
Abstract
Cerebral arterioles contribute critically to regulation of local and global blood flow within the brain. Dysfunction of these blood vessels is implicated in numerous cardiovascular diseases. However, treatments are limited due to incomplete understanding of fundamental control mechanisms at this level of circulation. Emerging evidence points to a key role of Rho-associated protein kinase in regulation of microvascular contractility. This study sought to decipher the mechanisms of Rho-associated protein kinase–mediated myogenic vasoconstriction in cerebral parenchymal arterioles. Here, we report that the Rho-associated protein kinase inhibitor H1152 strongly attenuated pressure-induced constriction, cytosolic [Ca2+] increases, and depolarization of isolated parenchymal arterioles. Further, the RhoA activator CN03 potentiated parenchymal arteriole myogenic constriction and depolarization, indicating important involvement of RhoA/Rho-associated protein kinase signaling in myogenic excitation-contraction mechanisms. Because of the well-established role of TRPM4 in pressure-induced depolarization, possible modulatory effects of Rho-associated protein kinase on TRPM4 currents were explored using patch clamp electrophysiology. TRPM4 currents were suppressed by H1152 and enhanced by CN03. Finally, H1152 elevated the apparent [Ca2+]-threshold for TRPM4 activation, suggesting that Rho-associated protein kinase activates TRPM4 by increasing its Ca2+-sensitivity. Our results support a novel mechanism whereby Rho-associated protein kinase–mediated myogenic vasoconstriction occurs primarily through activation of TRPM4 channels, smooth muscle depolarization, and cytosolic [Ca2+] increases in cerebral arterioles.
Introduction
Cerebral blood flow is precisely regulated through the vasomotor activity of surface pial arteries and penetrating arterioles. As a prime example, arterioles within the brain parenchyma contribute to as much as 40% of cerebrovascular resistance, playing a crucial role in global cerebral autoregulation. 1 Furthermore, these parenchymal arterioles (PAs) modulate local blood flow via dilation in response to increased neuronal activity, in the process known as neurovascular coupling. 2 Compared with upstream pial arteries, PAs exhibit unique functional properties and anatomical structure. Notably, distinct vasoconstrictor responses,3,4 altered ion channel functions, 5 and diverse Ca2+ signaling events 5 in PA smooth muscle all strongly suggest that mechanisms of vascular tone regulation might also be quite different from the signaling pathways in the pial vessels. Structurally, in contrast to the ramifying and anastomotic networks formed by surface arteries and capillaries, the topology of the PA network is more one-dimensional. 6 This creates a “bottleneck” effect in the cerebral microcirculation, which renders PAs and surrounding brain tissue especially vulnerable to changes in local blood flow. 6 Accordingly, arteriolar dysfunction is implicated in numerous cerebral vascular pathologies, including cerebral and coronary small vessel diseases, 7 ischemic and hemorrhagic stroke,8,9 hypertension, 10 and vascular dementia. 11 However, effective measures for disease prevention and treatment are limited due to incomplete understanding of the factors and mechanisms that regulate these blood vessels.
Myogenic tone represents one of most fundamental functions of resistance arteries in maintaining a relatively constant blood flow despite moment-to-moment fluctuations of arterial pressures. In the brain, myogenic tone sets the arteries and arterioles at a partially constricted background, so that blood flow can be tightly regulated by extracellular stimuli, particularly vasodilatory signals from neurovascular coupling and endothelial cell activation. Myogenic tone also contributes to controlling appropriate perfusion pressure and protecting downstream capillaries. The primary mechanism underlying pressure-induced vasoconstriction involves smooth muscle membrane depolarization and subsequent Ca2+ entry through voltage-dependent calcium channels (VDCCs). 12 Yet, how an extracellular mechanical stimulus is converted into intracellular membrane depolarization is not fully understood. Interestingly, compared with pial arteries, myogenic response plays a more prominent role in regulating PA reactivity, since these blood vessels exhibit significantly enhanced pressure-evoked depolarization, 8 intracellular [Ca2+] increase, 13 and contraction 13 versus pial arteries over the same range of vascular pressures. Hence, elucidating the mechano-stimulated cellular pathways is particularly important for the cerebral microcirculation. Previous studies from our laboratory have provided important inroads in this area. P2Y4 and P2Y6 receptors appear to be mechanosensors in PAs and mediate myogenic responses in a ligand-independent manner. 4 TRPM4 channels couple the mechanoactivation of P2Y4 and P2Y6 receptors to membrane depolarization and vasoconstriction in PAs. 14 However, the intracellular signaling cascades that mediate the stretch-induced, P2Y receptor-mediated activation of TRPM4 remain unknown.
Emerging evidence points to the dynamic role of the RhoA/Rho-associated protein kinase (ROCK) pathway in regulating cerebrovascular function, including myogenic control.9,15,16 Although it is generally accepted that ROCK promotes contraction by inhibiting myosin light chain phosphatase, i.e. via “Ca2+ sensitization” mechanisms, 17 recent findings have revealed that ROCK also regulates ion channel activity, including activation of epithelial Na+ channels (ENaC) 18 and inhibition of voltage-dependent K+ (Kv) channels, which modulates smooth muscle membrane potential, 19 suggesting ROCK may be involved in electromechanical coupling pathways. Following from these interesting findings, a major goal of the current study was to explore the roles of RhoA/ROCK signaling in myogenic reactivity of PAs. We hypothesized that RhoA/ROCK signaling mediates stretch activation of TRPM4 channels in the brain microcirculation. The results from this work support this hypothesis by demonstrating that pressure-induced and P2Y receptor agonist-triggered effects on TRPM4 activity, membrane potential, cytosolic [Ca2+] and vascular tone are all attenuated by ROCK inhibition and potentiated by RhoA activation, revealing a previously underappreciated contribution of ROCK signaling to myogenic regulation through depolarization and Ca2+-dependent pathways.
Materials and methods
Animals
All animal procedures were conducted in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines, and approved by the Institutional Animal Care and Use Committee at the University of Vermont and performed in accordance with the National Institutes of Health Policy on the care and use of laboratory animals. Male, Sprague-Dawley rats (15 to 20 weeks old; Charles River Laboratories, Saint Constant, QC, Canada) were used for all experiments. Rats were allowed free access to food and water, and maintained under temperature, humidity, and light-controlled conditions.
Tissue preparation
Rats were euthanized by an overdose of sodium pentobarbital (120 mg/kg, intraperitoneal injection) followed by exsanguination. The brain was rapidly removed and placed in cold (4℃) MOPS-buffered physiological salt solution (PSS) (in mmol/L): 3 MOPS, 145 NaCl, 5 KCl, 1 MgSO4, 2.5 CaCl2, 1 KH2PO4, 2 sodium pyruvate, 5 glucose, 1% albumin from bovine serum, pH 7.4. Middle cerebral arteries (MCAs) and attached PAs were isolated from the brain as previously described 20 and transferred to a small volume of MOPS-buffered PSS for further experimental use.
Pressure myography
Parenchymal arteriolar segments were cannulated on glass pipettes in an arteriograph chamber (Living Systems, Burlington, VT, USA) containing a bicarbonate-buffered artificial cerebral spinal fluid (aCSF) of the following composition (in mmol/L): 136 NaCl, 3 KCl, 15 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 4 glucose, 2 CaCl2 (temperature 37℃ and pH 7.3). To eliminate endothelial responses to P2Y receptor agonists, arterioles were stripped of endothelium by passing air bubbles through the arteriolar lumen. PAs were initially pressurized at 3 mmHg (with no flow), and superfused with warmed (37℃), gassed (20% O2/5% CO2/balance N2) aCSF. As an initial check of tissue viability, arterioles were reversibly constricted with the thromboxane receptor activator U46619 (100 nmol/L) (Enzo Life Sciences Inc., Farmingdale, NY, USA). Vessels were rejected for study if constrictions to U46619 were less than a 60% decrease in diameter. In experiments where myogenic constriction was not desired, e.g. agonist-induced vasoconstriction, intraluminal pressure was controlled at 3 mmHg. In experiments where steady myogenic tone was desired, PAs were pressurized to 40 mmHg. After myogenic tone developed, PAs were exposed to the IKCa and SKCa channel activator NS309 (1 µmol/L), which mediates endothelium-dependent vasodilator responses in these arterioles. 21 Absence of dilation by NS309 indicated successful endothelial cell removal.
Membrane potential measurements
The endothelium was removed from all vessels to eliminate any contribution of endothelial components to membrane potential. Arterioles were cannulated as described above and pressurized to 3 mmHg. In these experiments, the myosin light chain kinase inhibitor wortmannin (800 nmol/L) was included in aCSF prior to membrane potential measurements to prevent excessive vessel movement. Smooth muscle membrane potential was measured by repeated insertion of a sharp glass microelectrode (∼200 MΩ resistance when filled with 0.5 mol/L KCl) into the vessel wall. The criteria for successful impalement were (1) an abrupt negative potential deflection upon entry, (2) a stable membrane potential for at least 30 s, and (3) an abrupt positive potential deflection to 0 mV upon electrode withdrawal. Measurements were made with an electrometer (World Precision Instruments, Sarasota, FL, USA) and recorded via computer with AxoScope and Dataq software. At least three repeated impalements were made and the average membrane potential was calculated under each pharmacological treatment for every PA.
RhoA activation of isolated PAs
MCAs with PAs attached were isolated as previously described 20 and incubated with the selective RhoA activator CN03 (1 µg/mL, equivalent to 8.5 nmol/L) (Cytoskeleton, Inc., Denver, CO, USA) for 3 h in 2 mL DMEM/F-12 (serum-free) culture medium (37℃). Control arterioles were incubated for 3 h in DMEM/F-12 culture medium in the absence of CN03. Myogenic tone and membrane potential were then assessed as described above.
Intracellular Ca2+ concentration measurements
Freshly isolated PAs were cannulated on glass micropipettes and the endothelium was removed as described above. Cannulated arterioles were initially pressurized to 5 mmHg and equilibrated in aCSF for 1 h, and then loaded with the ratiometric Ca2+-sensitive dye fura-2 [acetoxymethyl ester (AM) membrane-permeant form] by incubating in aCSF containing fura-2 AM (5 µmol/L) (Invitrogen, Carlsbad, CA, USA) with pluronic acid (0.1%) (Invitrogen) for 1 h at room temperature (∼22℃). The myograph chamber was mounted on a Nikon TE2000-S inverted fluorescence microscope. Arterioles were washed with aCSF 3 times and de-esterification of fura-2 AM occurred by superfusing with warmed (37℃), gassed (20% O2/5% CO2/balance N2) aCSF for 20 min before recording. Bath pH was closely monitored and maintained at 7.30–7.35.
For pressure-response tests, studies were performed in which intravascular pressure was increased to 10, 20, 30, and 40 mmHg. Fluorescence ratio was obtained from the background-corrected ratio of the 510 nm emission from arterioles alternately excited at 340 and 380 nm with hardware and software developed by IonOptix (Milton, MA, USA). [Ca2+]i was estimated using the following equation: [Ca2+]i = Kd × β × (R − Rmin)/(Rmax − R). Rmin and Rmax, the ratios of emission signals under Ca2+-free and Ca2+-saturated conditions, respectively, were measured from a separate set of ionomycin-treated arterioles, and β was determined as the ratio of F380 intensities at Rmin and Rmax. Calibration values were pooled for calculation of intracellular [Ca2+] using an apparent dissociation constant (Kd) of 282 nmol/L of fura-2 for Ca2+.
Single cell isolation
Arterioles were placed in a cell isolation solution containing (in mmol/L): 55 NaCl, 80 Na Glutamate, 5.6 KCl, 2 MgCl2, 10 HEPES, 10 Glucose (pH 7.3). Arteriolar segments were initially incubated in 0.5 mg/mL papain (Worthington Biochemical Corp., Lakewood, NJ, USA) and 1.0 mg/mL dithioerythritol for 11 min at 37℃ and then in 1.0 mg/mL collagenase type F for 13 min at 37℃. The digested segments were washed 3 times in ice-cold cell isolation solution and incubated on ice for 30 min. Digested arterioles were triturated to liberate smooth muscle cells and stored in ice-cold cell isolation solution prior to use. Smooth muscle cells were studied within 5 h following isolation.
Patch clamp recordings
General methods
Isolated smooth muscle cells were placed into a recording chamber and allowed to adhere to glass coverslips for 20 min at room temperature. Currents were recorded with an AxoPatch 200B amplifier (Axon instruments) equipped with an Axon CV 2032wBU headstage (Molecular Devices). Recording electrodes (3–5 MΩ for perforated patch recordings, 5–8 MΩ for conventional whole-cell and inside-out patch recordings) were pulled from borosilicate glass (Sutter Instrument, Novato, CA, USA). All patch clamp experiments were performed at room temperature (22℃).
Perforated whole-cell patch recording
Gigaohm seals were obtained in bath solution of the following composition (in mmol/L): 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose (pH 7.4 adjusted with NaOH). The pipette solution contained (in mmol/L): 85 K-aspartate, 1 MgCl2, 30 KCl, 10 NaCl, 10 HEPES, 5 µmol/L EGTA (pH 7.2 adjusted with KOH). Nystatin (200 µg/mL) was included in the pipette solution to perforate the cell membrane. Perforation was deemed acceptable when series resistance was less than 50 MΩ. Currents were filtered at 1 kHz, sampled at 2 kHz, and stored for subsequent analysis. Clampex and Clampfit version 9.2 (Molecular Devices) were utilized for data acquisition and analysis, respectively. For all experiments, membrane potential was held at −70 mV, and all recordings were performed at room temperature (22℃). TRPM4 channel activity was calculated as the sum of the open probability (NPo) of multiple open states of 1.75 pA. This value was based on the reported unitary conductance of TRPM4 (25 pS). NPo was calculated using the following equation:
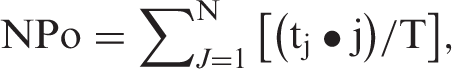
Conventional whole-cell patch recording for TRPM4 currents
Cells were initially held at a membrane potential (Vm) of 0 mV, and currents were recorded during voltage ramps between −120 and +80 mV. Voltage ramps were initiated as soon as whole cell conditions were established and were repeated every 2 s during the experiment. The bath solution contained (in mmol/L) 156 NaCl, 5 CaCl2, 10 HEPES, and 10 glucose (pH 7.4 adjusted with NaOH). The bath solution also contained a selective voltage-dependent Ca2+ channel (VDCC) blocker nimodipine (200 nmol/L) (Calbiochem, San Diego, CA, USA), and a selective large-conductance Ca2+-activated K+ (BKCa) channel blocker iberiotoxin (300 nmol/L). To prevent smooth muscle contraction under the condition of high intracellular [Ca2+], arteriolar myocytes were pretreated with the myosin light chain kinase inhibitor wortmannin (10 µmol/L) 5 min prior to patch clamp experiments. The pipette solution contained (in mmol/L) 156 CsCl, 1 MgCl2, 10 HEPES (pH 7.2 adjusted with CsOH). The Ca2+ concentration was adjusted between 1 and 100 µM by adding appropriate amounts of CaCl2 to 5 mmol/L EGTA calculated using the program Maxchelator (http://maxchelator.stanford.edu/). In some experiments, cells were treated with the ROCK inhibitor H1152 (1 µmol/L) or the TRPM4 channel blocker 9-phenanthrol (30 µmol/L) prior to patch recordings. For experiments with RhoA activation, the RhoA activator CN03 (0.1 ng/mL, equivalent to 0.85 pmol/L) was included in the pipette solution to allow for direct access to the inside of the smooth muscle cells and rapid activation of RhoA. Currents were filtered at 1 kHz, sampled at 5 kHz, and stored for subsequent analysis.
Conventional whole-cell patch recording for VDCC currents
The bath solution contained (in mmol/L): 125 NaCl, 10 BaCl2, 5 KCl, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 10 glucose (pH 7.4 adjusted with NaOH). The pipette solution contained (in mmol/L) 130 CsCl, 1 MgCl2, 10 HEPES, 10 EGTA, 10 glucose, 2 ATP, 0.5 GTP, 5 phosphocreatine (pH 7.2 adjusted with NaOH). Cells were initially voltage-clamped at −60 mV, and Cav channel currents were recorded by stepping the membrane potential to +10 mV. Ca2+ currents were compared in the same cell before and after application of the Rho kinase inhibitor H1152 (1 µmol/L). Currents were filtered at 0.5 kHz, sampled at 2 kHz, and stored for subsequent analysis.
Excised inside-out patch recording for TRPM4 channel currents in HEK cells
The bath solution contained (in mmol/L): 156 CsCl, 10 HEPES, 1 MgCl2, 0.1 CaCl2 (pH 7.2 adjusted with CsOH). The pipette solution contained (in mmol/L): 156 NaCl, 10 HEPES, 1 MgCl2, 5 CaCl2, 10 Glucose (pH 7.4 adjusted with NaOH). Patches were initially held at a holding potential of 0 mV during patching and excision. Single channel currents were recorded at a membrane potential of −60 mV. TRPM4 channel activity was calculated as the sum of the open probability (NPo) of multiple open states of 1.50 pA based on the reported unitary conductance of TRPM4 (25 pS). NPo was obtained using the method described above for perforated-patch recordings. Single TRPM4 currents were compared in the presence and absence of H1152 (1 µmol/L) and CN03 (0.1 ng/mL, equivalent to 0.85 pmol/L), respectively. Currents were sampled at 10 KHz, filtered at 0.5 KHz and stored for subsequent analysis.
HEK 293T cell culture and transient DNA transfection
Human embryonic kidney (HEK) 293 T cells were cultured in Dulbecco’s high glucose Modified Eagle Medium (Gibco, Grand Island, NY, USA) supplemented with 0.5% penicillin–streptomycin. Cells were maintained at 37℃ with 5% CO2, and sub-cultured when confluent using 0.25% trypsin. HEK cells were transiently transfected with a plasmid encoding a mouse TRPM4 protein (TransOMIC technologies Inc., Huntsville, AL, USA). Cells were cultured for 1–2 days prior to electrophysiology experiments.
Chemical and reagents
Buffer reagents, dissociation enzymes, NS309, nystatin, UDP, 9-phenanthrol, wortmannin, ionomycin, and iberiotoxin were purchased from Sigma-Aldrich (St Louis, MO, USA). UTPγS and H1152 were purchased from Tocris (Minneapolis, MN, USA). Nimodipine was dissolved in DMSO to a final solvent concentration of 0.1%. All other compounds were dissolved in the appropriate salt solution.
Statistical analysis
Values are expressed as Mean ± SEM, and n indicates the number of animals. Myogenic tone under different conditions was normalized as percentage tone, and calculated as follows:

Student’s t-test was used to compare two experimental groups. One-way analysis of variance (ANOVA) followed by the Tukey multiple comparison test was used in the comparison of multiple groups. Mean values were considered significantly different at P ≤ 0.05.
Results
ROCK is involved in pressure-induced and P2Y receptor-mediated vasoconstriction and membrane depolarization
We first examined the functional roles of ROCK in cerebral PAs. We found that a selective ROCK inhibitor (H1152) greatly attenuated pressure-induced tone with a half inhibitory concentration (IC50) of 0.24 µmol/L (Figure 1(a) and (b)), indicating a key role of ROCK in mediating myogenic response in the brain microcirculation, consistent with its contribution in pial arteries.16,22 We have previously established the essential involvement of P2Y4 and P2Y6 receptors in cerebral arteriolar myogenic regulation.
4
Therefore, the effects of ROCK inhibition on P2Y receptor-mediated responses were also explored. We found that H1152 (1 µmol/L) inhibited UTPγS- (P2Y4 receptor agonist) (0.5 µmol/L) and UDP (P2Y6 receptor ligand) (0.5 µmol/L)-induced vasoconstriction by 85% and 87%, respectively (Figure 1(c) and (d)).
ROCK inhibition suppresses pressure-induced and P2Y receptor-mediated vasoconstriction in endothelium-denuded PAs. (a, b) A representative diameter recording and summary data showing a selective ROCK inhibitor H1152 reduces myogenic constriction of PAs held at 40 mmHg (IC50 = 0.24 µmol/L). Lack of dilation by NS309 (1 µmol/L) indicates successful endothelium removal (n = 6). (c) A representative diameter recording showing H1152 (1 µmol/L) reduces P2Y4 receptor ligand (UTPγS: 0.5 µmol/L)-induced vasoconstriction in denuded PAs. (d) Summary data showing H1152 reduces P2Y4 (UTPγS: 0.5 mol/L) and P2Y6 (UDP: 0.5 mol/L) receptor ligand-initiated constriction of PAs (n = 6); **P < 0.01, ***P < 0.001 versus responses without H1152.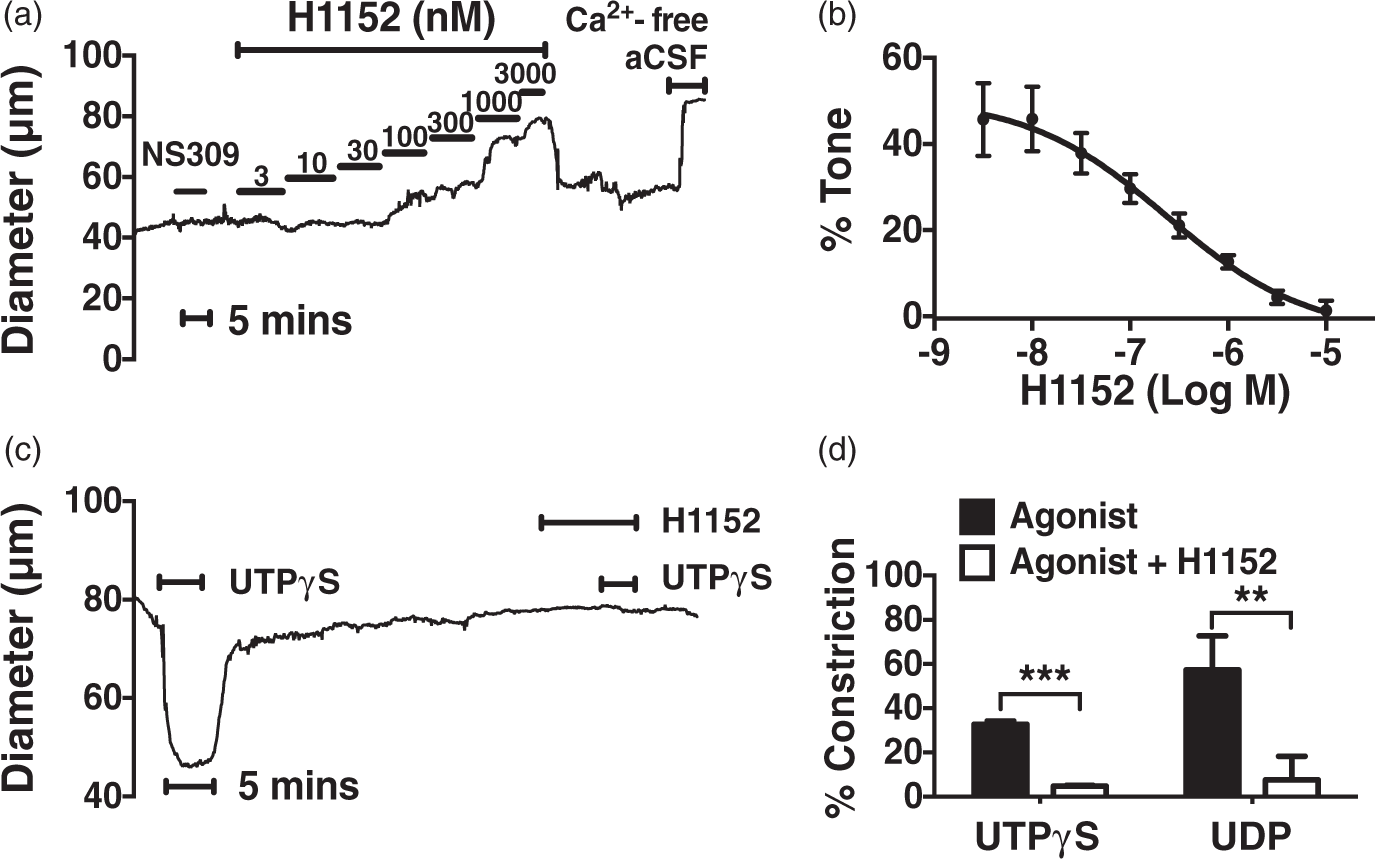
To determine whether ROCK-mediated myogenic constriction of PAs depends on cytosolic Ca2+, changes in intracellular Ca2+ concentration ([Ca2+]i) following stepwise increases in intraluminal pressure were measured. We found that H1152 (1 µmol/L) significantly suppressed [Ca2+]i elevations by 30–45% over a range of pressures from 5 to 30 mmHg (Figure 2(a) and (b)). [Ca2+]i oscillations observed at different pressures in control PAs were attenuated by H1152 (Figure 2(a)). To eliminate the possibility that these effects result from direct inhibition of H1152 on L-type VDCCs, whole-cell VDCC activity before and after applying H1152 was compared and no significant difference was found (Supplementary Figure 1). Further, the observation that myogenic [Ca2+]i elevation could be abolished by blocking VDCCs with nimodipine (100 nmol/L) (data not shown) suggests that this response is mediated by Ca2+ entry through VDCCs presumably due to membrane depolarization of PA smooth muscle.
ROCK inhibition attenuates myogenic [Ca2+]i elevation and depolarization in endothelium-denuded PAs. (a, b) Representative diameter recordings and summary data showing H1152 (1 µmol/L) inhibits [Ca2+]i elevation and oscillations induced by step-wise pressure increase (n = 5 for control, n = 4 for H1152). (c,d) H1152 (1 µmol/L) attenuates pressure-induced depolarization in arterioles held at 20 mmHg. This pressure is selected based on the greatest difference in [Ca2+]i observed in fura-2 Ca2+ measurement tests (n = 5); *P < 0.05, **P < 0.01 versus control.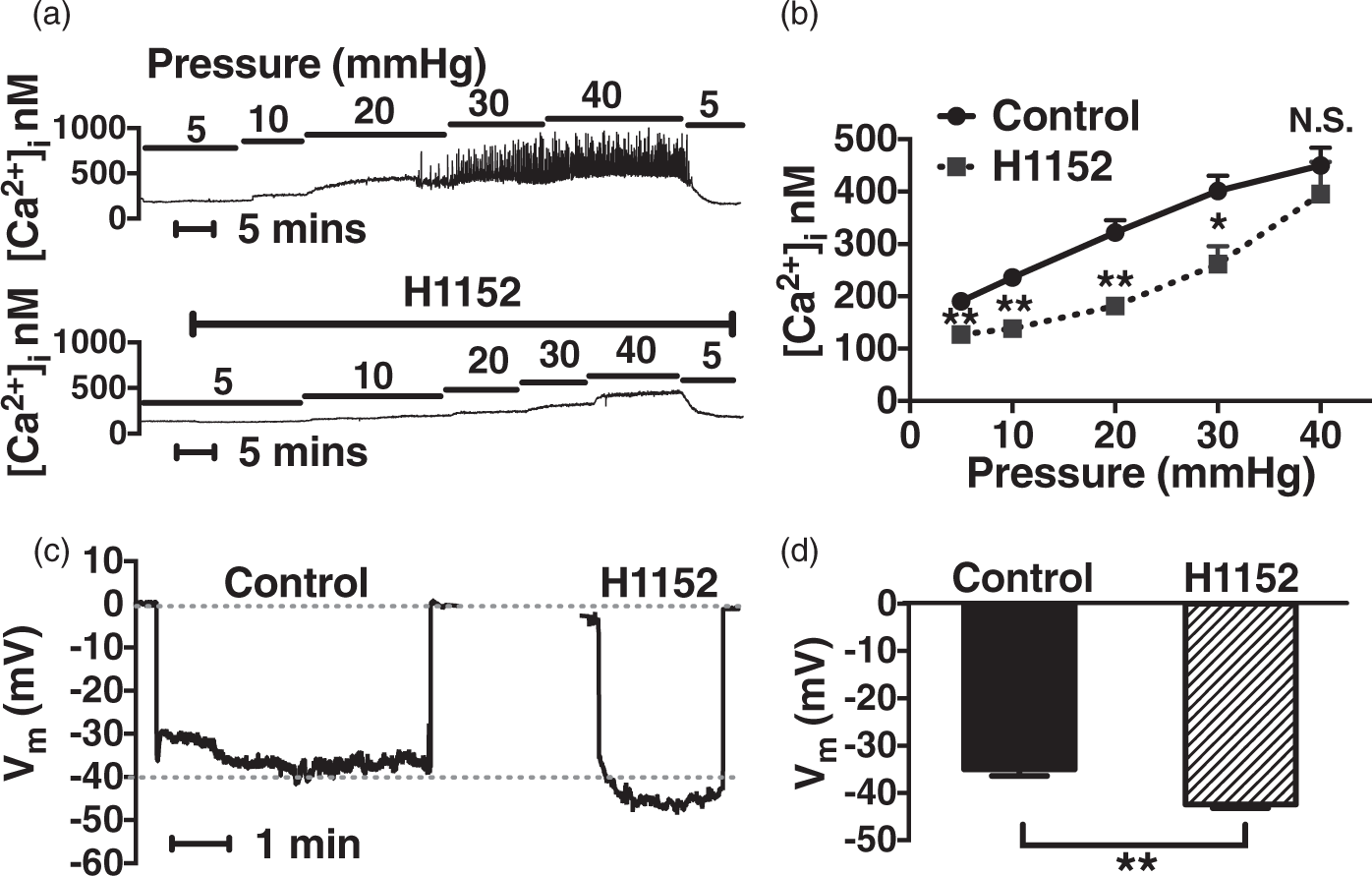
Therefore, we next examined the influence of ROCK inhibition on membrane potential. Consistent with our hypothesis, H1152 (1 µmol/L) hyperpolarized PAs held at 20 mmHg from −35 ± 1 mV to −43 ± 1 mV (Figure 2(c) and 2(d)), indicating that ROCK-mediated myogenic activation occurs in part through membrane depolarization and Ca2+ influx in PAs. To confirm this novel ROCK-related mechanism, P2Y receptor agonist-evoked changes in [Ca2+]i and membrane potential were studied. We found that H1152 (1 µmol/L) markedly attenuated UTPγS (0.5 µmol/L) and UDP (0.5 µmol/L) triggered [Ca2+]i increases by 61% and 50%, respectively (Figure 3(a) and (b)). H1152 also reversed agonist-induced smooth muscle depolarization in endothelium-denuded arterioles (Figure 3(c) and (d)). Thus, in contrast to the generally accepted view that ROCK influences smooth muscle contractility exclusively through Ca2+-sensitization,
17
these findings demonstrate that membrane depolarization and Ca2+ entry are also a crucial part of ROCK-associated modulation of vascular tone in the cerebral microcirculation.
ROCK inhibition suppresses P2Y4 and P2Y6 receptor-mediated [Ca2+]i increase and depolarization in endothelium-denuded PAs. (a) A representative recording of [Ca2+]i measurement showing H1152 (1 µmol/L) inhibits UTPγS (0.5 µmol/L)-induced [Ca2+]i increase in denuded PAs. (b) Summary data showing H1152 inhibit UTPγS (0.5 µmol/L) and UDP (0.5 µmol/L)-initiated [Ca2+]i elevation in PAs (n = 5); *P < 0.05 versus responses without H1152. (c) Representative membrane potential recordings showing H1152 (1 µmol/L) attenuates UTPγS (0.5 µmol/L)-induced membrane depolarization in denuded PAs held at 3 mmHg. (d) Summary data showing H1152 attenuate PA smooth muscle depolarization initiated by UTPγS (n = 5) and UDP (0.5 µmol/L) (n = 4); **P < 0.01 versus Control and Agonist.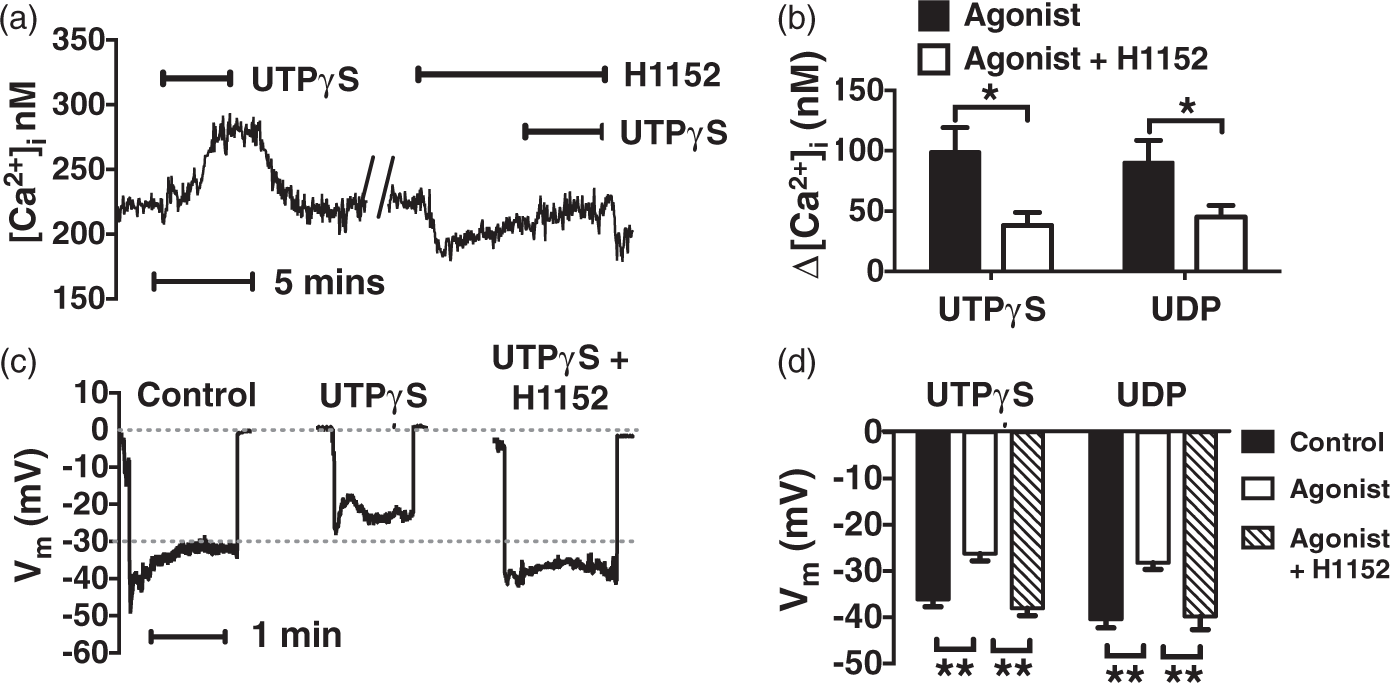
RhoA activation enhances myogenic tone and depolarization
To further elucidate the mechanisms of Rho signaling in PAs, we next investigated the effects of RhoA activation on tone and membrane potential. Arterioles pretreated with the RhoA activator CN0316,23 (1 µg/mL) for 3 h developed very high levels of myogenic tone (Figure 4(a) and (b)), which were diminished by H1152 (1 µmol/L) (data not shown). Control PAs (cultured for 3 h in the absence of CN03) also developed substantial myogenic tone, but significantly less than RhoA-activated arterioles. In addition, CN03-treated vessels exhibited a more depolarized membrane potential than control (−31 ± 1 mV versus −39 ± 0.5 mV), which was reversed by 1 µmol/L H1152 (−44 ± 1 mV) (Figure 4(c) and (d)), indicative of an important role of RhoA/ROCK signaling in modulating membrane potential. In light of the well-established contribution of TRPM4 channels to myogenic depolarization, we hypothesized that RhoA-stimulated depolarization is mediated by TRPM4. In accord with our prediction, the selective TRPM4 blocker 9-phenanthrol (30 µmol/L) hyperpolarized CN03-treated arterioles to −44 ± 3 mV (Figure 4(c) and (d)), strongly supporting the involvement of TRPM4 in Rho-mediated depolarizing mechanisms in PA myocytes.
RhoA activation enhances myogenic constriction and depolarization in endothelium-denuded PAs. (a, b) Representative recordings and summary data showing PAs pretreated with the RhoA activator CN03 (1 µg/mL) develop enhanced myogenic tone compared with vehicle-treated arterioles (n = 4); *P < 0.05 versus control. (c, d) CN03 (1 µg/mL)-treated arterioles exhibit depolarized membrane potential (n = 5) compared with control (n = 5), which is attenuated by H1152 (1 µmol/L) (n = 4) and the TRPM4 channel blocker 9-phenanthrol (30 µmol/L) (n = 4). Intraluminal pressure = 3 mmHg; **P < 0.01 versus CN03, ***P < 0.001 versus control and CN03.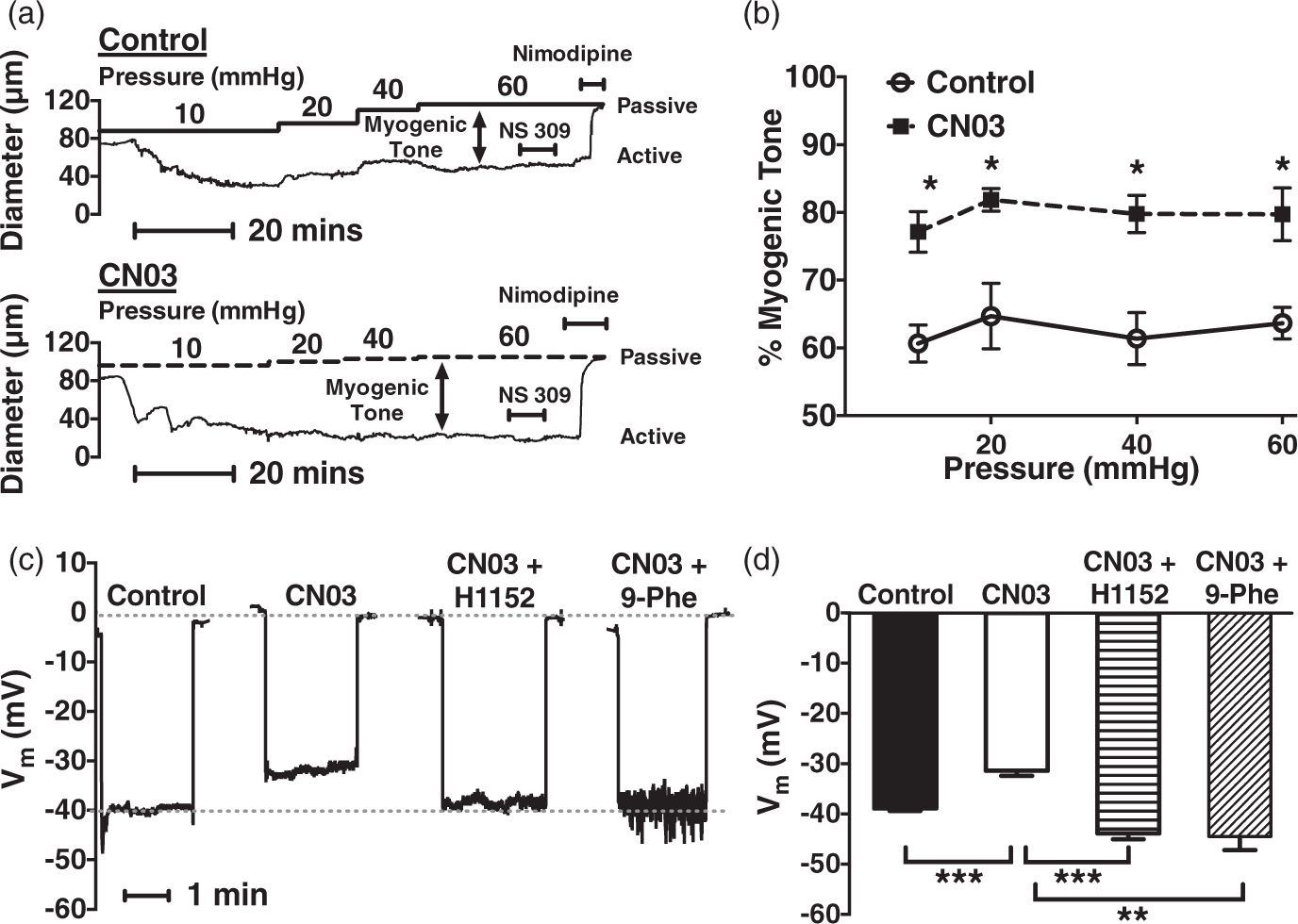
RhoA/ROCK-signaling activates TRPM4 channels
In this series of experiments, we studied the direct coupling between RhoA/ROCK signaling and TRPM4 channels using the perforated patch voltage-clamp technique. UTPγS (0.5 µmol/L) elicited a 3-fold increase in TRPM4 channel activity, which was diminished by H1152 (1 µmol/L) (Figure 5(a) and (b)). Similar inhibition was also observed for UDP-activated (0.5 µmol/L) TRPM4 currents (Supplementary Figure 2), pointing to an excitatory role of ROCK on TRPM4. To ensure that these results are not due to nonspecific effects of H1152 on the TRPM4 channel itself, we utilized the excised inside-out patch clamp configuration to study single TRPM4 channels expressed in HEK 293 cells. H1152 (1 µmol/L) had no effect on single TRPM4 currents in these membrane patches (Supplementary Figure 3), indicating no direct channel inhibitory action of H1152.
TRPM4 channel activity is reduced by ROCK inhibition and enhanced by RhoA activation in PA myocytes. (a, b) A representative nyastatin-perforated patch recording and summary data showing H1152 (1 µmol/L) inhibits UTPγS-activated (0.5 µmol/L) TRPM4 currents (n = 7); ***P < 0.001 versus control and UTPγS. (c, d) Representative conventional whole-cell recordings and summary data showing CN03 (0.1 ng/mL) potentiates TRPM4 currents (n = 7) versus control (n = 8), which is reversed by H1152 (1 µmol/L) (n = 8) and 9-phenanthrol (30 µmol/L) (n = 7). The representative currents are selected from cells with membrane conductance of 7–8 pF. [Ca2+]i = 1 µmol/L; ****P < 0.0001 versus control and CN03.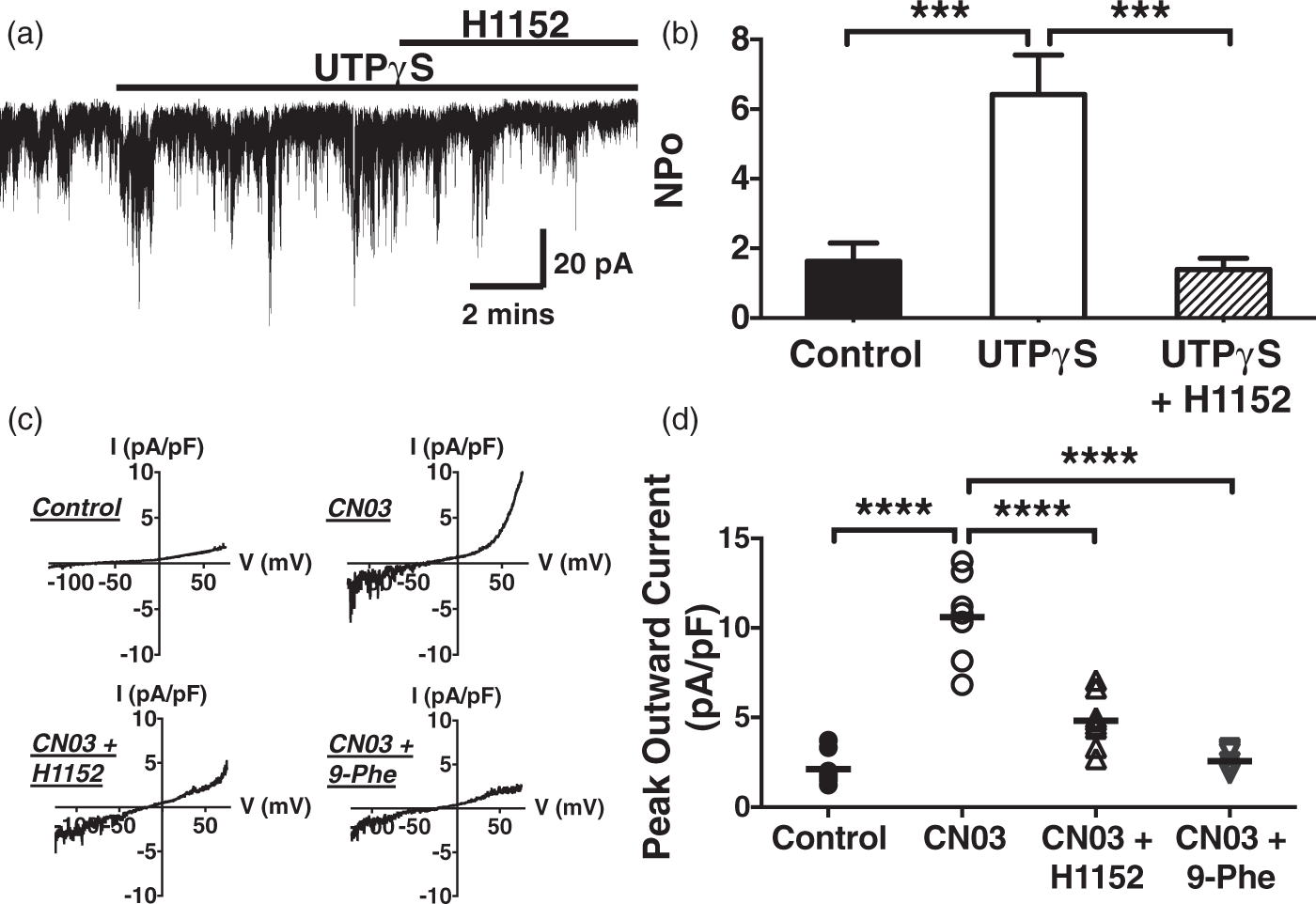
Next, effects of RhoA on TRPM4 activity were examined using the conventional whole-cell patch configuration, whereby RhoA activation was achieved by directly including CN03 (0.1 ng/mL) in the pipette solution. Whole-cell TRPM4 currents were potentiated by 4-fold in the presence of CN03 (Figure 5(c) and (d)). As CN03 had no direct influence on TRPM4 channel per se (Supplementary Figure 3), the RhoA activator-stimulated channel activity is potentially mediated by ROCK activation. Indeed, it was partially (68%) inhibited by H1152 (1 µmol/L) and nearly abolished (95%) by blocking TRPM4 channels with 9-phenanthrol (30 µmol/L) (Figure 5(c) and (d)). These data strongly support the proposal that RhoA/ROCK signaling couples the mechano-stimulation of P2Y receptors to TRPM4 channel opening, which leads to myogenic vasoconstriction in cerebral PAs.
ROCK increases the Ca2+-sensitivity of TRPM4 channels
Our laboratory previously established that protein kinase C (PKC) activates TRPM4 by increasing channel sensitivity to intracellular Ca2+ in pial arterial myocytes.
24
In the present study, we hypothesized that ROCK plays a similar role in the PAs, which appear to be highly dependent on RhoA/ROCK signaling for myogenic regulation.
13
In accord with past studies,24,25 TRPM4 activity was influenced by [Ca2+]i (Figure 6). We found that in 1 µmol/L [Ca2+]i, TRPM4 current activity levels were very low in both control (similar to our previous report
24
) and in H1152 (1 µmol/L)-treated myocytes with peak current density at 2.1 ± 0.3 pA/pF and 1.8 ± 0.3 pA/pF, respectively (Figure 6(a) and (b)). In 10 µmol/L [Ca2+]i, there was no apparent increase in TRPM4 activity in the presence of H1152 (2.1 ± 0.3 pA/pF), whereas there was near maximal current activation in the absence of H1152 (4.3 ± 0.9 pA/pF). In 100 µmol/L [Ca2+]i, currents were maximally activated in the presence and absence of H1152 with peak amplitude at 4.6 ± 1.3 and 4.9 ± 0.9 pA/pF, respectively (Figure 6(b)). These findings suggest that ROCK inhibition increased the apparent threshold of [Ca2+]i for TRPM4 activation, supporting the hypothesis that ROCK enhances the Ca2+ sensitivity of TRPM4 channels in PA smooth muscle cells.
ROCK inhibition decreases the Ca2+-sensitivity of TRPM4 channels in PA myocytes. (a) Representative current–voltage relationships from individual cells showing TRPM4 channel activity is Ca2+-activated under control conditions (left) and that H1152 (1 µmol/L) inhibits TRPM4 activation by Ca2+ (right). The representative currents are selected from cells with membrane conductance of 7–8 pF. (b) Summary data of peak outward currents showing H1152 increases the [Ca2+]i-threshold for TRPM4 activation. H1152 significantly inhibits TRPM4 currents at 10 µmol/L [Ca2+]i. (n = 5–9 cells from two to three rats per group); *P < 0.05 versus control.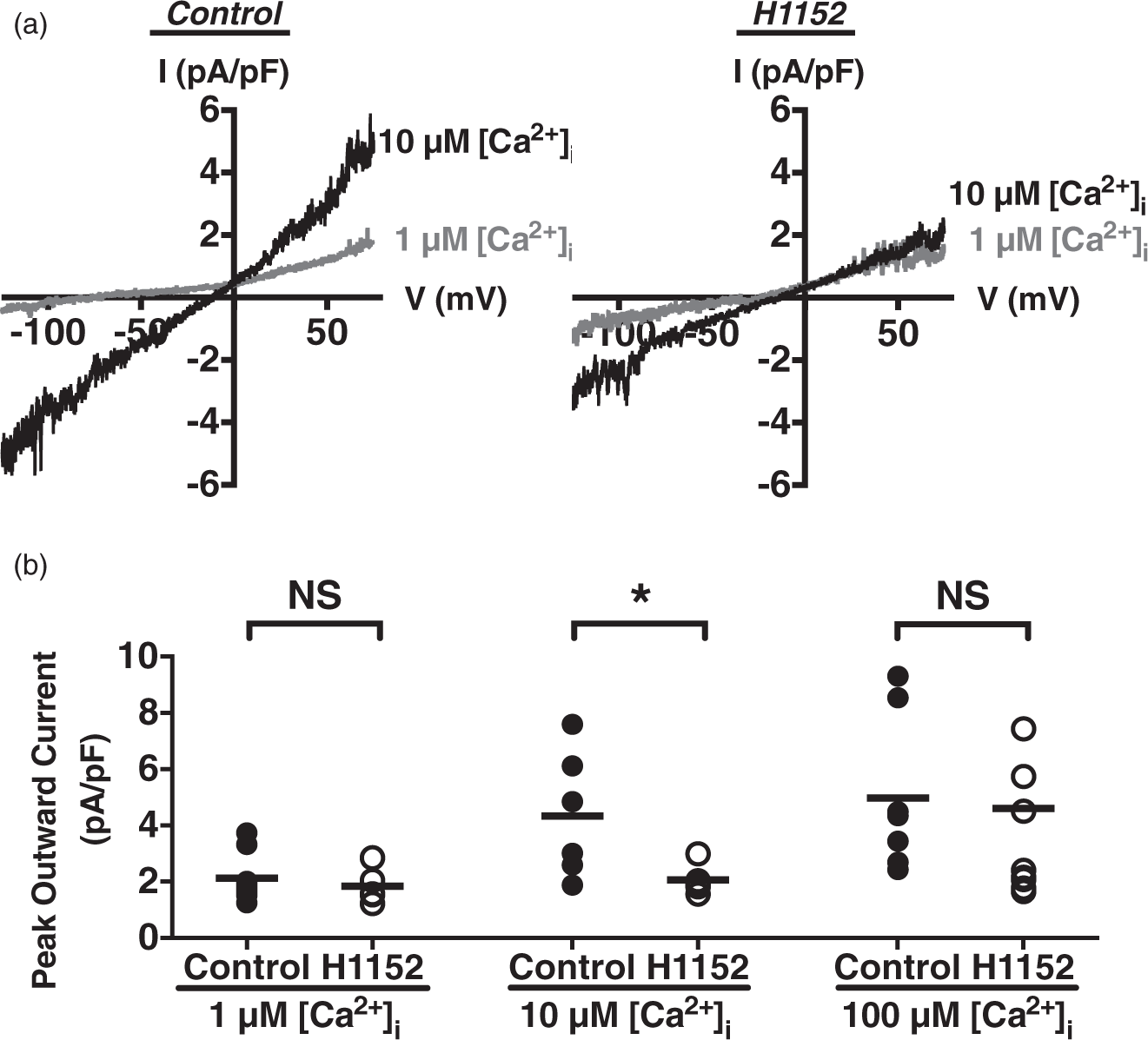
Discussion
Here, we investigated the contributions of the RhoA/ROCK signaling pathway to myogenic tone development in cerebral intraparenchymal arterioles. The major findings of the present study are (1) the RhoA/ROCK signaling is involved in pressure-induced and P2Y receptor-mediated smooth muscle depolarization, cytosolic [Ca2+] increase and vasoconstriction of cerebral PAs; (2) RhoA/ROCK signaling couples P2Y receptor activation to TRPM4 channel opening in PA myocytes; (3) ROCK enhances the apparent Ca2+-sensitivity of TRPM4 channels in PA smooth muscle cells. Collectively, these results demonstrate that in addition to the traditional Ca2+-sensitization pathway, the Rho signaling also facilitates mechano-activation of TRPM4 channels, leading to Ca2+-entry through VDCCs and associated myogenic responses in PAs (Figure 7).
Proposed mechanism of pressure-induced, P2Y-dependent activation of TRPM4 through the Rho signaling pathway in cerebral arterioles. Mechano-sensitive P2Y receptors are stimulated by increase in intravascular pressure, which initiates the activation of RhoA and ROCK. ROCK subsequently activates TRPM4 channels, and causes membrane depolarization, Ca2+ entry through VDCCs, and activation of MLCK, which phosphorylates MLC and results in smooth muscle contraction. Additionally, ROCK increases the Ca2+-sensitivity of the contractile machinery via inhibiting MLCP.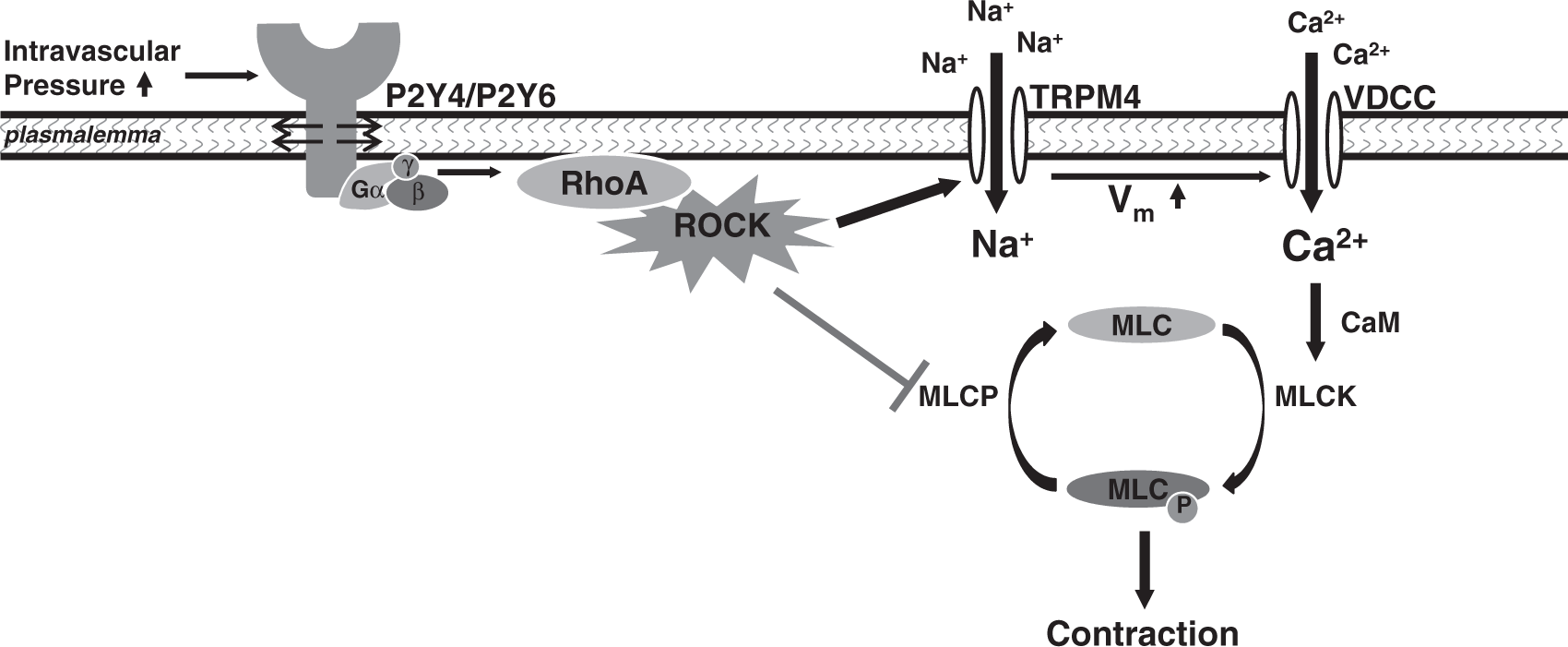
Mechanisms of RhoA/ROCK signaling in myogenic vasoconstriction
A correlation between ROCK function and myogenic reactivity has been previously reported in the cerebral circulation,9,22 with a generally accepted mechanism being a ROCK-mediated inhibition of myosin light chain phosphatase. This results in greater myosin light chain phosphorylation and activation, and enhanced smooth muscle contractility, which is not dependent on increased Ca2+ delivery to the contractile apparatus. 17 However, several recent studies point to a regulatory role of Rho signaling on the activity of ion channels, including ENaC, 18 Kv, 19 and inward rectifying K+ (Kir) channels. 26 ROCK has also been reported to mediate UTP-induced smooth muscle depolarization by inhibiting Kv channels, 19 further supporting an important yet underappreciated role of ROCK in regulating the excitation-contraction coupling in vascular smooth muscle. In the present study, we observed that the selective ROCK inhibitor H1152 significantly suppressed pressure-induced responses in membrane potential, cytosolic [Ca2+] and tone. Furthermore, the RhoA activator CN03 potentiated myogenic tone and membrane depolarization, both of which were sensitive to ROCK inhibition, providing the first evidence that the RhoA/ROCK signaling pathway participates critically in pressure-induced depolarizing mechanisms in cerebral PAs.
Our laboratory has recently established the coupling between mechano-sensitive P2Y4/P2Y6 receptors and TRPM4 channels, and its contribution in mediating myogenic depolarization and vasoconstriction in PAs.4,14 Consistent with ROCK-associated mechanisms in myogenic control, here we also observed that P2Y4 and P2Y6 receptor ligand-evoked constrictions, [Ca2+]i elevations and depolarization were attenuated by H1152. Furthermore, our prior study demonstrated that TRPM4 channels are stimulated by P2Y receptor agonists. 14 In the current work, UTPγS and UDP-activated TRPM4 currents were reduced by H1152. We also found that the RhoA activator CN03 elicited a substantial increase in TRPM4 activity in PA myocytes in a ROCK-dependent manner. These findings demonstrate that RhoA/ROCK signaling facilitates pyrimidine receptor-mediated activation of TRPM4 channels in PA smooth muscle.
Several mechano-sensitive Gq/11 protein-coupled receptors have been reported by us and others.4,27 P2Y4 and P2Y6 receptors appear to mediate stretch-evoked vasoconstriction in a ligand-independent manner, because myogenic behavior of PAs is not affected by altering ectonucleotidase activity, which regulates the local levels of purines and pyrimidines. 4 This is similar to the involvement of Angiotensin II (Ang II) receptors in cerebral pial arterial myogenic tone, which is sensitive to the Ang II receptor inhibitor losartan but not influenced by Ang II converting enzyme (ACE) inhibition. 27 Ang II receptor-mediated myogenic responses rely on Gq/11 protein-coupled, phospholipase C (PLC)-dependent signaling pathways.27,28 This is supported by the observations that the PLC inhibitor U73122 diminishes membrane stretch-induced responses in an Ang-II receptor/ion channel expression system, 27 and relaxes pressurized rat pial arteries. 29 In addition, molecular suppression of the γ1 isoform of PLC significantly attenuates myogenic depolarization and vasoconstriction, and also inhibits hypotonicity-stimulated TRPM4 currents. Interestingly, in our preliminary experiments, PLC did not appear to participate in myogenic regulation of PAs since U73122 had little effect on arteriolar diameter at a concentration that causes near maximal dilations in pial vessels (data not shown). Instead, current data illustrate the novel involvement of the Rho-associated mechanisms in P2Y receptor-mediated mechanical responses. Interestingly, in addition to Gq/11-protein, P2Y receptors are also coupled to G12/13 and initiate Rho signaling cascades in vascular myocytes. 30 Moreover, several apparent Gq/11 protein-coupled mechanosensors identified in expression systems, 27 such as ETA endothelin and V1A vasopressin receptors, also couple to G12/13 to stimulate RhoA/ROCK-dependent smooth muscle contraction. 31 Together with the key observations in the present study, these data strongly suggest that stretch-induced contraction of vascular myocytes is dynamically regulated not only by Gq/11-related signaling pathways but also by G12/13-meditated and Rho-dependent cellular mechanisms.
Contribution of RhoA to myogenic constriction of cerebral PAs
To further investigate the mechanistic involvement of RhoA in myogenic regulation, we utilized the commercially available RhoA activator CN03. This cell permeable recombinant protein functions by converting glutamine-63 to glutamate, thus disrupting both intrinsic- and GAP-stimulated GTPase activity, leaving RhoA constitutively active. 23 The effectiveness of CN03 has been confirmed in several studies, where RhoA activity in native smooth muscle cells is significantly increased following a 2 - to 4-h incubation with CN03 at concentrations ranging from 1 to 5 µg/mL.32,33 Functionally, we detected a significant augmentation of pressure-induced tone in arterioles cultured with 1 µg/mL (or 8.5 nmol/L) CN03 for 3 h, which was 25 to 30% higher than control, indicating the magnitude of responsiveness to pressure is directly associated with RhoA activity levels. This is consistent with a previous study on mouse cerebral arteries where CN03 (5 µg/mL, 4 h) induced a substantial elevation of myogenic tone. 16 In the present study, control PAs (cultured for 3 h in the absence of CN03) also developed significant amount of tone. This may be due to upregulation of unknown cellular factors during the culture process that potentiate myogenic reactivity of PAs. In addition to contractility, activation of RhoA also depolarized the smooth muscle membrane through an H1152-sensitive pathway, further supporting the notion that myogenic depolarization and vasoconstriction in PAs are regulated by upstream RhoA/ROCK signaling cascade.
A major finding of the present study is that Rho activates TRPM4 channels as illustrated with conventional whole-cell patch recordings. Channel activity detected with this configuration has been well characterized. These currents exhibit the hallmarks of TRPM4 channel properties: [Ca2+]i dependence, outward rectification, and time-sensitive decay.24,34 Additionally, these currents are diminished by suppressing TRPM4 expression with antisense. 24 We also found that the selective TRPM4 blocker 9-phenanthrol nearly abolished the channel activity, indicating they are TRPM4 currents. In the present series of experiments, CN03 (0.1 ng/mL or 0.85 pmol/L) was included in the pipette to obtain direct access to the inside of the cells and induce acute RhoA activation upon membrane rupture. TRPM4 currents were markedly activated by CN03. Because CN03 has no direct effects on TRPM4 itself (Supplementary Figure 3), this is indicative of an excitatory influence of Rho on the depolarizing mechanisms, leading to myogenic activation of PA myocytes.
Regulation of TRPM4 channels by ROCK
In the present study, ROCK regulated the responses of TRPM4 channels to Ca2+. Under control conditions, the EC50 of [Ca2+]i for TRPM4 activation appeared to lie between 1 and 10 µmol/L. Nevertheless, when myocytes were treated with H1152, the threshold for TRPM4 stimulation was elevated beyond 10 µmol/L [Ca2+]i, suggesting that ROCK regulates the Ca2+ sensitivity of the channel itself. The exact mechanisms underlying ROCK-associated TRPM4 activation are still unknown. However, several early studies showed that ROCK regulates Kv 35 and ENaC 18 channel trafficking and expression on the plasma membrane to modulate membrane excitability. Although it is not clear whether TRPM4 is subjected to this mechanism, the observations that TRPM4 currents can be acutely potentiated and attenuated by CN03 and H1152, respectively, point to immediate modulatory effects from ROCK, presumably through direct phosphorylation of the Ca2+-sensing sites on TRPM4. Importantly, PKC plays a similar role in pial artery myocytes, where the PKC activator PMA increases Ca2+ sensitivity of TRPM4 channels. 24 However, our preliminary experiments showed that PKC inhibition had little effect on PA vascular tone (data not shown), which is compatible with minimal contribution of PLC to myogenic regulation in cerebral PAs. Several prior studies concerning the roles of PKC and ROCK in the cerebral circulation reported similar observations that myogenic control in cerebral pial arteries is mediated by PKC and ROCK together,24,36,37 while only ROCK participates in pressure-evoked response in the penetrating arterioles. 9 Interestingly, both PKC and ROCK are serine/threonine kinases with similar consensus substrate phosphorylation sites, 38 further supporting the results that TRPM4 channels can be regulated by both enzymes. Although explanations for the apparent shift of functional kinases from large to small cerebral arteries requires further investigation, the unique contributions of ROCK in the cerebral microcirculation may provide important insights in preventing and treating microvascular diseases.
Conclusion
Collectively, our data reveal a novel role of RhoA/ROCK-signaling in cerebral vasoconstriction, which is determined primarily via activation of TRPM4 channels, smooth muscle depolarization and Ca2+ influx through VDCCs. As emerging evidence points toward important physiological and pathological contributions of ROCK in the cerebral circulation,9,16 a more complete understanding of the cellular mechanisms whereby RhoA/ROCK signaling is controlled may lead to development of innovative therapeutic strategies for microvascular pathologies in the brain.
Footnotes
Funding
This study was supported by NIH grant P01 HL095488, the Totman Trust for Medical Research, and an American Heart Association Predoctoral Fellowship (Yao Li)
Acknowledgments
We thank Drs. George Wellman and Anthony Morielli for their valuable advice, and Arsalan Syed and Sharath Madasu for their helpful technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Yao Li contributed to experimental design and performance, and data acquisition, interpretation and analysis, and manuscript preparation; Joseph E Brayden contributed to study design, data interpretation, critical manuscript revision, and final approval.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.