
Abstract
Abnormalities in the homeostasis of the renin-angiotensin system have been implicated in the pathogenesis of vascular disorders, including stroke. The authors investigated whether angiotensinogen (AGN) knockout mice exhibit differences in brain susceptibility to focal ischemia, and whether such differences can be related to special features of the collateral circulation. Wild-type and AGN-knockout mice were submitted to permanent suture occlusion of the middle cerebral artery (MCA). The collateral vascular system was visualized by systemic latex infusion, and the ischemic lesions were identified by cresyl-violet staining. The core and penumbra of the evolving infarct were differentiated by bioluminescence and autoradiographic imaging of A TP and protein biosynthesis, respectively. In wild-type mice, mean arterial blood pressure was 95.0 ± 8.6 mm Hg, and the diameter of fully relaxed anastomotic vessels between the peripheral branches of the anterior and middle cerebral arteries 26.6 ± 4.0 μm In AGN knockouts, mean arterial blood pressure was significantly lower, 71.5 ± 8.5 mm Hg (P <,01), and the anastomotic vessels were significantly larger, 29.4 ± 4.6 μm (P < .01). One hour after MCA occlusion, AGN-knockout mice exhibited a smaller ischemic core (defined as the region of ATP depletion) but a larger penumbra (the area of disturbed protein synthesis with preserved ATP). At 24 hours after MCA occlusion, this difference disappeared, and histologically visible lesions were of similar size in both strains. The observations show that in AGN-knockout mice the more efficient collateral blood supply delays ischemic injury despite the lower blood pressure. Pharmacologic suppression of angiotensin formation may prolong the therapeutic window for treatment of infarcts.
The renin-angiotensin system (RAS) is widely distributed throughout the body and plays an important role in maintaining blood pressure and ion homeostasis (Campbell, 1987). Angiotensinogen is broken down by renin to angiotensin I, and further hydrolyzed by angiotensin-converting enzyme (ACE) to produce the potent vasoconstrictor hormone angiotensin II which has both vascular and extravascular functions (Peach, 1977).
Angiotensinogen and ACE gene abnormalities have been recognized as risk factors for myocardial and brain infarction (Kamitani et al., 1995; Kario et al., 1996; Sharma, 1998). Deletion polymorphism in the ACE gene, which results in high serum levels of ACE, positively correlated with an increased risk and greater severity of ischemic lesions (Kario et al., 1996; Sharma, 1998). Furthermore, ACE inhibitors improved neurologic outcome (Werner et al., 1991), attenuated metabolic disorders (Sadoshima et al., 1993), and reduced infarct volume (Fujii et al., 1992; Slivka, 1991) after experimental focal cerebral ischemia. These facts suggest that the activity of RAS contributes to the pathophysiology of ischemic stroke.
A likely target of RAS in cerebrovascular disease is the collateral circulation of the brain. After middle cerebral artery (MCA) occlusion, stroke-prone spontaneously hypertensive rats (SHR-SP) suffer larger infarcts than Wistar-Kyoto rats (WKY) (Coyle and Feng, 1993). This has been explained by the smaller diameter and the less efficient blood supply via collateral vessels (Coyle and Heistad, 1986). Angiotensin II leads to hypertrophy of cerebral vessels and could therefore influence the size and vascular reactivity of collateral vessels (Griffin et al., 1991). We therefore studied whether RAS influences the severity of ischemic injury after MCA occlusion. To put the RAS out of action, we used angiotensinogen (AGN) knockout mice because AGN is the only known precursor of angiotensin peptides (Campbell, 1987). Our observations suggest that AGN does modulate the severity of ischemic injury and that manipulation of the RAS might be a useful approach for the treatment of acute stroke.
MATERIALS AND METHODS
Experimental animals
Experiments were performed according to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals, and approved by the local authorities. AGN knockout mice and their wild-type littermates were generated as described elsewhere (Tanimoto et al., 1994). Animals aged between 10 and 13 weeks were selected and housed under diurnal lighting conditions with free access to food and water before the experiment.
Visualization of cerebral angioarchitecture
Nine homozygous female AGN-knockouts and 10 wild-type littermates were used. Animals were anesthetized with 1.0% halothane in 70% N2O and 30% O2. The cerebral angioarchitecture was stained using the intravascular latex infusion technique of Coyle (Coyle and Jokelainen, 1982), as modified for use in mice (Maeda et al., 1998). A lethal dose of papaverine hydrochloride (40 to 50 mg/kg in sterile water) was injected intravenously to produce maximal vasodilation and to minimize cerebrovascular resistance. The thoracic aorta was clipped at the level of diaphragm and cannulated with polyethylene tubing (internal diameter 0.58 mm, Portex, England). Warm (38°C) undiluted Vultex (a white latex, Chicago Latex Production no. 563) was mixed with a small amount of carbon black (10 μl/g, Bokusai; Fueki, Tokyo, Japan) and injected into the ascending aorta. The volume injected was 0.4 mL, and the injection pressure was about 150 mm Hg.
Thirty minutes after the injection, animals were decapitated and the dorsal part of the skull and the dura removed. To prevent deformation of the brain, the entire head was fixed in 10% formalin for 4 weeks before brain removal. Thereafter, the dorsal side of the brain was inspected under the operating microscope and photographed at 15 times magnification. Three AGN knockouts and four wild-type mice were excluded from the study because of unstained pial vessels; the remaining animals, six in each group, were used for morphometric evaluation of anastomotic vessels.
Anastomoses on the dorsal surface of the hemispheres were localized by tracing the peripheral branches of the anterior cerebral artery (ACA) and the MCA to the anastomose points, defined as the narrowest part of the vessel or halfway between the nearest branching points of the ACA and the MCA branches (Coyle and Jokelainen, 1982). Adjacent anastomose points were connected by the line of anastomoses (Coyle, 1987), and the distance from the midline to the line of anastomoses was measured at coronal planes 2 mm, 4 mm, and 6 mm from the frontal pole on photographs taken from the dorsal brain surface. The number of anastomoses per hemisphere was counted, and the diameters of the three largest anastomoses in each hemisphere were measured using an image analyzer and NIH image 1.59 software (National Institutes of Health, Bethesda, MD, USA).
Middle cerebral artery occlusion
In 16 homozygous female AGN knockout mice and in 16 wild-type littermates the MCA was permanently occluded using a modification of the intraluminal filament technique (Hata et al., 1998b). Animals were anesthetized with 1.0% halothane in 70% N2O and 30% O2. Temperature was kept constant at 36.5 to 37.0°C, using an infrared lamp and a heating pad connected to a feedback-controlled power supply (YSI, Yellow Springs, OH, USA). The left femoral artery was cannulated for measurement of mean arterial blood pressure (MABP), Pao2, Paco2, and pH before and after MCA occlusion. After midline neck incision the left common and the left external carotid arteries were isolated and ligated. A microvascular clip (FE691; Aesculap, Tuttlingen, Germany) was temporarily placed on the left internal carotid artery. A 8–0 nylon filament (Ethicon; Ethicon Norderstedt, Germany) coated with silicon resin (Xantopren; Bayer Dental, Osaka, Japan) was introduced through a small incision into the common carotid artery, and advanced 9 mm past the carotid bifurcation for occlusion of the MCA.
Once the MCA had been occluded, animals were allowed to survive for either 1 or 24 hours (eight AGN knockouts and eight wild-type littermates for each group, respectively). In the 24-hour survival group, anesthesia and temperature control were discontinued 1 hour after vascular occlusion and resumed 1 hour before the end of the experiment. In the intervening period, animals were returned to their cages with free access to water and food.
At the end of experiments, heads were frozen in situ with liquid nitrogen. Brains were removed from the frozen head at −20°C in a cold temperature cabinet and cut at that temperature into 20 μm coronal cryostat sections for biochemical and morphologic investigation.
Imaging of protein synthesis and ATP content
Forty-five minutes before in situ freezing, animals received an intraperitoneal injection of 150 μCi L-[4,5−3H] leucine (specific activity 150 Ci/mmol; Amersham, Braunschweig, Germany). Incorporation of leucine into brain proteins was imaged in cryostat sections taken from a coronal plane 4 mm posterior to the frontal pole, using quantitative autoradiography as described by Mies et al., (1986). Cryostat sections were first incubated in 10% trichloroacetic acid to remove labeled free-leucine and metabolites other than proteins, and exposed for 14 days with 3H standards to tritium-sensitive radiographic film (Hyperfilm 3H, Amersham, Braunschweig, Germany). Autoradiograms were digitized with a CCD camera and processed using the NIH Image 1.59 software (National Institutes of Health, Bethesda, MD, USA). Inhibition of protein synthesis was defined as a reduction of amino acid incorporation below the lowest gray matter value of the opposite hemisphere, and the area of disturbed protein synthesis was outlined using this threshold. For quantification, the region of unimpaired protein synthesis was subtracted from the opposite hemisphere to correct for volume changes induced by ischemic brain swelling (Swanson et al., 1990).
Imaging of ATP was performed using a modification (Paschen et al., 1992) of the method described by Kogure and Alonso (Kogure and Alonso, 1978). Cryostat sections at a coronal plane 4 mm from the frontal pole, adjacent to those used for measurement of protein synthesis, were covered by the bioluminescence reaction mix and exposed at room temperature for 1 minute to photographic film (AGFAPAN APX100, Agfa Leverkusen, Germany). Film negatives were digitized and processed with the same imaging system as that used for autoradiograms.
The ATP depleted area was measured in the same way as the inhibition of protein synthesis, using a threshold of less than 30% of the mean value on the contralateral side. The penumbral area was calculated by subtracting the area of ATP depletion from that of disturbed protein synthesis (Hata et al., 1998a).
Histologic staining
In the 24-hour survival group, cryostat sections taken from coronal planes 2 mm, 4 mm, 6 mm, 7 mm, and 8 mm posterior to the frontal pole were stained with cresyl-violet and digitized for evaluation of infarct volume. Using the image analysis system, the border of infarcted tissue was outlined. The area of infarction was again corrected for brain swelling by subtracting the area of intact tissue from the opposite hemisphere.
Statistics
All results were expressed as mean ± SD. Differences between the distance from the midline to the line of anastomoses, and differences in the size of infarct areas were evaluated for statistical significance using one-way analysis of variance followed by Scheffé's post hoc analysis. Differences in infarct volume and the number and the diameter of anastomoses were compared using Mann-Whitney's U test. Differences in physiologic variables and differences in the areas of ATP depletion, protein synthesis suppression, and the peri-infarct penumbra were evaluated for statistical significance using two-way analysis of variance followed by Scheffé's post hoc analysis. A P value < .05 was considered to indicate statistical significance.
RESULTS
Cerebral angioarchitecture
Figure 1 shows representative photographs of the dorsal surface of the brain of an AGN knockout mouse and of the corresponding wild-type littermate after systemic infusion of colored latex. The peripheral branches of the ACA and MCA forming the pial vascular network of collateral vessels are clearly visible and allow precise identification of the line of anastomoses, as defined in the Materials and Methods section of this article.
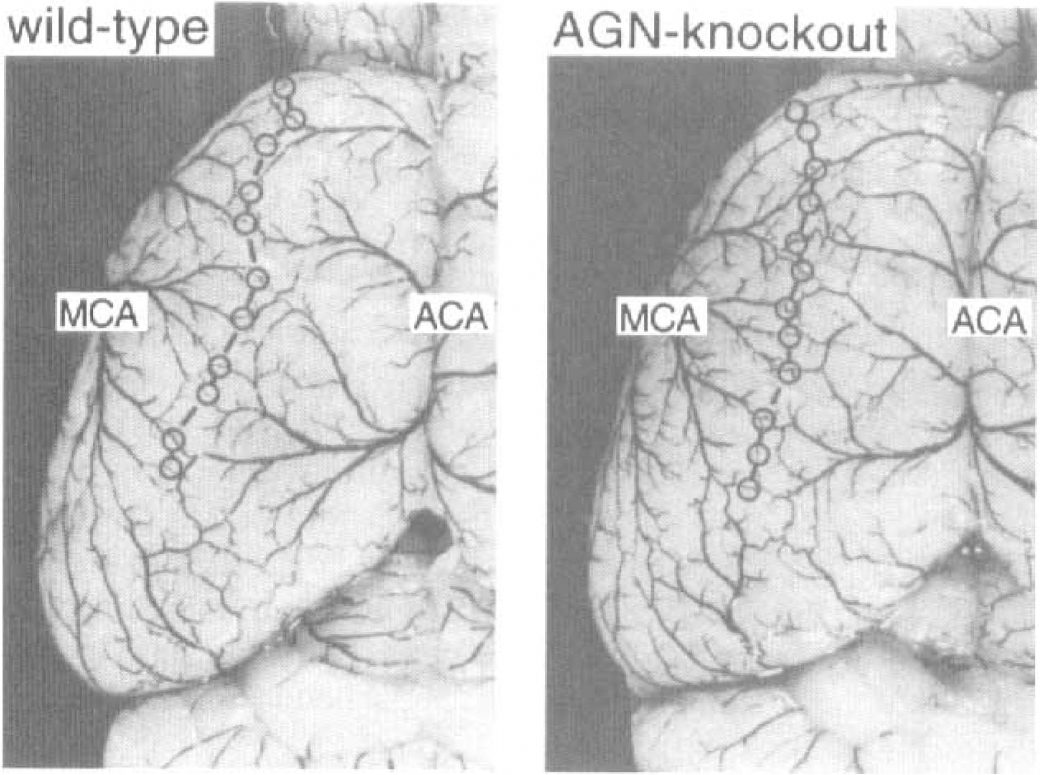
Cerebral angioarchitecture of Wild-type (left) and angiotensinogen-knockout mice (AGN-knockout, right). Vessels on the dorsal brain surface were stained by systemic infusion of colored latex. The points of anastomoses between the peripheral branches of the anterior and middle cerebral arteries are marked by circles and interconnected by the line of anastomoses. Note similar angioarchitecture in both strains.
Quantitative evaluations of the distance of the line of anastomoses from the midline are shown in Fig. 2. There were no statistical differences between the AGN knockout mice and wild-type littermates at any of the three levels investigated, 2 mm, 4 mm, and 6 mm from the frontal pole. The number and the diameter of papaverinedilated ACA-MCA anastomoses are given in Table 1. The number of anastomoses did not differ between wild-type and mutants but the diameter of fully relaxed anastomoses was significantly larger in AGN knockout mice (P < .01).
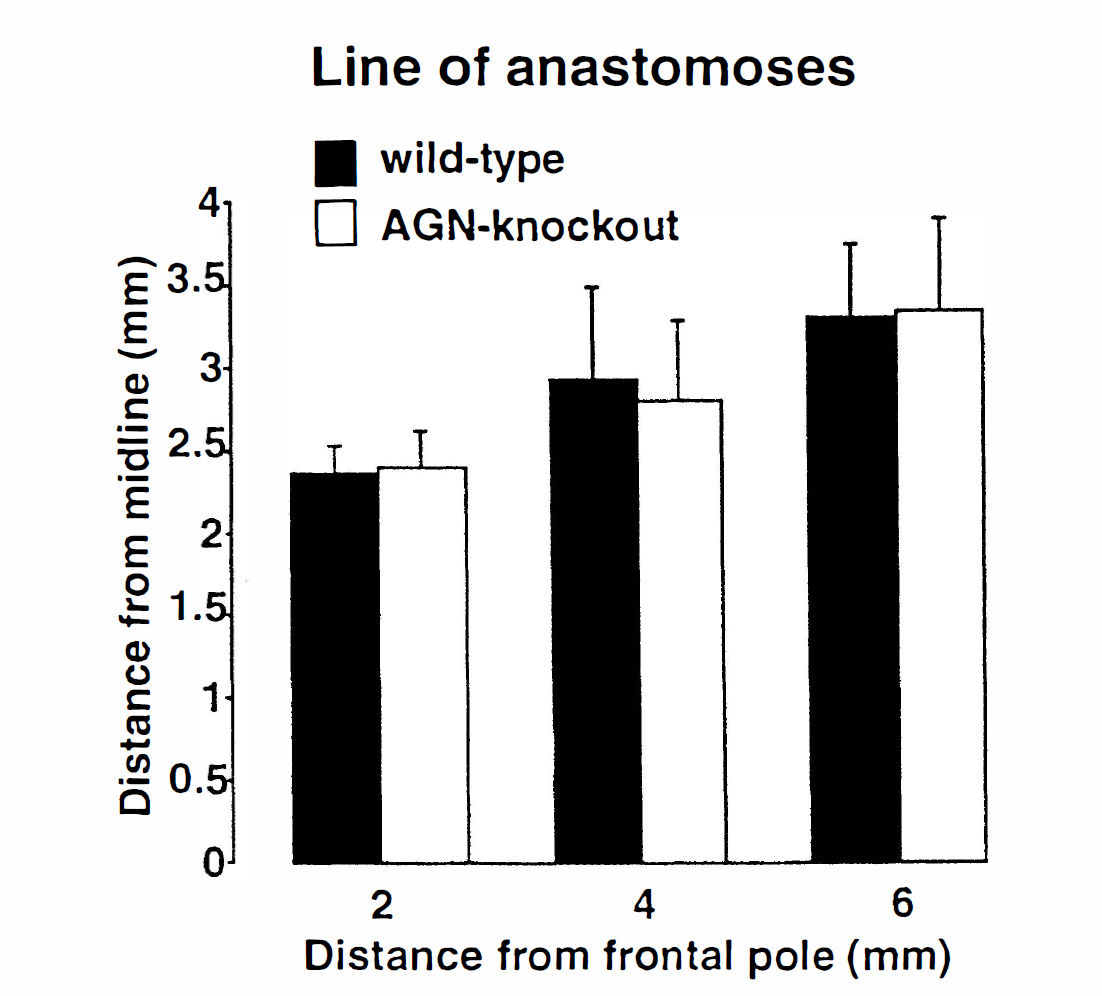
Distance of the line of anastomoses from the midline in wild-type (black bars) and AGN-knockout mice (white bars). Measurements were made at the distances indicated from the frontal pole. Note absence of strain differences (values are mean ± SD; n = 12 for each group).
Number and diameter of anastomoses between the peripheral branches of the anterior and middle cerebral arteries
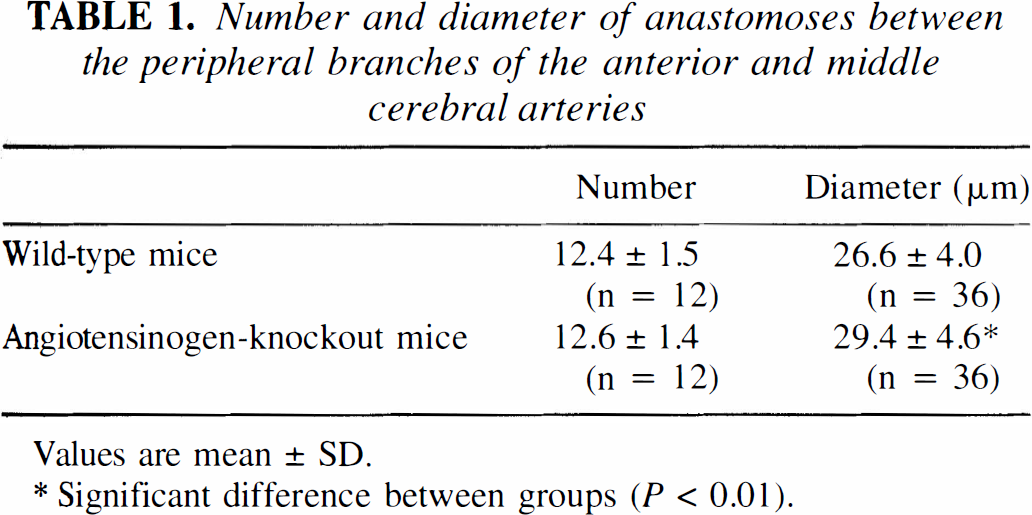
Values are mean ± SD.
Significant difference between groups (P < 0.01).
Effect of MCA occlusion
The physiologic parameters are given in Table 2. In AGN knockout mice, MABP was about 20 mm Hg lower than that of wild-type animals. This difference persisted after MCA occlusion (P < .01). Blood gases and arterial pH, in contrast, were similar in the two groups.
Physiologic variables before and 15 minutes after middle cerebral artery occlusion
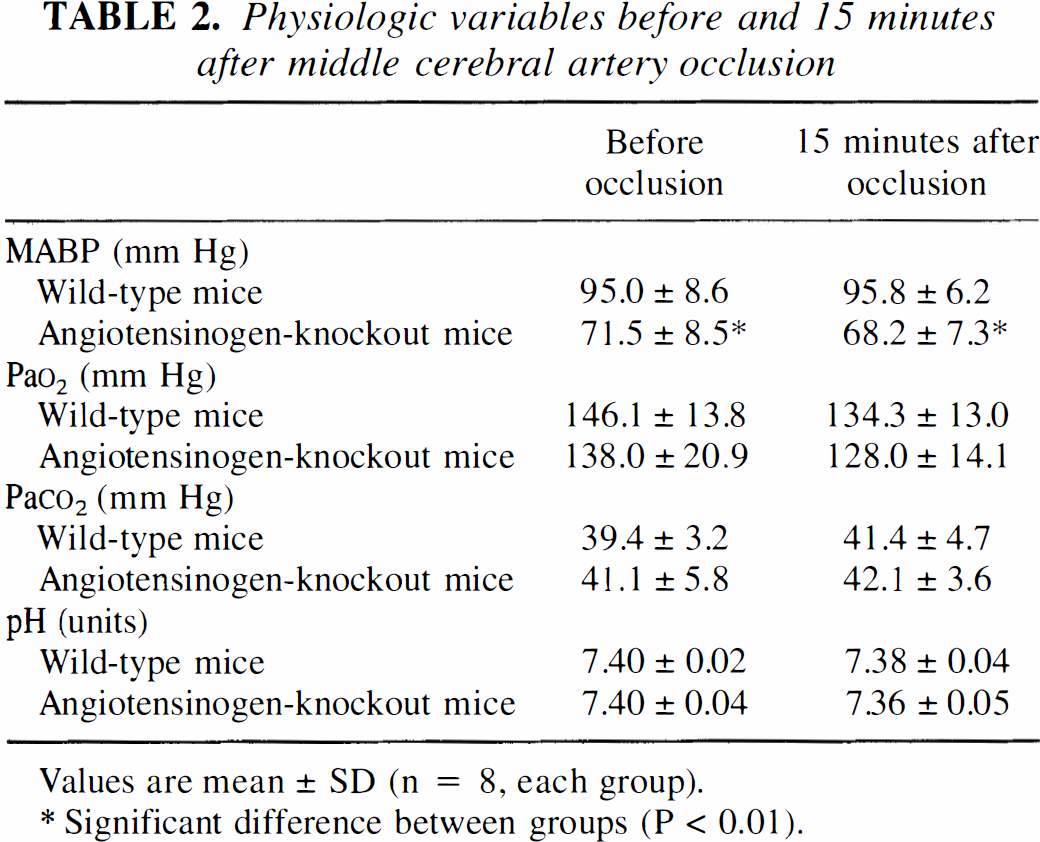
Values are mean ± SD (n = 8, each group).
Significant difference between groups (P < 0.01).
Imaging of A TP content and protein synthesis revealed a remarkable difference between AGN knockout mice and wild-type littermates at 1 hour after MCA occlusion (Fig. 3) but not after 24 hours (Fig. 4). At the early time point, the area of ATP depletion was significantly smaller in AGN knockout mice, indicating that vascular occlusion resulted in less severe primary ischemic injury. The area of protein synthesis inhibition, in contrast, did not differ at this time. Because of the smaller ATP depletion area, the penumbra—defined as the area in which protein synthesis is impaired but A TP is not depleted—was significantly larger in AGN knockout mice (Fig. 5).
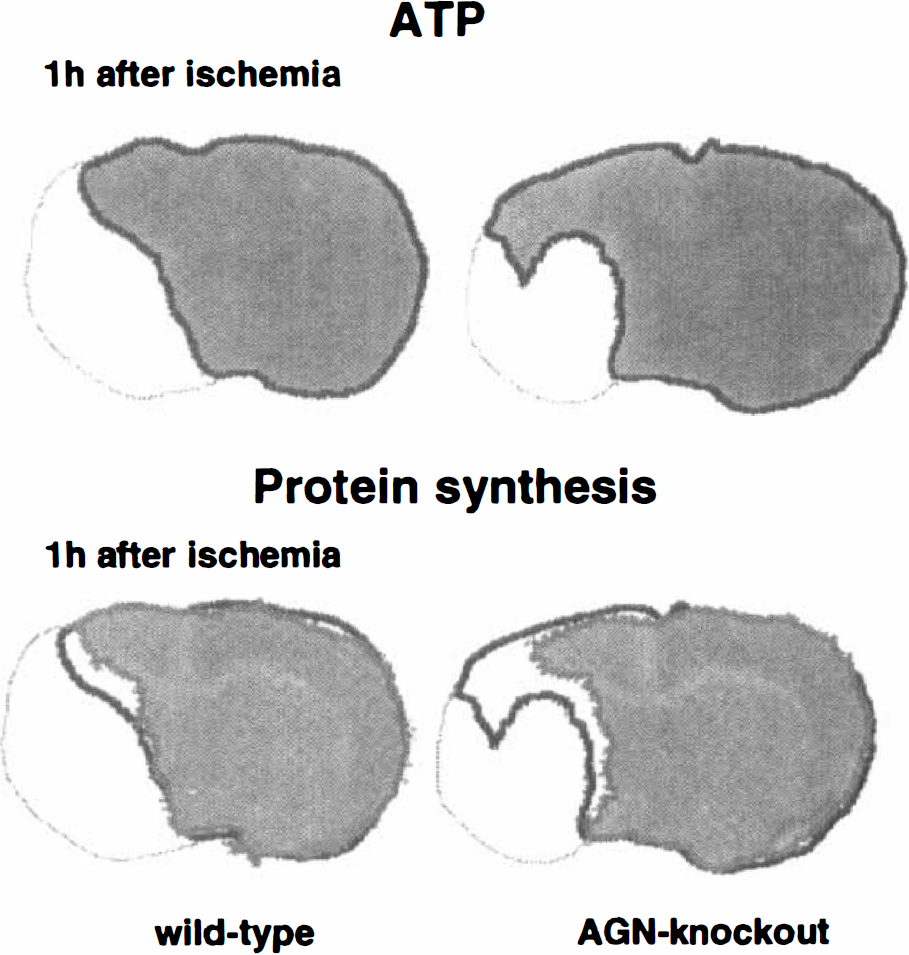
Imaging of brain ATP content and of protein synthesis in wild-type and angiotensinogen-knockout mice at 1 hour after middle cerebral artery occlusion. Measurements were made on coronal cryostat sections passing through the centre of the middle cerebral artery territory (4 mm from frontal pole). The outlining of the ATP-preserved area is superimposed on the image of protein synthesis to show the peri-infarct penumbra (defined as the area of suppressed protein synthesis but maintained ATP). Note smaller ATP-depleted region and larger penumbra in the angiotensinogen-knockout animal.
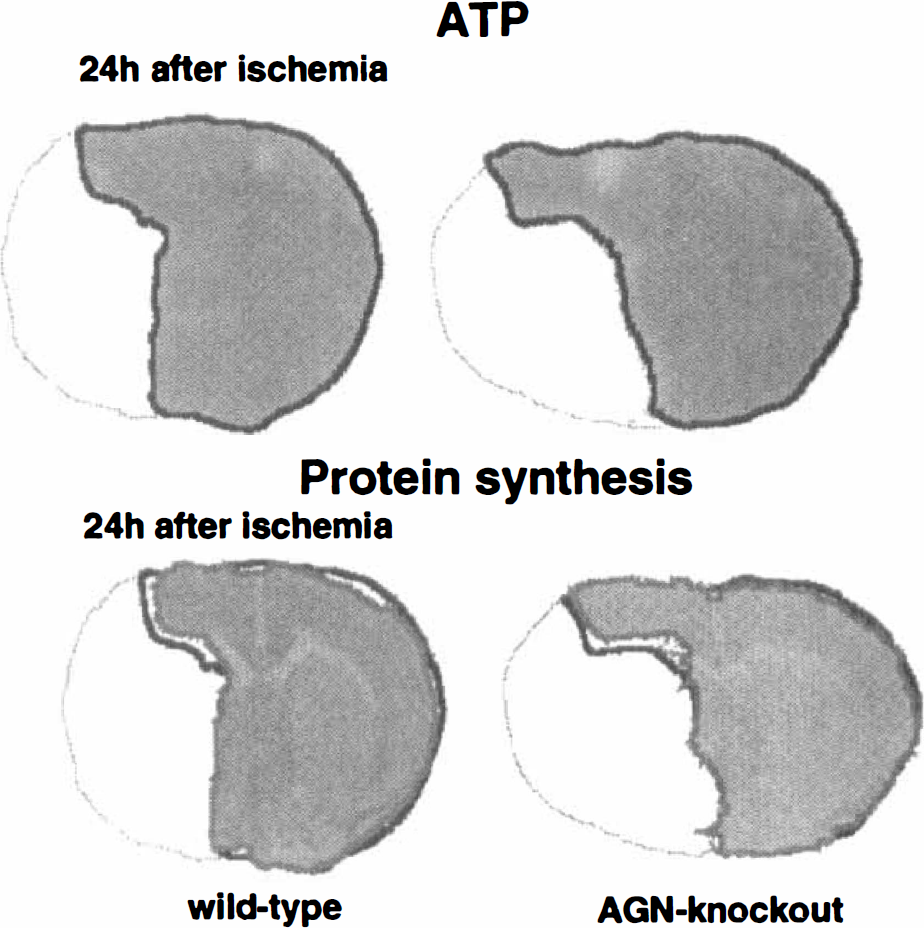
Imaging of brain ATP content and of protein synthesis in wild-type and angiotensinogen-knockout mice at 24 hours after middle cerebral artery occlusion. Same measurements as in Fig. 3. Note disappearance of penumbra and similar size of ATP-depleted area in wild-type and angiotensinogen-knockouts at this time point.
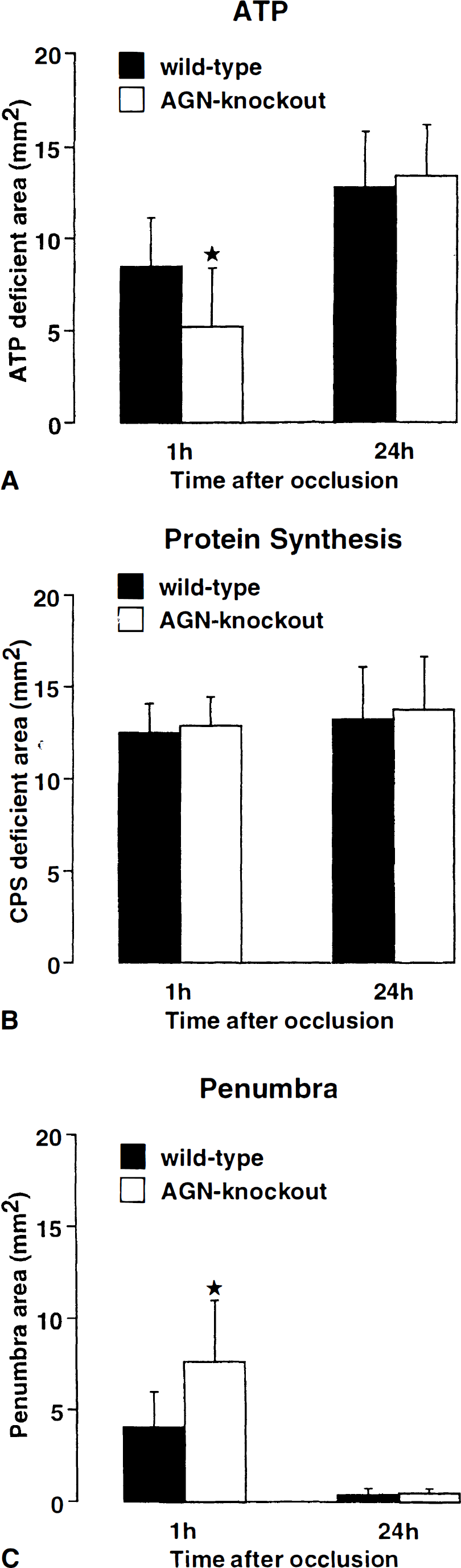
Mean size of areas with ATP depletion, suppressed protein synthesis, and the calculated penumbra after middle cerebral artery (MCA) occlusion in wild-type and angiotensinogen (AGN)-knockout mice. Measurements were made at 1 hour and 24 hours after vascular occlusion on coronal cryostat sections passing through the centre of the MCA territory (4 mm from frontal pole; mean ± SD; n = 8 for each group). Note similar changes of protein synthesis but significantly smaller area of ATP depletion and significantly larger penumbra in AGN-knockouts at 1 h after MCA occlusion (P < .05). The disappearance of this difference at 24 hours shows that AGN deficiency ameliorates ischemic injury only transiently.
At 24 hours after MCA occlusion, the area of suppressed protein synthesis only slightly changed in both strains but the area of ATP depletion markedly increased and in both strains approached that of disturbed protein synthesis (Fig. 4). As a consequence, the strain differences that were visible at the earlier timepoint of ischemia disappeared, and the penumbra was no longer detectable at 24 hours in either of the two strains (Fig. 5).
The histologic evaluation of infarct volume at 24 hours after MCA occlusion revealed slightly less injury in the mutant strain (AGN knockout mice: 87.3 ± 18.5 mm3, wild-type: 106.0 ± 30.3 mm3) but this difference was not statistically significant. The planimetric measurements of the edema-corrected infarct areas suggested a slight reduction of infarct size in AGN knockout mice at the posterior level of the brain, but these data do not show significant difference either.
DISCUSSION
Evidence from genetic investigations in human stroke victims suggest that abnormalities of the RAS might influence the incidence and severity of ischemic brain injury (Kario et al., 1996; Sharma, 1998). These observations are supported by the present experimental study which shows that deletion of the AGN gene in mice reduces early metabolic disturbances after MCA occlusion, despite significant lowering of blood pressure.
The obvious reason for this improvement is the significantly larger diameter of anastomotic vessels, leading to an improved collateral blood supply to the ischemic territory. This finding is not surprising in view of the known functional effects of the RAS in the brain. Angiotensin II, the vasoconstrictor hormone derived from angiotensin I by the ACE, stimulates growth of vascular smooth muscle cells (Dzau et al., 1991) and results in hypertrophy of the vascular wall (Griffin et al., 1991), presumably by induction of smooth muscle growth factors (Delafontaine and Lou, 1993; Naftilan et al., 1989). The deleterious effect of vascular hypertrophy on focal vascular reactivity has been previously demonstrated in SHR-SP. In this strain, hypertrophic pial vessels dilate less in response to pharmacologic (Yang et al., 1993) or pathophysiologic stimuli (Coyle and Heistad, 1986), which explains the larger infarct volume after MCA occlusion in SHR-SP, as compared to rat strains with normal blood pressure and normotrophic cerebral angioarchitecture. Inhibitors of ACE or angiotensin receptor antagonists prevent vascular hypertrophy (Clozel et al., 1989; Vacher et al., 1996). Therefore, a relief of trophic influence on vascular smooth muscle cells in combination with the lowering of blood pressure, as observed in the AGN knockout mice investigated here, provides a plausible explanation for the observed reduction of metabolic injury.
The ablation of the AGN gene not only depletes mice from agiotensin II but also from all other metabolites of the protein. In particular interesting for brain physiology is angiotensin IV for which specific receptors have been found in the CNS and which has been shown to influence regional hemodynamics and behavior (Wright et al., 1995). Furthermore, the impaired blood-brain barrier function observed in AGN-deficient mice can be rescued not only by the administration of angiotensin II but also of angiotensin IV (Kakinuma et al., 1998). Therefore, we cannot exclude that this or any other AGN metabolite different from angiotensin II may play a role in the susceptibility to focal cerebral ischemia.
The protective effect of the larger anastomotic vessels in AGN knockout mice was only of limited duration. One day after the onset of MCA occlusion, the severity of ATP depletion and the volume of histologically visible ischemic lesions, were similar in AGN knockout mice and their wild-type littermates. Pictorial measurements of A TP and protein synthesis revealed that this was due to the expansion of the infarct core into the peri-infarct penumbra, the outer margin of which was similar in both strains. This phenomenon can be explained by the threshold concept of metabolic disturbances (Hossmann, 1994). Protein synthesis is suppressed essentially in the whole supplying territory of the MCA whereas energy metabolism fails only in the central parts where blood flow declines below a threshold of 15% to 20% of control levels (Mies et al., 1991). Variations in the efficiency of the collateral blood supply will therefore lead to a more marked difference in the area of ATP depletion than that of impaired protein synthesis.
However, as ischemia proceeds, the threshold of ATP depletion slowly increases and eventually merges with that of suppressed protein synthesis, leading to the expansion of the infarct core into the peri-infarct penumbra (Mies et al., 1991). The reason for this expansion is only partly understood. Peri-infarct depolarizations (Iijima et a., 1992), free radical mediated injury (Siesjö et al., 1989), release or activation of neurotoxic mediators (Takagi et al., 1993), and programmed cell death (Li et al., 1995) have all been discussed as possible contributors to this pathophysiology, but the respective role of each of these factors is difficult to establish.
As shown in this study, the functional elimination of the cerebral RAS in AGN knockout mice alleviates only the early hemodynamically induced metabolic disorders but does not prevent the secondary infarct expansion. A possible explanation could be impaired blood-brain barrier function in angiotensin-deficient mice (Kakinuma et al., 1998) but positive evidence of this pathophysiology is not yet available. On the other hand, RAS-dependent mediators of tissue injury, such as nitric oxide (Pueyo et al., 1998), bradykinin (Textor et al., 1981), vasopressin (Sterling et al., 1980), prostaglandin (Toda and Miyazaki, 1981) or the release of catecholamines and excitatory amino acids (Phillips, 1987) are probably of lesser importance because AGN knockout does not improve the final outcome. However, RAS may nevertheless be a target for therapeutical interventions because amelioration of the initial metabolic disorder by angiotensin receptor antagonists may extend the time slot for additional therapeutic interventions.
In conclusion, AGN knockout mice have larger anastomotic vessels and suffer less initial metabolic injury after MCA occlusion than their wild-type littermates. However, owing to the progression of ischemic injury into the peri-infarct penumbra, this effect is only temporary. Pharmacologic suppression of angiotensin formation may prolong the therapeutic window for treatment of infarcts but requires combination with other therapeutic approaches to achieve lasting effects.
Footnotes
Acknowledgments
Prof. A. Fukamizu (Tsukuba University) kindly supplied AGN-knockout mice. The authors thank Mrs. U. Beckmann for excellent technical assistance, Mr. B. Huth and Mrs. I. Mühlhöver for the artwork, and Mrs. D. Schewetzky for the careful preparation of the manuscript. The authors also thank Mrs. F. Wharton for editing the English.