
Abstract
Psychostimulants and nicotine are the most widely abused drugs with a detrimental impact on public health globally. While the long-term neurobehavioral deficits and synaptic perturbations are well documented with chronic use of methamphetamine, cocaine, and nicotine, emerging human and experimental studies also suggest an increasing incidence of neurovascular complications associated with drug abuse. Short- or long-term administration of psychostimulants or nicotine is known to disrupt blood-brain barrier (BBB) integrity/function, thus leading to an increased risk of brain edema and neuroinflammation. Various pathophysiological mechanisms have been proposed to underlie drug abuse-induced BBB dysfunction suggesting a central and unifying role for oxidative stress in BBB endothelium and perivascular cells. This review discusses drug-specific effects of methamphetamine, cocaine, and tobacco smoking on brain microvascular crisis and provides critical assessment of oxidative stress-dependent molecular pathways focal to the global compromise of BBB. Additionally, given the increased risk of human immunodeficiency virus (HIV) encephalitis in drug abusers, we have summarized the synergistic pathological impact of psychostimulants and HIV infection on BBB integrity with an emphasis on unifying role of endothelial oxidative stress. This mechanistic framework would guide further investigations on specific molecular pathways to accelerate therapeutic approaches for the prevention of neurovascular deficits by drugs of abuse.
Keywords
Introduction
Drug abuse, a devastating neuropsychiatric disorder, remains one of the somber public health concerns worldwide. Especially, increased incidence of psychostimulant and nicotine use inflicts significant rates of mortality and morbidity with an alarming increase in health care costs. 1 It is also evident that chronic abuse of psychostimulants (such as methamphetamine (METH) and cocaine) causes significant level of neurotoxicity and inflammation leading to persistent neurochemical abnormalities and loss of synaptic integrity.2–5 Pronounced levels of oxidative stress and mitochondrial dysfunction, due to uncontrolled increase in cellular reactive oxygen (ROS) and nitrogen species (RNS), were identified as major pathological mechanisms underlying psychostimulant or smoking-induced neurodegeneration.6–12 In fact, clinical studies suggest a diminished anti-oxidative stress response in cocaine or methamphetamine abusers compared with normal subjects. 13 Thus, altered “redoxomics” predisposes the long-term neurotoxic sequelae and secondary brain injuries in drug abusers.
Recently, the interaction of drugs of abuse with cerebrovascular function has received a significant interest14–17 and has emerged as a critical facet of drug abuse-related neuropathology. Overarching evidence suggests that psychostimulant abuse and smoking have long-term influence on brain vascular rheology and dynamics18–21 with an increased risk for ischemic stroke, cerebral infarction, intracerebral hemorrhage, and vascular dementia.22–24 It is now well evident that psychostimulants and tobacco smoking critically impair the structure and function of “blood-brain barrier” (BBB) within the dense microcapillary network of brain.
BBB is a specialized neuroanatomical interface composed by unique endothelial phenotype at the brain microvascular beds and strictly enforces CNS homeostasis.25–29 As shown in Figure 1, pericytes, astrocyte end feet processes, and neurons within the neurovascular unit (NVU) induce and regulate the barrier functions of BBB.30–32 Structurally, the inter-endothelial tight junction complexes comprising occludin, claudins, and membrane-directed scaffolding proteins (such as zonula occludentes-1 (ZO-1)) contribute to physical barrier nature of BBB and strictly limit the molecular/cellular influx from circulation.33,34 Furthermore, the uniqueness of the BBB endothelium is endorsed by abundant expression of efflux pumps and stereospecific solute transporters.35,36 For a comprehensive understanding, the readers are referred to excellent reviews on BBB structure and function.26,27,29
Structural and functional aspects of the BBB constituted by brain microvascular endothelial cells surrounded by pericytes and astroglial end feet processes. As shown in the figure, BBB permeability to paracellular markers is strictly limited due to the expression of interendothelial TJ complexes. Also, BBB endothelium is equipped with a number of nutrient and efflux transporter systems that tightly regulate the transmembrane passage of endogenous and xenobiotic substrates including nutrients.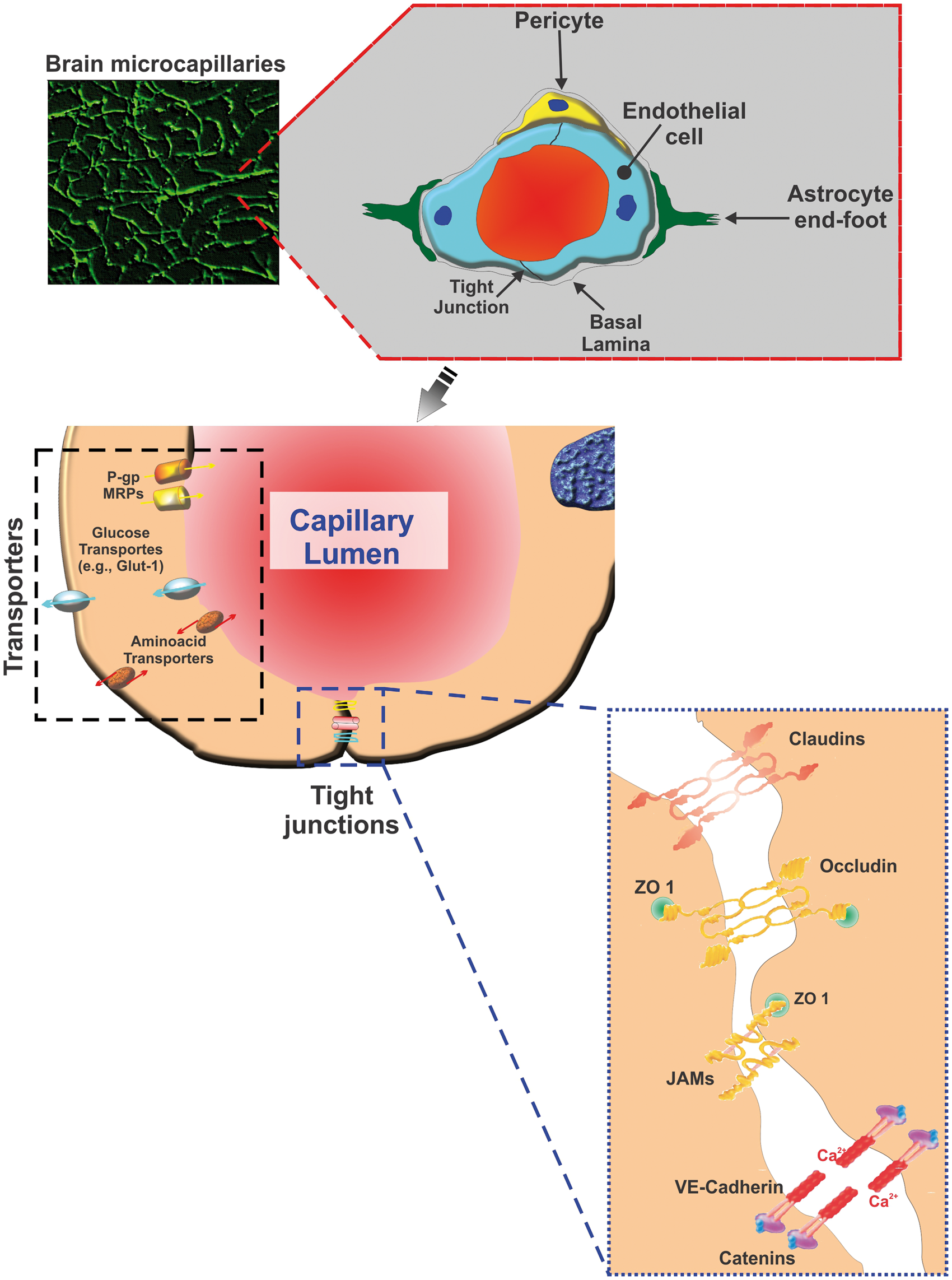
A host of vascular and neuro-glial substrates are known to regulate the physical, metabolic, and transport barrier properties of BBB via endocrine, paracrine, or autocrine signaling mechanisms.37–39 Brain microvascular endothelial cells are highly sensitive to imbalance in redox homeostasis40–42 and increased endothelial oxidative stress significantly compromises the stringency of BBB with marked reduction in the immunological privilege of CNS.28,43–45 To this end, compelling evidence suggests that psychostimulants as well as tobacco smoking could have profound pro-oxidant effects and significantly activate endothelial oxidative stress responses, thus, leading to a compromised BBB integrity.
Therefore, the objective of this review is to provide a comprehensive overview of the pathological impact of drugs of abuse, limited to psychostimulants (METH and cocaine) and tobacco smoking, on BBB structure and function with an emphasis on the oxidative stress pathways. We have also summarized the potential interactions between psychostimulants and human immunodeficiency virus (HIV) infection at BBB interface, and how METH or cocaine exacerbates the neuropathogenesis of HIV encephalitis. Mainly, we have integrated a host of cellular and molecular mechanisms under the canopy of oxidative/inflammatory stress at BBB endothelium.
METH abuse and blood-brain barrier dysfunction
As mentioned in earlier sections, recreational and/or long-term METH abuse precipitates a wide spectrum of neurotoxic and inflammatory adversities, mainly, stemming from increased oxidative stress and its corollary of molecular cascades.2,6,16 Importantly, these pathological manifestations are strongly associated with various cerebrovascular abnormalities such as ischemia or hypoxia in METH abusers.17,18,46 For instance, METH abuse, independent of the route of administration, is strongly associated with stroke pathogenesis and hypoxia in humans47,48 and rats.46,49 Notably, binge use of METH in humans elicits a sustained reduction in global and regional cerebral blood flow that is prominent even after 2 years of abstinence (Chung et al. 2010).50 In this line, METH-induced neurotoxicity and astroglial reactivity accentuates the loss of BBB integrity within the NVU,17,51,52 an ultimate outcome of METH abuse-related cerebrovascular dysfunction. 53
Following a series of initial demonstrations of mammalian BBB disruption by amphetamines,54,55 it is now widely established that METH abuse/addiction directly impairs various structural and functional facets of BBB resulting in loss of barrier integrity.17,56 Both acute/binge and long-term (chronic) paradigms of METH administration promotes a region-specific BBB disruption in a concentration and time-dependent manner.57,58 For example, acute high dose of METH (40 mg/kg) in mice, reflecting a binge intoxication model, was shown to elicit a rapid and transient BBB disruption primarily in the hippocampus within 3-h post-injection that is associated with severe hyperthermia and seizures. 57 This region-specific pattern of BBB disruption was also demonstrated in rats 49 within 2-h exposure to METH (3 or 9 mg/kg, i.p) and in mice after 24 h following 30 mg/kg dose. 58 However, no changes in BBB permeability were observed in mouse midbrain or striatum at 3–24 h following repeated dosing paradigm of METH (4 mg/kg, i.p.). 60 Intriguingly, the magnitude of BBB damage was significantly elevated following spontaneous withdrawal of METH. 53 This phenomenon is possibly caused by altered neurochemical profiles underlying negative behaviors associated with drug withdrawal. 61 Moreover, clinical findings from abstinent METH abusers suggest a long-lasting loss of BBB integrity and cerebrovascular functions. 61 Additionally, Kousik et al. 49 have documented a sustained increase in rat striatal endothelin-1 (a vasoconstrictor implicated in stroke pathogenesis) even after discontinuation of METH self-administration for 10 days. Thus, BBB dysfunction is a long-term cerebrovascular complication that is evident during various phases of METH abuse/addiction in humans and rodent models. It should also be noted that at high doses of METH intoxication in rodents, the BBB breakdown is initiated rapidly within a short kinetic frame that is reversible with progression of time. 59
A growing body of evidence strongly suggests that BBB TJ protein complexes are highly sensitive to the detrimental effects of METH challenge in various dosing paradigms. As illustrated in Figure 2, acute or binge-like METH administration in rodents or in vitro models of BBB was shown to significantly perturb endothelial TJ assembly by down-regulation, fragmentation, or re-distribution of major TJ proteins such as occludin, claudin-5, and/or ZO-1, resulting in reduced endothelial barrier tightness (decreased TEER) and increased BBB paracellular permeability.63–67 Another binge model of METH administration in mice (4 × 10 mg/kg, i.p. with 2-h interval on a single day) resulted in altered and weakened structure of brain microvessels with increased permeability to plasma proteins.
68
In addition, acute (10 mg/kg, i.p.) or chronic METH exposure (15 mg/kg, i.p.) for 5–6 weeks in mice contributed to a robust suppression of occludin and ZO-1 in intact brain microvessels.67,69 This effect was associated with a concomitant increase in albumin flux into brain parenchyma.
69
Methamphetamine interaction with the BBB. METH directly affect the endothelial physiology by eliciting a significant inflammatory and oxidative stress response through release of pro-inflammatory cytokines, MMPs, and increased nuclear translocation of NF-κB. A subsequent activation of MLCK/Rho-A leads to down-regulation of TJ proteins such as ZO-1, claudin-5, and occludin. Also, METH deregulates glucose transport and cellular metabolism by abrogation of GLUT-1. Increased expression of inflammatory cell adhesion molecules at the BBB endothelium (ICAM, VCAM) facilitates endothelial-leukocytes interaction and WBC transmigration across the BBB.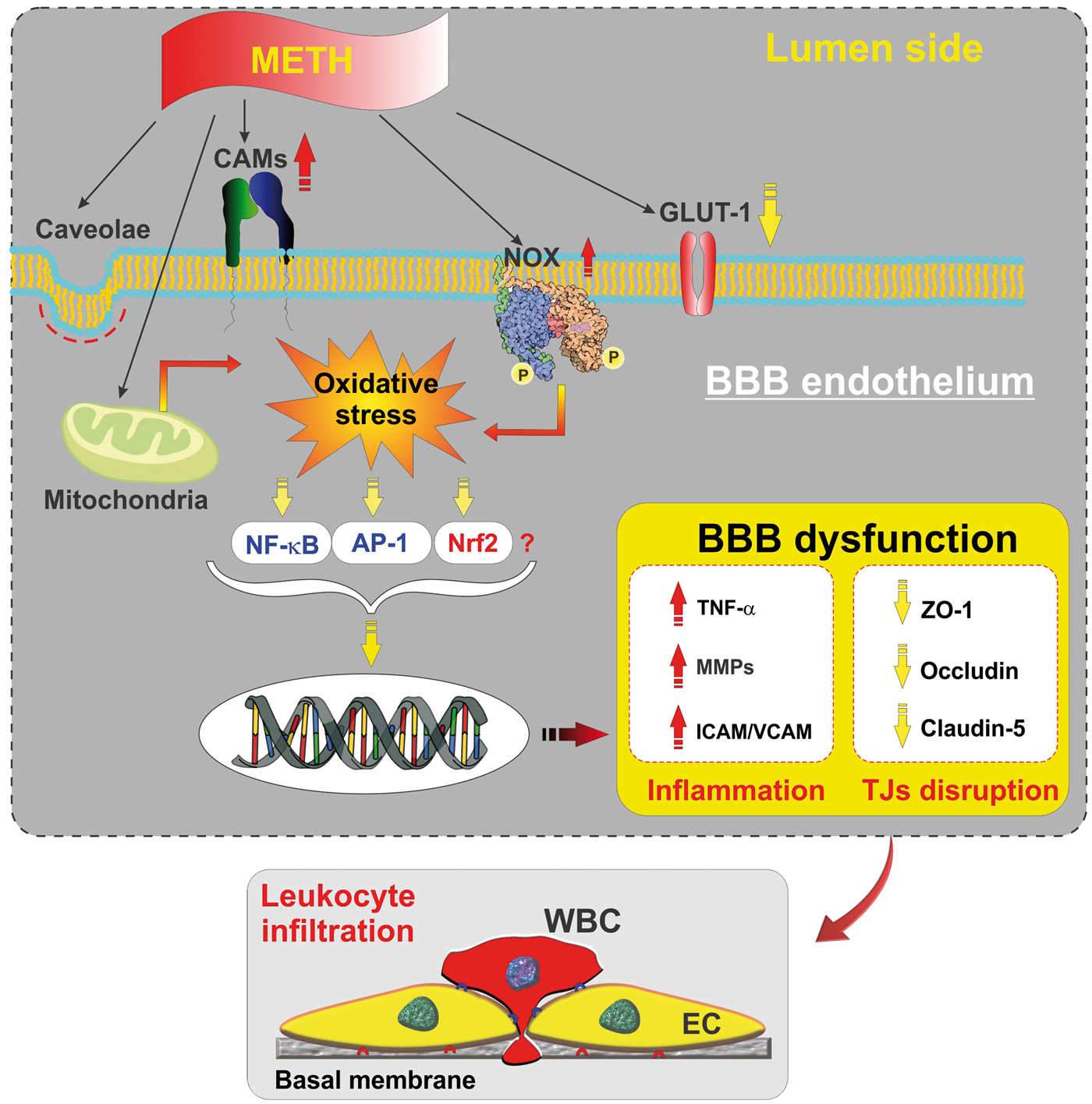
In contrast, no discernable changes were observed in TJ protein continuity at cell–cell borders, particularly claudin-5 69 and BBB hyperpermeability to dextrans by METH exposure in vitro.60,71 For example, at relatively higher concentrations above the physiological range, METH challenge (10 µM) resulted in rapid decline in occludin levels (starting at 30 min), whereas claudin-5 was relatively unchanged even after 24-h exposure. 70 The conundrum of METH-induced impact on BBB integrity was further highlighted by mouse brain regional differences in susceptibility to METH-induced alterations in TJ proteins and BBB hyper-permeability, as characterized by a bell-shaped response with progression of time from 1- to 72-h window. 59 Collectively, these studies point out that the magnitude, selectivity, and the kinetic window of METH-induced loss of BBB integrity directly correlate with the model, concentration, and dosing regimen of the drug. 64,66,69,70 Taken together, we speculate that these divergent effects of METH on TJ protein and BBB integrity as reported in various studies can be explained by differences in a number of experimental variables including dosing paradigms (e.g. very high concentrations of METH in preclinical experiments in rodents lead to neurotoxic stress, hyperthermia, and death, which was accompanied by BBB breakdown), kinetic, and concentration-dependent methods of administration. Only a few of these studies controlled the effective plasma concentrations of METH during the observation period. 72
Various molecular pathways have been proposed and their cross talk implicates oxidative stress as a central pathogenic mechanism that may contribute to METH-induced endothelial dysfunction and BBB damage.63,64,67,70 Importantly, METH challenge elicited an intense oxidative and inflammatory stress response by increased generation of ROS, pro-inflammatory cytokines, and depletion of inherent anti-oxidant systems (such as glutathione and related enzymes) in BBB endothelium. 64,65,67 For example, repeated intravenous administration of METH (3 × 10 mg/kg with 2-h interval) led to significant down-regulation of TJ proteins that is possibly associated with GSH depletion and increased ROS following 24-h exposure. 66 Reported molecular framework suggests that METH-induced endothelial ROS levels trigger actin polymerization possibly through activation of Arp2/3 complex 73 or myosin light chain kinase and its downstream target, RhoA.64,65 These effects can lead to TJ redistribution and BBB hyper-permeability (Figure 2), notably occurring at physiologically relevant low concentrations of METH. Furthermore, METH-induced oxidative stress evoked post-translational oxidative modifications of TJ proteins in mouse brain capillaries through molecular adducts with ROS and RNS 66 or increased endocytosis and degradation of occludin in endothelial cultures, 73 which was blocked by pretreatment with anti-oxidants. Another pathological outcome of METH-induced inflammatory stress at BBB is an increased expression and activity of matrix metalloproteinases (MMPs) that are extensively known to degrade the basal lamina and TJ proteins leading to barrier collapse.58,74 Additionally, impaired BBB endothelial cell metabolism caused by METH-induced down-regulation of glucose transporter (GLUT1) and glucose uptake is a potential causative factor for loss of BBB integrity in mice. 69
It is also imperative to understand the molecular mechanisms upstream to METH-induced endothelial oxidative stress for devising efficient therapeutic strategies to prevent cerebrovascular complications and neurotoxicity in METH abusers. In this context, substantial evidence from rodent and in vitro studies has revealed the potential role of NADPH oxidase (NOX), a significant source of cellular ROS, and its regulatory subunits in METH-induced TJ dysregulation and BBB disruption.67,70 Moreover, a robust and transient increase in c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) phosphorylation in brain capillary endothelial cells 60 could be a possible mechanism underlying METH-induced increase of active MMP levels and BBB dysfunction. 75 Striking lines of evidence suggest that METH dose-dependently (50–200 µM) increases the transcriptional activities of inflammatory factors such as activator protein-1 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in human BBB endothelial cells. 76 This was shown to accompany a marked reduction in GSH levels within a short time frame (3 h) following METH exposure. In addition, acute METH injection (10 mg/kg, i.p.) in mice initiates a rapid nuclear factor E2-related factor-2 (Nrf2) response in brain capillaries, 67 a master regulator of anti-oxidant gene expression. 45 However, with progression of METH exposure, we propose that the molecular “switch on/off” functions of Nrf2 turn out to be unresponsive, thus, culminating in loss of redox balance and increased BBB endothelial stress.
An important downstream effect of METH-induced NF-κB activity is a rapid and pronounced inflammatory response through secretion of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNFα) in BBB endothelial cells,68,76 which critically down-regulate TJ proteins and compromise the BBB integrity.77,78 In this inflammatory landscape where cytokines activate NADPH oxidase-dependent oxidative stress, 79 TNFα release mediates both paracellular and transcellular (vesicular) permeability of BBB following acute METH exposure at relatively low doses (1 µM), which was blocked by NF-κB inhibitor. 68
METH administration was also shown to differentially modulate the expression of BBB gatekeepers belonging to ABC superfamily of efflux transporters through stimulation of apolipoprotein E signaling at BBB endothelium. 60 Mainly, METH exposure rapidly elevated luminal expression of ABCB1 (P-glycoprotein), while ABCC1 (MRP1) levels were down-regulated. An important functional implication of these findings is that METH may exacerbate the neurotoxicity of other blood-borne substrates including xenobiotics. This is an important issue that needs to be further investigated. A closer look at these studies also suggests that the pharmacological responses of the brain microvasculature to METH exposure recapitulate (to some extent) the BBB dysfunction mechanisms observed during stroke. This may not be entirely surprising given that METH administration causes ischemic-like hypoxia. 49
The consequences of METH-induced BBB disruption are many-fold and, as mentioned in later sections, chronic METH exposure in escalating dosing regimen (2.5–10 mg/kg over 3 weeks) aggravates CNS infiltration of viral and microbial pathogens, resulting in decreased survival rates of mice. 80 Strikingly, METH (10 nM–10 µM range) potentiates CNS infiltration of leukocytes across the BBB in various in vitro studies either by disruption of TJ proteins64,65,73 or enhancement of transcytosis, 71 possibly through increased production of cytokines such as TNFα.68,76 In addition, low doses of METH significantly up-regulated the expression of endothelial adhesion molecules (such as ICAM-1) that are engaged in endothelial-leukocyte interactions. By contrast, ElAli et al. 60 did not observe an increase in leukocyte trafficking following METH administration in mouse midbrain. This probably suggests region-specific interactions of METH with BBB.
Furthermore, METH-induced pro-oxidant effects at other NVU cellular components are also shown to compromise the BBB functions through impaired crosstalk mechanisms. 68 For example, METH increased the expression of glial fibrillary acidic protein (GFAP), an exclusive phenotypic marker of reactive astrocytes. 81 Additionally, METH at concentrations relevant to binge use/acute intoxication potentiated neuroinflammation through increased expression of σ1 receptors 82 and release of TNFα, interleukin (IL)-6, and IL-8 from mouse and rat astrocytes. This elicited a significant reduction in TEER and paracellular permeability across endothelial monolayers.68,83 It is also possible that METH-induced inflammatory phenotype of microglial cells with elevated TNFα release can activate the BBB endothelium resulting in increased transmigration of circulating leukocytes. 84
Taken together, a retrospective assessment of these findings suggests that METH causes neurotoxicity by (1) significant disruption of BBB integrity and function through elevated oxidative stress, and (2) enhanced inflammatory stress responses triggered by the invading immune cells through leaky BBB.
Cocaine abuse and blood-brain barrier impairment
Cocaine abuse is one of the growing neuropsychological health problems in the United States reaching pandemic proportions. Acute or chronic cocaine abuse precipitates various systemic and central effects mediated through immunomodulatory and neuroinflammatory responses, which result in wide spread neurodegeneration and toxicity. 4 It is well evident that cocaine elevates inflammatory and oxidative stress signaling cascades in vascular and neuronal tissues by stimulating the release of proinflammatory cytokines.13,85,86 Like METH and related amphetamines, cocaine was also shown to significantly impact the neurovascular function in humans and experimental animal models.14,19 Decreased cerebral blood flow is generally observed in cocaine abusing subjects.20,87 Acute or chronic administration of cocaine also increases the risk for ischemic stroke, hypoxia, and brain microvascular pathologies including vasculitis and associated complications.14,19,22,88
Cocaine-induced brain microvascular deficits have received significant attention in the recent years.51,89,90 Emerging evidence suggests a complex pathological impact of cocaine on BBB structure and function that involves both direct pro-apoptotic effects on the endothelial cells and indirect paracrine responses manifested by increased pro-inflammatory stimuli.85,86 It is widely demonstrated that elevated levels of oxidative/inflammatory stress pathways and endothelial activation are the primary pathological processes implicated in cocaine-mediated BBB disruption.85,89 Indeed, cocaine exhibits cell-specific and concentration sensitive multimodal interactions with endothelial, immune, and neuroendocrine cells that strongly converge on BBB dysfunction and increased brain extravasation of reactive molecules and immune cells.91,92 It should also be noted that intense hyperthermia associated with cocaine administration can also evoke potential BBB damage and brain edema. 89
Assessment of the complex pathophysiological effects of cocaine on BBB integrity revealed a spectrum of molecular pathways linked to endothelial dysfunction, as depicted by the schematic in Figure 3. Although the effects of cocaine on BBB TJ components remain little explored, existing studies reveal an adverse impact on TJ integrity. For example, in primary brain endothelial cultures, micromolar concentrations of cocaine were shown to rapidly disrupt the inter-endothelial TJ complexes. This well correlates with decreased TEER values across the BBB endothelial monolayers.85,93,94 Cocaine exposure in vitro and in vivo produced a progressive decline of steady state ZO-1 mRNA and protein expression leading to BBB hyper-permeability.93,95 These studies also postulate the existence of a putative cocaine binding site at BBB endothelium since pretreatment with tyrosine kinase inhibitor neutralized cocaine-induced endothelial activation and BBB disruption.
85
Specifically, cocaine activates and up-regulates σ1 receptors at BBB endothelium with subsequent stimulation of the Egr1 pathway-dependent release of endothelial platelet-derived growth factor (PDGF)-BB.90,95,96 Pharmacological or genetic manipulation of this signaling axis reversed cocaine-induced BBB breakdown.
95
Cocaine induces BBB dysfunction. Cocaine interacts with σ1 receptors on the BBB endothelium and elicits activation of p38 MAPK signaling pathways and NF-κB nuclear translocation. Downstream to the activation of p38 MAPK, cocaine up-regulates the expression of PDGF-β. This leads to compromised TJ integrity through down-regulation of ZO-1 and claudin-5. Additionally, cocaine promotes the release of pro-inflammatory cytokines (e.g. IL-6, TNF-α) from circulating WBCs and expression of BBB endothelial adhesion molecules that facilitate endothelial-leukocyte interaction and infiltration into the brain parenchyma.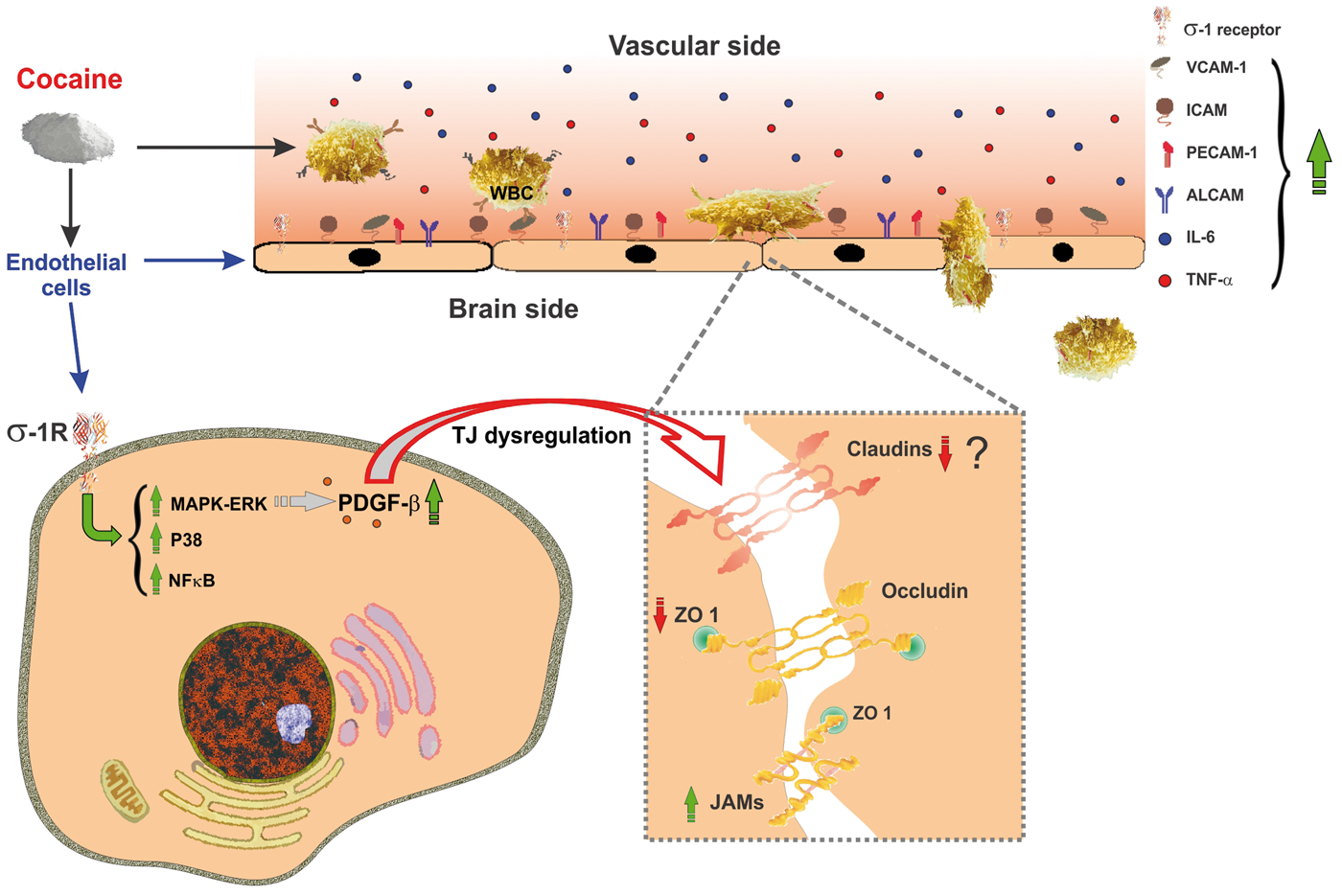
A major consequence of cocaine-induced BBB inflammatory response and loss of barrier integrity is an increased infiltration of circulating immune cells.91,93,94,96 It is well established that cocaine potentiates endothelial-monocyte interactions through dose-dependent up-regulation of inflammatory mediators and cell adhesion molecules (such as ICAM-1, VCAM, and ALCAM) at the BBB endothelium.86,91,96 Interestingly, cocaine-induced ALCAM expression involves the activation of σ1 receptor-PDGF-β signaling axis. 96 In addition, increased transcription of inflammatory signaling molecules by NF-κB activation and nuclear translocation could mediate cocaine-induced endothelial activation and monocyte transmigration across BBB.85,96
Although the pathological impact of cocaine on the extravascular cellular elements of NVU is not well established, it is evident that cocaine exposure potentiates aberrant astroglial responses in cellular and animal models,97–99 which would further compromise BBB integrity and function. Histochemical analyses following acute or repeated daily injections of cocaine (20 mg/kg) in mice revealed a significant increase in the expression of GFAP at the site of neuronal injury. 98 Detailed mechanistic studies suggested the involvement of Egr-1 and MAPK signaling cascades possibly through engagement of σ1 receptor activation in acute cocaine-induced reactive astrogliosis at physiologically relevant concentrations. 97 Furthermore, cocaine at doses that exceed the physiological range was shown to rapidly elevate ROS levels with concomitant reduction in cellular GSH leading to increased oxidative stress in astroglia-like cells. 100 However, the clinical relevance of these latter findings needs further investigation. Notably, in addition to its direct inflammatory responses at BBB endothelium, cocaine administration (5 mg/kg) in rats was also shown to stimulate induction and release of pro-inflammatory cytokines (such as TNFα and IL-1β) in reward regions of the brain 101 that would further aggravate regional BBB damage.43,102 Brain microglial cells that secrete a plethora of cytokines, chemokines, and other neurotoxic factors upon activation are also shown to be critical cellular targets for cocaine-induced neuroinflammation and ensuing BBB disruption.99,103
Thus, it is plausible to hypothesize that cocaine can initiate a number of molecular events leading to BBB endothelial inflammation and oxidative injury as characterized by activated adhesion signals and disruption of TJ complexes. This ultimately leads to the local recruitment and infiltration of circulating immune cells, which further aggravate the BBB damage.
Smoking and cerebrovascular complications: Does nicotine have a role?
Tobacco use is a major public health hazard worldwide and continues to be a leading cause of premature mortality and morbidity rates with a substantial toll on health care costs (>$289 billion/year in USA) and personal life (CDC, 2014). Cigarette smoking (including smokeless tobacco) is an independent and preventable risk factor for a spectrum of vascular morbidities at various target sites because of its proatherogenic and proinflammatory effects. Main and side stream tobacco smoke (TS) were shown to elicit microvascular endothelial dysfunction through activation of oxidative/inflammatory and immune responses.104–106 In this line, accumulating evidence revealed an adverse impact of TS on cerebrovascular system leading to a substantial increase of the risk for ischemic stroke, silent cerebral infarctions, vascular dementia, and incidence of other major CNS disorders (including Alzheimer’s disease).107–110
Effect of TS exposure and nicotine on BBB endothelial physiology and function. Schematic illustration describing oxidative stress and inflammatory pathways involved in TS-derived nicotine and free radicals-induced BBB endothelial dysfunction. Up-regulation of pro-inflammatory cytokines and MMPs including endothelial cell adhesion molecules triggers BBB endothelial activation resulting in loss of BBB integrity and facilitate leukocytes infiltration.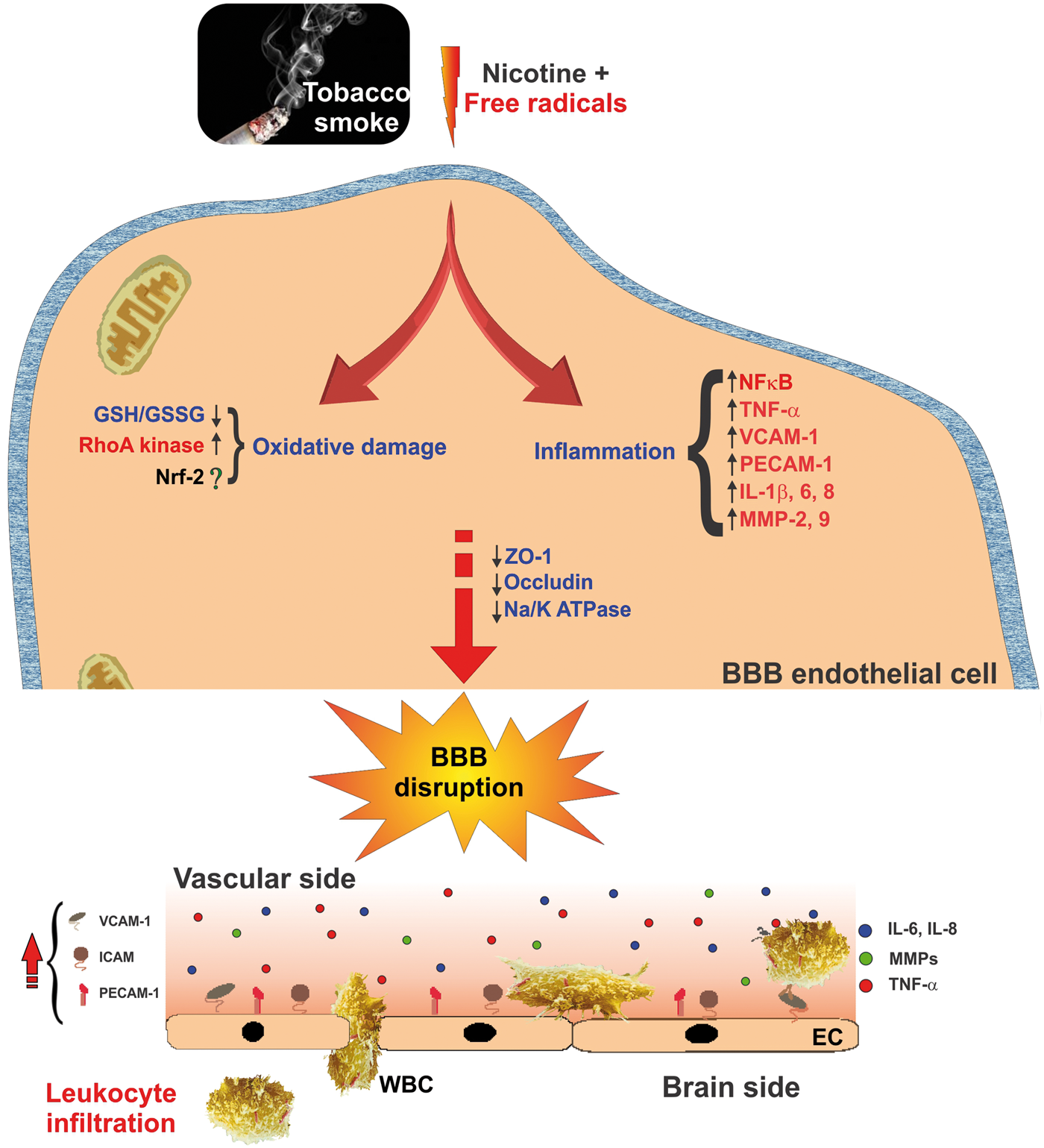
Striking lines of evidence have established a significant link between smoking and pathophysiology of BBB endothelial dysfunction.111–113 As shown in Figure 4, a acute or chronic TS exposure significantly elevates endothelial oxidative and nitrosative stress responses resulting in a progressive loss of BBB integrity. 21 For example, using static or flow-based in vitro BBB models, we previously demonstrated a marked increase in intracellular ROS/RNS and secretory profile of various pro-inflammatory markers accompanied by alterations in BBB TJ protein expression and re-distribution following TS exposure.43,114 In addition, DNA microarray-based global transcriptome profiling indicated a rapid and transient up-regulation of Nrf2 and its downstream anti-oxidant molecular networks in response to prolonged TS exposure in brain microvascular endothelial cultures. 115 Interestingly, anti-oxidant supplementation toned down TS-induced oxidative stress thus preventing BBB damage. 114
An interesting argument posed by multiple studies is the extent to which nicotine contributes to the pathogenesis of smoking-related cerebrovascular complications and BBB dysfunction. This is not an easy conundrum to solve given the many discrepancies in the published literature (Hawkins et al., 2002). First, nicotine is known to directly affect the BBB endothelial physiology116–119 and earlier studies suggest that chronic, but not acute, nicotine exposure decreased ATP dependent ion transporters in isolated brain microvessels. 120 In addition, nicotine alone (≥plasma concentrations in chronic smokers) in vitro or in vivo induced a differential down-regulation/redistribution of TJ proteins and BBB disruption. The underlying mechanism may involve oxidative stress dependent activation of RhoA and cytoskeleton reorganization.116–118,121 The pro-oxidant effects of nicotine were further signified by increased BBB endothelial activation and leukocyte-endothelial interactions. 122 Notably, these BBB endothelial responses were blocked by nicotinic receptor antagonists.116,123
Moreover, a multitude of studies assessing the independent effects of nicotine in experimental stroke models strongly indicate that nicotine plays a significant role in TS-enhanced risk for stroke and worsened neurological outcome in post-ischemic injuries. For instance, chronic nicotine exposure aggravated TJ disruption and ionic imbalance within the BBB microenvironment following ischemic hypoxia116,124,125 and intensified stroke-associated brain edema and neuronal injury.124,126,127 Strikingly, chronic nicotine administration in mice subjected to ischemic stroke exacerbated immune cells infiltration in ischemic regions, leading to aggravated post-ischemic neuroinflammation. 127
Conversely, BBB function remained intact following acute nicotine exposure. 128 Adding to the bimodal effects of nicotine, Uzum et al. 129 have demonstrated that chronic low dose injections of nicotine prevent the BBB disrupting effects of a single high-dose injection. It is plausible that repeated low doses might desensitize the nicotinic receptors at the BBB endothelium and thus produce a diminished response to acute challenges (Rahman et al., 2008). On the other side, chronic nicotine infusion significantly up-regulated BBB endothelial nutrient transporters (such as GLUT1) ensued by increased local utilization and metabolism of cellular energy substrates.130,131 Taken together, the effects of nicotine and its contribution to TS-induced BBB impairment directly correlate with the duration and concentration of nicotine exposure. It would be equally interesting to examine how nicotine affects the BBB endothelium in the presence of other smoke constituents.
Methamphetamine, cocaine, and HIV encephalitis: From the BBB perspective
Human immunodeficiency virus encephalitis (AIDS dementia) is a significant neurological complication in advanced HIV-infected patients manifested by severe cognitive and motor deficits accompanying profound neuronal damage.132,133 Increased risk and prevalence of HIV infection in chronic drug abusers globally suggests that this clinical subset is more susceptible to the incidence of AIDS dementia and neurological syndrome. 99 Supporting these epidemiological studies, existing clinical and preclinical data have revealed that concomitant or previous history of psychostimulant abuse accelerates CNS invasion by HIV and the rapid development of neuro-AIDS with severe neuropathological manifestations.96,134,135 Substantial evidence suggests that METH or cocaine potentially interacts with viral encoded proteins at the BBB interface and primes various pro-inflammatory responses to HIV (or vice-versa), resulting in additive or synergistic BBB disruption.91,134,135 For example, severe alterations in BBB TJ composition are rapidly initiated by the combined exposure to HIV-1 encoded proteins (TAT and gp-120) and METH or cocaine, resulting in a synergistic loss of barrier resistance.65,94 Importantly, METH exposure exacerbated gp-120 or TAT-induced endothelial oxidative stress responses in vitro and in vivo, leading to potential down-regulation, dissociation, and/or oxidative modification of TJ proteins and BBB disruption. 66 In this context, it is also speculated that psychostimulants may also worsen TAT-induced BBB disruption by exacerbating the functional loss of pericytes (perivascular cells that critically maintain the BBB integrity) possibly through activation of NF-κB and PDGF-BB signaling axes. 136 In addition, METH and cocaine significantly increased endothelial-leukocyte interactions80,96 and transmigration of HIV-infected monocytes from circulation across the BBB; a secondary portal of entry for HIV neuroinvasion.
Recent studies also highlight the neuroinflammatory role of astrocytes and microglia in psychostimulant-induced onset and progression of HIV-neuropathogenesis. For example, concomitant exposure to METH and TAT led to a significant (additive) down-regulation of the neuroprotective Wnt/β-catenin signaling in astroglial cells, albeit through different mechanisms, thus possibly altering the BBB homeostatic function of astrocytes. 137 Importantly, METH and gp120 synergistically interact at astrocytes resulting in potential induction of pro-inflammatory genes such as IL-6 through NF-κB signaling mechanisms. 138 Acute or repeated cocaine (10 µM or 20 mg/kg) modulates the immune functions of microglial cells and elicits a pro-inflammatory phenotype resulting in increased expression of cytokines and chemokines (e.g. monocyte chemotactic protein-1) that facilitate HIV-infected monocyte extravasation across rodent BBB.101,103 Moreover, cocaine-induced inflammatory activation of microglial cells directly correlated with increased activity of NADPH oxidase and NF-κB, likely through engagement of σ1 receptor. 103 Substantial evidence also suggests that cocaine exposure potentiates HIV envelope protein (gp120) induced oxidative stress and mitochondrial dysfunction in rat primary astrocytes by increased inflammatory gene transcription through NF-κB signaling. 139 The clinical relevance of these findings was further demonstrated by an exacerbated reactive astrogliosis in cerebral cortex of HIV-infected patients with cocaine abuse history. 97 Nonetheless, the implication of psychostimulant-induced astroglial responses in BBB dysfunction is a rising horizon within the context of HIV-related neurological syndrome in drug abusers.
Thus, an impaired neurovascular barrier inflicted by psychostimulants, alone or mutually in the presence of HIV, could be a principal mechanism underlying augmented viral access to the brain parenchyma and neuropathogenesis of AIDS dementia in METH and cocaine abusers. On an additional note, there is an increasing prevalence of drug abuse in patients with mild or moderate traumatic brain injury, but their comorbid effects on neurovascular functions remain largely unknown. Neuroimaging studies demonstrated a reduced pericontusional cerebral blood flow following METH administration in patients prior to sustaining TBI, 140 while cigarette smoking has been associated with a significant risk factor for hemorrhage and microvascular bleeding after TBI. 141 It was previously shown that METH-induced neurotoxicity and conditions such as traumatic brain injury (TBI) share common cellular and molecular neuropathological substrates. 142 In fact, compelling evidence from clinical and preclinical studies suggest that BBB dysfunction is an important neurovascular complication following TBI that involves the activation of oxidative and inflammatory stress signaling cascades.143–145 Thus, it can be envisioned that chronic psychostimulant abuse would aggravate the BBB disruption and secondary brain damage by TBI resulting in a worsened neurological outcome in this population.
Conclusions and future perspectives
This review summarized and evaluated recent data from genetic, physiological, cellular, and morphological studies addressing the signaling mechanisms involved in drug abuse-related BBB dysregulation. Collectively, these studies suggest that increased endothelial oxidative stress has a focal role in psychostimulant or TS-induced BBB dysfunction. Activation of oxidative/inflammatory stress signaling cascades by drugs of abuse is the primary mechanism for increased endothelial activation and leukocyte migration across the BBB into the brain. Recent studies from our group (Sajja et al., 46 Naik et al., 115 and Prasad et al. 146 ) and other groups (Alfieri et al. 147 and Sandberg et al. 148 ) have highlighted the critical neuroprotective role of Nrf2 that includes regulation and maintenance of BBB TJ integrity and function. However, how psychostimulants may impact Nrf2-dependent redox hemostasis at the BBB endothelial level is unknown, thus providing an excellent opportunity for future studies to address this critical gap in our knowledge and promote the basis for development of novel antioxidant therapeutics.
Given the fact that BBB dysfunction is aggravated during spontaneous “drug withdrawal,” it would be an interesting argument if restoration of BBB integrity could reverse the withdrawal symptoms during abstinence. Although this scientific question has not been explored, we predict that blockade of neuroinflammatory cascades within the NVU could partially alleviate the negative reinforcement, given the evidence that cocaine and METH evokes an increased expression of IL-1β and TNFα in reward enriching brain regions,68,101 while ROS scavengers attenuated cocaine-seeking behaviors in rodents. 149 In fact, astrocytic GFAP expression was increased during the withdrawal phase following a repeated dosing paradigm of cocaine in rats suggesting the role of astrocytic inflammatory responses in drug-induced synaptic plasticity. Thus, these findings demonstrate that alterations in immune response and activation of inflammatory cascades by drugs of abuse in the NVU could play a critical role in psychostimulant addiction. 150 However, further investigations are required to critically evaluate the causal link between BBB dysfunction and “drug withdrawal” behaviors. It would also be interesting to examine whether BBB function could be used to index neurobehavioral deficits and recovery during drug withdrawal.
One major challenge among drug abusers is the frequent use of more than one drug (poly-drug abuse). It is apparent that cigarette smoking is highly prevalent in psychostimulant abusers. Therefore, additional investigations into the cerebrovascular impairments caused by concomitant exposure of more than one drug might possibly delineate shared molecular pathways. Additionally, the current knowledge on effects of METH or cocaine abuse on the BBB integrity is mainly limited to acute toxicity or binge-like administration model and thus the long-term effects of these psychostimulants remain to be further elucidated for clinical translation. Overall, a detailed mechanistic insight into BBB dysfunction associated with drug abuse would reveal important molecular targets for development of rational and effective therapeutic interventions.
Currently, various tobacco products are being commercially marketed such as “reduced exposure,” “light,” or “nicotine-free” products including electronic cigarettes with different levels of nicotine, nitrosamines, and other toxic chemicals, claiming a reduced health hazard compared with regular brands. However, recent studies from our laboratory and others assessing the differential BBB toxicity of various cigarette products have challenged the claimed safety of these reduced or low-exposure cigarette brands. Interestingly, our data revealed a potential oxidative/inflammatory stress response at the BBB endothelium (affecting the Nrf2 antioxidant response system 115 ) by low-nicotine and high tar-containing cigarettes higher than or comparable to conventional cigarettes. 43 Therefore, rigorous analysis of the safety (or toxicity) profiles of various tobacco products on cerebrovascular function in experimental and clinical studies is critically needed for the regulatory control of these new smoking alternatives.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Manuscript preparation was supported, in part, by NIH/NIDA R01-DA029121-01A1, and Alternative Research Development Foundation grants received by LC.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.