
Abstract
Methamphetamine (METH), a potent stimulant with strong euphoric properties, has a high abuse liability and long-lasting neurotoxic effects. Recent studies in animal models have indicated that METH can induce impairment of the blood–brain barrier (BBB), thus suggesting that some of the neurotoxic effects resulting from METH abuse could be the outcome of barrier disruption. In this study, we provide evidence that METH alters BBB function through direct effects on endothelial cells and explore possible underlying mechanisms leading to endothelial injury. We report that METH increases BBB permeability in vivo, and exposure of primary human brain microvascular endothelial cells (BMVEC) to METH diminishes the tightness of BMVEC monolayers in a dose- and time-dependent manner by decreasing the expression of cell membrane-associated tight junction (TJ) proteins. These changes were accompanied by the enhanced production of reactive oxygen species, increased monocyte migration across METH-treated endothelial monolayers, and activation of myosin light chain kinase (MLCK) in BMVEC. Antioxidant treatment attenuated or completely reversed all tested aspects of METH-induced BBB dysfunction. Our data suggest that BBB injury is caused by METH-mediated oxidative stress, which activates MLCK and negatively affects the TJ complex. These observations provide a basis for antioxidant protection against brain endothelial injury caused by METH exposure.
Introduction
Methamphetamine (METH) is a synthetic drug that is easily abused because of its powerful psychostimulant and addictive properties. Abuse of METH is a growing problem in the United States; epidemiologic studies have indicated that ∼4.9% of Americans have tried METH at least once in their life (Tata and Yamamoto, 2007). In 2004, statistics from the DAWN (Drug Abuse Warning Network) showed that METH-related emergency department admissions accounted for 8% of all admissions or more than 150,000 cases that year, underscoring the fact that short- and long-term abuse of METH leads to deleterious health effects. In the brain, METH toxicity has been characterized by the disruption of monoamine production and synaptic integrity of the dopaminergic system. Furthermore, METH neurotoxicity also generates a strong and lasting glial response, resulting in astrogliosis and release of inflammatory mediators. Recently, the brain endothelium has also been shown to be a target of METH toxicity. Methamphetamine exposure in vivo has been shown to disrupt the function of the blood–brain barrier (BBB) (Sharma and Kiyatkin, 2009).
Methamphetamine-induced pathophysiological changes (hyperthermia, hyponatremia, and hypertension) can result in increased BBB permeability (Persidsky et al, 2006b). Quinton and Yamamoto (2006) showed that administration of the METH derivative, 3,4-methylenedioxymethamphetamine (MDMA; ecstasy), produced long-lasting disruptions of BBB permeability in the striatum and dorsal hippocampus. Similarly, Bowyer et al (2008) showed BBB-increased permeability in the limbic region (the medial and ventral amygdala and the hippocampus). The underlying mechanism of METH-induced BBB impairment remains poorly understood.
Human immunodeficiency virus-1 (HIV-1) seroincidence associated with METH use is 5.8 times higher than among nonabusers (Mansergh et al, 2006). Clinical studies indicate that METH dependence has an additive effect on cognitive deficits associated with HIV-1 infection (Chana et al, 2006). The causes of HIV-1-associated neurotoxicity include excitotoxic effects of glutamate, secretory products of chronically activated glial cells, and oxidative stress (Ellis et al, 2007), which are very similar to the factors mediating METH-induced neuronal injury (O’Dell and Marshall, 2005; Quinton and Yamamoto, 2006; Sekine et al, 2008; Stephans and Yamamoto, 1994). Blood–brain barrier dysfunction is a common feature of HIV-1 neurodegeneration (Persidsky et al, 2006b). Taken together, METH abuse and HIV-1 central nervous system (CNS) infection could lead to combined injury, resulting in enhanced neural compromise and cognitive dysfunction.
Multiple studies indicate that METH-induced neurotoxicity involves the production of both reactive oxygen species (ROS) and nitrogen species (Flora et al, 2003). The role of oxidative stress is further supported by the finding that ROS scavengers and antioxidants attenuate the neurotoxic effects of METH (Fukami et al, 2004). Considering that oxidative stress has a significant role in METH-mediated neurotoxicity, we hypothesized that ROS generation mediated by METH also occurs in the brain endothelium, resulting in BBB compromise by the activation of myosin light chain kinase (MLCK) which increases BBB permeability through modifications of tight junctions (TJs) and cytoskeleton (Haorah et al, 2005). In addition, we posed the question whether alterations in the barrier by METH promotes monocyte transendothelial migration, to support the notion that METH BBB disruption is a contributing factor to HIV-1 neuropathogenesis. Using primary human brain microvascular endothelial cells (BMVEC), we showed that METH exposure led to ROS generation, decreased expression of TJ proteins, increased BBB permeability, and enhanced monocyte migration across BMVEC monolayers. Antioxidant administration prevented METH-induced effects in the brain endothelium, suggesting both an underlying mechanism and a potential treatment approach to drug-induced BBB dysfunction.
Materials and methods
Animals and Drug Administration
Four-week-old male NOD/C.B-17 SCID mice purchased from Jackson Laboratory (Bar Harbor, ME, USA) were maintained in sterile microisolator cages under pathogen-free conditions in accordance with the institutional ethical guidelines for care of laboratory animals, NIH (National Institutes of Health) guidelines, and the Institutional Animal Care Use Committee. Animals were weight-matched and randomly assigned to various treatment groups. A common pattern in most recreational METH abusers is initial use of lower doses progressively increasing to higher doses and eventually engaging in multiple daily administrations. Therefore, to simulate this pattern of METH exposure, we adopted a safe escalating dosing regimen (Schmidt et al, 1985) that did not produce potentially lethal hyperthermia in METH-treated animals, while simultaneously producing similar stimulant effects observed in human METH abusers who binged. The mice received seven subcutaneous injections of 1.5 to 10.0 mg/kg METH or sterile 0.9% sodium chloride. Injections were administered in incremental doses on alternating days. On day 9, once the animals reached a total dose of 10 mg/kg, 4 doses of 10 mg/kg every 2 h were injected. A single dose of Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid; 50 mg/kg, Cayman Chemicals, Ann Arbor, MI, USA) was administered intraperitoneally on alternate days (Diaz et al, 2007).
Cell Culture
Human BMVEC were cultured from the resection path tissue recovered from the removal of epileptogenic cerebral cortex material during surgical treatment of epilepsy. The isolation of brain microvessels and the subsequent expansion of the endothelial culture were performed and provided by Michael Bernas and Dr Marlys Witte (University of Arizona Health Science Center, Tucson, AZ, USA). The transfer and use of BMVEC cultures were approved by the Institutional Review Board at Temple University. The BMVEC were maintained in Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F-12) media containing 10% fetal bovine serum, ECGS (endothelial cell growth supplement, BD Biosciences, Franklin Lakes, NJ, USA), heparin (1 mg/mL, Sigma, St Louis, MO, USA), amphotericin B (2.5 μg/mL, Invitrogen, Carlsbad, CA, USA), penicillin (100 U/mL, Invitrogen), and streptomycin (100 μg/mL, Invitrogen). Brain microvascular endothelial cells of low passages and from different donors were used in repeated experiments. Confluent monolayers of BMVEC were replaced by media containing the above formulation but lacking ECGS and heparin. To confirm the presence of barrier formation, transendothelial electrical resistance (TEER) was measured and the presence of typical brain endothelial markers was verified. For certain studies (as indicated in the figures), a telomerase-immortalized human brain endothelial cell line was used. This hCMEC/D3 cell line shows similar properties to those seen in primary BMVEC and are routinely used in in vitro modeling of the BBB (Weksler et al, 2005) Experiments were conducted in both primary cells and on the cell line hcMEC/D3; all results shown are obtained from primary endothelial cells except for those in Figure 2, which shows results from hCMEC/D3. Preliminary experiments showed that METH at concentrations 5 μmol/L to 1 mmol/L did not affect the viability of brain endothelial cells after 48 h of exposure (data not shown).
Primary human monocytes isolated by countercurrent centrifugal elutriation were obtained from the Human Immunology Core at the University of Pennsylvania (Philadelphia, PA, USA). The cells were maintained in DMEM containing heat-inactivated 10% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 U/mL), and
Evaluation of Blood–Brain Barrier Permeability
Changes in BBB permeability were assessed using the fluorescent tracer, sodium-fluorescein (Na-F); the procedure performed was a modification of previously described methods (Lenzser et al, 2005; Phares et al, 2007). Briefly, animals were injected intraperitoneally with 100 μL of 2% Na-F in phosphate-buffered saline. The tracer was allowed to circulate for 30 mins. The mice were anesthetized and then transcardially perfused with phosphate-buffered saline until colorless perfusion was visualized. The animals were then decapitated and the brains quickly isolated. After removal of the meninges, cerebellum, and brain stem, the tissue was weighed and homogenized in 10 times the volume of 50% trichloroacetic acid. The homogenate was centrifuged for 10 mins at 13,000 × g, and the supernatant was neutralized with 5 mol/L NaOH (1:0.8). Measurement of Na-F fluorescence was determined at excitation/emission wavelengths of 440/525 nm using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Fluorescent dye content was calculated using external standards with a range of 10 to 200 ng/mL, and the data are expressed as amount of tracer per gram of tissue.
Measurement of Transendothelial Electrical Resistance
To determine the integrity of brain endothelial monolayers, TEER measurements were performed using the 1600R ECIS system (Applied Biophysics, Troy, NY, USA). The ECIS system provides real-time monitoring of changes in TEER. In brief, BMVEC at 1 × 105 per well were plated on collagen type I-coated 8W10E electrode arrays (Applied Biophysics). The cells were then allowed to form monolayers reaching stable TEER values. After 7 days (with media change every 3 days), the monolayers were exposed to various concentrations of METH as indicated. The readings were acquired continuously for 12 h at 400 Hz and at 10-min intervals. Confluent BMVEC monolayers showed baseline TEER readings between 1,500 and 2,400 Ω/cm2. The data are shown as percentage change of baseline TEER along with the s.e.m. of condition replicates.
Immunofluorescence Staining
Assessment of TJ protein distribution and expression was performed by indirect immunofluorescence. Using standard immunohistochemistry methods, monolayers of hCMEC/D3 on type I collagen-coated coverslips (BD Biosciences) were fixed with 3% (v/v) formaldehyde (Polysciences, Warrington, PA, USA) and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline. Primary antibodies and dilutions that were used included polyclonal antibodies to occludin (1:100, Invitrogen) and monoclonal antibodies to zonula occludens-1 (ZO-1) (1:100, Invitrogen). The cells and primary antibodies were incubated overnight at 4°C. For occludin staining, the cells were preextracted for 2 mins on ice with ice-cold 0.2% Triton X-100 in phosphate-buffered saline before fixation. The secondary antibodies used included Alexa-fluor 488-conjugated donkey anti-rabbit antibodies (1:400, Invitrogen) and Alexa-fluor 594-conjugated donkey anti-mouse antibodies. After removal and rinsing of the primary antibodies, the secondary antibodies were incubated with the cells at room temperature for 1 h. Cells were then washed and mounted onto slides with ProLong Antifade plus DAPI (4’,6-diamidino-2-phenylindole) (Invitrogen). Immunofluorescence was visualized using a Nikon Eclipse 80i fluorescent microscope (Nikon, Tokyo, Japan).
Image Analysis of Tight Junction Protein Alteration
Tight junction abnormalities were evaluated by dual labeling of ZO-1 and occludin followed by measurement of pixel intensity across the TJ. Images were captured at × 20 magnification from monolayers with or without METH exposure with a camera setting kept uniform (i.e., exposure time) throughout all experimental sets. Line histograms, to identify values of intensity/pixel, were placed perpendicular to the TJ and the intensity was measured.
As endothelial cells have various contact points with adjacent cells, only one contact point per cell selected at random was used in the analysis. TJ intensity profiles from each condition were grouped into the following categories: intensity profiles that exemplified sharp TJ or no abnormality (a single peak), gaps (multiple peaks), discontinuity (low or absent peaks), or both gaps and discontinuity. Visualization of immunofluorescence was performed using an Eclipse 80i microscope (Nikon), fitted with a CoolSnap-EZ digital camera (Photometrics, Tucson, AZ, USA). Image acquisition/analysis was performed using NIS Elements R (Nikon) imaging software. The data represent the analysis of at least five images from each replicate condition (w/o METH) and from three independent experiments.
Protein Preparation and Immunoblotting
Confluent monolayers of BMVEC were prepared for whole cell lysate or for membrane and cytosolic fractions. Cells were lysed with CelLytic-M (Sigma) for preparation of whole cell lysates. Cellular fractions were collected using the ProteoExtract kit (Calbiochem, La Jolla, CA, USA) as outlined by the manufacturer's protocol and described by Stamatovic et al (2006). The protein concentrations in whole lysates or fractions were estimated using the BCA method (Thermo Scientific, Rockford, IL, USA). Cellular fractions or whole cell lysates were loaded at 50 μg per lane and resolved by SDS-PAGE on 4% to 20% precast gradient gels (Thermo Scientific). Proteins were transferred into nitrocellulose membranes and incubated overnight with polyclonal antibodies against occludin (1:500, Invitrogen), claudin-5 (1:500, Invitrogen), myosin light chain, (MLC 2) (1:100, Cell Signaling Technologies, Danvers, MA, USA) and phospho-MLC 2 (S19, 1:100, Cell Signaling Technologies), and β-tubulin (1:1,000, Abcam, Cambridge, MA, USA). Secondary antibodies conjugated to HRP (horseradish peroxidase) were then added for 1 h and subsequently detected using Supersignal West Pico chemiluminescence substrate (Thermo Scientific). Chemiluminescence signal detection was performed using the gel documentation system G:Box Chemi HR16 (Syngene, Frederick, MD, USA). Densitometry analysis was carried out using GeneTools software package (Syngene).
Reactive Oxygen Species Measurement
Detection of ROS generation was measured in BMVEC monolayers with the fluorescence probe, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Invitrogen). After 24 h treatment of the experimental conditions (as shown in Figure 4), the cells were washed thrice with serum-free media and then loaded with 10 μmol/L of CM-H2DCFDA in the serum-free media for 45 mins at 37°C. Once inside the cell, the cell-permeable H2DCFDA becomes fluorescent after the removal of the acetate groups and oxidation of the indicator. After loading of the indicator, the cells were washed once with Hank's buffered salt solution (HBSS), and fluorescence was measured at excitation/emission wavelengths of 495/525 nm using kinetic mode settings of 5-min intervals on a SpectraMax M5 microplate reader (Molecular Devices). When present, the concentration of the antioxidant Trolox was 25 μmol/L, consistent with previous reports by other investigators (Martin et al, 2005; Yatin et al, 2002). The data for the 30-min time point are shown in the Figure 4 and expressed as relative fluorescence.
Migration Assay
To quantitatively measure the transendothelial migration of monocytes in vitro, a fluorescence-based assay was used as previously described (Ramirez et al, 2008). In brief, BMVEC at 2.5 × 104 cells per insert were plated on type I collagen-coated FluoroBlok tissue culture inserts (with 3-μm pores, BD Biosciences). As the cells on these inserts cannot be visualized by microscopy, TEER measurements taken using a voltmeter (EVOM, World Precision Instruments, Sarasota, FL, USA) confirmed that the monolayers showed typical barrier formation (Persidsky et al, 2006a). The endothelial cells were then treated (as indicated in Figure 5) for 24 h. Before initiation of the migration assay, all treatments were ceased and media replaced lacking the experimental compounds. Monocytes were labeled with the cell tracker, Calcein-AM (Invitrogen), according to the manufacturer's instructions and then introduced into the endothelial monolayers at 1 × 105 cells per insert. Monocytes were transferred in the absence or presence of recombinant human monocyte chemotactive protein-1 (CCL2/MCP-1, 30 ng/mL, R&D Systems, Minneapolis, MN, USA) placed into the lower chamber of the insert system. The cells were incubated under normal tissue culture conditions, and the migration of fluorescently labeled monocytes was measured at 2 h. Measurement of relative fluorescence was acquired from the lower chamber using the M5 fluorescence plate reader. Calculations for the actual number of migrated monocytes were derived from external standards of labeled monocytes. The data are shown as the fold difference of migrated cells compared with the background migration of cells in inserts without chemoattractant or experimental treatments.
Data Analysis
The results were collected from at least three independent experiments. Within an individual experiment, each data point was determined from three to five replicates. Apart from the densitometry analysis that is expressed as the mean±s.d., all other values are expressed as the mean±s.e.m. Statistical analysis of the data was carried out using Graphpad Prism v5 (Graphpad Software, Sorrento Valley, CA, USA). Comparisons between samples were made by unpaired Student's t-test; multiple group comparisons were made by one-way ANOVA (analysis of variance) with Dunnett's post hoc tests. Differences were considered significant at P-values ≤0.05.
Results
Increase in BBB Permeability and BBB Integrity Disruption by Methamphetamine
Although neuronal injury and glial activation has been well documented in METH-associated CNS injury, few studies have focused on the effect of METH on the cerebral microvasculature (Sekine et al, 2008). Recent reports have suggested that an increase in BBB permeability occurs in animals exposed to high doses of METH. Bowyer et al (2008) found accumulation of IgG (immunoglobulin G) around microvessels, indicating significant BBB disruption. To evaluate barrier dysfunction in a quantitative manner, we treated mice with escalating doses of METH (1.5 to 10.0 mg/kg METH on days 1 to 7) or with sterile 0.9% sodium chloride. Thirty minutes before killing, the animals were injected with the small molecular tracer Na-F(376 Da) and the Na-F tissue content was measured. Methamphetamine exposure led to a seven-fold increase in BBB permeability as compared with vehicle controls (Figure 1A). Next, we evaluated whether METH directly affects BBB integrity. Primary human BMVEC were cultured on ECIS array electrodes, and TEER was monitored continuously over the course of 12 h. Formation of confluent monolayers achieved typical basal-level TEER readings of at least 1,500 Ω/cm2. After METH application, average TEER measurements showed a 20 to 50% decrease in resistance in a dose-dependent manner (Figure 1B). The lower concentration of METH (50 μmol/L) induced a gradual decrease in TEER that rebounded slightly, whereas the higher METH dose (250 μmol/L) produced a quicker and sustained decrease in TEER values, indicating a partial loss of monolayer integrity. Therefore, both in vitro and in vivo data point to a rapid BBB compromise after drug exposure.
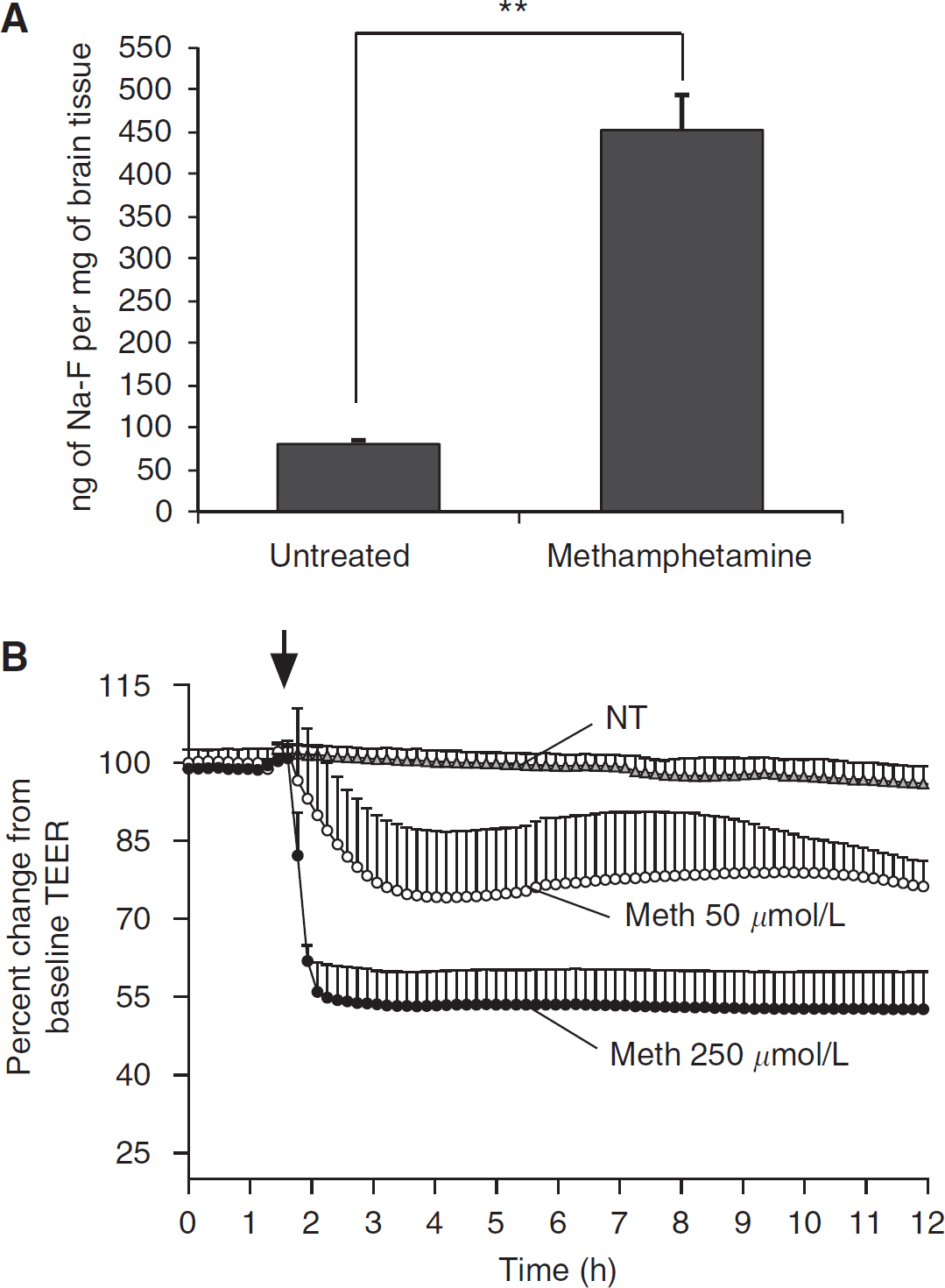
METH induction of BBB leakiness in vivo and in vitro. (
Appearance of Tight Junction Abnormalities in Methamphetamine-Treated Endothelial Cells
Barrier tightness and high electrical resistance are mediated by the level and distribution of the TJ proteins linking brain endothelial cells (Persidsky et al, 2006b). Consequently, the diminished barrier function correlates with the downregulation of TJ proteins or with their redistribution from the membrane to the cytosol (Aghajanian et al, 2008). To evaluate whether the effect of METH on BBB permeability may be explained by morphologic differences in TJ formation, we examined the expression and distribution of occludin, ZO-1, and claudin-5. Indeed, microscopy analysis of immunofluorescence-labeled occludin showed that BMVEC exposed to METH had increased numbers of altered TJs showing low occludin staining and gap formation (Figure 2A). These changes are in stark contrast to the untreated control cells, where occludin staining showed sharp and continuous TJ. To evaluate TJ abnormalities, semi-quantitative image analyses were carried out using pixel intensity measurements across cellular junctions throughout METH-treated and METH-untreated monolayers (Figure 2B). The results showed progressive TJ alterations over the period of METH exposure (Figure 2B). Staining for claudin-5 showed similar results (data not shown). However, no noticeable distribution or expression changes were evident in the TJ anchoring protein, ZO-1 (data not shown), suggesting the involvement of differential effects of METH on different TJ proteins.
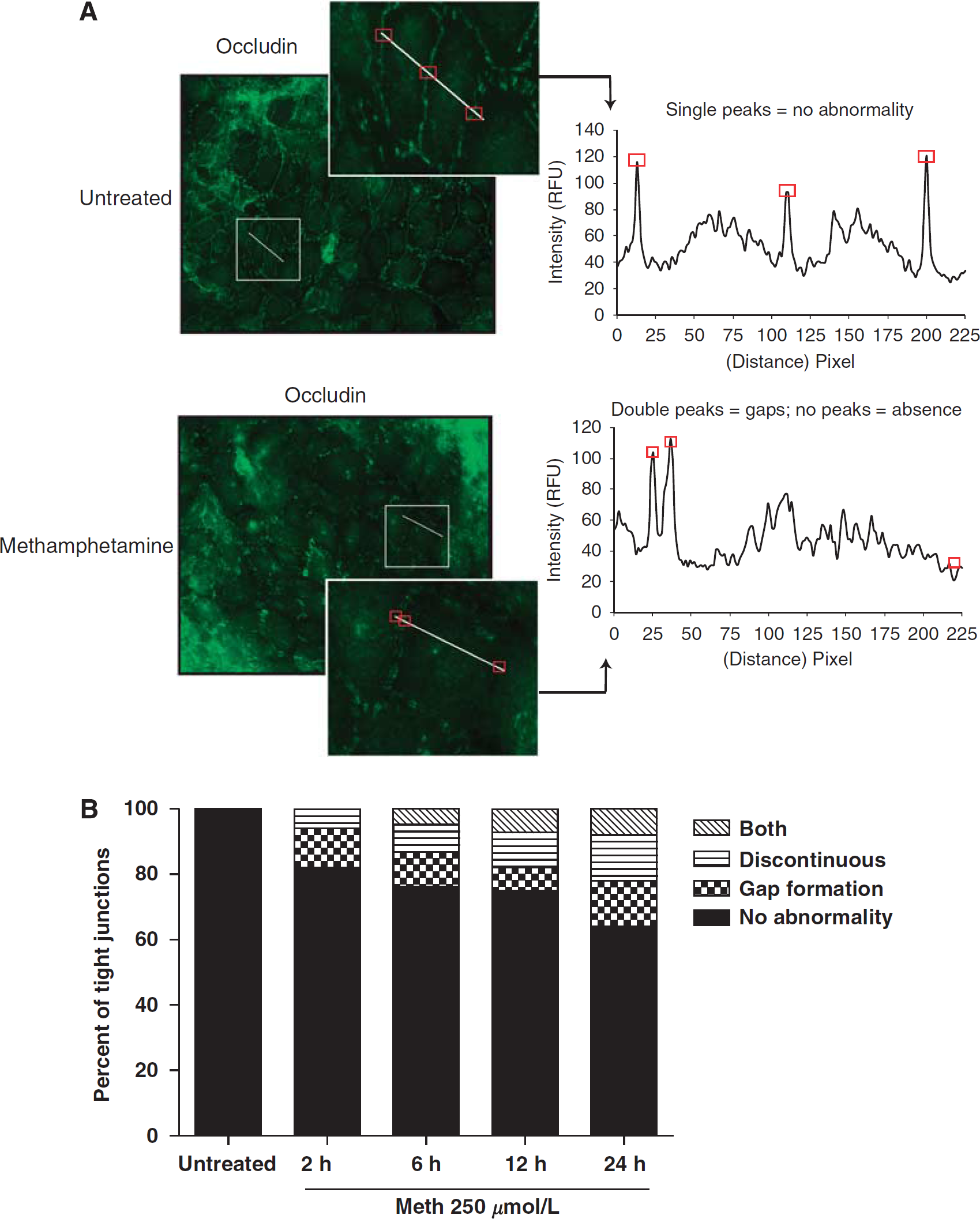
Altered TJ appearance in METH-treated BMVEC. HCMEC/D3 cells were used for the results shown herein. (
Methamphetamine Downregulates the Expression of Tight Junction Proteins
Tight junction changes after METH treatment were further evaluated by western blot. We analyzed levels of occludin and claudin-5 in membranous and cytoplasmic fractions of BMVEC. Figure 3A indicates a dose- and time-dependent decrease in occludin content at the membrane by 25% to 67%. While at the 6-h time point, higher doses (50 and 250 μmol/L) caused more prominent reduction in occludin as compared with 5 μmol/L. Longer exposure with low-dose METH also resulted in further loss of TJ protein at the membrane. Similar results were also found for claudin-5 levels after METH application (Figure 3B). Western blot analyses showed a decrease in claudin-5 expression (20% to 68%) in membranous fractions of BMVEC exposed to different concentrations of METH as compared with untreated controls. In contrast to occludin, claudin-5 did not appear to follow a time-dependent decrease in expression within a given METH concentration; thus, suggesting that claudin-5 is more stable than occludin after METH insult. Interestingly, no redistribution or increased cytosolic content of occludin and claudin-5 was observed, suggesting that it is not redistribution but an inhibition in protein expression that results from METH exposure.
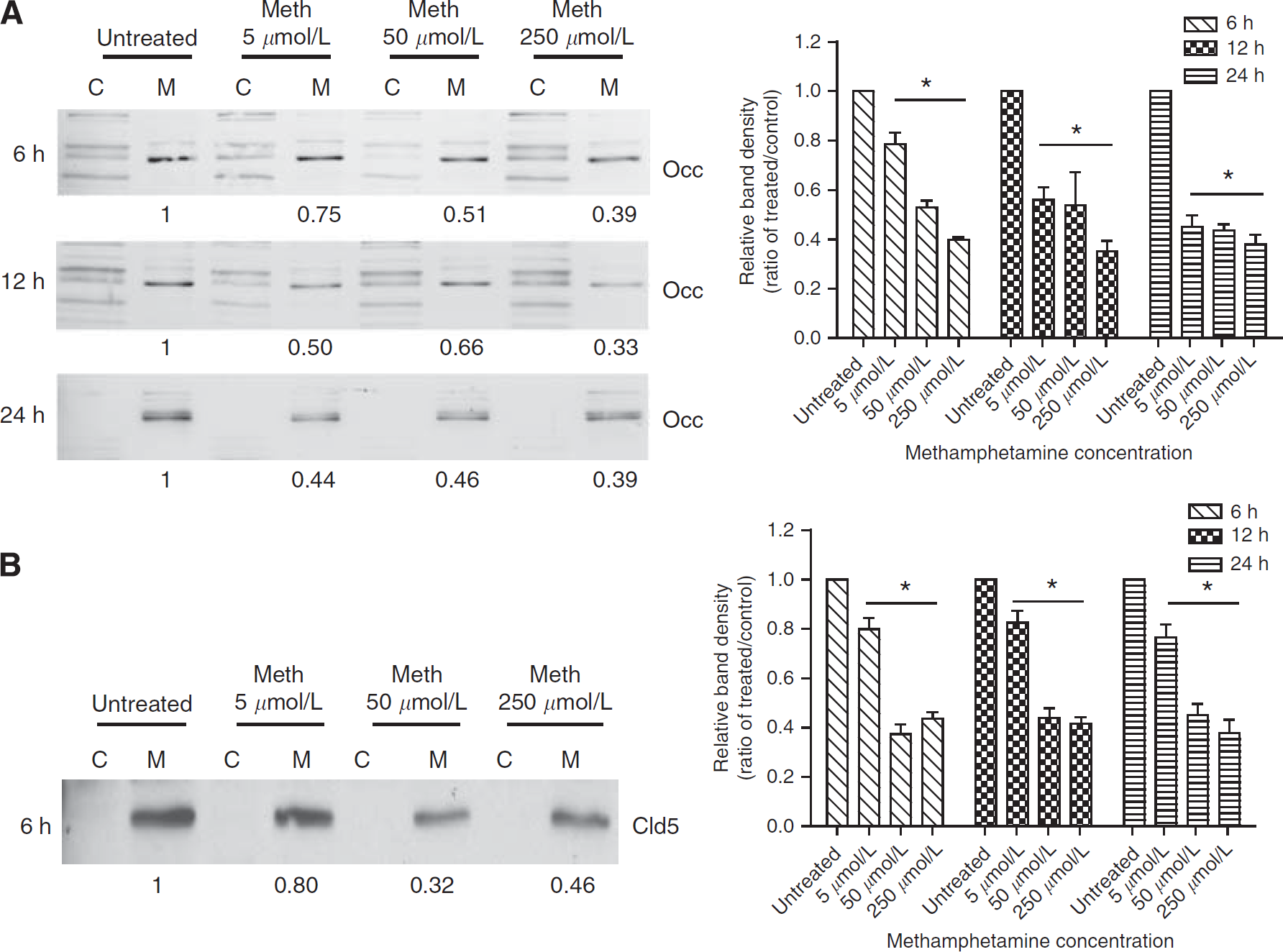
METH downregulates levels of TJ proteins in BMVEC. Representative western blots of occludin (Occ) (
Reactive Oxygen Species Generation in METH-Exposed BMVEC
Considering the fact that oxidative stress has an important role in neurotoxic effects of METH, we sought to determine whether METH could enhance ROS production in BMVEC at nontoxic concentrations. Using the ROS-sensitive dye, DCF, BMVEC were exposed to increasing concentrations of either H2O2 (control), METH or the antioxidant, Trolox. In parallel, the cells were exposed to both increasing concentrations of METH and Trolox (25 μmol/L). The results show the readings acquired at 30 mins after the start of treatments. Methamphetamine generated a dose-responsive increase in the amount of ROS production, shown by the increase in fluorescence from the DCF indicator. However, METH in the presence of Trolox showed significant inhibition of ROS production (P<0.005 when compared with METH-exposed cells, Figure 4A). We next assessed the potential protective effects of the antioxidant, Trolox, on TJ protein expression. As seen earlier, exposure of 50 μmol/L METH for 24 h downregulated occludin. Addition of Trolox together with METH partially restored occludin levels (Figure 4B). Similar to occludin, claudin-5 levels were preserved by Trolox (data not shown). Taken together, these results suggest a potential contribution of oxidative stress to brain endothelial alterations elicited by METH.
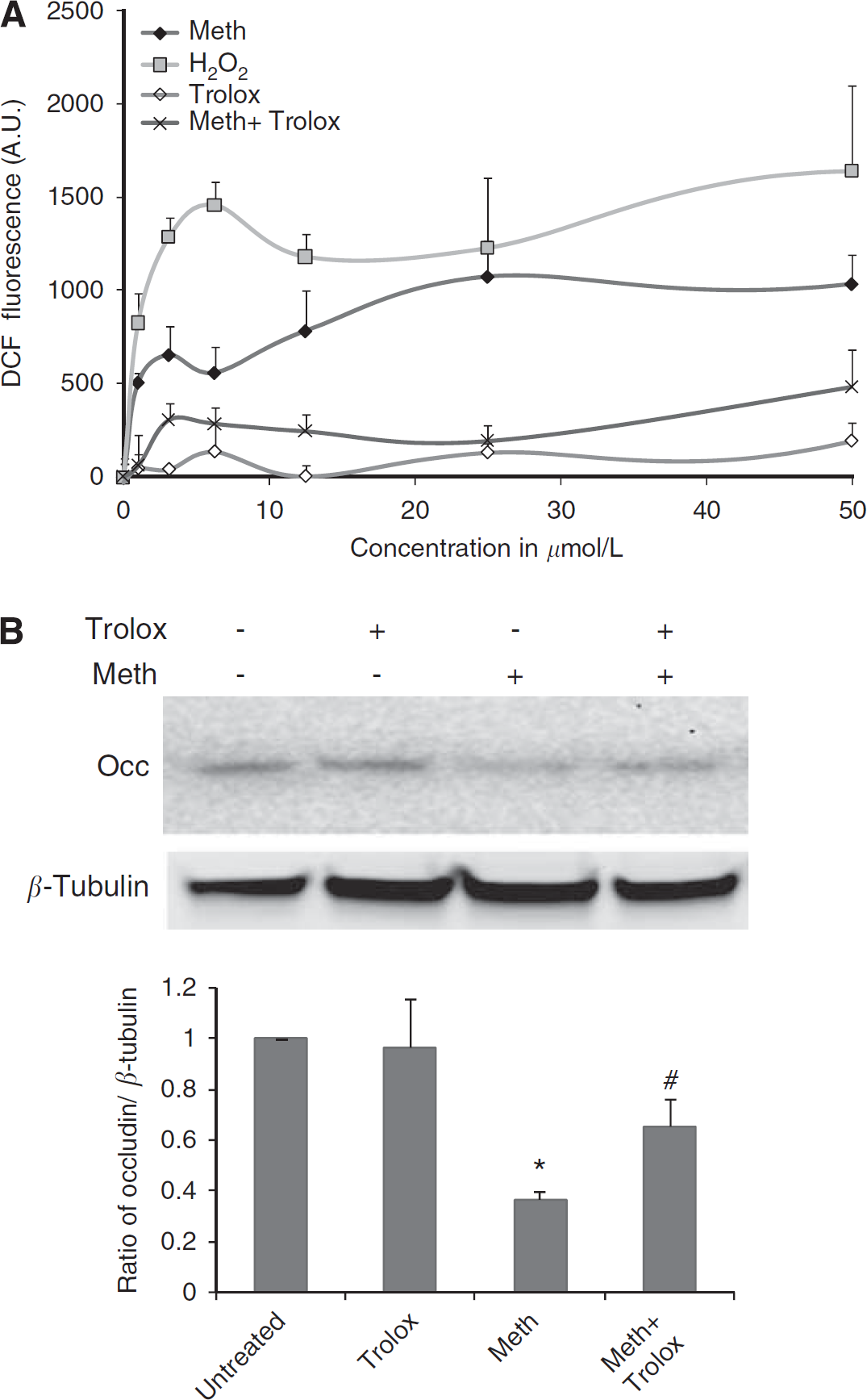
METH generates ROS in the BMVEC. (
Trolox Restores Blood–Brain Barrier Integrity In Vivo and In Vitro
We next studied whether leakiness in the BBB could be prevented in animals exposed to escalating doses of METH. Mice received escalating injections of either METH or sterile 0.9% sodium chloride (reaching a final concentration of 10 mg/kg) over a period of 7 days. Trolox groups received a single dose of antioxidant (50 mg/kg, intraperitoneally) every other day from the start of METH administration. On day 9, when the animals reached a total dose of 10 mg/kg, they were administered the permeability tracer, Na-F. Blood–brain barrier permeability was measured by detection of Na-F in the brain tissue. As before, METH increased BBB permeability approximately sevenfold. In contrast to METH-only-administered animals, mice treated with METH and the antioxidant (Trolox) showed significant reduction in the amount of the fluorescence tracer in the brain tissue (twofold, P<0.005) when compared with METH-treated animals (Figure 5A). It is of note that mice treated with only Trolox (used as controls) showed no change in BBB permeability. In parallel, we measured BMVEC monolayer integrity by TEER. Methamphetamine application led to a significant decrease in TEER (Figure 5B). Addition of antioxidant together with METH rendered complete protection and thereby maintained TEER at control levels (Figure 5B). Therefore, multiple lines of evidence indicate that antioxidants could ameliorate barrier demise after METH exposure.
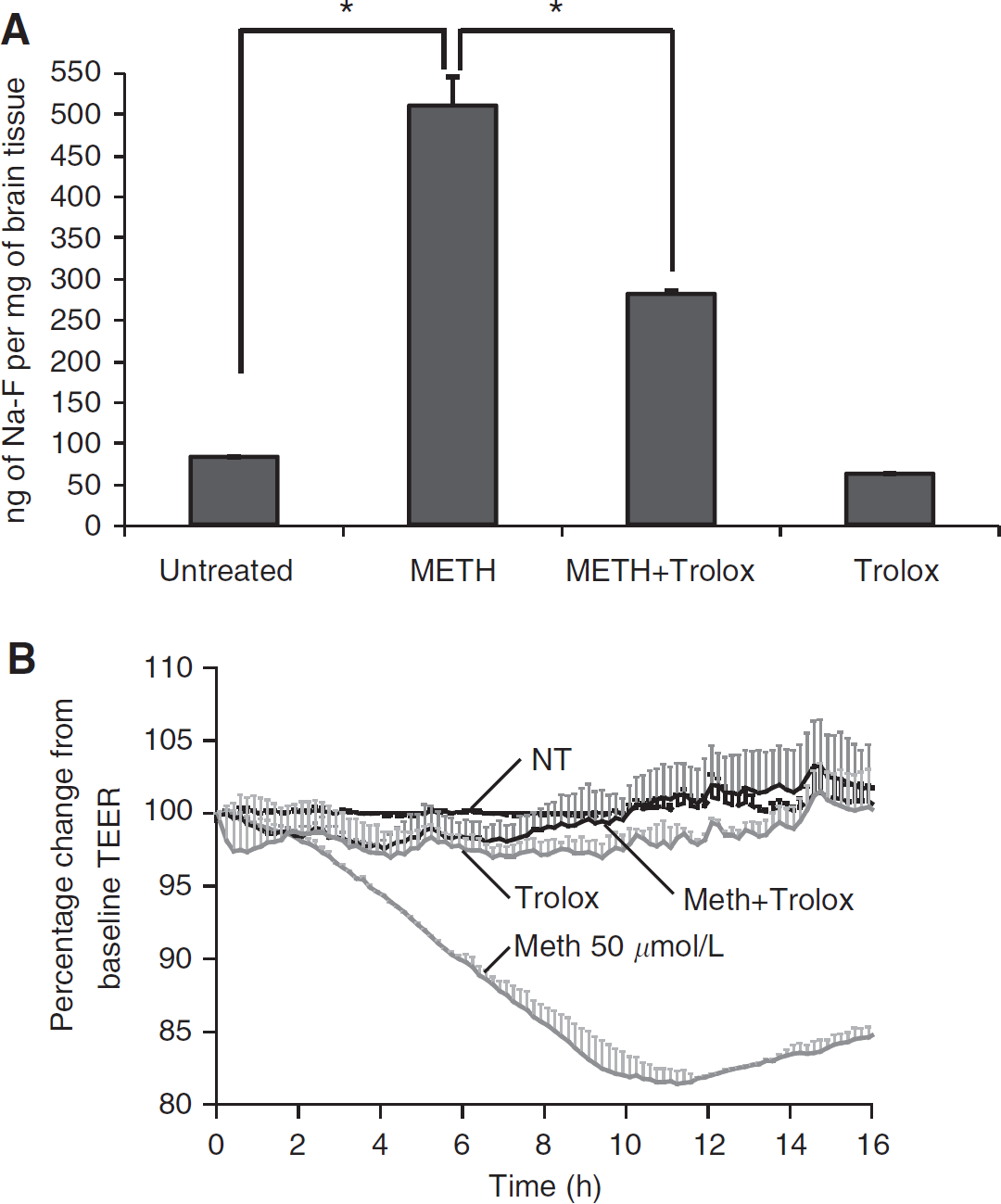
Antioxidant treatment prevented METH-induced barrier dysfunction in vivo and in vitro. Four-week-old male NOD SCID mice received seven subcutaneous injections of METH, sterile 0.9% sodium chloride, Trolox (50 mg/kg administered intraperitoneally every other day) or in combination with METH and Trolox. Na-F administration was used to evaluate BBB permeability. Animals were then perfused to eliminate tracer in the vessels. The data shown in (
Methamphetamine Augments Monocyte Transendothelial Migration
Human immunodeficiency virus-1 infection of the CNS is associated with enhanced migration of monocytes across the BBB. Such cell trafficking is mediated by enhanced production of the β-chemokine, CCL2 (MCP-1) in the brain (Persidsky et al, 1999). Given the high prevalence of HIV-1 infection in METH abusers, we explored the possibility that METH injury of the BBB could increase monocyte transendothelial migration. We investigated monocyte migration after BMVEC treatment with METH (18 h) using a model previously established from our laboratory (Ramirez et al, 2008). Drug treatment resulted in 25% to 50% augmentation of monocyte passage through the BBB model. There was a two-fold increase in monocyte migration in response to CCL2. The combination of METH and β-chemokine resulted in a significant enhancement in monocyte migration, which was dose-dependent and synergistic (Figure 6A, 100% and 150% by 50 or 250 μmol/L METH treatments, respectively, as compared with CCL2 alone, P<0.05). Next, we tested the ability of antioxidant to prevent METH-induced monocyte migration. Indeed, when added together with METH, Trolox attenuated monocyte migration in a dose-dependent manner, and at high concentrations, the antioxidant decreased monocyte migration induced by CCL2 and METH to control levels (Figure 6B). Our previous work related to BBB dysfunction associated with oxidative stress pointed to the possible downstream activation of MLCK by ROS. Pretreatment of BMVEC monolayers with MLCK inhibitor (ML-7) prevented the effects of METH/CCL2 on monocyte migration (Figure 6C). To further confirm the involvement of the MLCK pathway in METH-treated cells, we looked into the status of MLC phosphorylation, which is a downstream effector of MLCK. In fact, METH application did enhance the levels of p-MLC with a significant return to control levels by the addition of Trolox introduced together with METH (Figure 6D). Taken together, these data strongly support the role of oxidative stress in BBB impairment with MLCK possibly mediating the effects of ROS in the endothelium.
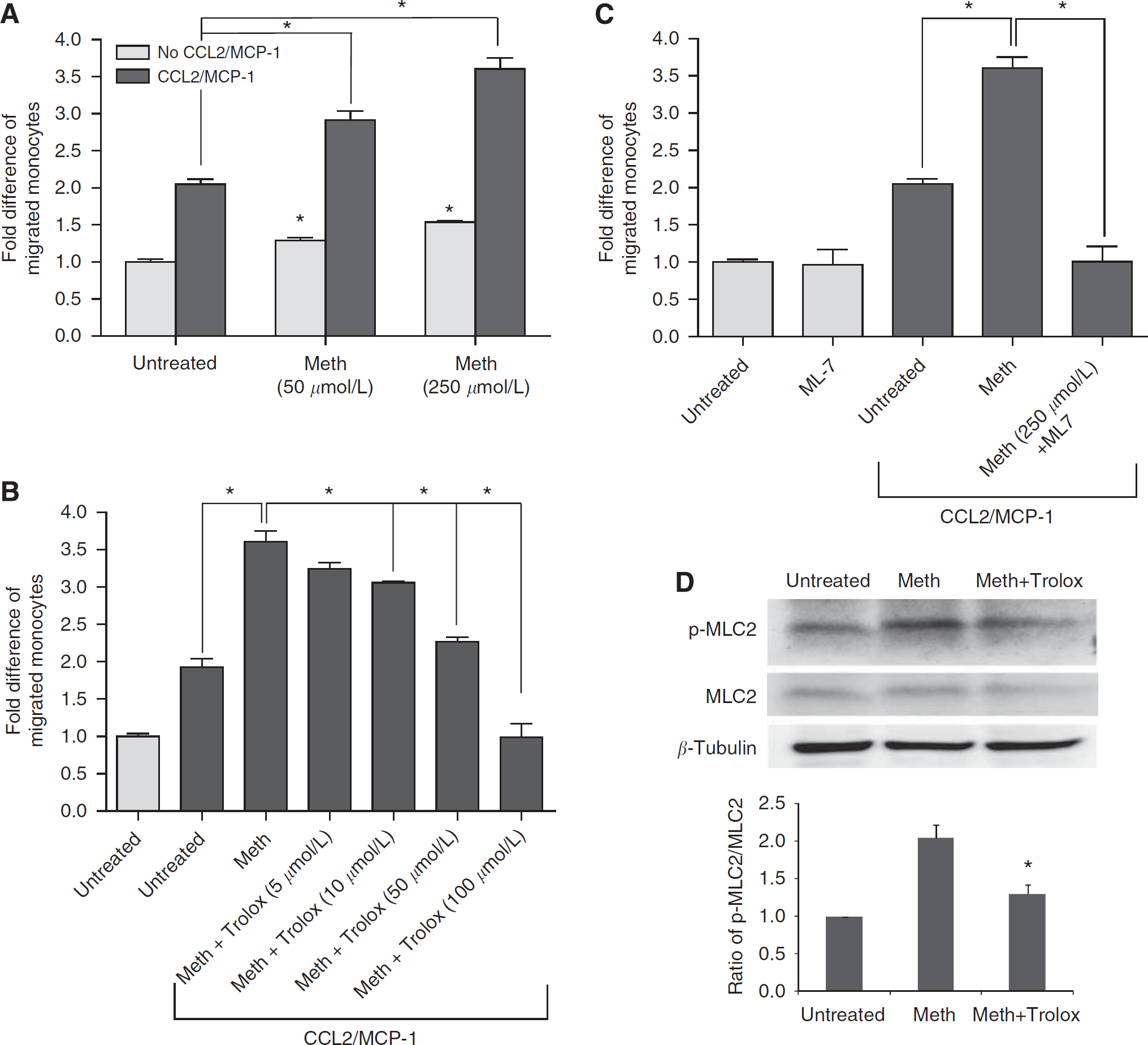
METH increased monocyte transendothelial migration by the activation of MLCK in endothelial cells. Monolayers of primary BMVEC were left untreated or were exposed to METH at the concentrations shown for 12 h (
Discussion
Short- and long-term METH abuse damages multiple organ systems; however, none is more prominent than in the brain. METH neurotoxicity disrupts normal neuronal communication in the dopaminergic system and induces neuroinflammation due to glial activation (Sekine et al, 2008). METH neurotoxicity manifests as severe cognitive deficits, such as memory loss and psychotic behavior. As METH use also occurs by intravenous injection and as behavior changes include a propensity to engage in precarious sexual behavior, METH users are more at risk of contracting HIV-1 (HIV & Drugs. Meth Use Develops Stronger Link to HIV Risk, 2005). One of the consequences of HIV-1 infection is the development of HIV-1 encephalitis characterized by monocyte migration across the BBB. Increased HIV-1 infection among METH users creates an additional layer of complexity in understanding the harmful effects that METH has on the brain. In recent years, METH is increasingly being recognized as a damaging agent not only to the neuronal environment but also to the cerebral vasculature.
Although much is known about the devastating effects of METH on neuronal and glial function, the effect of METH on the brain endothelium remains largely under-characterized. Interestingly, BBB compromise present in HIV-1 encephalitis (Avison et al, 2004) was recently shown in vivo after METH administration (Quinton and Yamamoto, 2006); however, the underlying mechanisms were not addressed. The main goal of this study was to provide evidence that METH can alter BBB function through direct effects on endothelial cells and to explore possible underlying mechanisms leading to endothelial injury. Our experiments showed enhanced BBB permeability, diminished tightness of BMVEC monolayers, decreased expression of cell membrane-associated TJ proteins, ROS production, and increased monocyte migration across METH-treated endothelial monolayers. These functional alterations were accompanied by the activation of MLCK. Importantly, antioxidant treatment attenuated or completely reversed all tested aspects of METH-induced BBB dysfunction.
Methamphetamine-induced hyperthermia and hypertension may lead to increased BBB permeability (Persidsky et al, 2006b). Indeed, two groups (Bowyer and Ali, 2006; Quinton and Yamamoto, 2006) showed that the administration of METH or its derivative MDMA (ecstasy) led to leakiness of the BBB in vivo. These reports proposed that breaches in the BBB permit the entry of glutamate from the blood into the brain parenchyma, thus generating a condition of glutamate excitotoxicity. However, enhanced BBB permeability was found after exposure to high concentrations of METH and the mechanism leading to the actual disruption in the BBB was not addressed in these studies. Sharma and Kiyatkin (2009) found increased BBB permeability using Evans blue and documented brain endothelial cell abnormalities in rats (exposed to 9 mg/kg of METH). They concluded that these alterations are secondary to hyperthermia.
Using BMVEC cultures, Lee et al (2001) showed that BMVEC respond in an inflammatory manner to METH insult by the activation of nuclear factor-κB/activator protein-1 transcription factors and tumor necrosis factor-α gene upregulation; however, the investigators did not correlate these findings with BBB functional alterations (permeability, barrier integrity, or leukocyte migration). Mahajan et al (2008) showed some marginal decreases in claudin-3 and claudin-5 mRNA levels with no change in occludin levels after 48-h treatment with METH. However, the study did not extend the analysis of claudin-3, claudin-5, and occludin to protein expression levels or distribution. In light of this report, it is possible that our significant decrease in claudin-5 and occludin may occur by the effects of METH on protein turnover rather than gene expression.
Toxic effects of METH depend primarily on the dosing, the route of administration (Riviere et al, 1999), and several other factors (reviewed by Itzhak and Achat-Mendes (2004)). In our in vivo and in vitro experiments, we used doses relevant to those seen in drug abusers. There is a significant range of concentrations in the blood of METH abusers because of the pattern of administration (single dose versus binge) and development of tolerance. Methamphetamine blood levels measured in individuals detained by the police in California were 2.0 μmol/L on average but as high as 11 μmol/L (Melega et al, 2007). Controlled studies indicated that a single 260 mg dose reached a level of 7.5 μmol/L (Melega et al, 2007). However, abusers tend to self-administer METH in binges, and with a METH half-life of 12 h such administration leads to significantly higher levels (Cho et al, 2001; Harris et al, 2003). Studies modeling binge pattern showed that the fourth administration of 260 mg during a single day produces blood levels of 17 μmol/L, reaching 20 μmol/L on the second day of such a binge (Melega et al, 2007). Binge doses of 260 mg to 1 g resulted in 17 to 80 μmol/L blood METH levels, and these estimates are consistent with blood levels detected after fatal injuries (Logan et al, 1998; Wilson et al, 1996).
In animal studies, high-dose acute METH administration can lead to severe METH toxicity (Harvey et al, 2000). The major lethal effect of METH exposure in animal studies is the development of hyperthermia. However, through the use of relatively continuous escalating dosing, it has been shown that animals develop tolerance to the anorectic and lethal effects of METH (Fischman and Schuster, 1974). The escalating dosing regimen adopted in this study produces tolerance to the dopaminergic and serotonergic neurotoxicity which otherwise would be induced by multiple high-dose administrations of METH, while simultaneously producing stimulant effects similar to those seen in human METH abuse. To that end, using escalating nontoxic concentrations of METH, we found that the in vivo administration of METH resulted in enhanced BBB permeability to the small molecular tracer Na-F. Continuous TEER measurements in vitro showed a marked decrease in TEER after METH administration, which occurred rapidly and was also sustained. Barrier compromise at the level of the TJ was evident in our morphologic analysis of occludin on BMVEC monolayers exposed to the drug. The altered morphologic appearance of the TJ after METH exposure was such that the TJ appeared discontinuous with gap formation. Expression of occludin and claudin-5 in the membranous fractions was reduced as a function of time and METH concentration, suggesting that TJ alteration may be the cause of METH-induced BBB permeability. It has been previously shown that depletion of occludin as a result of siRNA knockdown (Kevil et al, 1998) or by addition of vascular endothelial growth factor decreased transendothelial resistance and increased microvascular endothelial cell permeability (Wang et al, 2001).
Multiple studies suggest that METH-induced neurotoxicity involves the production of ROS and nitrogen species (such as nitrotyrosine) (Stephans and Yamamoto, 1994). Therefore, we explored the possibility that ROS generation results in BBB dysfunction in METH abuse. Indeed, METH induced ROS in primary brain endothelial cells, and antioxidant prevented the production of oxidative radicals and restored monolayer integrity. Furthermore, we showed that increased permeability caused by METH in vivo (mice exposed to escalating doses of drug) was partially prevented by the antioxidant, Trolox. Association of oxidative stress with enhanced endothelial permeability was documented in vitro and in vivo (reviewed in (Mehta and Malik, 2006). Endothelial monolayers exposed to H2O2 (Alexander et al, 2001) or hyperoxia (Phillips et al, 1988) showed increased transendothelial permeability of albumin. Human BMVEC treated with ROS donors or alcohol (causing ROS production) showed reduced TEER, redistribution of TJ proteins (occludin, claudin-5, ZO-1), and increased permeability with the antioxidant reversing these changes (Haorah et al, 2005). Therefore, oxidative stress induced by diverse mechanisms results in barrier compromise.
The exact mechanism underlying ROS production after BMVEC exposure to METH is not clear. Mitochondria are common targets for oxidative species and mitochondrial dysfunction and increased energy utilization has an important role in mediating the prooxidant and neurotoxic effects of METH (Bachmann et al, 2009). Methamphetamine treatment of cultured astrocytes increased ROS, adenosine triphosphate, and changed the mitochondrial membrane potential (Lau et al, 2000). The role of oxidative stress is further supported by the findings that ROS scavengers and antioxidants attenuated the neurotoxic effects of METH (Fukami et al, 2004) and resulted in significant behavior improvement associated with drug exposure (Coccurello et al, 2007). Thus, further analysis would need to be performed to understand the cause(s) and origin of oxidative stress induced by METH in the brain endothelium.
Disruption of the endothelial barrier results in augmented permeability and can enhance transendothelial leukocyte migration causing subsequent tissue damage (Aghajanian et al, 2008). To address this possibility, we studied the migration of primary human monocytes across confluent BMVEC monolayers after pretreatment with METH. β-Chemokine (CCL2) applied on the opposite side of BBB constructs to mimic neuroinflammatory conditions (such as HIV-1 encephalitis; Persidsky et al (1999)) increases cell migration twofold, whereas METH alone augmented monocyte migration by 30% to 50%; METH in combination with CCL2 led to synergistic 3- to 3.5-fold increases in monocyte passage. This finding is significant because monocytes carry HIV-1 across the BBB, and their accumulation correlates with clinical signs of cognitive decline and neuronal injury in the CNS of infected patients (Ellis et al, 2007). As antioxidants prevented barrier impairment in vitro and in vivo, we applied it to METH/CCL2-exposed BBB constructs and studied monocyte migration. Trolox diminished monocyte egress in a dose-dependent manner to levels comparable with untreated control constructs. As the generation of ROS could activate the MLCK pathway, which in turn could cause changes to the TJ, we therefore explored the effects of METH on MLCK. Our previous work indicated that oxidative stress activates MLCK in BMVEC leading to reduced integrity of the BMVEC monolayer promoting monocyte migration across the BBB (Haorah et al, 2005). Indeed, MLCK inhibition in BMVEC monolayers blocked METH/CCL2-induced monocyte migration. Activated MLCK phosphorylates MLC at Ser-19 or at Ser-19/Thr-18 (Goeckeler and Wysolmerski, 1995), produces endothelial cell contraction, and results in barrier dysfunction in response to inflammatory mediators (Dudek and Garcia, 2001). Our finding showed that METH exposure increased the levels of phosphorylated MLC indicative of MLCK activation; conversely, antioxidant prevented this increase.
MLCK inhibition induced by oxidative stress prevents TJ modifications, MLC phosphorylation, monocyte migration, and changes in TEER (Haorah et al, 2005). Results of this study suggest that many of the signaling pathways leading to BBB disruption overlap and converge on the same targets (such as MLCK or TJ modifications). Our findings provide evidence that show the direct effects of METH on the BBB, along with a possible underlying mechanism for this injury. Future investigations will address how METH causes ROS generation, decreases expression of TJ proteins, identifies pathways involved in MLCK activation, and whether leukocyte–endothelial interactions (such as adhesion, expression of adhesion molecules, etc.) are affected by the drug. The results of this study also indicate that METH can exacerbate BBB dysfunction, which may promote HIV-1 infection in the brain. Our results also point to a potential use for antioxidants as a therapeutic strategy to protect the brain endothelium against the effects of METH toxicity.
Footnotes
Acknowledgements
We thank Dr Ronald Tuma for critical reading of this manuscript
The authors declare no conflict of interest.