 1988). Values were, for microvessels, whole brain, and ratio microvessels–whole brain, 9,822 ± 623, 1,058 ± 89, 10.2 ± 1.0, and 8,191 ± 949, 896 ± 69, 9.1 ± 1.0, nmol/mg protein for WKY rats and SHR, respectively.
Reverse transcription–polymerase chain reaction for endothelial and inducible nitric oxide synthases
To study the expression of eNOS and iNOS mRNA, we isolated brain microvessels, common carotid arteries, and circle of Willis from animals anesthetized and perfused with saline as described herein, froze the tissues in isopentane on dry ice, and stored them at −80°C. We extracted total RNA after disruption of each vessel segment in liquid nitrogen and homogenization using 1 mL TRIzol reagent containing 200 μg glycogen at 4°C, followed by purification using an RNeasy minikit (Qiagen GmbH). We performed reverse transcription–polymerase chain reaction by standard methods with 0.5 μg of total RNA. We synthesized first-strand complementary DNA by using SuperScript II first-Strand Synthesis System and performed PCR amplification with synthetic gene-specific primers for eNOS (upstream primer, 5′-AGA GGA GTC CAG CGA ACA GCAG-3′; downstream primer, 5′-GGC AGC CCC AAA CAC CAA AGT CAT-3′; product length, 505 base pairs [bp]), iNOS (upstream primer, 5′-ACA CTA CAC TTC CAA CGC AAC A-3′; downstream primer, 5′-GGC AGC AGG CAC ACG CAA TGA T-3′; product length, 595 bp), GAPDH (upstream primer, 5′-TCC ATG ACA ACT TTG GCA TC-3′; downstream primer, 5′-CAT GTC AGA TCC ACC ACG GA-3′; product length; 255 bp) using platinum PCR supermix for 30 cycles of denaturation at 95°C for 30 seconds, annealing at 56°C for 30 seconds, and elongation at 72°C for 1 minute. We optimized the reaction conditions to obtain reproducible and reliable amplification within the logarithmic phase of the reaction. The reaction was linear to 35 cycles with use of the ethidium bromide detection method. We separated the PCR products by electrophoresis on 2% agarose gel containing ethidium bromide and visualized the products by ultraviolet-induced fluorescence.
Western blot analysis of endothelial and inducible nitric oxide synthases
We homogenized brain microvessels and common carotid arteries, obtained from animals perfused with saline as described earlier, in buffer containing protease inhibitor cocktail (Complete Mini) and 2 mg/mL aprotinin at 4°C, centrifuged the homogenates at 1,000g for 5 minutes at 4°C and used the supernatant as a postnuclear fraction. We determined protein concentrations with the Bradford protein assay. We subjected the postnuclear fraction (eNOS: 20 μg of protein, iNOS: 50 μg of protein) of each sample to sodium dodecyl sulfate–polyacrylamide gel electrophoresis using 10% gels and transferred the proteins electrophoretically to polyvinyl difluoride sheets for 1 hour at 2 mA/cm2. The sheets were immunoblotted with anti-eNOS or anti-iNOS antibodies in a buffer containing 10 mmol/L Tris/HCl, pH 7.5, 100 mmol/L NaCl, 0.1% Tween 20, and 5% skim milk followed by peroxidase-conjugated goat anti–mouse immunoglobulin G. We detected the eNOS and iNOS proteins using the ECL immunoblotting detection system. We quantified the amount of each protein using a Microsoft-based image processing system (ScionImage software; Scion Corporation) and expressed as a relative percentage of vehicle-treated WKY rats.
Statistical analysis
Results are expressed as mean ± SD, and compared by oneway analysis of variance followed by post hoc analysis using the Newman-Keuls multiple comparison test. Differences of P < 0.05 were considered statistically significant.
RESULTS
Blood pressure
Before treatment, systolic blood pressures were higher in SHR (156.0 ± 4.0 mm Hg) when compared with their WKY controls (112.5 ± 4.5 mm Hg, P < 0.05). Treatment with an AT1 receptor antagonist for 28 days reduced the blood pressure in SHR to 113.5 ± 6.0 mm Hg (P < 0.05 vs. untreated SHR) without significantly affecting the blood pressure in WKY rats (99.5 ± 4.0 mm Hg) (P > 0.05 vs. untreated WKY rats).
First experiment
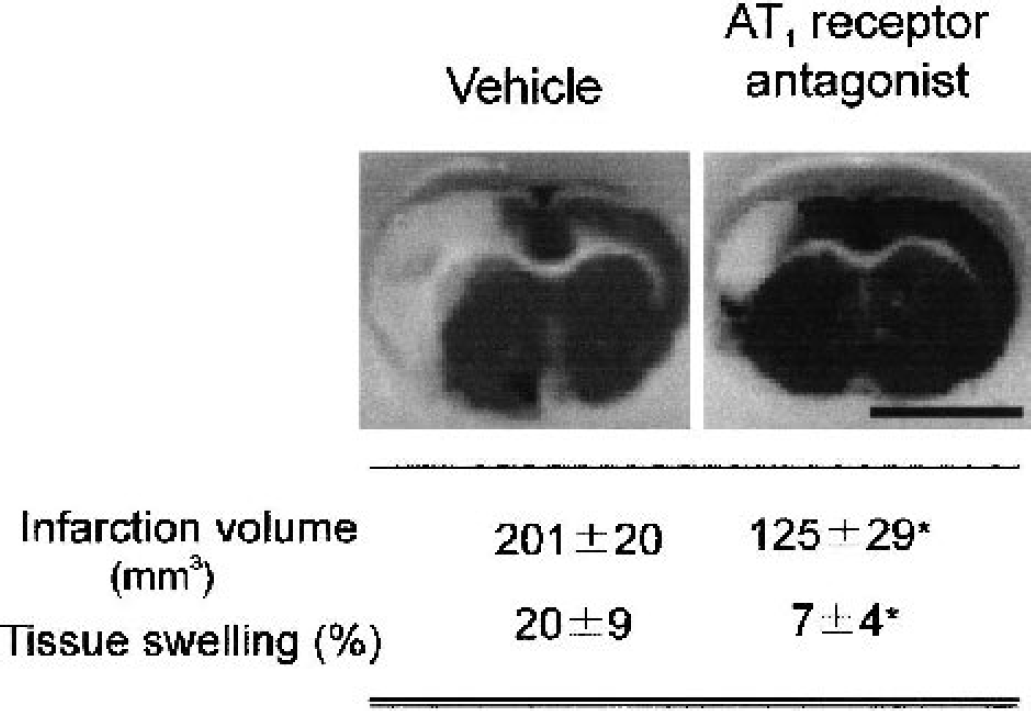
Prevention of brain ischemia by angiotensin II AT1 receptor antagonism. Infarction volume and tissue swelling decreased in treated spontaneously hypertensive rats after occlusion of the middle cerebral artery when animals were pretreated with the AT1 receptor antagonist. Ischemic tissue appears in white. Results are means ± SD, N = 5. *P < 0.05, treatment with the AT1 antagonist versus treated with vehicle. Bars represent 1 cm.
Second experiment
Characteristics of middle cerebral and common carotid arteries
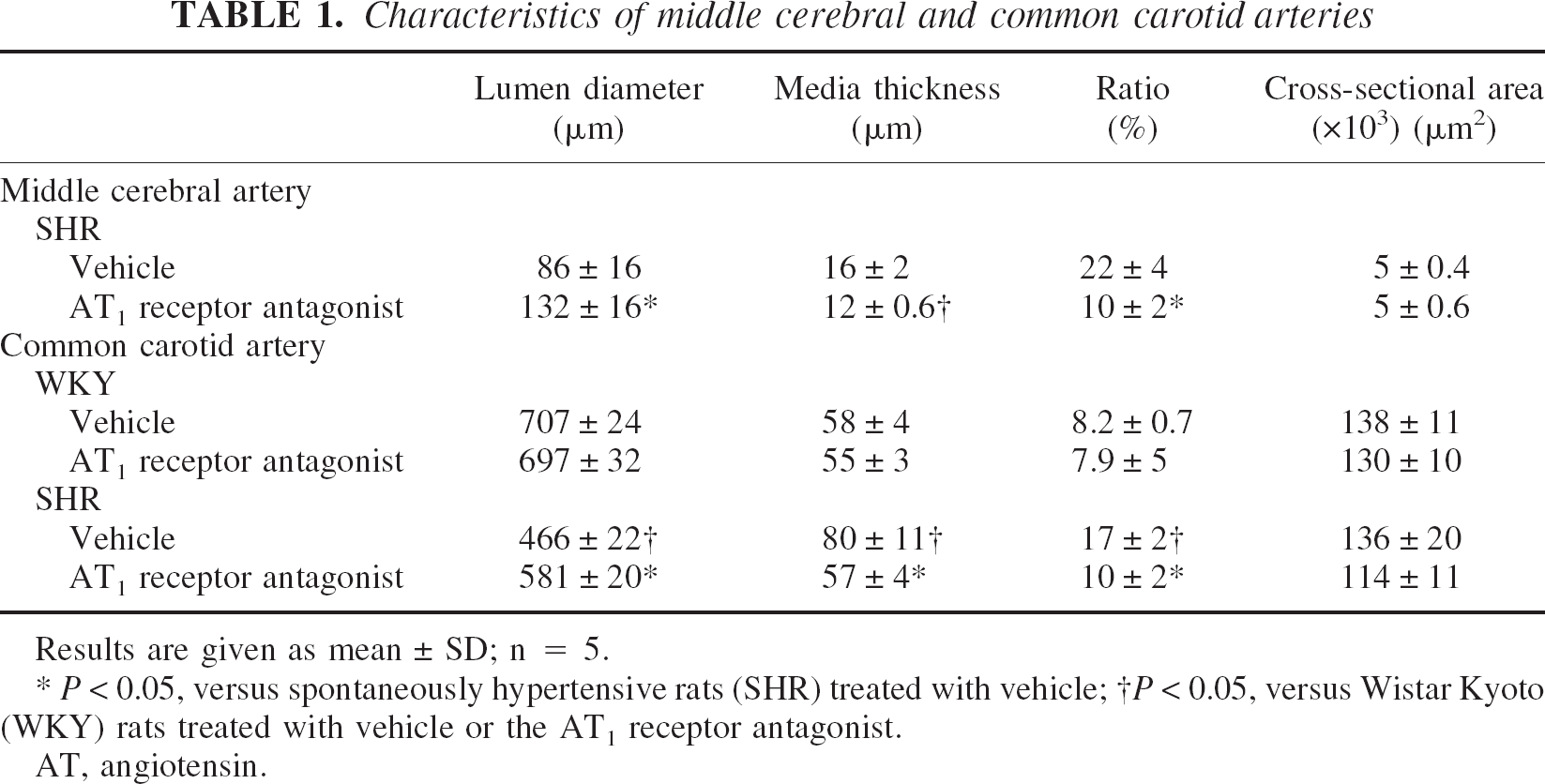
Results are given as mean ± SD; n = 5.
P < 0.05, versus spontaneously hypertensive rats (SHR) treated with vehicle;
P < 0.05, versus Wistar Kyoto (WKY) rats treated with vehicle or the AT1 receptor antagonist.
AT, angiotensin.
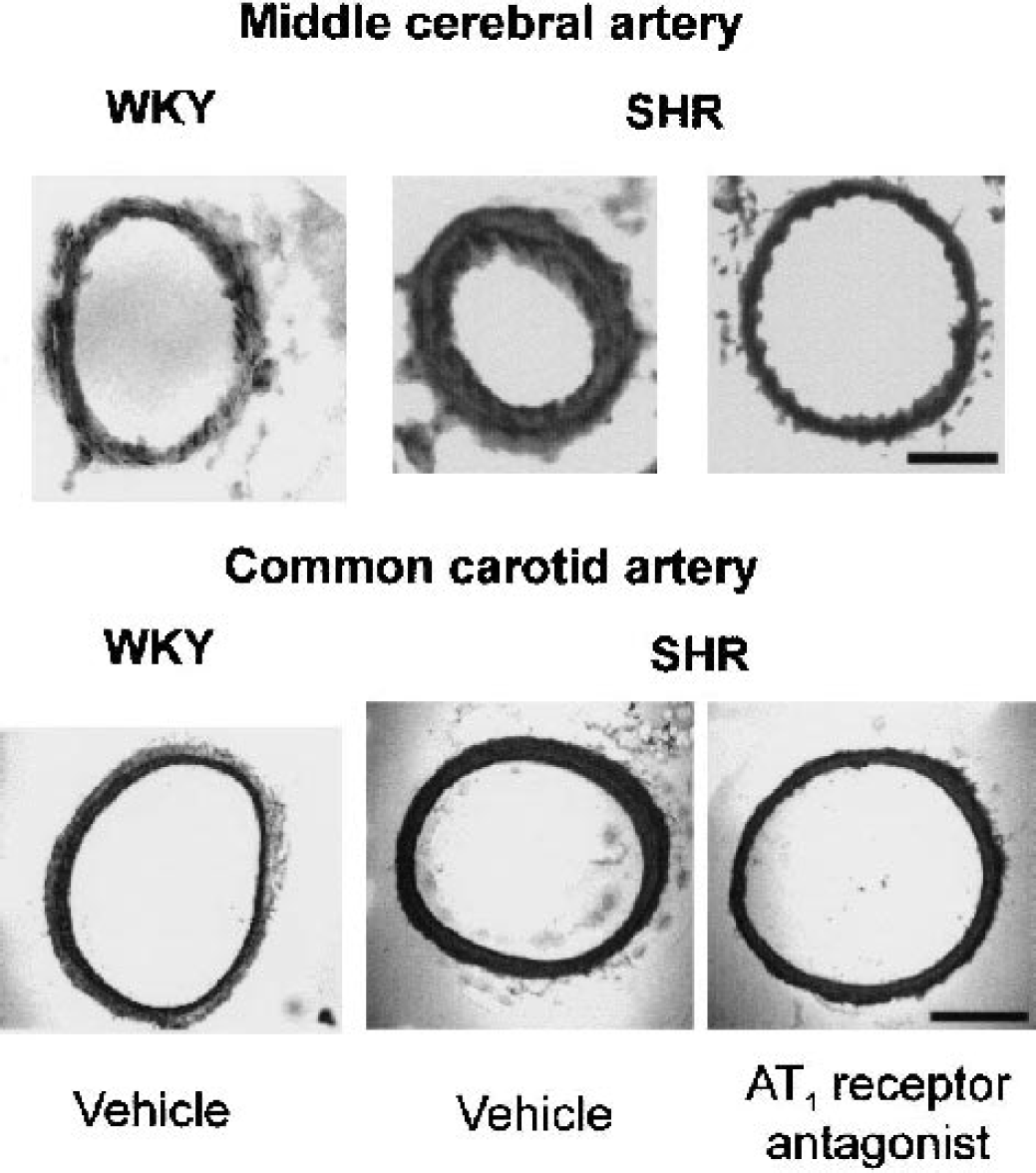
Inhibition of hypertension-induced remodeling in middle cerebral arteries and common carotid artery after treatment with an AT1 receptor antagonist. Bars represent 50 μm (upper panel) and 200 μm (lower panel). WKY, normotensive control Wistar Kyoto rats; SHR, spontaneously hypertensive rats.
In the common carotid artery, SHR treated with vehicle exhibited lower lumen diameter and increased media thickness and media–lumen ratio than normotensive WKY rats did (Fig. 2, Table 1). There was no significant difference in the cross-sectional area between SHR and WKY rats (Fig. 2, Table 1). Treatment with the AT1 receptor antagonist increased the lumen diameter and decreased the media thickness and the media–lumen ratio in SHR without affecting the cross-sectional area, in a manner similar to that of the MCA (Fig. 2, Table 1). Conversely, AT1 receptor blockade did not affect any of these measures in the common carotid artery of WKY rats (Fig. 2, Table 1).
Third experiment
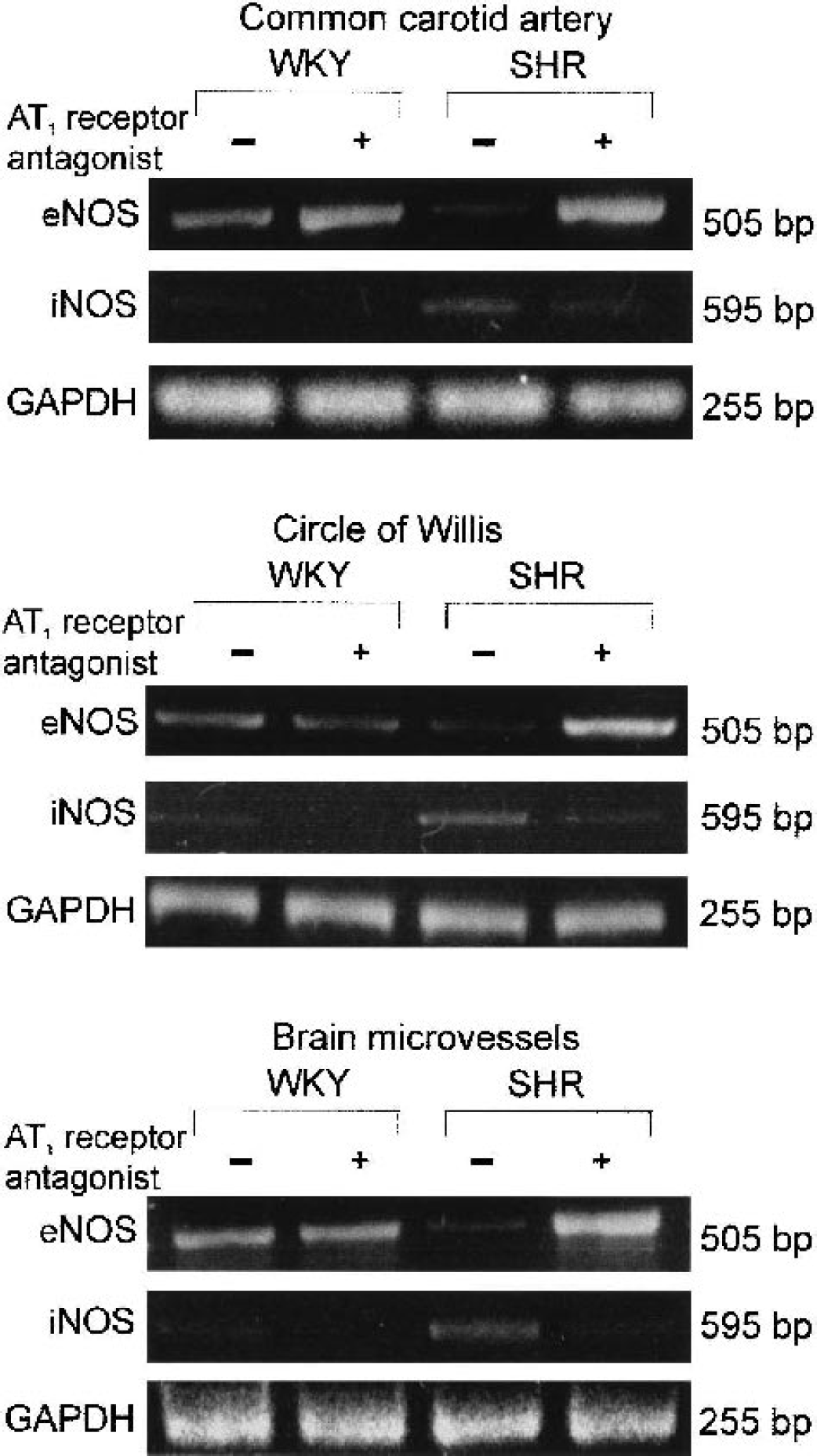
Normalization of endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) messenger RNA in common carotid artery, circle of Willis, and brain microvessels of spontaneously hypertensive rats (SHR) after treatment with an angiotensin II AT1 antagonist. Results represent experiments performed with material obtained from pools of five rats per sample. WKY, normotensive control Wistar Kyoto rats; bp, base pair.
AT1 receptor blockade increased eNOS mRNA in all brain vessels from SHR, to values higher than those in WKY rats (Fig. 3). After treatment, there were no major changes in eNOS mRNA expression in the circle of Willis and brain microvessels from WKY rats, and a relatively minor increase in eNOS mRNA in the common carotid artery from WKY rats (Fig. 3). The overall result of treatment was one of reversal of the decreased eNOS mRNA expression in brain vessels of untreated SHR, when compared with untreated WKY rats. Instead, after AT1 receptor blockade, eNOS mRNA expression in brain vessels of SHR was actually higher than that of WKY rats.
AT1 receptor antagonism decreased iNOS mRNA expression in brain microvessels, circle of Willis, and common carotid artery of SHR, and in the circle of Willis of WKY rats (Fig. 3). Thus, treatment with the AT1 receptor antagonist eliminated the increased expression of iNOS mRNA that occurred in brain microvessels, circle of Willis, and common carotid artery of untreated SHR when compared with untreated controls (Fig. 3).
Endothelial and inducible nitric oxide synthase protein in the common carotid artery and brain microvessels
In parallel with the expression of isoenzyme mRNAs, there were significant and opposite differences in the eNOS and iNOS protein expression between untreated SHR and WKY rats, both in the common carotid artery and in the brain microvessels, as determined by Western blotting.
Untreated SHR expressed decreased eNOS protein and increased iNOS protein when compared with untreated WKY rats, both in the common carotid artery and brain microvessels (Fig. 4). In SHR, the eNOS/iNOS protein ratio was significantly lower than in WKY rats (Fig. 4).
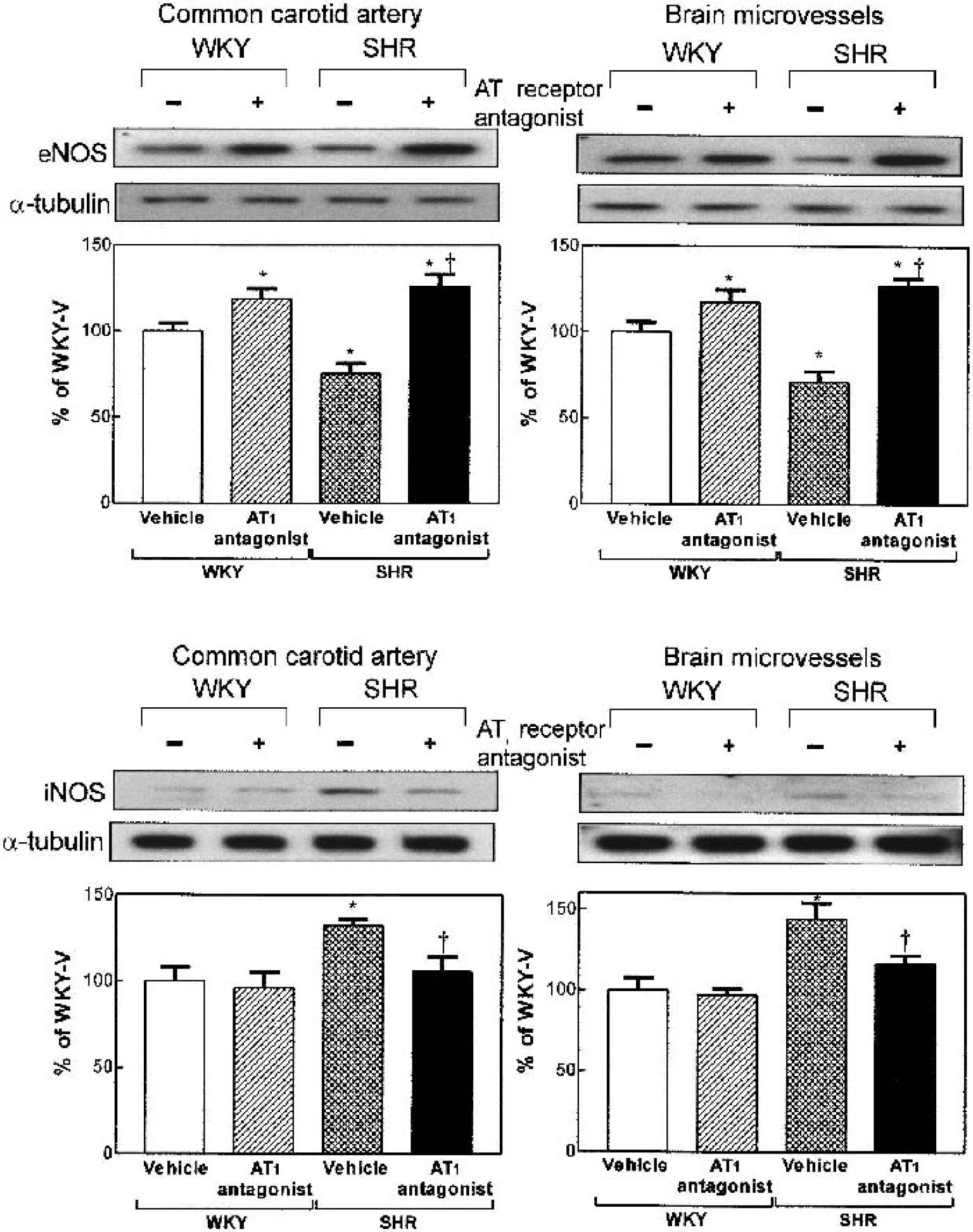
Normalization of endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) protein in common carotid artery and brain microvessels of spontaneously hypertensive rats (SHR) after treatment with an angiotensin II AT1 antagonist. Results are given as mean ± SD, N = 5, each animal measured individually. *P < 0.05, versus Wistar Kyoto (WKY) rats treated with vehicle, †P < 0.05 versus SHR treated with vehicle.
Treatment with the AT1 antagonist increased eNOS expression in the common carotid artery and brain microvessels in both strains, but the effect was more pronounced in SHR (Fig. 4). After treatment, eNOS expression in SHR was actually higher than that in untreated WKY rats (Fig. 4). The result of the administration of the AT1 receptor antagonist was to eliminate the difference in eNOS expression between untreated SHR and untreated WKY rats (Fig. 4).
Conversely, AT1 receptor antagonism decreased iNOS protein expression in SHR without affecting iNOS expression in WKY rats (Fig. 4). Again, after treatment with the AT1 inhibitor, the difference in iNOS expression between untreated SHR and untreated WKY rats was eliminated, and the iNOS expression in treated SHR was not different from that of untreated WKY rats (Fig. 4).
The net result of the treatment was the elimination of the differences in eNOS and iNOS protein expression between SHR and WKY rats (Fig. 4). Thus, the eNOS/iNOS protein ratio in treated SHR was not different from that found in untreated normotensive WKY controls (Fig. 4).
DISCUSSION
Confirming recent findings, we report that long-term pretreatment with the Ang II AT1 receptor antagonist candesartan protects genetically hypertensive rats from ischemia after permanent occlusion of a major brain artery. In addition, we present here our first study intended to clarify the molecular mechanisms of protection from ischemia by AT1 receptor inhibition. Candesartan was administered to SHR not subjected to ischemia and to normotensive controls in doses identical to those protecting from stroke. This treatment reversed the hypertension-induced brain and carotid arterial remodeling and abnormal arterial and brain microvessel eNOS and iNOS expression. We hypothesize that these results could help explain part of the protective mechanism of AT1 receptor antagonism against brain ischemia.
Our results are important for several reasons. First, they highlight the role of the Ang II system in the cerebrovascular response to ischemia. AT1 receptor blockade protects against brain ischemia (Nishimura et al., 2000b; Ito et al., 2002, and present results) by normalizing the capacity of cerebral arteries from hypertensive animals to dilate, improving collateral flow and reducing the loss of CBF in the periphery of the zone of ischemia (Ito et al., 2002).
Second, we demonstrate that normalization of brain arterial compliance, requiring prolonged AT1 receptor blockade (Ito et al., 2002), parallels the reversal of the decreased lumen diameter and increased medial thickness characteristic of hypertension-induced pathologic remodeling in peripheral and brain arteries (Intengan and Schiffrin, 2001; Baumbach and Heistad, 1992; Mulvany et al., 1996). Reversal of remodeling requires prolonged treatment, and short-term treatment is not effective (Ito et al., 2002). Thus, the inhibition of cerebrovascular vasoconstrictor tone and systemic blood pressure decrease early during treatment are not sufficient to protect against ischemia, and the improved cerebrovascular compliance in response to cerebral ischemia is a function of the normalization of cerebrovascular morphometry that follows long-term inhibition of AT1 receptors.
Third, our results may help to clarify the controversial role of NOS isoenzymes, intimately associated with the Ang II system (Briones et al., 2002; Chou et al., 1998; Bennai et al., 1999; Barsotti et al., 2001; Kobayashi et al., 2001; Zhuo et al., 2002) in cerebrovascular control during hypertension. In peripheral vessels of hypertensive animals, decreased constitutive, calcium-dependent eNOS has been linked to decreased endothelium-dependent relaxation and abnormal remodeling through nonhemodynamic processes (Chou et al., 1998; Rudic et al., 1998; Rudic and Sessa, 1999). In cerebral vessels, however, eNOS expression and activity in hypertension was found to be similar (Briones et al., 2002) or decreased (Bennai et al., 1999) when compared with that of normotensive animals, whereas recombinant eNOS expression improved vasodilatation (Tsutsui et al., 2000). Overexpression of the cytokine-inducible isoform iNOS in inflammatory cells in the tunica adventitia (Yogo et al., 2000) is associated with increased arterial inflammation, enhanced oxygen species formation and increased NO scavenging, and unfavorable effects on vascular remodeling (Intengan and Schiffrin, 2001). Other investigators, however, did not find changes in vascular iNOS during hypertension (Barsotti et al., 2001) or suggested, instead, a protective role for iNOS-generated NO (Zhuo et al., 2002; Yogo et al., 2000). In hypertension, iNOS activity in cerebral vessels was found to be similar (Briones et al., 2002), whereas its expression was reported to be higher (Bennai et al., 1999). We focused our study on eNOS and iNOS because of their demonstrated involvement in vascular function and physiopathology. In addition, neuronal NOS is expressed in cerebral arteries (Briones et al., 2002). Further studies will be needed to clarify whether AT1 receptors modulate the expression of this NOS isoenzyme.
Fourth, the present study advances our understanding of the controversial effects of Ang II and Ang II system inhibition on the NO system. Ang II increases reactive oxygen species, enhancing scavenging of NO (Intengan and Schiffrin, 2001). Treatment with AT1 receptor antagonists or ACE inhibitors induces eNOS overexpression (Barsotti et al., 2001; Kobayashi et al., 2001) and decreases the number of perivascular macrophages in brain perforating arteries (Bennai et al., 1999), whereas inhibition of the Ang II system was reported to decrease (Chou et al., 1998) or increase (Zhuo et al., 2002) iNOS expression in peripheral arteries. These contradictory results depended on the vascular bed and animal model studied and can be explained either as deficient NO production leading to hypertension or as increased NO synthesis as a compensatory mechanism in hypertension.
We have found that, in brain microvessels and common carotid artery of SHR, eNOS mRNA and protein expression are higher and iNOS mRNA and protein expression are lower, when compared with normotensive WKY rats, and similar findings were found for eNOS and iNOS mRNA expression in large cerebral arteries of the Willis polygon. We hypothesize that these changes are the expression of a role switch in cerebrovascular NOS isoenzymes, from physiologic and protective to deleterious, and that such a switch is a consequence of Ang II AT1 receptor stimulation. Our results support the hypothesis (Samdani et al., 1997) that overproduction of NO from iNOS during hypertension may lead to neurotoxicity, whereas NO production from eNOS may protect the brain by contributing to maintaining regional brain flow. Decreased cerebrovascular eNOS expression during genetic hypertension, an expression of endothelial dysfunction (Heistad and Baumbach, 1992), might be responsible for the decreased capacity to vasodilate in response to ischemia (Fujii et al., 1992) and might influence the development of pathologic remodeling by nonhemodynamic actions (Chou et al., 1998; Rudic et al., 1998; Rudic and Sessa, 1999). Increased iNOS expression, however, may result in upregulation of NO production, leading to induced generation of reactive oxygen species and inflammation (Yogo et al., 2000; Cromheeke et al., 1999).
In parallel to the protection against ischemia and the reversal of changes in cerebrovascular morphology, AT1 receptor antagonism normalizes the hypertension-induced alterations in cerebrovascular expression of NOS isoenzymes. These results agree with the hypothesis of a balanced interaction between the Ang II and NO systems (Cahill et al., 1995; Millatt et al., 1999). By restoring the balance in the expression of NOS isoenzymes, AT1 receptor antagonism may contribute to the normalization of NO function. Restoration of eNOS expression may stimulate NO production by the endothelium, thus improving vasodilatation and reversing the pathologic arterial remodeling, an important mechanism for neuroprotection during ischemia (Yogo et al., 2000; Amin-Hanjani et al., 2001; Leker et al., 2001). Inhibition of iNOS may reduce Ang II–induced generation of reactive oxygen species, decreasing NO scavenging, cellular damage, and inflammation (Rajagopalan et al., 1996; Griendling et al., 1994).
AT1 receptor inhibition in SHR results in parallel changes in mRNA and protein expression in NOS isoenzymes, indicating the possibility of a role of Ang II AT1 receptors in the regulation of transcriptional mechanisms or mRNA stability. The regulation of NOS isoenzymes by Ang II and by blockade of the Ang II system is a complex process. Ang II induces a number of growth factors that may upregulate eNOS expression, such as transforming growth factor-β1 (Li et al., 2002; Stouffer and Owens, 1992; Yoshizumi et al., 1993). In cultured smooth vascular cells from SHR, however, these factors are higher than those in cells from WKY rats, and their expression is decreased by AT1 receptor blockade (Satoh et al., 2001). These findings are not in agreement with our results, and the molecular mechanisms of in vivo regulation of eNOS gene expression in hypertension remain an open question. Growth factors are also involved in the regulation of iNOS. For example, tumor necrosis factor-α stimulates transcription of iNOS, and AT1 receptor blockade decreases tumor necrosis factor-α expression in peripheral arteries (Zhang et al., 1998; Wu et al., 2001). It is possible that a similar regulatory mechanism occurs in cerebral arteries, and this may explain the decrease in iNOS mRNA and protein expression and the cerebral vasculature after AT1 receptor blockade.
Fifth, we have found that hypertension-induced alterations in vascular morphometry and/or expression of NOS isoenzymes, and their normalization after AT1 receptor blockade, occur not only in the brain arteries of the Willis polygon and the MCA, but also in brain microvessels. This supports a role for both large and resistance cerebral arteries in the regulation of blood flow (Faraci et al., 1987) and in the increased sensitivity to ischemia during hypertension. We have found that the hypertension-induced morphologic changes, the alteration in expression of NOS isoenzymes, and their response to AT1 receptor blockade occur not only in the cerebral arteries and microvessels but also in the common carotid artery. This is of interest because the degree of carotid stenosis is one element in the prediction of stroke risk (Eugene et al., 1999). Thus, the reversal of the eNOS/iNOS ratio, a restoration of the balance lost during hypertension, occurs throughout the entire cerebrovascular system and is associated with the normalization of brain arterial morphology and with protection against ischemia. Our results, however, do not exclude the possible participation of additional mechanisms in the protective effect of AT1 antagonists against brain ischemia. For example, inhibition of the Ang II system has been associated with increased plaque stability and reduction of intraplaque inflammation in arteriosclerosis (Gorelick, 2002; Schieffer et al., 2000).
Our findings that hemodynamic, morphologic, and biochemical alterations in hypertension associated with increased vulnerability to brain ischemia can be fully reversed by AT1 receptor blockade indicate that activation of these receptors is crucial for the maintenance of the pathologic alterations in cerebrovascular circulation during hypertension, and that their blockade may be of significant therapeutic advantage and end-organ protection.
Footnotes
Acknowledgments:
The authors thank Dr. Gustavo Baiardi, Section on Pharmacology, N.I.M.H., for his help in the preparation of the figures; and Dr. Maria Deli, Laboratory of Molecular Neurobiology, Institute of Biophysics, Biological Research Center, Szeged, Hungary, for help with the microvessel preparation protocols.