
Abstract
Mannose-binding lectin is present in the contusion area of traumatic brain-injured patients and in that of traumatic brain-injured mice, where mannose-binding lectin-C exceeds mannose-binding lectin-A. The reduced susceptibility to traumatic brain injury of mannose-binding lectin double knock-out mice (mannose-binding lectin−/−) when compared to wild type mice suggests that mannose-binding lectin may be a therapeutic target following traumatic brain injury. Here, we evaluated the effects of a multivalent glycomimetic mannose-binding lectin ligand, Polyman9, following traumatic brain injury in mice. In vitro surface plasmon resonance assay indicated that Polyman9 dose-dependently inhibits the binding to immobilized mannose residues of plasma mannose-binding lectin-C selectively over that of mannose-binding lectin-A. Male C57Bl/6 mice underwent sham/controlled cortical impact traumatic brain injury and intravenous treatment with Polyman9/saline. Ex-vivo surface plasmon resonance studies confirmed that Polyman9 effectively reduces the binding of plasma mannose-binding lectin-C to immobilized mannose residues. In vivo studies up to four weeks post injury, showed that Polyman9 induces significant improvement in sensorimotor deficits (by neuroscore and beam walk), promotes neurogenesis (73% increase in doublecortin immunoreactivity), and astrogliosis (28% increase in glial fibrillary acid protein). Polyman9 administration in brain-injured mannose-binding lectin−/− mice had no effect on post-traumatic brain-injured functional deficits, suggestive of the specificity of its neuroprotective effects. The neurobehavioral efficacy of Polyman9 implicates mannose-binding lectin-C as a novel therapeutic target for traumatic brain injury.
Keywords
Introduction
Traumatic brain injury (TBI) remains a major cause of death and disability worldwide 1 and is known to be associated with primary biomechanical damage followed by pathogenic secondary molecular and cellular cascades, activated within minutes following injury, that interact in a complex network leading to cell death or recovery.2,3 In patients surviving TBI, this secondary injury cascade is believed to account for the majority of brain damage. Because traumatic brain damage evolves over days after the impact, there exists an opportunity for pharmacological therapeutic intervention. Although multiple preclinical studies have shown beneficial effects of a number of pharmacological compounds,4,5 no specific agent has been proven effective in attenuating the neurologic sequelae of TBI patients in the clinical setting.
It is well established that inflammatory responses contribute to secondary brain injury following TBI. The complement system is a powerful arm of innate immunity and is known to be involved in the pathogenesis of brain injury. 6 This system includes several circulating and cell-associated proteins that, upon activation, contribute to the pathophysiological sequelae of brain injury by: (1) exacerbation of the inflammatory response via C3a and C5a-mediated blood–brain barrier (BBB) leakage; (2) promoting an increase in leukocyte infiltration into injured brain with subsequent free radical production; (3) induction of neuronal and glial cell apoptosis via C3a and C5a receptor binding and (4) induction of neuronal death through membrane attack complex (MAC) – mediated cell lysis.6–8
Complement system activation occurs through (1) classical, (2) alternative, and/or (3) the lectin pathways, each comprised specific initiators and enzymes. The lectin pathway is activated by mannose-binding lectin (MBL) and ficolins, which are pattern-recognition molecules known to circulate as complexes together with MBL-associated serine proteases (MASPs). After binding to terminal mannose groups located on the surface of bacteria (pathogen-associated molecular patterns, PAMPs) or to the surface of damaged cells (damaged associated molecular patterns, DAMPs), MBL and ficolins complex with MASPs to initiate complement system activation.9–11
Our laboratory has reported that administration of C1 inhibitor (C1-INH) – an endogenous inhibitor of the complement system endowed with several anti-inflammatory effects – is neuroprotective in experimental models of focal ischemic injury12,13 and TBI. 14 The neuroprotective effect of C1-INH in CNS injury models has been attributed, in part, to its binding with MBL. 15
MBL has been documented in the brain tissue after both experimental and clinical TBI. Of the two murine MBL isoforms, MBL-A and MBL-C, the latter represents the prevalent isoform found in the injured cortex of mice following TBI. 16 Moreover, in this study, MBL double knock-out mice (MBL−/−) subjected to TBI showed attenuated sensorimotor deficits from 2 to 4 weeks post injury and reduced neuronal cell loss assessed at five weeks post injury. 16 These observations led us to hypothesize that MBL may play a deleterious role following TBI and that compounds that selectively bind to and inhibit the physiological effects of MBL-C may represent a promising novel therapeutic strategy for the treatment of TBI. In the present study, we evaluated the effects of Polyman9 (named glycodendrimer 13.3 in Varga et al. 17 ), a novel polymannosylated compound performing as a selective ligand of the carbohydrate recognition domain of MBL on neurobehavioral function and neurogenesis in a well-characterized experimental model of TBI.
Materials and methods
Binding studies with surface plasmon resonance (SPR)
Polyman9 was chemically synthesized by Dr. Bernardi and collaborators at the Department of Chemistry, University of Milan. Further details about its chemical structure and synthesis are available in Varga et al. 17 (where Polyman9 was named glycodendrimer 13.3). A recently reported SPR assay 18 was used to evaluate the binding of Polyman9 to murine MBL isoforms. Briefly, this methodology measures the binding of plasma MBL to mannose residues immobilized on an SPR sensor surface, thereby permitting the identification of MBL-A and MBL-C using the corresponding antibodies. Analysis of plasma samples pre-incubated with putative MBL antagonists permits to identify those compounds capable of preventing MBL-A/C interaction with mannose residues and to determine their potency (i.e. the concentration that inhibits MBL-A/C binding by 50% = IC50).
Binding studies were carried out using a ProteOn XPR36 Protein Interaction Array system (Bio-Rad Laboratories, Hercules, CA, USA) based on SPR technology. Mannosylated bovine serum albumin (Man-BSA, Dextra Laboratories, West Berkshire, UK) and bovine serum albumin (BSA) (Sigma-Aldrich, St Louis, MO, USA) were covalently immobilized onto two parallel flow cell surfaces of the same GLC sensor chips (Bio-Rad) using amine coupling chemistry, 18 with immobilization levels of 3000 and 4000 resonance units (RU, 1RU = 1 pg protein/mm2), respectively. Mouse plasma was diluted 50-fold with phosphate-buffered saline (150 mM NaCl in 10 mM phosphate buffer, PBS), preincubated for 30 min at room temperature with or without different concentrations of Polyman9 (10–200 µM) and injected over the immobilized ligands, followed by an injection of an anti-MBL-A antibody (10 µg/ml, Hycult Biotech, Uden, The Netherlands), or an anti-MBL-C antibody (10 µg/ml, Hycult Biotech). The running buffer was PBS containing 0.005% Tween 20 (PBST). All assays were performed at 25℃. The “sensograms” (time course of the SPR signal in RU) were normalized to a baseline value of “0.” The signal observed from the surfaces immobilizing Man-BSA was corrected by subtracting the nonspecific response observed from the reference surface (BSA only). For these studies, blood samples were collected from the vena cava of control (uninjured) C57Bl/6 mice, in 10 mM ethylendiaminetetracetic acid (EDTA) and 0.125% polybrene (Sigma-Aldrich). As previously demonstrated, polybrene has an anticoagulant activity. 19 Plasma, was separated by centrifugation for 15 min at 2000 g at 4℃ and immediately stored at −80℃ until use.
Animals
The IRCCS-Istituto di Ricerche Farmacologiche Mario Negri adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorization n.19/2008-A issued 6 March 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for those conducting animal experiments (quality management system certificate – UNI EN ISO 9001:2008 – Reg. N° 6121); the NIH Guide for the care and use of laboratory animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE). The statement of compliance (assurance) with the public health service (PHS) policy on human care and use of laboratory animals has been recently reviewed (9 September 2014) and will expire on 30 September 2019 (animal welfare assurance #A5023-01). All procedures regarding the study design, animal experiments, statistical analysis, and data reporting fulfil the criteria of the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (http://www.nc3rs.org.uk/page.asp?id=1357) as detailed in supplementary information. Before commencing all procedures, mice were housed (n = 5/cage) for at least one week in their home cages at a constant temperature on a 12-h light-dark cycle with ad libitum access to food and water in a specific pathogen free (SPF) animal facility.
Eight-week-old C57Bl/6 mice (WT, 20–25 g; Harlan Laboratories, Cambridgeshire, United Kingdom) and C57Bl/6 mice with a targeted mutation of both MBL-A and MBL-C genes (MBL−/−, 20–25 g; purchased from Jackson Laboratories (Bar Harbor, ME USA) and colonized at the Mario Negri Institute) were used. Mice were anesthetized with an intraperitoneal (i.p.) injection of sodium pentobarbital (65 mg/kg) and placed in a stereotactic frame.
Experimental brain injury
Mice were subjected to a craniectomy followed by induction of controlled cortical impact (CCI) brain injury at a velocity of 5 m/s and depth of 1 mm as previously described. 14 Sham (uninjured) control mice received identical anesthesia and surgery without brain injury. At the conclusion of the procedure, the craniectomy was covered with a cranioplasty and the scalp sutured. During and following all surgical procedures mice were kept on a heating pad and maintained at a body temperature of 37℃ degrees. This model of severe TBI is typically associated with minimal/no mortality. 16
Treatments
For in vivo experiments, Polyman9 was dissolved in 0.9% NaCl at a concentration of 900 µM. One hundred microliters of this solution (300 µg/mouse) of Polyman9 was injected intravenously (i.v.) 10 min post injury in order to attain circulating levels corresponding to the IC50 (see Results). For ex-vivo SPR studies, mice were sacrificed 20 min following treatment, blood collected, and plasma prepared as described above. To evaluate potential toxicity, in conformity with OECD Guideline for Testing of Chemicals, and Directive 75/318/EEC, single-dose toxicity studies were conducted on C57Bl/6 male mice. Polyman9 at the dose of 440 µg/mouse was administered intravenously to three mice. Intravenous administration was the unique route considered, since identical to that intended for human use. Mice were accurately observed from the earlier stages up to two months after the treatment. Physical appearance, body weight, behavior, and mortality were recorded. After two months from Polyman9 administration, a macroscopic examination of autopsy was conducted and necrotic tissue presence evaluated.
Assessment of sensorimotor deficits
Sensorimotor deficits were evaluated weekly for four weeks following TBI using the composite neuroscore and the beam walk tests. The neuroscore evaluated (1) forelimb function, (2) hindlimb function, and (3) resistance to lateral right and left pulsion. The maximum (best) score an animal could attain was 12. 16 The beam walk test evaluated the number of footfaults of the right hindlimb, while the mouse walks along a 5-mm wide, 1 -m long beam (60 steps). 20 Better motor performance using this test is associated with a lower score.
Contusion volume
At five weeks after TBI, mice were sacrificed for histological analysis and perfused brains were removed, frozen and cryosectioned at 20 µm. 14 Eight coronal sections from bregma +0.6 to −4 mm were stained with cresyl violet (Sigma-Aldrich, St. Louis, MO). The analytical image system (Imaging Research Inc., Brock University, St Catharines, Ontario, Canada) was used for image acquisition and contusion volume was calculated as previously described. 21
Neuronal cell count
The neuronal cell count was performed at five weeks after TBI. Three 20 µm-thick coronal sections at 0.4, 1.6, and 2.8 mm posterior to bregma and stained with cresyl violet (Sigma-Aldrich, St. Louis, MO) were selected from each mouse brain to quantify neuronal cell loss. The entire sections were acquired at 20× by an Olympus BX-61 Virtual Stage microscope, with a pixel size of 0.346 µm. Acquisition was done over 10 µm thick stacks, with a step size of 2 µm. The different focal planes were merged into a single stack by mean intensity projection to ensure consistent focus throughout the sample. Neuronal count was performed by segmentating the cells over the entire cortex and excluding the round-shaped signal sized below the area threshold of 25 µm2 that is known to be associated with glial cells (Figure S1) as reported previously. 16 Quantification was performed by Fiji software and neuronal cell loss was expressed as the ratio of neurons between ipsi- and contralateral cortices. For anatomic damage, cresyl violet stained sections were quantified as previously reported. 20
Immunohistochemistry
Immunohistochemistry was performed on 20 µm-thick coronal sections from perfused mouse brains. The sections were incubated overnight at 4℃ with primary polyclonal antibody goat anti-mouse doublecortin (DCX, 1:200, Santa Cruz Biotechnology, Dallas, TX, USA) or with primary monoclonal antibody mouse anti-mouse glial fibrillary acid protein (GFAP, 1:2000, Millipore, Billerica, MA, USA). Biotinylated secondary antibodies (1:200, Vector Laboratories) were used. DCX and GFAP immunopositive cells were identified by reaction with 3,3 diaminobenzidine-tetrahydrochloride (DAB, Vector Laboratories, Burlingame, CA, USA) as previously described. 22 Negative control studies, without the primary antibody, were performed in parallel. One coronal section per mouse (obtained from bregma) was selected to quantify the DCX-stained area in order to ensure unbiased, operator-independent sampling.22,23 An Olympus BX61 microscope equipped with a motorized stage and managed with AnalySIS software (Olympus) was used for sampling of the region of interest (ROI). Twelve 40× magnification fields were positioned in the ipsilateral hemisphere along the entire boundary of the ventricle with no vertical gap or overlap between each field. Immunostaining for DCX was quantified by segmentation of stained area using Fiji software. 24 To subtract the background signal, a minimum threshold was applied based in the highest grayscale (0–256) value of background. For GFAP quantification, three brain coronal sections per mouse (at 0.4, 1.6, and 2.8 mm posterior to bregma) were acquired at 20× by Olympus BX-61 Virtual Stage microscope, with a pixel size of 0.346 µm. Acquisition was done over 6 µm thick stacks, with a step size of 2 µm. The different focal planes were merged into a single stack by mean intensity projection to ensure consistent focus throughout the sample. The ipsilateral cortex was analyzed over an area included within a 350 µm radius from the contusion edge. Images were analyzed using Fiji software. DCX and GFAP immunostained area were expressed as positive pixels/total assessed pixels and reported as the percentage of total stained area as previously described.25,26
Western blot analysis
Blood samples were collected from the vena cava and plasma separated by centrifugation as described for SPR (vide supra). Equal amounts of plasma proteins (10 µg/sample) were electrophoresed onto a 12% sodium dodecyl sulfate polyacrylamide gel and transferred to polyvinylidene fluoride membranes. A rabbit anti-C4 polyclonal antibody (1:100, Hycult Biotech) followed by a rabbit peroxidase-conjugated antibody (1:2500; Santa Cruz Biotechnology, Dallas, TX, USA) were used. Quantification was carried out using Quantity One Software (Bio-Rad). For each sample, the C4b band or the Ponceau lane was selected and the corresponding optical density was automatically calculated by Quantity One Software. The resulting optical density of the C4b band was then normalized for that obtained by the Ponceau lane.
Immunofluorescence and confocal analysis
Immunofluorescence was performed on 20 µm-thick coronal sections according to the previously described method.15,22 The sections were incubated overnight at 4℃ with the primary antibody anti-GFAP (1:2000, Millipore, Billerica, MA, USA). Fluorconjugated secondary antibody Alexa 488 anti-mouse (1:500, Invitrogen, Carlsbad, CA) was used. Brain vessels were stained with Alexa 647 fluoro-conjugated isolectin (IB4, 1:200, Invitrogen, Carlsbad, CA). Cell nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI, 1 µg/ml, Invitrogen, Carlsbad, CA). Appropriate negative controls without the primary antibodies were performed. None of the immunofluorescence reactions revealed unspecific fluorescent signal in the negative controls. Immunofluorescence was acquired using a scanning sequential mode to avoid bleed-through effects by an IX81 microscope equipped with a confocal scan unit FV500 with three laser lines: Ar-Kr (488 nm), He-Ne red (646 nm), and He-Ne green (532 nm) (Olympus, Tokyo, Japan), and a UV diode. Three-dimensional images were acquired over a 10 µm z-axis with a 0.30 µm step size and processed using Imaris software (Bitplane, Zurich, Switzerland) and Photoshop CS2 (Adobe Systems Europe Ltd).
Physiology
Blood pressure measurement and blood gas analysis were performed on mice 10 min after TBI and saline or Polyman9 administration. Blood pressure was measured noninvasively by tail cuff using a computerized system (BP-2000 series II, blood pressure analysis system; visitech system physiological research instruments). Blood gas analysis was performed using the i-STAT 1 analyzer (Oxford Instruments S.M., Burke & Burke, Menfis Biomedica) as previously described.14,16
Study design and blinding for in vivo studies
Mice were randomly allocated to surgery (uninjured controls versus TBI) and to treatment (saline versus Polyman9). The following experimental groups were included: (1) WT sham or brain-injured mice treated with saline or Polyman9 10 min post injury or (2) MBL−/− brain-injured mice treated with saline or Polyman9 10 min after TBI. The number of animals for each group was established a priori in order to distribute them equally across experimental days and batches to avoid systematic errors. All experimental procedures including surgery, drug treatment, physiology, behavioral testing, lesion volume quantification, neuronal cell counting, DCX and GFAP quantification, and Western blot analyses were performed by investigators blinded to the experimental conditions.
Statistical analysis
Data are presented as mean +SD or box and whiskers (min, max, and median). The sample size for our analysis was predetermined based on power analyses performed using raw data from previous studies. GraphPad Prism 6 software was used for statistical analysis. Data from the physiology studies were analyzed using a Mann–Whitney test. Comparisons between groups for neuroscore and beam walk tests were performed using a two-way repeated-measurement analysis of variance (ANOVA) followed by Bonferroni post hoc test. Data from studies evaluating the inhibitory effect of Polyman9 on MBL-A/-C binding to mannose residues, anatomic damage, neuronal cell counts, and neurogenesis were checked for normal distribution using the Kolmogorov–Smirnov (KS) test and analyzed using a “t” test. Comparisons between groups for circulating C4b levels were performed using two-way ANOVA followed by a Sidak correction post hoc test. A “p” value of less than 0.05 was considered statistically significant.
Results
Polyman9 decreases the binding of MBL-C selectively over that of MBL-A to mannose residues as assessed by in vitro and ex-vivo SPR assay
A novel in vitro SPR-based assay was employed to screen polymannosylated, dendrimeric compounds as potential inhibitors of MBL-induced effects.
18
Using this assay, we measured the potency of these compounds to inhibit the binding of plasma MBL-A or MBL-C to mannose residues immobilized on the sensor surface. Figure 1 shows the results obtained with one of these compounds, Polyman9, which prompted its use in subsequent in vivo studies. Murine plasma, diluted 1:50, was pre-incubated in the absence or presence of 10–200 µM Polyman9 and the solutions were injected over a surface of exposed mannosylated residues. The subsequent injection of an anti-MBL-A antibody resulted in a non-saturating binding signal, indicating that plasmatic MBL-A had been captured by mannose residues. The amount of MBL-A bound to mannose residues was not affected by pre-incubation of plasma with Polyman9, up to 200 µM (Figure 1(a)). Subsequent injection of an anti-MBL-C antibody resulted in a saturating binding signal, indicating that also plasma MBL-C had been captured by mannose residues. The amount of MBL-C bound to mannose was dose-dependently reduced in plasma samples pre-incubated with Polyman9, demonstrating a specific inhibitory effect of the compound on MBL-C, with an IC50 of 65 µM (Figure 1(b) and (c)).
In vitro inhibitory effect of Polyman9 on the binding of plasma MBL-C, but not MBL-A, to mannose residues. A three-step surface plasmon resonance (SPR) assay was carried out. First, murine plasma, preincubated with different concentrations of Polyman9, was flowed onto a surface coated with mannose residues. Panels a and b show representative sensograms obtained following subsequent injections with an anti-MBL-A or an anti-MBL-C antibody, for 2 min (bar). Panel c: concentration-dependent inhibition by Polyman9 of the SPR signal observed with the anti-MBL-C antibody (i.e. binding of MBL-C to mannose residues). Values from three independent experiments are shown (open, filled circles and triangles).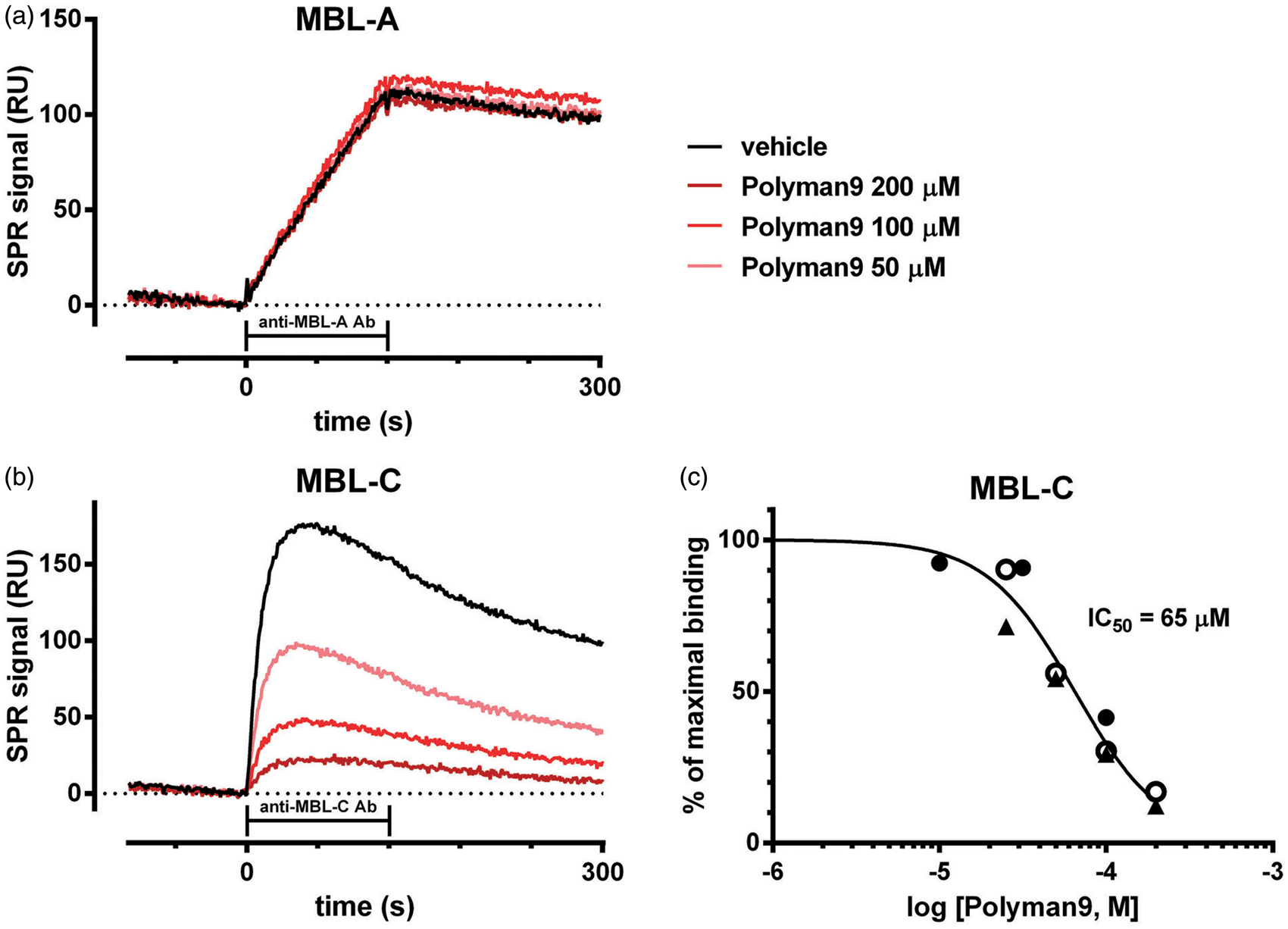
For ex-vivo studies, mice were intravenously treated with Polyman9 (300 µg/mouse) or saline, 10 min after induction of TBI. Twenty minutes after the treatment, all animals were sacrificed and blood was collected to obtain plasma for SPR analysis. The amount of plasma MBL-C and MBL-A bound to mannose was determined as the SPR signal observed in subsequent injections with the corresponding antibodies. Polyman9 inhibited the binding of plasma MBL-C, but not that of MBL-A to mannose residues (Figure 2).
Ex-vivo inhibitory effect of Polyman9 on the binding of plasma MBL-C, but not MBL-A, to mannose residues. Ten minutes following TBI, mice were treated with Polyman9 (300 µg/mouse) or saline. Twenty minutes later all animals were sacrificed and blood collected to obtain plasma which was injected into the surface plasmon resonance (SPR) device over immobilized mannose residues. The amount of plasma MBL-C and MBL-A bound to mannose was determined as the SPR signal observed following subsequent injections with the corresponding antibodies. Data are presented as box and whiskers: min, max, and median (n = 6). Unpaired t-test; *p < 0.05 (Student’s T test).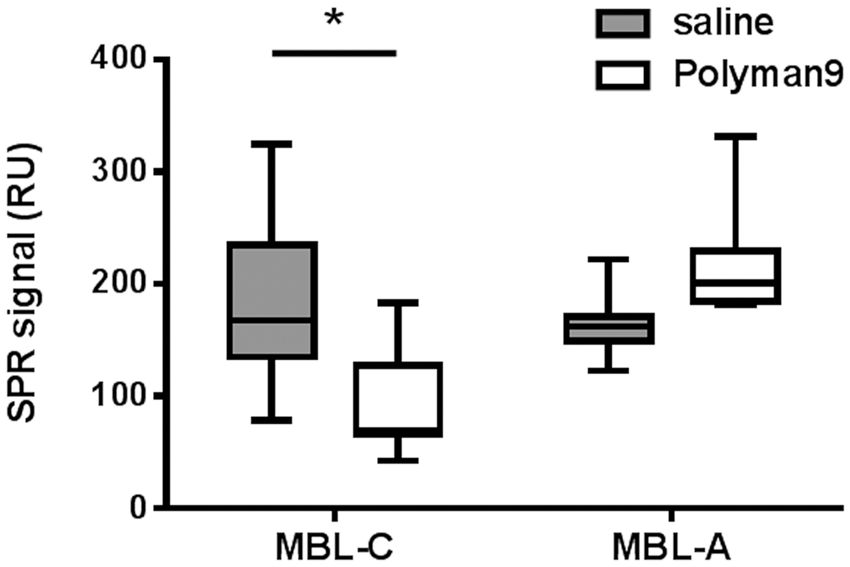
Polyman9 administration does not significantly alter physiological parameters in vivo
Physiological data from saline- versus Polyman9-treated mice 10 min after injection.
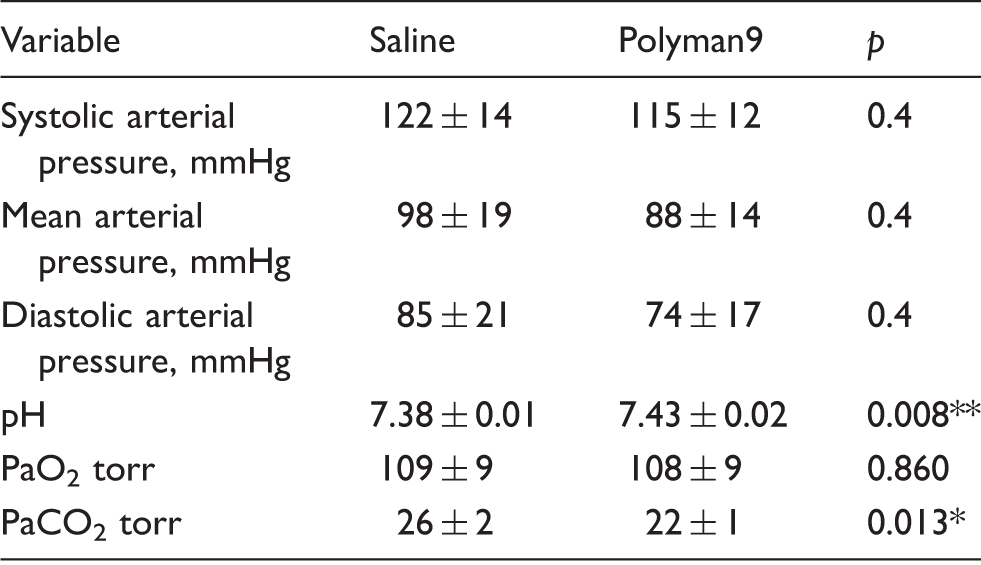
Note: The effect of Polyman9 treatment on physiological variables was assessed 10 min after the injection. Data are presented as mean ± SD (n = 5).
p < 0.01; *p < 0.05: saline-versus Polyman9-treated mice, Mann–Whitney test.
Polyman9 attenuates sensorimotor deficits after TBI
The mean neuroscores of all experimental groups of brain-injured mice improved over the four-week observation period, as previously described.14,16 At one-week post injury, mice subjected to TBI and receiving either saline or Polyman9 showed a similar performance on the composite neuroscore test. However, from the second up to the fourth week post injury, Polyman9 treatment significantly attenuated TBI-induced functional deficits compared to saline-treated brain-injured animals (Figure 3(a)). The beam walk test showed a similar pattern of improvement in motor function at two and three weeks post injury following Polyman 9-treatment when compared to saline-treated mice (Figure 3(b)). At five weeks after TBI, brain-injured WT mice subjected to saline or Polyman9 treatment showed comparable anatomic damage (Figure 3(e)).
Polyman9 attenuates TBI-induced sensorimotor deficits in WT but not MBL−/− mice. Functional analysis (a–d). Sensorimotor function was assessed over four weeks after sham injury (n = 6) or TBI (n = 12) using the composite neuroscore test (a, maximal performance score = 12) and beam walk test (b, maximal performance score = 0) in mice receiving saline or Polyman9 (300 µg/mouse, i.v.) 10 min after surgery/TBI. Polyman9 was effective in attenuating post-traumatic sensorimotor deficits beginning two weeks post-treatment. Data are presented as mean + SD. Two-way ANOVA for repeated measures (p < 0.001) followed by Bonferroni post hoc test were used to evaluate injury effect (sham vs. injury, p < 0.001, data not shown) and treatment effect (brain injury + Polyman9 vs. brain injury + saline; *p < 0.05, ***p < 0.001) for each of the four weeks. Composite neuroscore test (c) and beam walk test (d) were applied to MBL−/− mice receiving saline (n = 9) or Polyman9 (300 µg/mouse, i.v., n = 7) 10 min after TBI. Data are presented as mean + SD. Two-way ANOVA for repeated measures followed by Bonferroni post hoc test were used to evaluate the effect of treatment, ns. Histopathological analysis (e–h). At five weeks after TBI, Polyman9 (blue) did not affect the lesion volume compared to saline (red), either in WT (n = 12, e) or MBL−/− (n = 7–9, f) mice. Unpaired t-test: ns. At the same time point, saline and Polyman9 WT (n = 11–12, g) and MBL−/− (n = 7–9, h) brain-injured mice present less neurons in ipsi- compared to contra-lateral hemisphere with no treatment effect. Data are presented as box and whiskers: min, max, and median. °°°p < 0.001: ipsilateral versus respective contralateral hemispheres, two-way ANOVA followed by Sidak post hoc test.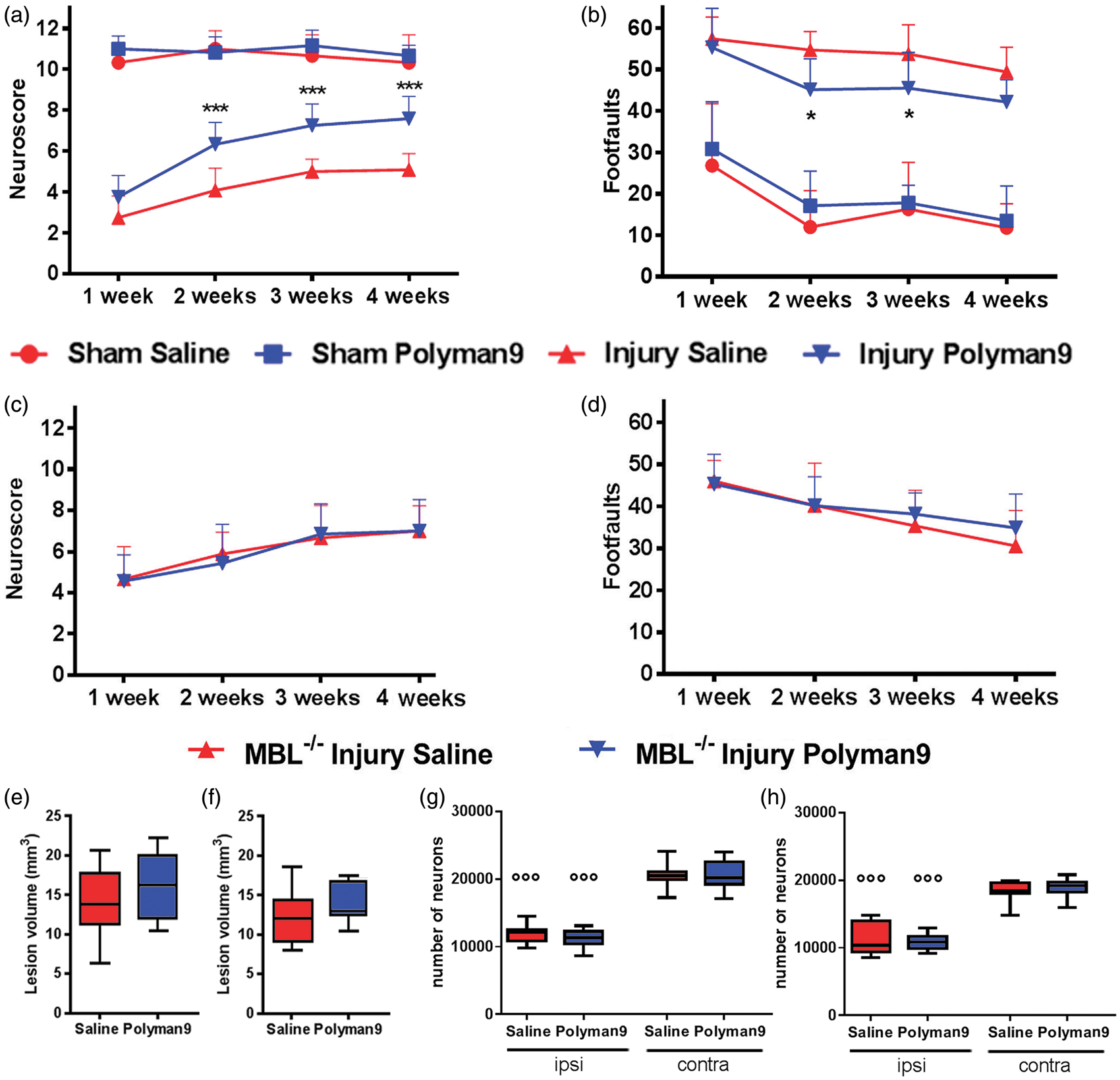
Polyman9 administration does not affect TBI-induced neurobehavioral deficits in MBL−/− mice
To further explore whether the beneficial effect of Polyman9 treatment observed in wild-type mice following TBI is driven specifically by MBL inhibition, MBL−/− mice were subjected to TBI or surgery without TBI and post injury treatment with either saline or Polyman9 (identical dose, timing and route of injection used for wild type mice). Sensorimotor deficits were evaluated weekly from post injury week 1 through week 4. Mean neuroscores of all groups of brain-injured mice treated with either saline or Polyman9 improved over the four-week observation period as previously reported.14,16 The neuroscore values of brain-injured MBL
Polyman9 enhances neurogenesis and astrogliosis without affecting neuronal cell loss
By five weeks post injury, an extensive macroscopic area of cortical tissue loss extending rostrocaudally from bregma +0.6 to −4 mm was present in brain-injured mice treated with either saline or Polyman9. The cell count of healthy neurons was performed on the entire cortex of ipsilateral and contralateral hemispheres to calculate neuronal loss. WT (Figure 3(g)) and MBL−/− (Figure 3(h)) mice subjected to TBI and saline or Polyman9 treatment showed comparable neuronal cell loss. Endogenous neurogenesis was measured by quantifying the area of DCX-immunopositivity in the subventricular zone (SVZ) of the ipsilateral hemisphere (Figure 4(a) and (b)). Brain-injured mice treated with Polyman9 showed a significant increase in the total area of DCX-immunopositive staining (1.32 ± 0.60) when compared to saline mice (0.76 ± 0.45, p = 0.0203 Figure 4(c)).
Polyman9 enhances neurogenesis and astrogliosis in TBI mice. Immunohistology was done five weeks after brain injury in mice receiving saline or Polyman9 (300 µg/mouse, i.v.) 10 min after TBI. Doublecortin (DCX) staining was quantified in the SVZ, in the frames located as shown in panel a. Representative immunostaining is shown in panel b, scale bar = 20 µm. Polyman9-treated mice showed increased DCX expression (positive/total pixels expressed as % of the stained area) when compared to saline-treated animals, c. LV = left ventricle. Glial fibrillary acid protein (GFAP) immunostained area was quantified in the ipsilateral cortex, over the area indicated by the red dotted line as presented in panel d. Representative immunostaining is shown in panel e, scale bar = 50 µm. Polyman9-treated mice showed increased GFAP immunostained area than saline-treated animals, (f). Higher magnifications of the areas indicated in (e) suggest close contacts between astrocytes and vessels in Polyman9- (g′) but not saline-treated mice (g). This observation is further confirmed by three-dimensional confocal analysis and renderings of astrocytes (GFAP, green), vessels (IB4, red) and nuclei (Hoechst, blue) in TBI mice treated with Polyman9 or saline, (h′) and (h), respectively. Scale bars = 20 µm Data are presented as box and whiskers: min, max, and median; n = 11–12. Unpaired t test; *p < 0.05; **p < 0.01.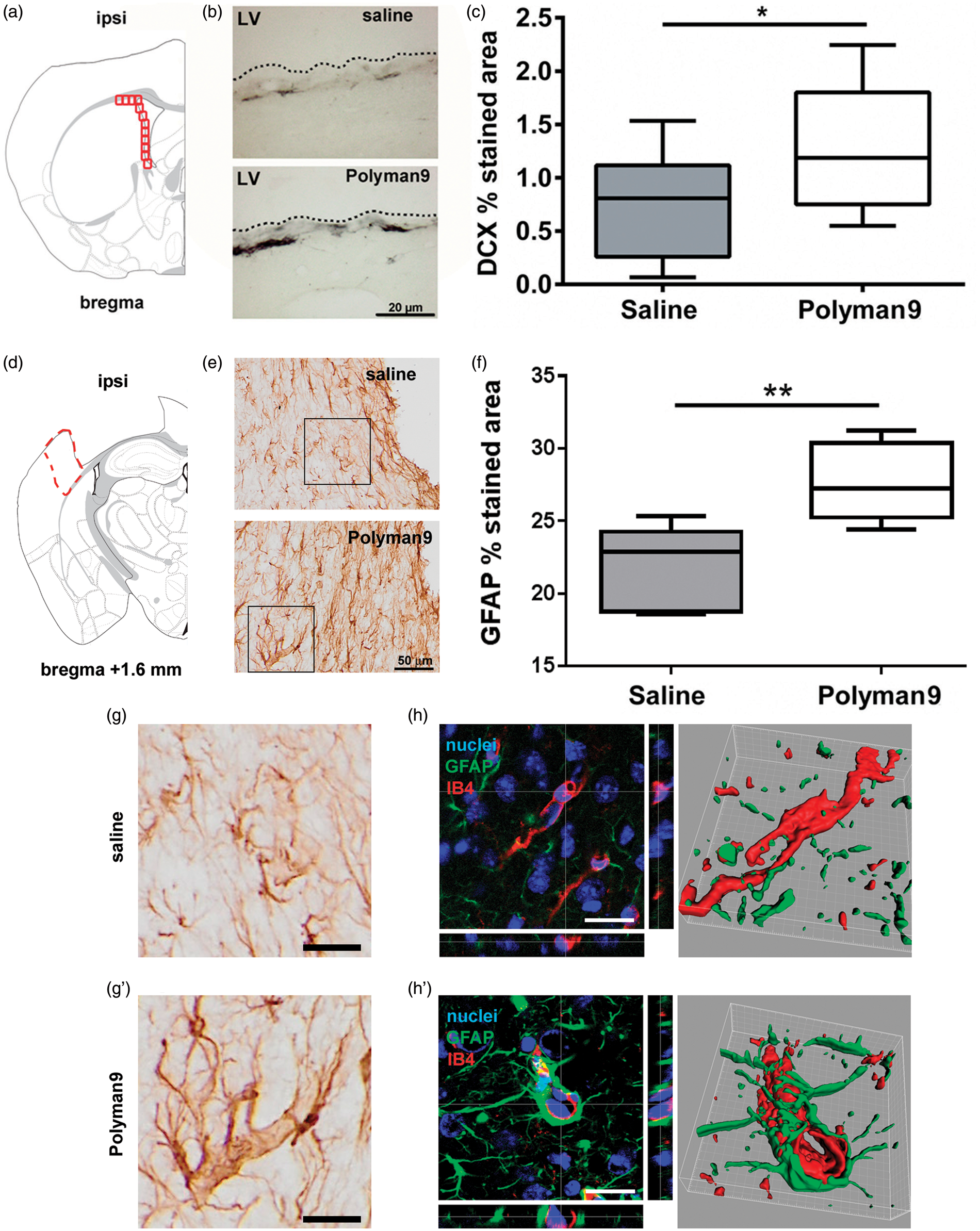
At the same time point, astrogliosis was measured quantifying GFAP immunopositive area at the edge of the contusion area in the ipsilateral cortex (Figure 4(d)). Brain-injured mice treated with Polyman9 showed a significant increase in the total GFAP positive immunostained area (27.6 ± 2.7% stained area ± sd) compared to saline mice (21.8 ± 2.9, p = 0.007 Figure 4(e) and (f)). At variance with saline-administered mice (Figure 4(g) and (h)), in Polyman9-treated TBI mice, GFAP was mostly present in relation to brain vessels (Figure 4(g′) and (h′)), as clearly shown by three-dimensional confocal microscopy and 3D renderings.
Effect of Polyman9 treatment on circulating C4b
We further investigated whether Polyman9-mediated inhibition of MBL-C affected complement activation following TBI by measuring plasma levels of C4b that reflects the activation degree of the classical and lectin pathways. Plasma samples from naïve or brain-injured mice were collected 24 h after treatment with saline or Polyman9. We found that C4b was significantly increased in saline-treated, brain-injured mice when compared to controls, suggesting that the lectin pathway was activated after TBI. Furthermore, we found that Polyman9 administration significantly reduced C4b levels in brain-injured mice when compared to saline-treated, brain-injured animals (Figure 5).
Polyman9 reduces circulating C4b levels 24 h after TBI. Mice subjected to TBI with saline treatment exhibited significantly increased C4b levels compared to non-injured mice. Treatment with Polyman9 following TBI was effective in reducing the post-traumatic increase in circulating C4b. Representative blot is reported below the graph: the bands correspond to different samples obtained in the same Western blot but not necessary adjacently. Data are presented as box and whiskers: min, max, and median; n = 6. Two-way ANOVA followed by Sidak post hoc test for the effect of injury (naive versus injury, **p < 0.01) or that of treatment (injury Polyman9 versus injury saline; *p < 0.05).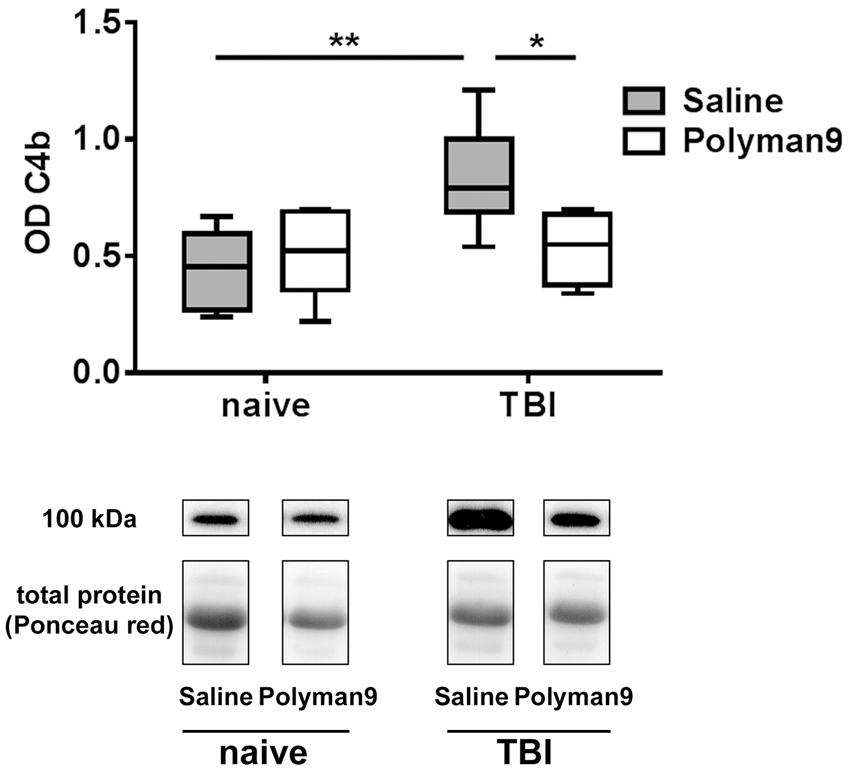
Discussion
We have previously reported that MBL is present in the brain tissue from surgically excised human contused tissue and after experimental TBI in mice. Furthermore, TBI in mice genetically engineered to be deficient in MBL is associated with attenuated functional deficits and tissue damage, suggesting that MBL modulation might be a potential therapeutic target after TBI. 16 The results of the present study demonstrate that the selective pharmacological inhibition of MBL, and, in particular of the MBL-C isoform, improves functional neurobehavioral outcome following TBI. We found that Polyman9, a polymannosylated molecule: (1) selectively inhibits the ability of plasma MBL-C to bind mannose residues in vitro and ex-vivo; (2) improves sensorimotor function and increases neurogenesis following experimental TBI; (3) reduces circulating post-traumatic C4b levels; and (4) is not effective in improving neurobehavioral function in brain-injured MBL−/− mice, suggesting that its effects on functional outcome and neurogenesis are dependent on the presence of MBL.
While a single isoform of MBL has been described in humans, 27 two isoforms have been characterized in mice. 28 Although little is known about the functional similarities and carbohydrate specificity of human versus murine MBL proteins, structural analysis indicates that human MBL is the homologue of mouse MBL-C.28,29 We have reported that, following experimental TBI in mice, MBL-C expression is markedly increased when compared with MBL-A in the contused tissue and is elevated from 30 min to one-week post injury. 16 Based on these previous observations, we have focused on the identification of a therapeutic molecule capable of targeting MBL-C. The C-terminal carbohydrate recognition domain (CRD) of circulating MBL recognizes mannosylated glycoproteins expressed on the surface of pathogens or “altered” self-cells and is known to trigger the complement system,30,31 thereby activating the associated serine proteases (MASPs).6,32 MBL assembly into oligomeric structures allows formation of multiple CRD domains (clusters of 2-6 CRDs) which lead to an efficient binding complex with multivalent properties. Although each individual interaction between the CRD and the mannosylated glycoproteins has a characteristic low binding affinity, the MBL self-assembly into high-order oligomers provides a means to recognize repetitive arrays of its carbohydrate targets with high avidity. Thus, to effectively interfere with these multivalent interactions, the development and characterization of multivalent inhibitors are critical. To this end, we have selected Polyman9, a hexavalent pseudo-dimannoside, capable of selectively targeting MBL-C. 17 The results obtained with our novel SPR-based assay indicate that pre-incubation of plasma with Polyman9 results in a dose-dependent decrease of MBL-C binding to a mannosylated surface, with an IC50 of 65 µM. No effect of the compound up to 200 µM on the binding of MBL-A to mannose residues was observed.
After demonstrating that Polyman9 inhibits MBL-C binding, we then assessed its potential therapeutic effect in experimental TBI in mice. Initially, ex-vivo SPR studies confirmed that i.v. treatment with Polyman9 (300 µg/mouse) significantly decreased the binding of plasma MBL-C to the mannosylated surface, suggestive of an effective in vivo interaction of Polyman9 with MBL-C. We then assessed the potentially beneficial effects of Polyman9 on neurobehavioral outcome following experimental TBI. Post injury Polyman9 administration induced significant improvement in motor function performance as assessed by the composite neuroscore and beam walk tests, beginning two weeks after TBI. The extent and temporal pattern of functional improvement were identical to those previously observed in brain-injured MBL−/− mice, suggesting a similar mechanism of therapeutic efficacy. 16 Polyman9-treated mice subjected to TBI failed to show evidence of decreased neuronal cell loss. The observation that a protective treatment can affect functional but not histopathological outcome after experimental TBI is widely reported and debated33–38 and may be due to the fact that functional improvement may also depend on neuroplasticity and reorganization events not directly reflected by cell death or survival. 39 Specifically, complement system components are involved in synaptic pruning during development or disease and their inappropriate activation in neuroinflammatory conditions may lead to synaptic loss. 40 We hypothesize that the protected phenotype associated to Polyman9 administration is related to the preservation of neuronal wiring and to the reduction of synapse loss, with no effect on structural damage.
Polyman9-induced behavioral improvement was, instead, associated with increased neurogenesis in the SVZ of the hippocampus. Whether the neurobehavioral effects of this compound are due to the remodeling of neuronal circuits through the migration of newly born neurons to the damaged area remains to be elucidated. Our results are in line with previous data showing that an increased expression of DCX in the SVZ was associated with an improvement of sensorimotor function and enhanced rescue after TBI. 26 A more prolonged assessment of neuronal cell counts following TBI and treatment with Polyman9 is therefore warranted, particularly in light of current evidence indicating that neurogenesis is involved in functional recovery after TBI. 41 In addition to its effects on neurogenesis, we also demonstrate that Polyman9 significantly decreased circulating C4b, suggestive of an effect on the lectin pathway of the complement system. Previous studies have shown that mice with depleted C4 expression subjected to TBI exhibited reduced motor deficits and decreased anatomical damage compared to brain-injured WT mice. 42 Moreover, since C4 fragments may interact with the complement receptor 2 (CR2) localized on the surface of DCX-positive cells, thereby inhibiting neurogenesis, and since CR2−/− mice exhibit an increased number of neuronal progenitor cells compared to WT mice,42,43 we suggest that Polyman9 may affect neurogenesis via the reduction of circulating C4 fragments. Interestingly Polyman9 administration significantly increased astrogliosis at cortical contusion site. In particular, we observed a dramatic increase in contacts between astrocytic endfeet and brain vessels, suggestive of a trophic action at the neurovascular unit. 44
Although Polyman9 has the ability to prevent the binding of MBL-C to mannose residues as assessed by SPR analysis, we cannot exclude the possibility of it interacting with other primary targets. Our observation that Polyman9 administration in MBL−/− mice subjected to TBI is ineffective in improving functional outcome suggests that its beneficial properties may be due to its interaction with MBL.
Yager et al. 45 previously reported that MBL gene deficiency increased cerebral cell death 6 h after TBI with no difference in sensorimotor deficits one-week post injury suggesting a potential neuroprotective role of MBL post-TBI. While methodological differences can possibly account for differential results, as discussed previously, 16 it should be underlined that the authors did not evaluate longer times after injury. In our present work, we found that Polyman9-dependent inhibition of MBL attenuates sensorimotor deficits from the second week post injury onwards. We hypothesize that MBL inhibition attenuates mechanisms of secondary brain damage and promotes neuroreparative processes at longer times post-TBI compared to those assessed by Yager et al., 45 therefore justifying the apparent discrepancy between the two studies.
While preclinical studies have shown beneficial effects of a number of pharmacological compounds in the treatment of experimental TBI,4,5 no specific agent has been found effective in attenuating the neurologic sequelae of TBI in humans. 4 This discrepancy has been attributed, in part, to various factors such as selection of inappropriate target mechanisms, 5 lack of selectivity of drug targeting, and administration of molecules that need to cross the blood–brain barrier (BBB) to demonstrate efficacy. 4 The results of the present study suggest that Polyman9 may be of significant benefit in the treatment of TBI since MBL is a circulating protein and systemic administration of Polyman9 does not need to cross the BBB in order to show therapeutic efficacy.
In the present study, we provide evidence that MBL-C plays an important role in the activation of pathogenic cascades after TBI. We have developed a novel and specific pharmacological tool to show that MBL inhibition improves neurobehavioral outcome, increases hippocampal neurogenesis in the SVZ, and astrogliosis following TBI. From a clinical perspective, MBL is present in contused tissue removed from patients within several hours up to days after TBI. 16 In addition, we have recently shown that Polyman9 is effective in inhibiting the binding of recombinant human MBL to mannose residues with an IC50 of 136 µM, therefore validating its potential use in humans. 18 In view of these all data, the polymannosylated molecule Polyman9 may represent a novel therapeutic strategy for the treatment of TBI. Further studies of this molecule are warranted.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support including Drs De Blasio and Fumagalli fellowship was provided by Fondazione Cariplo (grant number 2012-0590).
Acknowledgement
The authors thank Dr Tracy K. McIntosh for English editing of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
DDB, SF, and FO planned and performed the experiments, collected the data, and participated to the ms draft. MDGS planned the experiments, analyzed the data and drafted the ms. ERZ performed the experiments, analyzed the data and participated to the ms draft. AB and MG planned the experiments and analyzed the data. LL, AP, MS, GV performed the experiments.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.