
Abstract
During brain damage and ischemia, the cytokine interleukin-1ß is rapidly upregulated due to activation of inflammasomes. We studied whether interleukin-1ß influences cortical spreading depolarization, and whether lipopolysaccharide, often used for microglial stimulation, influences cortical spreading depolarizations. In anaesthetized rats, cortical spreading depolarizations were elicited by microinjection of KCl. Interleukin-1ß, the IL-1 receptor 1 antagonist, the GABAA receptor blocker bicuculline, and lipopolysaccharide were administered either alone or combined (interleukin-1ß + IL-1 receptor 1 antagonist; interleukin-1ß + bicuculline; lipopolysaccharide + IL-1 receptor 1 antagonist) into a local cortical treatment area. Using microelectrodes, cortical spreading depolarizations were recorded in a non-treatment and in the treatment area. Plasma extravasation in cortical grey matter was assessed with Evans blue. Local application of interleukin-1ß reduced cortical spreading depolarization amplitudes in the treatment area, but not at a high dose. This reduction was prevented by IL-1 receptor 1 antagonist and by bicuculline. However, interleukin-1ß induced pronounced plasma extravasation independently on cortical spreading depolarizations. Application of lipopolysaccharide reduced cortical spreading depolarization amplitudes but prolonged their duration; EEG activity was still present. These effects were also blocked by IL-1 receptor 1 antagonist. Interleukin-1ß evokes changes of neuronal activity and of vascular functions. Thus, although the reduction of cortical spreading depolarization amplitudes at lower doses of interleukin-1ß may reduce deleterious effects of cortical spreading depolarizations, the sum of interleukin-1ß effects on excitability and on the vasculature rather promote brain damaging mechanisms.
Introduction
In the brain, numerous cytokines are synthetized and released, e.g. tumor necrosis factor (TNF), interleukin-6 (IL-6), interleukin-1ß (IL-1ß). These cytokines are mainly produced by glial cells, but some of them are also synthetized by neurons and other cells. 1 Since glial cells have different functions such as controlling the extracellular milieu, modifying synaptic strength and plasticity as well as immune functions,2,3 it is a major challenge to identify the role of cytokines in the brain. E.g. TNF has physiological functions, and in brain diseases it may promote brain damage but also provide neuroprotection. 4 Recently, we found that TNF reduces the amplitude of cortical spreading depolarization (CSD) by acting on TNF receptor 2 which are expressed on GABAergic inhibitory interneurons. 5
CSDs are mass depolarizations.6–8 They are observed as single events in transient brain dysfunctions such as migraine aura.9–12 However, a series of CSDs, e.g. evoked in the aftermath of stroke, may promote brain damage because CSDs are very energy demanding and cause severe hypoperfusion finally aggravating hypoxic conditions.13–15 The reduction of the CSD amplitude by TNF, resulting from GABAergic inhibition, may therefore represent a neuroprotective mechanism.
Another major cytokine in the brain is IL-1ß. It is physiologically expressed at low concentrations but strongly upregulated in pathological conditions such as stroke, Alzheimer’s disease, and others.2,16–19 As a major effector molecule in innate immunity, 2 IL-1ß is produced by inflammasomes which have a pathogenic role in these diseases. 19 According to the available literature, IL-1ß at elevated concentrations is neurotoxic. In stroke, IL-1ß is enhanced within the first hours and exacerbates ischemic damage.16,18 Focal ischemia in rat cortex induced the expression of IL-1ß mRNA in the infarct zone and remote areas of the ipsilateral hemisphere. 20 Early application of the antagonist, IL-1 receptor 1 antagonist (IL-1RA) reduces the infarct volume and is, therefore, neuroprotective. 18
Interestingly, activation of the inflammasome and the release of IL-1ß is stimulated by high extracellular potassium concentration, 19 a stimulus which can elicit CSD. 21 A functional interaction between potassium, CSD, and IL-1ß is conceivable. Within 4 h after KCl-induced CSD, the expression of IL-1ß mRNA increased by 24-fold. 22 In general, receptors for IL-1ß (IL-1R1) are expressed in neurons, astrocytes, microglia, and endothelial cells.18,19,23 Thus, IL-1ß influences neuronal functions 18 and vascular functions such as the modification of the blood-brain barrier 16 and cerebral blood flow. 18 In addition, IL-1ß causes plasma extravasation and perivascular edema, e.g. in lung tissue 24 and in the brain. 25
The different patterns of TNF, having both brain damaging as well as neuroprotective effects, and of IL-1ß, having virtually only neurotoxic effects, caused us to study whether effects of IL-1ß on CSDs are different than those of TNF. Furthermore, we studied whether TNF and IL-1ß have a different effect on the vasculature, with respect to their ability to evoke plasma extravasation. Finally, we studied whether lipopolysaccharide (LPS) stimulation which is used to stimulate the release of cytokines, e.g. TNF or IL-1ß26,27 influences CSD similarly as IL-1ß and whether, correspondingly, neutralization of IL-1ß prevents the effect of LPS stimulation on CSDs.
Materials and methods
The present study was performed according to the Protection of Animals Act of the Federal Republic of Germany (Tierschutzgesetz der Bundesrepublik Deutschland) and was approved by the Thuringian State Office for Consumer Protection (Thüringer Landesamt für Verbraucherschutz, TLV, Reg. No. 02-005/12). The animals were treated in accordance with the declaration of Helsinki and the guiding principles in the care and use of animals. Data sampling, evaluation, and presentation complied with the ARRIVE guidelines.
Surgical preparation of the rats
Adult male Wistar rats (n = 46; 350–450 g, aged older than 90 days, housed in the Animal Facility of University Hospital Jena) were deeply anesthetized with sodium thiopental (Trapanal; Inresa, Freiburg, Germany; initially 100 mg/kg intraperitoneally [i.p.]). During dissection, depth of anesthesia was regularly assessed by testing the corneal blink reflex and reflexes to noxious squeezing of the tail tip. During the experiments, supplemental doses of 20 mg/kg Trapanal i.p. maintained the absence of the corneal blink reflex. The trachea and the right femoral vein and artery were cannulated. The mean arterial blood pressure was continuously monitored. Body temperature was kept at 37℃ using a feedback-controlled heating system.
After stereotactic fixation of the head, two trephinations (Figure 1(a)) were made over the left hemisphere (one frontal, spanning from 2 mm in front of bregma over a length of 5–6 mm, 3–4 mm wide, and one caudal, with a diameter of 3–4 mm in front of lambda) using a minidrill and cooling with artificial cerebrospinal fluid (ACSF). The composition of ACSF was in millimols per liter: NaCl 138.4, KCl 3.0, CaCl2 1.3, MgCl2 0.5, NaH2PO4 0.5, urea 2.2, and glucose 3.4, warmed to 37℃ and equilibrated with 5% CO2 in O2. The underlying dura and arachnoidea were removed, and the exposed cortex was kept moist with ACSF. A wall was built with dental acrylic on the skull around the large frontal trephination thereby forming a trough with a capacity of 150 to 200 µL.
Display of experimental approach and effect of IL-1ß on CSD. (a) Schematic view of the adult rat skull (not to scale) with the two trephinations. The frontal trephination is surrounded by a wall made from dental acrylic (grey oval) forming the “treated area” with recording electrodes DC3 and DC4. The rear trephination contained recording electrodes DC1 and DC2 glued to the stimulating electrode, in the “untreated area.” Electrode arrays were lowered to a depth of 1200–1400 µm, approximately cortical layer V for the deepest electrode tips. Recording electrodes in an array had vertical and horizontal tip separations of 800 µm. (b) Representative samples of CSD from the untreated cortex and from treated cortex before and after application of 50 ng IL-1ß showing the direct current recording (top rows, respectively) and the bandpass (0.01–45 Hz) filtered signal with depression of electroencephalographic activity in the phase of negative DC shifts (bottom rows, respectively). (c) CSDs in the untreated and treated areas after application of 100 ng IL-1ß. Same presentation as in (b).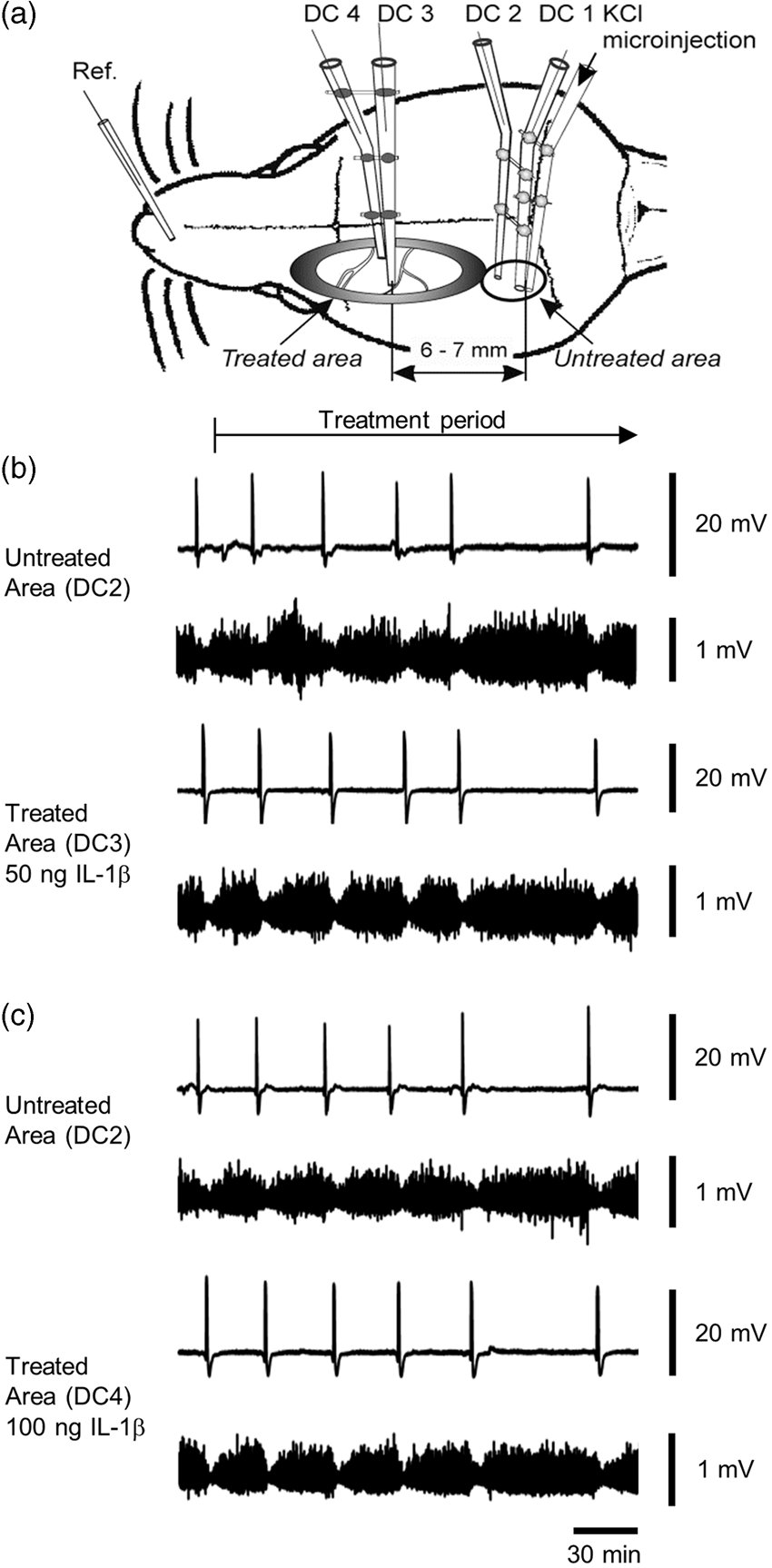
Recording of intracortical direct current potentials and data processing
Experiments lasted for 4 to 5 h, and CSDs were elicited by the microinjection of KCl at intervals of 30 min in the first 2 h after application of a test substance and every hour in the last 2 h, respectively. We used an Ag/AgCl reference electrode (containing 2 mol/L KCl) on the nasal bone. The electrode arrays for elicitation of CSD and recordings of intracortical direct current (DC) potentials at sites 1 to 4 (DC1–4) are shown in Figure 1(a). Electrodes for DC recordings had tip diameters of approximately 5 µm, resistance <10 MΩ, and were filled with 150 mmol/L NaCl. The electrode for CSD elicitation contained 1 mol/L KCl; SD waves were elicited by injection of 0.5 µL KCl with a pressure of 100 kPa for 1 s using a microinjector (picoinjector PLI-100; Harvard Apparatus, Holliston, MA). The signals were recorded using a 4-channel high-impedance amplifier (Meyer, Munich, Germany) and stored on a personal computer (sampling rate 2048 Hz). CSD was accepted if they had a steep onset, exceeded amplitudes >5 mV, and migrated over the whole recording area.
CSDs were evaluated regarding occurrence in the treated areas, maximal amplitudes related to baseline before the steep onset of the depolarization, duration at half-maximal amplitude and propagation time from the site of elicitation (electrode DC1) to the deepest electrode in the treated area (electrode DC3).
In order to analyze depression patterns of the electrocorticographic activity associated with spreading depolarization, DC recordings were resampled with a sample rate of 205 Hz and first detrended by appropriate adaptive filtering, followed by band pass filtering (bandpass 0.01–45 Hz). In order to reveal electrocorticographic activity, the signals were high-pass filtered with a lower frequency limit of 0.5 Hz. In addition, power of bandpass filtered recordings (0.5–45 Hz) and the integral of power of bandpass filtered recordings were calculated.
Application of compounds
Compounds were administered topically onto the exposed cortex into the trough for treatment. We applied recombinant rat IL-1ß (R&D Systems Inc., Minneapolis, MN) [10.0; 50.0; 100.0 ng in 100 µL, dissolved with maximal 1% bovine serum albumin in phosphate-buffered saline (PBS)] for 4 h alone. In additional experiments, we applied either 50.0 ng IL-1ß together with the recombinant rat IL-1RA (Biotrend, Cologne, Germany) (500 ng in 100 µL, dissolved in PBS) for 4 h, or 50.0 ng IL-1ß together with the GABAA receptor antagonist bicuculline (at 1 µM, Sigma-Aldrich, Taufkirchen, Germany) for 4 h. In these experiments, either IL-1RA or bicuculline were applied alone for 30 min before they were co-applied with IL-1ß. In a subset of rats, we applied IL-1RA alone for 4 h. To test the effects of LPS on CSD, we applied 2 µg LPS (Sigma-Aldrich), dissolved in PBS at an amount of 200 µL for 4 h topically to the rat cortex. In another group of rats, we pretreated with 500 ng IL-1RA for 30 min, and then 2 µg of LPS were co-applied together with 500 ng IL-1RA for 4 h.
At the end of the experiment, the animals were sacrificed using an overdose of Trapanal (100 mg per rat, intravenously).
Data are reported as means ± Std. dev. For statistics we performed tests within groups (Wilcoxon matched pairs signed rank test) and between groups (one-way ANOVA). When required, Bonferroni adjustment was performed. Significance was accepted at p < 0.05.
Assessment of plasma extravasation
For the assessment of plasma extravasation, animals were anesthetized with Trapanal and received the same preparation (see above) with and without insertion of the electrodes and elicitation of CSD, respectively. The same amounts of substances (50 ng IL-1ß, 5 ng TNF, 2 µg LPS) were applied to the treated areas of the cortices and left there for 4 h. Thirty minutes before perfusion the animals received 0.5 mL of 4% Evans blue (Sigma-Aldrich, Saint Louis, USA) intravenously. Under normal conditions, Evans blue is bound to albumin and will not permeate from blood vessels into the surrounding tissue. 28 Then, the rats were transcardially perfused with 4% ice-cold phosphate-buffered paraformaldehyde (Sigma-Aldrich) under deep anesthesia. Brains were directly removed, post-fixed for 24 h in 4% paraformaldehyde, equilibrated in 30% sucrose, cut in sequential 40 µm transversal sections using the cryostat Leica CM3050S (Leica Biosystems, Nussloch, Germany) and embedded in Aqua Poly/Mount (Polysciences, Inc., Warrington, USA). Images of plasma extravasation were recorded using the confocal laser scanning microscope TCS SP5 (Leica, Wetzlar, Germany). Evans blue was excited with a diode-pumped solid-state laser at 561 nm. Fluorescence signals were recorded between 570 and 700 nm using a 20 × 0.7 dry objective.
Results
Effect of local application of IL-1ß on CSD amplitude and propagation velocity
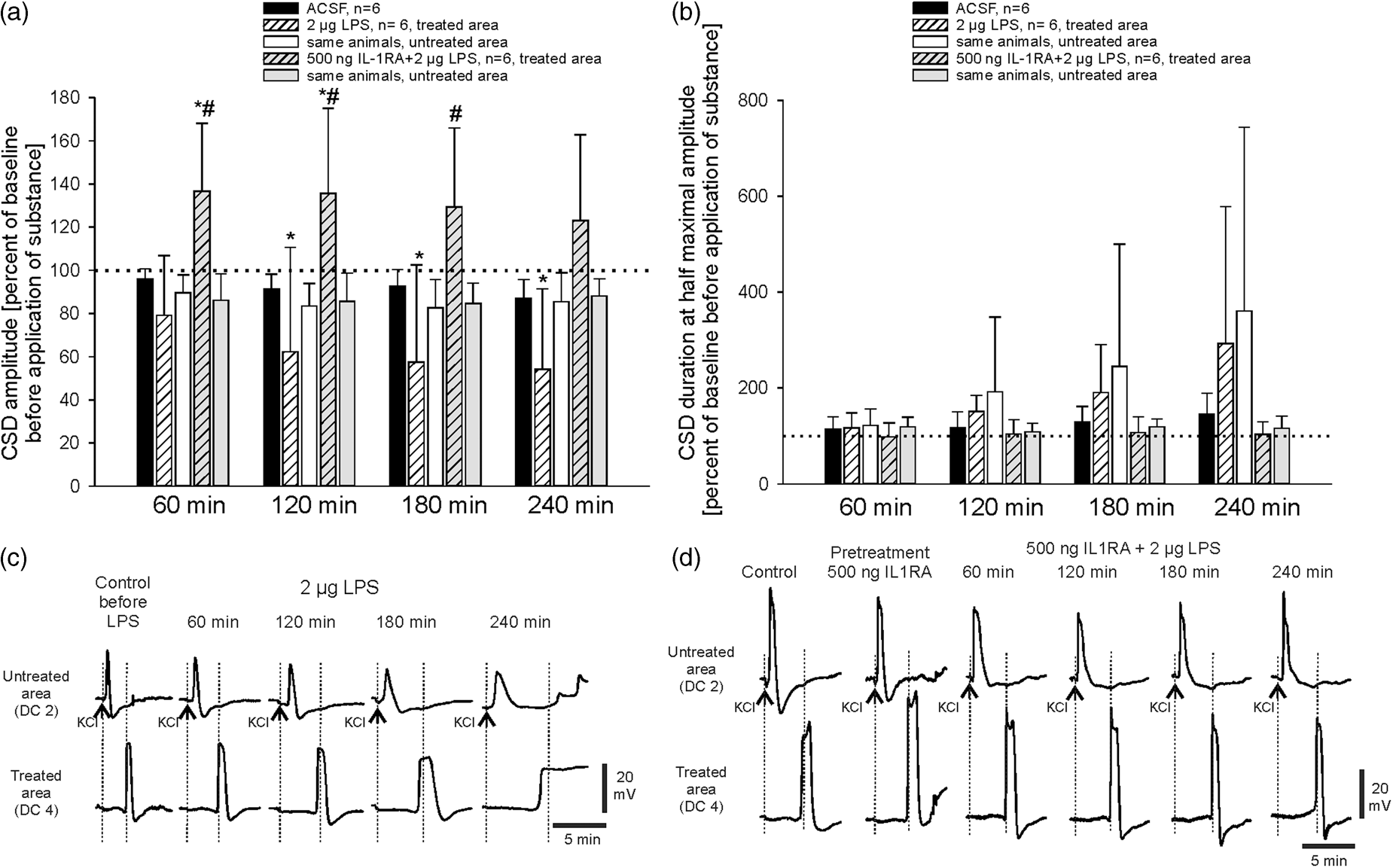
CSDs were elicited at intervals of 1 h by the microinjection of KCl through the electrode attached to the recording electrodes DC1 and DC2 in the “untreated area.” From there CSDs propagated to the recording electrodes DC3 and DC4 located in the “treated area” (Figure 1(a)). In the experiments displayed in Figures 1(b) and 2(a), 50 ng IL-1ß were applied to the treatment area. CSD amplitudes at DC2 showed a small decrease over time, whereas CSD amplitudes in the treated area showed a pronounced decrease. Evaluation of EEG activity showed that the depolarization waves elicited spreading depression of EEG activity, and it showed that the cortex remained electrically active after IL-1ß application. In all six experiments the CSD amplitudes at DC2 in the untreated area showed a small decrease over time from 18.8 ± 2.9 mV to 15.1 ± 2.6 mV, and CSD duration at half-maximal amplitude in the same time range increased by up to 159.6 ± 65.6% of controls. Simultaneously, the CSDs recorded at DC3/DC4 in the treated area showed a progressive decrease of their amplitudes within 240 min (from 24.1 ± 1.6 mV to 13.8 ± 6.5 mV at a depth of 1200 µm, and from 21.1 ± 2.2 mV to 11.5 ± 4.4 mV at a depth of 400 µm) and CSD duration at half-maximal amplitude increased as well (up to 139.9 ± 73.8% at 1200 µm, up to 159.4 ± 77.9% at 400 µm). Such a decrease of the CSD amplitudes was not observed when 100 ng IL-1ß were administered to the treatment area (Figure 1(c)).
Comparison of the effects of different doses of IL-1ß on CSD. (a) CSD specimens from one experiment with application of 50 ng IL-1ß. The typical shape of CSD curves is maintained 240 min after IL-1ß but the amplitude is markedly reduced in the treated area. (b) Percentaged changes of CSD amplitudes at different time points after topical application of either ACSF, or 10 ng, or 50 ng, or 100 ng IL-1ß to the cortical surface. The diagram shows the changes in the treated cortical areas only. The asterisks mark statistically significant differences to the control values (p < 0.05, Wilcoxon matched pairs test). (c) Percentaged change in CSD propagation velocity from the untreated into the treated area in the same animals as displayed in panel B.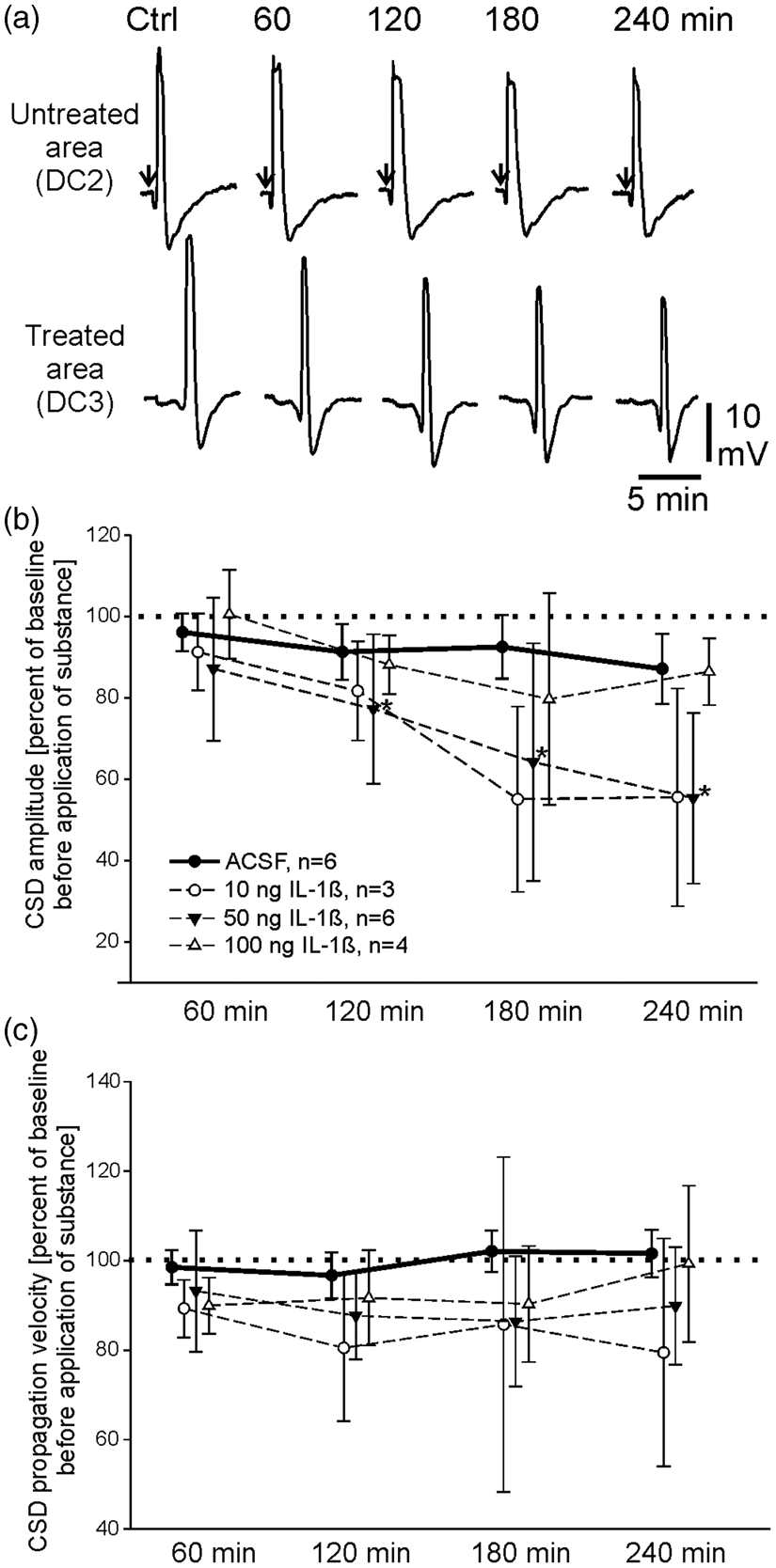
The normalized data from all experiments with IL-1ß application to the treatment area are displayed in Figure 2. Figure 2(a) shows specimens of CSD before and after topical application of 50 ng IL-1ß. The typical shape of the CSD waves is still present 4 h after the IL-1ß administration. The data points in Figure 2(b) show the average CSD amplitude in the treatment area. Upon ACSF application to the treatment area the amplitudes decreased to about 85% of control (the initial CSD amplitude in the treatment area was set 100%). By contrast, after application of IL-1ß at 10 or 50 ng to the treatment area the CSD amplitudes showed a significant decrease. However, after 100 ng IL-1ß the CSD amplitudes did not decrease. In none of the experiments the electrodes in the non-treatment area showed a reduction of CSD amplitudes below 85% of control (see Figure 3). The propagation velocities of the CSD declined from 2.2 ± 0.1 mm/min before IL-1ß to 1.8 ± 0.6 mm/min after 4 h of application of 50 ng IL-1ß. This slowing of propagation, however, did not reach statistical significance (Figure 2(c)).
The effect of IL-1ß on CSD amplitudes can be blocked by IL-1RA. The bars show mean CSD amplitudes at different time points after application of either ACSF, or IL-1ß alone, or after simultaneous application of IL-1ß together with IL-1RA. The asterisks mark statistically significant differences to the control values (p < 0.05, Wilcoxon matched pairs test), the # marks statistically significant differences to the untreated areas in the same animals (p < 0.05, paired t-test, Student).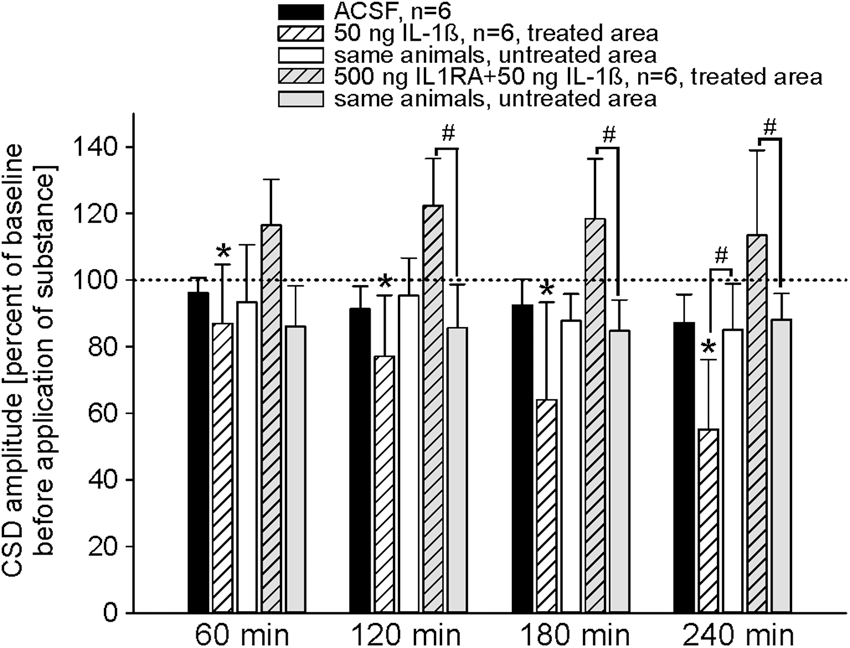
Next, we tested whether the coapplication of IL-1ß and of IL-1RA changes the CSD amplitudes. Figure 3 displays the changes of CSD amplitudes related to initial control values in the non-treatment and the treatment area, either after application of 50 ng IL-1ß, or after coapplication of 50 ng IL-1ß and 500 ng of the IL-1RA to the treatment area. After coapplication of IL-1ß and IL-1RA to the treatment area, the CSD amplitudes in the treatment area exhibited an increase above the initial CSDs, but CSD amplitudes in the non-treatment area showed a small decrease. Two hours after coapplication of IL-1ß together with IL-1RA CSD amplitudes in the treated area were significantly larger than in the untreated area (1200 µm control 22.9 ± 2.4 mV, after 2 h 25.2 ± 2.7 mV; 400 µm control 19.1 ± 5.9 mV, after 2 h 21.5 ± 6.5 mV). Thus, IL-1RA antagonized the IL-1ß effect.
Application of IL-1RA alone also increased the CSD amplitude in the treatment area (1200 µm control 22.1 ± 4.4 mV, after 1 h 23.4 ± 5.4 mV; 400 µm control 23.3 ± 4.6 mV, after 1 h 24.8 ± 2.2 mV) and even elicited few spontaneous CSDs (3 to 5 within 30 min). However, after coapplication of IL-1ß and IL-1RA no further spontaneous CSDs were observed.
Effect of the GABAA receptor antagonist bicuculline on CSD in the presence of IL-1ß
In order to test whether GABA is involved in the reduction of CSD amplitude after IL-1ß, the GABAA receptor antagonist bicuculline was coadministered in a pretreatment protocol with IL-1ß to the treatment area. Figure 4 shows the average CSD amplitudes in the non-treatment area and in the treatment area after coapplication of bicuculline and IL-1ß to the treatment area. In the non-treatment area the CSD amplitudes showed a slight reduction, but in the treatment area the CSD amplitudes were enhanced already after application of bicuculline. Within the pretreatment time of 30 min with bicuculline, we observed in all animals series of 3–7 subsequently occurring CSD waves that were not induced by microinjection of KCl. After coapplication of bicuculline together with 50 ng of IL-1ß, CSD amplitudes in the treated area were still significantly larger than in the untreated area, and IL-1ß did not reduce the CSD amplitudes.
The IL-1ß effect on CSD can be prevented by the GABAA receptor antagonist bicuculline. The bars show mean CSD amplitudes at different time points with simultaneous application of IL-1ß together with bicuculline. The # marks statistically significant differences to the untreated areas in the same animals (p < 0.05, paired t-test, Student).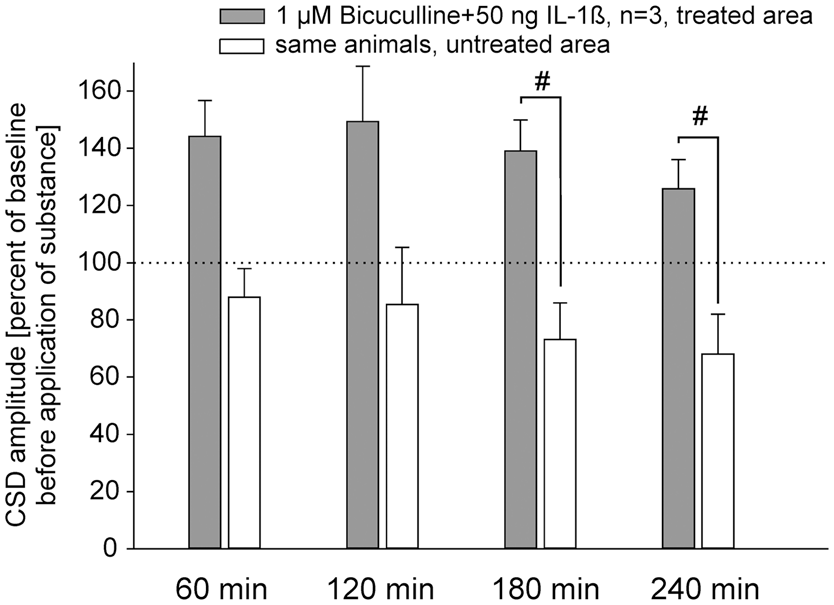
Effect of local LPS stimulation on CSD amplitudes
In Figure 5(a), the typical time course of an experiment with topical administration of LPS is displayed. Application of LPS to the treatment area reduced CSD amplitudes significantly (1200 µm control 24.9 ± 2.3 mV, after 4 h 13.6 ± 9.5 mV; 400 µm control 23.2 ± 2.6 mV, after 4 h 14.0 ± 8.4 mV) in the treatment area but not in the non-treatment area (control 18.2 ± 2.1 mV, after 4 h 15.5 ± 2.5 mV) (Figure 6(a)). This decrease resembled the effects of 50 ng IL-1ß. However, in two of the six animals, CSDs could still be elicited in the non-treatment area, but they did not propagate to the recording electrodes in the treated area starting 90 min after topical application of LPS. Both in untreated and in LPS-treated cortical areas there was still electrocorticographic activity by the end of the experiment showing that the cortex remained electrically active (Figure 5(b) to (d)). Within 4 h of LPS application, the CSD propagation velocity declined from 2.6 mm/min to 2.1 mm/min. In addition, a prolongation of CSD duration was observed in both the treatment and the non-treatment area that was more than three times longer than in the animals that were superfused with ACSF only (Figure 6(b) and (c)). Such a prolongation did not occur after topical application of IL-1ß. Neither local depolarizations nor propagating CSD waves originated in the LPS-treated area. All effects of LPS were prevented by the coapplication of IL-1RA (Figure 6(a), (b) and (d)). The pretreatment with IL-1RA induced an increase in CSD amplitudes as seen before. For the first 2 h of coapplication of LPS together with IL-1RA CSD amplitudes were significantly larger than at the beginning. This enlargement, however, was seen only in the treated area, where amplitudes were significantly larger than in the untreated area for the first 3 h of coapplication (Figure 6(a) and (d)). These data show that LPS evoked effects beyond those of the application of IL-1ß, but importantly, the development of these changes was prevented by IL-1RA.
Time-dependent effects of LPS administration on CSD development in the rat cortex. The left panels show the data from the cortex area superfused with ACSF (“untreated”), the right ones data from the cortex area treated with PBS containing LPS (2 µg/200 µL), treatment onset is indicated by the black arrow. (a) Recordings of spreading depolarizations in the DC recordings after filtering (bandpass 0.01–45 Hz) The dots below the upper traces accentuate the time points when CSD was elicited by microinjection of KCl at electrode DC1. Note the missing CSD waves that did not migrate into the treated area (electrodes DC3 and DC 4). (b) High-pass filtered electrocorticographic data with a lower frequency limit of 0.5 Hz of the respective recordings. (c) Power of bandpass filtered recordings (0.5–45 Hz) and (d) Integral of power of bandpass filtered recordings in c (0.5–45 Hz), respectively, which allow the visualization of the spreading electrocorticographic depressions. Note the reduction of CSD amplitude early after LPS administration with complete disappearance at the later stage of this experiment despite maintained electrocorticographic activity. Comparison of the effects of LPS alone and of LPS together with IL-1RA on CSD in rat cortex. (a) Percentaged changes of CSD amplitudes at different time points after topical application of either ACSF, of LPS alone, or of LPS together with IL-1RA to the cortical surface. The diagram shows the changes in the untreated and in the treated cortical areas. The asterisks mark statistically significant differences to the control values (p < 0.05, Wilcoxon matched pairs test), the # marks statistically significant differences to the untreated areas in the same animals (p < 0.05, paired t-test, Student). (b) The prolongation of CSD duration at half-maximal amplitude in the same animals as in panel A was only seen after LPS, but was prevented by IL-1RA. Due to some sustained depolarizations after LPS not all CSDs could be evaluated, and a statistical analysis failed. (c) Representative specimens of CSD propagating from electrode DC 2 to electrode DC 4 (treated area) in adult rat cortex before and after application of 2 µg LPS. The dotted lines accentuate the slowing of CSD propagation; arrows indicate the time points of KCl microinjection to elicit CSD. (d) Representative specimens of CSD propagating from electrode DC 2 to electrode DC 4 (treatment area) in adult rat cortex before and after application of 2 µg LPS together with 500 ng of IL-1RA. Note that one CSD is displayed during the 30 min pretreatment with 500 ng IL-1RA alone. Dotted lines and arrows as above.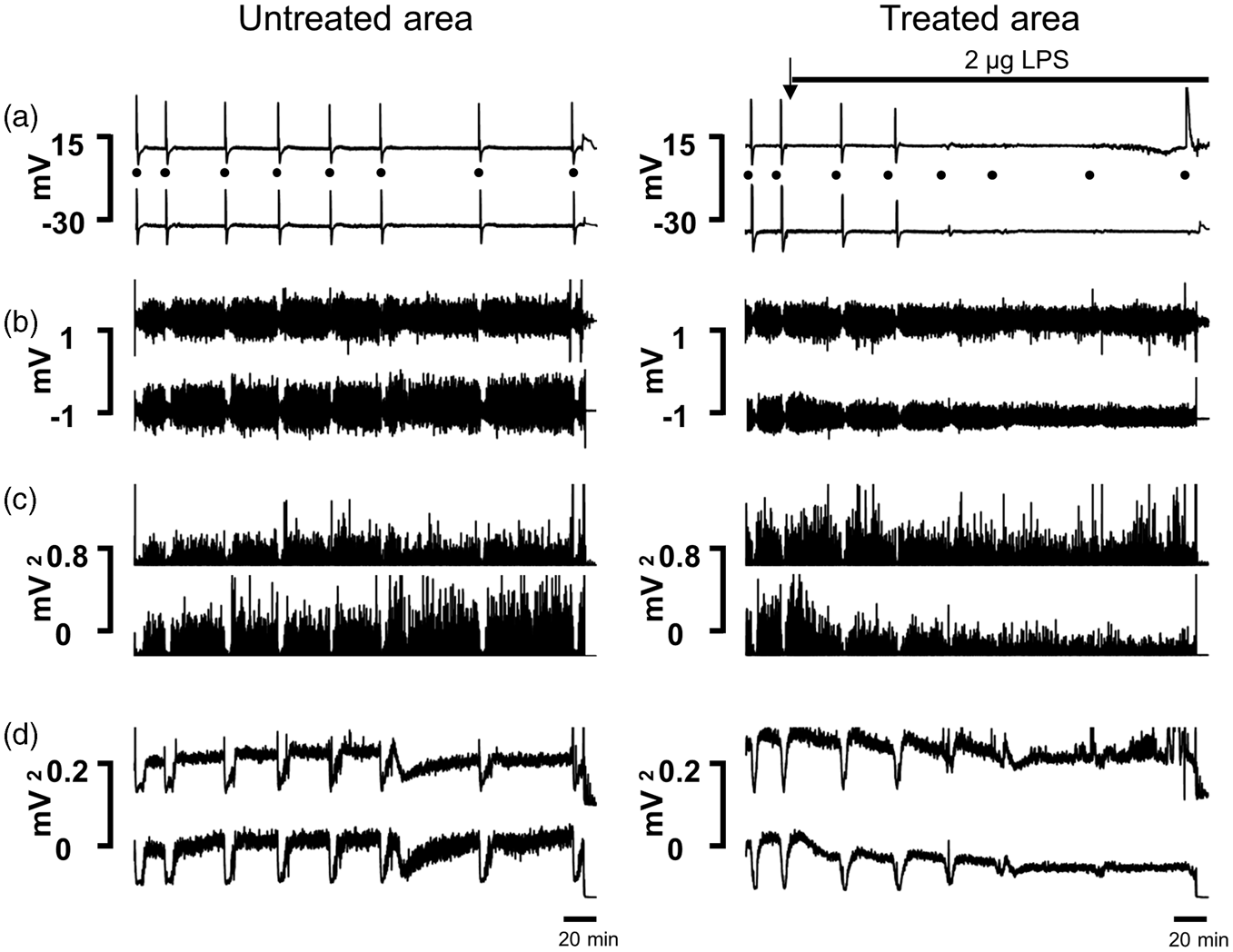
Effect of IL-1ß on plasma extravasation
We assessed plasma extravasation in the treatment and non-treatment area, and either we elicited CSDs after IL-1ß application or we just applied IL-1ß. For comparison, we also applied TNF to the treatment area because one aim of the study was to explore, whether induction of plasma extravasation was a common effect of proinflammatory cytokines. Representative images of plasma extravasation in cerebral cortex caused by IL-1ß, TNF, and LPS are displayed in Figure 7. IL-1ß caused pronounced plasma extravasation in the treatment area when CSDs were elicited (Figure 7(a)) but not in the untreated area (Figure 7(b)). A similar strong plasma extravasation was evoked in the treatment area in experiments in which no electrodes were introduced into the cortex, and no CSDs were elicited (Figure 7(c) and (d)). Thus, plasma extravasation was not a result of penetration of the brain tissue with recording electrodes or a consequence of propagating CSD waves. The topical application of 5 ng TNF (a dose sufficient to abolish CSD propagation
5
) for 4 h did not cause plasma extravasation (Figure 7(e)). After application of LPS plasma extravasation similar to the effect of IL-1ß was observed (Figure 7(f)).
Evans blue staining of cortical slices. (a) Plasma extravasation in the cerebral cortex after 4 h of topical application of 50 ng IL-1ß and propagating CSD waves. (b) No plasma extravasation in the remote (untreated) cortical area. (c) Plasma extravasation in the cerebral cortex after 4 h of topical application of 50 ng IL-1ß without insertion of recording electrodes and without eliciting CSD. (d) No plasma extravasation in the remote (untreated) cortical area (same section). (e) No plasma extravasation in a cortical area that was treated for 4 h with 5 ng TNF. (f) Plasma extravasation in a cortical area that was treated for 4 h with 2 µg LPS.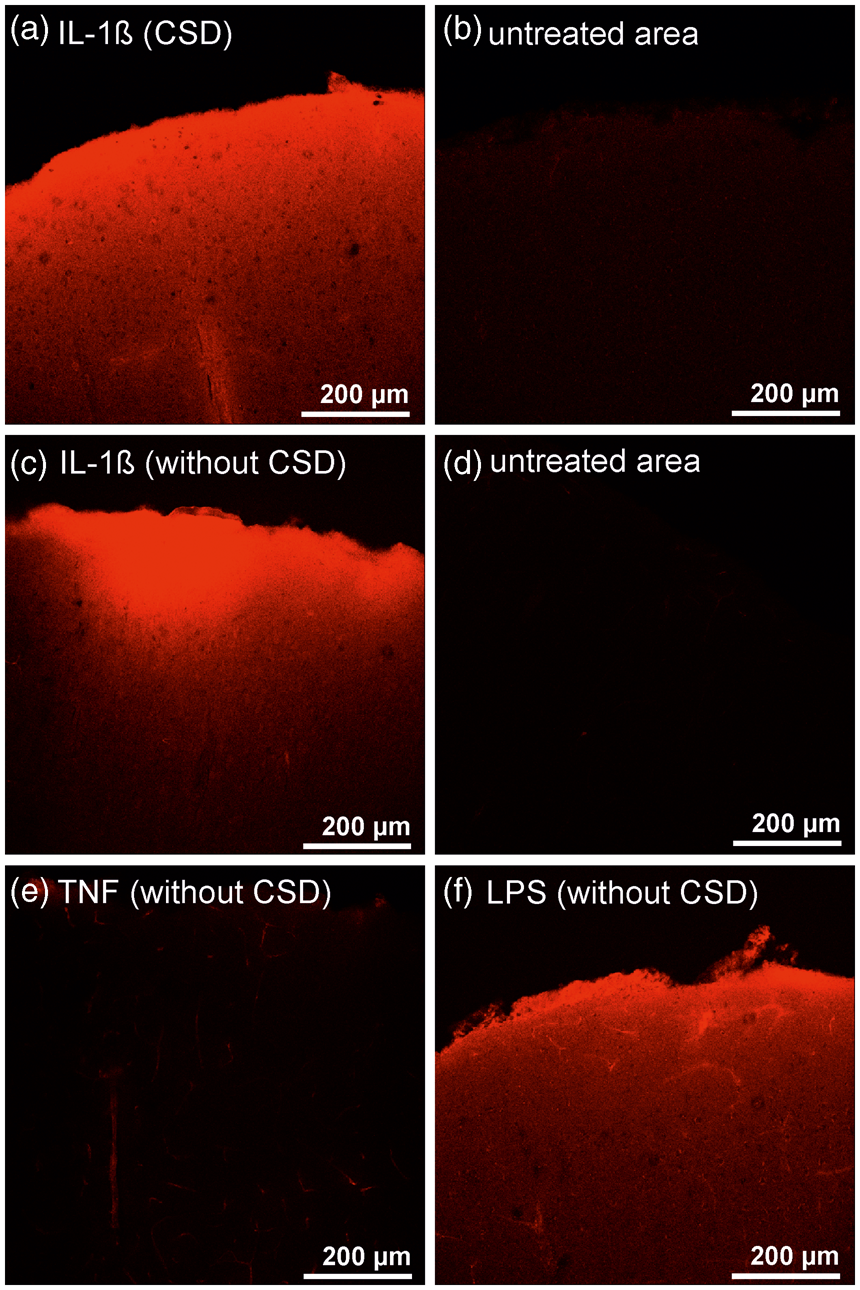
Discussion
The data show pronounced effects of IL-1ß and LPS on CSD amplitudes and on plasma extravasation. IL-1ß reduced CSD amplitude except at the highest concentration tested. The effect was prevented either by an IL-1 receptor antagonist, or by bicuculline, a GABAA receptor antagonist. IL-1ß induced pronounced plasma extravasation, unrelated to CSD. Application of LPS evoked effects beyond those of IL-1ß (prolongation of CSD duration), but these effects were also prevented by IL-1RA.
Previous studies have identified IL-1ß as a cytokine which is involved in the pathogenesis of brain diseases such as stroke. An intriguing question is by which mechanisms IL-1ß generates brain damage. Since CSD is an important pathophysiological mechanism contributing to brain damage in compromised tissue we asked whether IL-1ß induces or modifies CSD. Furthermore, we asked whether IL-1ß induces plasma extravasation, another pathophysiological mechanism in brain diseases. Because we recently investigated the effects of TNF on CSD, we could also address the question whether effects of IL-1ß and TNF are similar (potentially overlapping) or different.
As shown previously, cytokines diffuse from the cortical surface into local brain tissue. 5 The recording of CSD in a non-treatment and a treatment area allowed us to assess specific IL-1ß effects. Similar as topical application of TNF 5 IL-1ß reduced the CSD amplitude in the treated area. However, two distinct differences were found between effects of TNF and IL-1ß. TNF reduced the CSD amplitudes dose-dependently, and at the highest dose CSDs disappeared completely. By contrast, IL-1ß reduced the amplitudes to about 60% of the maximum, and the highest concentration had no effect on CSD amplitudes at all. This suggests that mechanisms of TNF and IL-1ß differ at least in part. Another striking difference is that IL-1ß elicited pronounced plasma extravasation whereas TNF evoked at best a small effect.
The prevention of the reduction of CSD amplitude by IL-1RA strongly indicates that the reduction of the CSD amplitudes by IL-1ß was mediated by the receptor IL-1R1. The occurrence of some spontaneous CSDs and the increase in CSD amplitudes after application of IL-1RA suggests that some IL-1ß was present under the base line conditions of the experiments, and that IL-1RA counteracts such inhibitory effects of IL-1ß.
By which mechanism does IL-1ß reduce CSD amplitude? A strong candidate mechanism is GABAergic inhibition. IL-1ß enhanced GABAergic presynaptic inhibition of synaptic transmission and neuronal hyperpolarization. 29 In mouse hippocampal slices, IL-1ß enhanced tonic inhibitory currents generated by GABAA receptors. 30 GABA inhibits CSD in the cortex 31 and in the isolated retina.32,33 In mouse hippocampal slices, GABAA receptor activation significantly reduced the SD propagation rate in the CA1 region. 34 Enhanced GABAergic transmission was proposed to reduce over-excitation after an ischemic insult. However, the effects of GABA are not uniform and may depend on the brain structure as well as on the experimental condition. After anoxia, GABA facilitated cell swelling in hippocampal slices and induced deleterious effects to neurons. 35
In the present experiments, the GABAA receptor antagonist bicuculline prevented the reduction of CSD amplitudes by IL-1ß. This strongly suggests that GABA releasing interneurons are involved in CSD inhibition by IL-1ß. However, other mechanisms cannot be excluded because receptors for IL-1ß were found on neurons and on glial cells throughout the cortex. 23 Interestingly, the effects of bicuculline and IL-1RA were quite similar because both compounds increased the CSD amplitudes and prevented the reduction of CSD amplitudes by exogenous IL-1ß. This may indicate that there is some tonic inhibitory activity of endogenous IL-1ß involving GABAergic inhibition. Some basal expression of IL-1ß in the normal brain was reported. 16
However, at high doses of IL-1ß CSD amplitudes were not reduced. The literature indicates that effects of IL-1ß on neurons may differ between brain structures, and that they may also depend on the experimental preparation.36–39 The latter arguments are unlikely to explain the failure of a high doses of IL-1ß to alter CSD amplitudes because the site of the recordings and the preparation were the same in experiments using the low and the high concentrations of IL-1ß. We rather assume that the higher doses of IL-1ß caused additional effects. Either the inhibition was inhibited, or additional excitatory mechanisms were evoked. In contrast to TNF receptor 2 which was only localized on neurons (many of which were inhibitory 5 ), IL-1R1 is localized on numerous structures including neurons, glial cells, and endothelial cells. 23 Thus, low and high concentrations of IL-1ß may activate different types of cells and thus evoke a different spectrum of effects. We could not further explore the potential cellular effects of high IL-1ß concentrations because it is difficult to define the mechanisms of “zero effects.”
IL-1ß receptors were described in endothelial cells.18,23 IL-1ß was reported to reduce cerebral blood flow 18 and also to cause breakdown of the blood–brain barrier. 2 Here, we tested, whether IL-1ß causes plasma extravasation with the dye Evans blue 28 that is often used to visualize plasma extravasation in traumatic brain injury 40 or in stroke models, 41 in particular after induction of IL-1ß release. 42 We found that IL-1ß caused pronounced plasma extravasation in the treated area suggesting that IL-1ß caused vascular leakage. Such an effect may contribute to ischemic injury in areas of reduced blood flow.
The damage of the blood–brain barrier is also involved in other pathological events, but so far the results are heterogenous. In the rat brain slice, the disruption of the blood brain barrier by application of deoxcholic acid evoked epileptic discharges, and this hyperexcitability was induced by a reduction of inward-rectifying K+ currents.43,44 In rat cortex in vivo, disruption of the blood–brain barrier increased the threshold for CSD and reduced the number of recurrent CSD. 45 In the long term, the damage of the blood–brain barrier also induces neurodegeneration. The epileptogenesis and neurodegeneration are thought to be due to disturbed buffering of potassium through TGF-β-mediated uptake of albumin into astrocytes.43–45 However, the development of hyperexcitability lasted at least 24 h, and structural changes were seen only after at least one month. These data suggest that plasma extravasation per se is a pathogenic factor. Whether the plasma extravasation under such conditions is related to IL-1ß, has not been explored.
Because LPS is used to activate microglia,46,47 and thereby to stimulate the release of IL-1ß from inflammasomes, 48 we also tested the effects of LPS on CSD. Similar as IL-1ß the application of LPS reduced CSD amplitudes, but the effect of LPS was “stronger” as LPS finally caused a prolongation of CSDs and even long-lasting depolarizations resembling terminal spreading depolarizations. Interestingly, however, these effects were inhibited by IL-1RA. We believe, therefore, that LPS induces a cascade of reactions in which IL-1ß plays a pivotal role. There are several explanations for the prolongation of CSD by LPS. One possibility is a reduction of Na/K-ATPase function causing long-lasting depolarizations. It is known that compromising the energy supply in the cortex, e.g. after stroke, results in long-lasting or anoxic depolarizations that do not recover.49,50 There is some indication that LPS interacts with the Na/K-ATPase and inhibits pumping activity in hippocampus of rat pups 51 or down-regulates the Na/K-ATPase in astrocytic cell cultures.52,53 Another possibility is the activation of P2X7 receptors at the microglia by LPS (cf. 54 ) that induce the release of other cytokines beyond IL-1ß, e.g. TNF. 55 We did not measure changes in local cerebral blood flow or tissue partial pressure of oxygen after LPS treatment, therefore, we cannot definitely state which mechanism caused the long-lasting depolarizations in our experiments.
How are the present findings related to the present knowledge on the pathogenic role of IL-1ß? This question has to be related to the functional importance of CSD in pathological brain conditions. A CSD is a mass depolarization in the cortex which occurs under pathological conditions such as stroke or traumatic brain injury.13,15 CSD waves are very energy demanding, since recovery of ionic homeostasis after the depolarization requires activation of the ATP-driven membrane pumps, e.g. the Na/K-ATPase. Furthermore, under pathological conditions, inverse neurovascular coupling during CSD can lead to severe hypoperfusion in tissue at risk of progressive damage. 13 As a consequence, during hypoxia reversible CSDs may develop into sustained hypoxic or anoxic depolarizations. 56 Thus, CSDs may be part of a vicious circle in which pathological conditions involving reduced oxygen supply elicit CSDs which by themselves are oxygen- and energy-demanding and may thus further aggravate the unfavorable conditions. Such a significant contribution of CSD to the pathophysiological processes during ischemia is suggested by the fact that repetitive CSD can evoke pathological changes even in the normal brain. During repetitive CSD pathophysiological metabolic processes develop in the brain tissue, neurons swell and morphologic changes such as beading and loss of spines occur. 57 In normoxic brain tissue, CSD (as well as high extracellular K+, a well-known trigger of CSD) can induce astrogliosis thus causing a further restriction of volume transmission in the affected brain area.58,59 Indeed, under ischemic conditions astroglial volume changes were observed that were either induced by acute osmotic stress or by increased extracellular potassium concentration, 60 suggesting a contribution of CSD to the development of these changes. It was directly shown that repetitive CSD in the ischemic penumbra zone caused neuronal dendritic beading and spine loss. 61
On the background of such a pathogenic role of repetitive CSDs, a reduction of neuronal excitability causing a reduction of CSD may be neuroprotective. Thus, at lower concentrations, IL-1ß may be neuroprotective concerning the effects on CSD. However, the plasma extravasation induced by IL-1ß may counteract those effects. Plasma extravasation is deleterious to neurons and to glial cells because it is associated with cell swelling, swelling of mitochondria together with a loss of energy supply and eventually membrane ruptures. 62 Furthermore, at high concentrations IL-1ß did not reduce anymore the CSD amplitudes, and thus the putative neuroprotective effect of low IL-1ß concentrations by reduction of CSD amplitudes is lost. The differences between the effects of IL-1ß and of TNF on both CSD and the vasculature may explain why TNF can exert neuroprotective effects (in addition to damaging effects) whereas IL-1ß is mainly “neurotoxic” at higher concentrations. Thus, we believe that the present data contribute to the understanding why IL-1ß exacerbates ischemic damage, 16 why IL-1ß/IL-1alpha knockout mice exhibit a smaller infarct volume than wild type mice, 18 why mice deficient in caspase-1 are protected from neural injury by ischemia, 19 and why the application of an IL-1 receptor antagonist within the first hours of stroke reduces the ischemic injury.16,18
Interestingly, inflammasomes such as NPLRP1 and NPLRP3 are activated by high extracellular potassium. 19 High potassium is thought to change the excitability of neurons within a few seconds. During CSD waves, the extracellular potassium concentration further increases. 21 Thus, the CSDs which are elicited locally and propagate in the brain may contribute to brain pathology by activating inflammasomes at distant sites. The activation of caspase-1 and the ensuing release of IL-1ß and IL-18 from microglia may promote pathogenic reactions in the brain. In this respect, it is interesting that LPS application which stimulates inflammasomes, causes a reduction of CSD amplitudes (which may be protective), but causes in addition a prolongation of CSD reaching finally persistent depolarization which is a strong damaging effect. It is known that LPS causes the release of IL-1ß from microglia. 63 Furthermore, the loss of inhibitory action of IL-1ß on CSD amplitudes at high concentration may further aggravate the pathogenic mechanisms. Thus, there may be an important relationship between CSDs and brain damage under different pathological conditions.
In sum, this study reveals a complex spectrum of effects of IL-1ß in the brain, and it indicates pronounced differences between the effects of IL-1ß and TNF. Although small amounts of IL-1ß reduced CSD amplitudes, greater amounts of IL-1ß do not inhibit CSD amplitudes. In addition, IL-1ß causes pronounced plasma extravasation. Thus, in sum, the cytokine IL-1ß mainly furthers pathogenic mechanisms and has to classified, therefore, as a cytokine with a potent pathogenic role in the brain.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors thank the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung) for funding the positions of Annett Eitner and Johannes Leuchtweis.
Acknowledgment
The authors thank K. Ernst for excellent technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FR designed the study, carried out the electrophysiological experiments, analyzed the data and wrote the manuscript. AE and JL carried out the microscopical studies, analyzed the data and contributed to the writing of the manuscript. RB carried out the EEG data analysis, analyzed the data and contributed to the writing of the manuscript. AL designed the study and contributed to the writing of the manuscript. HGS designed and coordinated the study, analyzed the data and wrote the manuscript.