
Abstract
Synaptic bioenergetic deficiencies may be associated with early Alzheimer's disease (AD). To explore this concept, we assessed pre-synaptic mitochondrial function in hemizygous (+/−)TgMcGill-R-Thy1-APP rats. The low burden of Aβ and the wide array of behavioral and cognitive impairments described in 6-month-old hemizygous TgMcGill-R-Thy1-APP rats (Tg(+/−)) support their use to investigate synaptic bioenergetics deficiencies described in subjects with early Alzheimer's disease (AD). In this report, we show that pre-synaptic mitochondria from Tg(+/−) rats evidence a decreased respiratory control ratio and spare respiratory capacity associated with deficits in complex I enzymatic activity. Cognitive impairments were prevented and bioenergetic deficits partially reversed when Tg(+/−) rats were fed a nutritionally complete diet from weaning to 6-month-old supplemented with pyrroloquinoline quinone, a mitochondrial biogenesis stimulator with antioxidant and neuroprotective effects. These results provide evidence that, as described in AD brain and not proven in Tg mice models with AD-like phenotype, the mitochondrial bioenergetic capacity of synaptosomes is not conserved in the Tg(+/−) rats. This animal model may be suitable for understanding the basic biochemical mechanisms involved in early AD.
Introduction
The amyloid hypothesis proposes that high levels of amyloid β (Aβ) culminate in neuronal damage and Alzheimer's disease (AD). 1 Positron emission tomography (PET) with 18F-fluoro-2-deoxy-D-glucose has shown that in AD, glucose metabolism changes over time indicating that it could serve as a predictive marker of neurodegeneration. 2 Brain hypometabolism in AD cases is consistent with a decreased metabolic demand 3 which may be partially explained by a lower expression of mitochondrial genes in specific brain areas. 4 Mitochondrial accumulation of Aβ (mitAβ) 5 is a pathogenic event only described by ultrastructural analysis in AD brains and in transgenic (Tg) mice models of AD. MitAβ seems to induce mitochondrial dysfunction, one of the earliest and most prominent alterations in AD brain. 6 Recently, we showed that the catabolism of mitAβ 7 is pharmacologically modulated by pyrroloquinoline quinone (PQQ). PQQ is an anionic, water-soluble compound that has been shown to be an essential nutrient in mice. 8 PQQ can cross the blood–brain barrier 9 and has neuroprotective effects. 10 In several AD mouse models, mitochondria function has been studied, overall showing deficits in respiration and changes in morphology and transport.11,12 In addition to Aβ accumulation and mitochondrial dysfunction, it was proposed that bioenergetic defects in large nerve terminals (synaptosomes) are associated with progression of AD. However, this hypothesis has not been proven in commonly used Tg mice models with a complete amyloid phenotype (including dense fibrillar extracellular Aβ deposition) characteristic of late AD. 13 Recently, novel murine models of AD pathology have been developed including TgMcGill-R-Thy1-APP rats. Homozygous animals display the full amyloid pathology (mature plaques, dystrophic neurites and peri-plaque inflammation) while hemizygous (Tg + /−) rats show intra-neuronal accumulation of Aβ (iAβ) throughout their entire life and occasionally generate amyloid plaques. Moreover, they show a wide array of behavioral and cognitive impairments at pre-plaque stages. 14 To determine whether Tg(+/−) rats imitate the hippocampal bioenergetic defects observed in early AD brain, brains of 6-month-old Tg(+/−) and wild type (WT) controls were analyzed. By in situ respirometry, we detected in Tg(+/−) decrements in synaptosomal respiratory control ratio and spare respiratory capacity as compared to WT rats in agreement with a reduced mitochondrial complex I enzymatic activity. By contrast, neither complex II nor α-ketoglutarate dehydrogenase complex (α-KGDHC) activities showed significant differences between Tg(+/−) and WT rats. Cognitive defects characteristic of these animals at 6 months of age were not related to any ultrastructural change of the hippocampal CA1 region but were prevented, and mitochondrial function partially recovered when Tg(+/−) rats were fed a nutritionally complete diet supplemented with PQQ. These findings suggest that at 6 months of age Tg(+/−) rats display core behavioral features of early AD possibly due to initial bioenergetic alterations of hippocampal synaptosomes and provide further validation for its usage as a valuable tool to understand the underlying early mechanisms of this neurodegenerative disorder.
Materials and methods
Ethical statements
The study was carried out in strict accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) and the OLAW-NIH (Office Laboratory Animal Welfare) guidelines. The protocol was approved by the local Animal Care Committee of Fundación Instituto Leloir (FIL) Assurance# A5168-01.
Reagents
BioPQQ™ (pyrroloquinoline quinone disodium salt) was kindly provided by Mitsubishi Gas Chemical Company (Tokyo, Japan). Diamidino-2-phenylindole (DAPI) was purchased from Life Technologies. Oligomycin, carbonyl cyanide m-chlorophenyl hydrazone (m-CCCP), carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP), 2,6-dichlorophenol indophenol (DCPIP), antimycin, horseradish peroxidase, superoxide dismutase, scopoletin,
Animals and nutritional protocols
Hemizygous transgenic McGill-R-Thy1-APP (Tg(+/−)) rats harboring the human APP751 transgene with the Swedish and Indiana mutation under the control of the murine Thy1.2 promoter were generated using HsdBrI:WH Wistar strain. 14 Animals were provided to Fundación Instituto Leloir (FIL) by The Royal Institution for the Advancement of Learning/McGill University, Montreal, Quebec (Canada). An in-house colony was established at FIL as previously described. 15 Tg(+/−) rats and littermates wild type (WT) controls were maintained in polycarbonate cages in a temperature-controlled animal facility with a 12-h dark/light cycle and allowed to consume standard diet and water ad libitum. For separate experiments, BioPQQ was added to water at 2 mg/kg. 16 Rats were fed with BioPQQ diet from weaning up to 6 months of age. Thirty days before the end of the experiment, 5-month-old animals received a supplement of BioPQQ (20 mg/kg/day) given once a day via oral gavage (10 mL/kg body weight). Mixed gender (approximately half male and half females) was used for hippocampal bioenergetic assessment. By contrast, for behavioral performance, only male rats were used to avoid any potential effects of female estrus cycle on this paradigm.
Behavioral and cognitive testing
Neurological reflexes, exploratory activity, emotional behavior, recognition and spatial working and reference memory were assessed as previously reported. 15 Description of each test is given online in the Supplementary Information (SI).
Mitochondrial oxygen consumption studies
Subcellular fractions enriched in mitochondria were obtained from hippocampal homogenates of 6-month-old rats by differential centrifugation following standard procedures. 17 Oxygen consumption studies to evaluate mitochondrial respiration were performed using high-resolution respirometry (Hansatech Oxygraph, Hansatech Instruments Ltd, Norfolk, UK). 18 Mitochondria were incubated in respiration buffer supplemented with 6 mM malate and glutamate or 7 mM succinate as substrates. An initial rest state respiration in the absence of ADP (state 4) was established under these conditions, which was then switched to active state respiration (state 3) by the addition of 1 mM ADP. State 3 is defined as the maximum physiological ATP production rate and O2 consumption, and it is considered to be responsible for the physiological O2 consumption. Results are expressed as ng-at O/min/mg protein.
Mitochondrial oxidative metabolism
Hydrogen peroxide (H2O2) generation was assessed in hippocampal mitochondria of 6-month-old rats by the scopoletin-horseradish peroxidase method, following the decrease in fluorescence intensity at 365–450 nm (λexc–λem) at 30℃. 19 H2O2 production was triggered by the addition of 6 mM malate and 6 mM glutamate. Results were expressed as nmol H2O2/min/mg protein. Oxidative damage to phospholipids was evaluated in hippocampal mitochondria by measuring thiobarbituric acid reactive substances (TBARS) with a fluorometric assay. 20 Results are expressed as nmol TBARS/mg protein.
Synaptosomes isolation, biochemical and microscopical characterization
Six-month-old WT and Tg(+/−) rats were decapitated and brains were quickly removed. Hippocampi were dissected in ice-cold MSTE buffer (0.23 M mannitol, 0.07 M sucrose, 10 mM Tris-HCl, 1 mM EDTA, pH 7.4) and homogenized with a manual Teflon glass homogenizer in 2 mL of MSTE. Homogenates were centrifuged at 600 × g for 10 min to discard cell debris and nuclei and supernatants were centrifuged again at 8000 × g to obtain crude mitochondrial pellets. Pellets were washed, resuspended in 2 mL of MSTE, and layered on 13%, 8%, and 3% Ficoll step-gradients. Gradients were centrifuged at 11,500 × g for 30 min. The fraction at 8% was recovered, diluted in HBS buffer (20 mM HEPES, 10 mM
ATP production rate
A chemiluminescence assay based on the luciferin–luciferase system was used to evaluate ATP production rate in hippocampal mitochondria of 6-month-old rats or in freshly purified and permeabilized (0.01% digitonin) synaptosomes. Methodology is described online in the Supplementary Information. The number of phosphorylated ADP molecules per oxygen atom (P/O ratio) was calculated as the mean value of mitochondrial ATP production/mean value of state 3 oxygen consumption rate.
Synaptosomal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) assays
Synaptosomes were rapidly thawed, diluted in HBS and centrifuged at 11,500 × g for 5 min. Pellets were resuspended in HBS and protein concentration was determined with a Pierce BCA Protein Assay Kit (Thermo Scientific). Twenty-five µg of protein were plated into 20 wells of a XFe24 V7 cell culture microplate (Seahorse Bioscience) previously coated with polyethyleneimine (1:15 000 dilution of a 50% v/v stock) and centrifuged at 3600 × g for 1 h at 4℃ to attach synaptosomes. HBS medium was replaced with 600 µL of incubation medium (3.5 mM KCl, 120 mM NaCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1.2 mM Na2SO4, 2 mM MgSO4, 15 mM
Enzyme assays
Methodology to assess mitochondrial complex I, complex II, citrate synthase and α-ketoglutarate dehydrogenase enzyme complex (α-KGDHC) activities is described online in the Supplementary Information.
Quantitation of synaptic density and mitochondrial morphology by transmission electron microscopy
Methodology to estimate the synaptic density and the size and shape of mitochondria is described online in Supplementary Information.
Estimation of iAβ levels and oxidative damage in the hippocampus affected by iAβ accumulation
SDS-soluble Aβ40 peptide quantitation was performed by ELISA as previously described. 15 Oxidative stress was estimated by immunohistochemistry using a monoclonal antibody against 8-hydroxy-2′-deoxyguanosine (8-OHdG) (a marker of nuclear and mitochondrial DNA oxidation). The methods are described online in the Supplementary Information.
Laser cell microdissection and Quantitative PCR (qPCR) to evaluate mitochondrial content and mitochondrial DNA common deletion in hippocampal neurons
Rats were decapitated and brains were quickly removed and flash-frozen in isopentane. The tissue blocks were then cut into 20 µm sections, fixed in xylol and sandwiched between the metal-framed polyethylene naphthalate (PEN) membrane slides (Applied Biosystems, Life Technologies, Naerum, Denmark) and a supporting glass slide, and placed in the ArcturusXT™ LCM System instrument (Applied Biosystem, USA) equipped with an infrared (IR) capture laser (150 mW, maximum output at 804–813 nm) and a UV cutting laser (1W, 250 µJ, 15 ns pulse, maximum output at 349 nm) for LCM. Then, after the subiculum, CA1 and CA2 hippocampal neurons were identified, the thermoplastic ethylene vinyl acetate (EVA) membrane of the cap was melted and glued with the PEN membrane specifically over the area of interest by applying IR laser pulses. The targeted samples were released from the remainder of the tissue and the PEN membrane by cutting with the UV laser. The caps were then lifted, and following microscopic verification for the efficiency of the procedure, the captured cells were processed for the RNA or DNA extractions. Three cryosections were dissected from each rat brain and recovered in a single cap. DNA was isolated with Arcturus® PicoPure® DNA extraction kit following the manufacturer's instructions. The amount of mitochondria and the presence of mtDNA deletion were quantified by qPCR with gene-specific primers shown in the Supplementary Information (Table S1). ND1 is a mitochondrial gene located out of the common deletion region (∼4977 bp) and it is used to target total mtDNA; ND4 is a mitochondrial gene located in the region of the common deletion and cystic fibrosis transmembrane conductance regulator (CFTR) is a nuclear gene used as an indicator of genomic DNA. The ratios ND1/CFTR and ND4/ND1 are indicative of the amount of mitochondria and mtDNA deletions, respectively. One to 50 µg of DNA recovered from cells isolated by LCM was used per well. Samples were plated in triplicate. SYBR-Green qPCR was performed by using the KAPA SYBR® FAST Universal 2X qPCR Master Mix (Kapa Biosystems). Reactions were run in an Mx3005P cycler (Stratagene) and results analyzed by the MxPro software in a Comparative Quantitation mode.
Evaluation of energy metabolism and Aβ catabolism genes expression in hippocampi of 6-month-old Tg(+/−) rats
Two alternative procedures for RNA isolation were performed. RNA from iAβ containing neurons obtained by LCM was isolated with PicoPure RNA Isolation Kit (Applied Biosystems) and from hippocampal homogenates, a standard protocol was used. 7 RNA quality control and quantification were performed with a bioanalyzer (Agilent 2100). Samples with a RIN (RNA Integrity Number) value lower than 6 were discarded as indicative of degraded RNA. One microgram of total RNA was reverse transcribed using oligo(dT) primer and SuperScript II reverse transcriptase (Invitrogen). PGC-1α, NRF-1, and IDE-Met1 were targeted to assess the status of the energetic metabolism (PGC-1α, NRF-1) and Aβ catabolism (IDE-Met1) genes. Gene-specific primers are shown in Supplementary Information (Table S1). cDNA was amplified by SYBR Green RT-qPCR with a Mx3005P cycler (Stratagene). Cycling conditions were as follows: 95℃ for 10 min, 95℃ for 30 s, annealing for 30 s at 58℃, and 72℃ for 30 s for 40 cycles. Melt curve analysis and agarose gels verified the formation of a single desired PCR product. The relative amount of transcripts to TATA-binding protein (TBP) was quantified by the 2−ΔΔCt method using MxPro software.
Statistical analysis
All data are shown as the mean ± SEM of at least three independent experiments. Data were analyzed by unpaired two-tailed Student's t-test, one and two-way ANOVA, or by two-way mixed ANOVA tests followed by Tukey's HSD post hoc tests for multiple comparisons, unless noted otherwise. The significance level was set at p = 0.05. SPSS 15.0 for Windows software (Chicago, IL, USA) was used to perform all statistical analyses.
Results
Hippocampal mitochondria of Tg(+/−) rats have lower complex I-dependent respiration, produce more H2O2, and accumulate more TBARS than WT rats
Metabolic and oxidative parameters in isolated hippocampal mitochondria of 6-month-old rats.
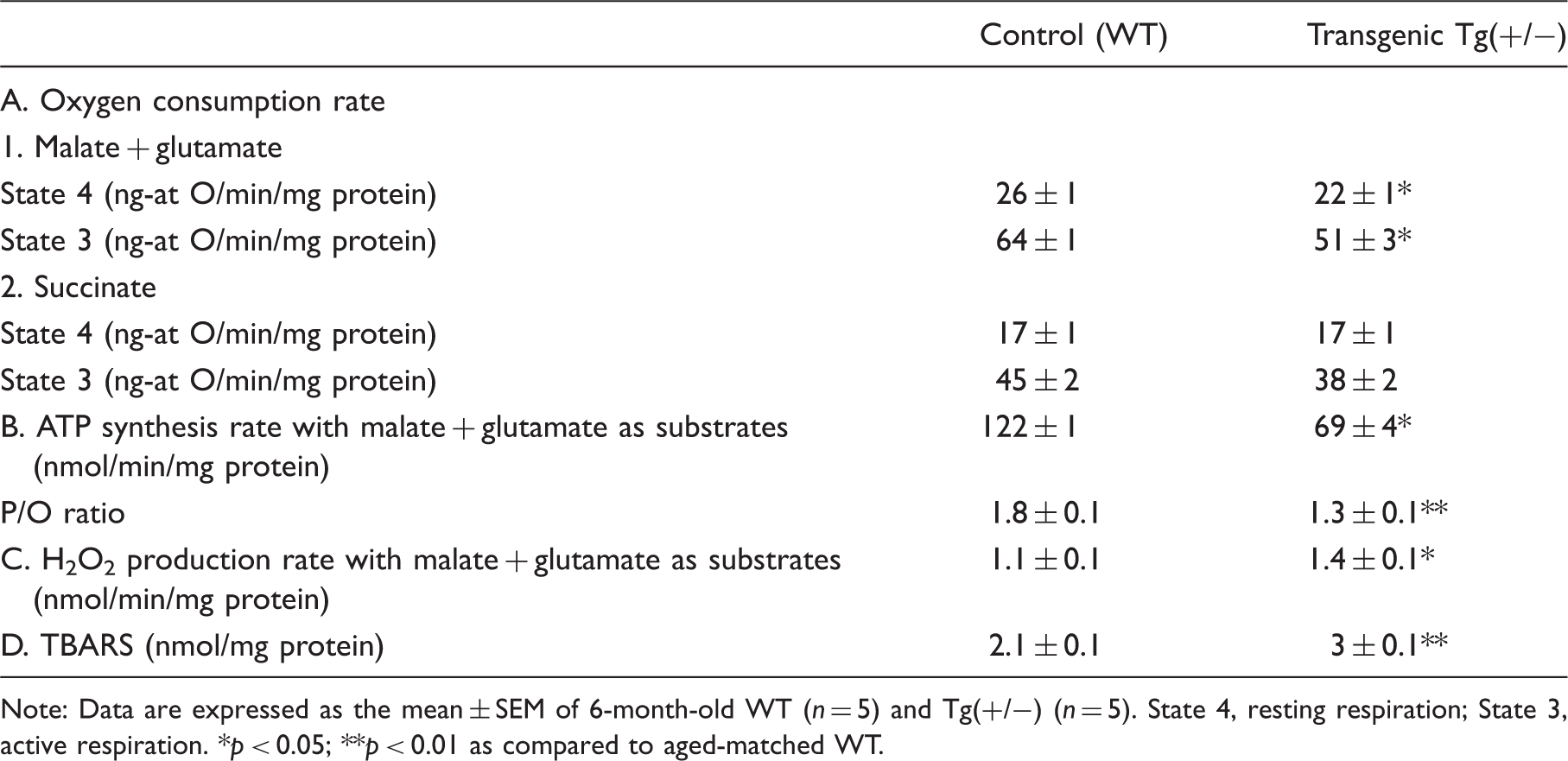
Note: Data are expressed as the mean ± SEM of 6-month-old WT (n = 5) and Tg(+/−) (n = 5). State 4, resting respiration; State 3, active respiration.
p < 0.05;
p < 0.01 as compared to aged-matched WT.
Hippocampal synaptosomes from Tg(+/−) rats have dysfunctional mitochondria
To fully evaluate mitochondrial function in synaptosomes isolated from hippocampi of 6-month-old rats, we used an extracellular flux analyzer. The enrichment in synaptophysin (Figure 1(a), upper panel) and the morphology and size of particles obtained after the biochemical fractionation (Figure 1(a), lower panel) were used as criteria of quality control of the preparation of synaptosomes. As previously reported for guinea-pig and mouse synaptosomes, rat synaptosomes utilize exogenous glucose and pyruvate as substrates for active respiration.22,23 Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) profiles of 6-month-old WT rat synaptosomes incubated with 15 mM glucose or 15 mM glucose plus 10 mM pyruvate were determined. Under these conditions, typical OCR and ECAR patterns were obtained measuring basal values and after the addition of oligomycin, FCCP and antimycin. Basal OCR (Figure 1b) is significantly increased in the presence of pyruvate, yet oxygen consumption with only glucose as substrate evidences the existence of glycolytic enzymes in synaptosomes. Coupling efficiency estimated after the addition of oligomycin in glucose/pyruvate medium (∼13%) was improved (∼30%) using glucose medium, reinforcing the concept that pyruvate translocation across mitochondrial membrane is proton coupled.
24
The addition of FCCP to WT synaptosomes uncoupled oxygen consumption from ATP synthesis, allowing determination of the maximum respiration and spare respiratory capacity. As expected, the presence of pyruvate highly increased the maximum respiration rate, and the spare respiratory capacity was ∼3-fold higher than the basal respiration, suggesting that synaptosomal mitochondria can respond to an increase in energy demand. Moreover, the extracellular acidification rate (ECAR) in glucose/pyruvate medium increased after the addition of oligomycin and FCCP (Figure 1c), in agreement with the activation of anaerobic glycolysis in response to the inhibition of mitochondrial ATP synthesis.21,22 In addition, we determined ATP synthesis rate in permeabilized WT synaptosomes in the presence of complex I substrates, malate and glutamate, before and after the addition of oligomycin or antimycin, respectively. Significant decrements were detected in ATP synthesis rate with oligomycin or antimycin (Figure 1d), suggesting that respiration in our synaptosomal preparation is coupled to ATP synthesis.
Biochemical and metabolic characterization of synaptosomes in 6-month-old WT rats. (a) Representative Western blotting of mitochondrial (Mit.) and synaptosomal (Synap.) enriched fractions immunoreacted with the mitochondrial ATP synthase β-subunit (ATP syn-β) and synaptosomal synaptophysin (Synaptoph.) protein markers antibodies (upper panel); frozen-thawed plate-attached synaptosomes were imaged using an Olympus CKX41 microscope (lower panel). Synaptosome with the characteristic ‘doughnut’ shape is indicated by arrowhead. Inset, typical autofluoresence of synaptosomes aggregates imaged using an Olympus BX50 microscope. Scale bars, 10 µm; inset, 50 µm. (b), Representative profile of the oxygen consumption rate (OCR) for hippocampal synaptosomes from 6-month-old WT rats. OCR was determined in the presence of 15 mM glucose (Glc) or 15 mM glucose plus 10 mM pyruvate (Pyr) as substrates. Graph shows basal OCR, OCR after ATP synthase inhibition by 5 µM oligomycin and maximum OCR after the addition of 1 µM FCCP. Data are means ± SEM. (c) Graph shows the extracellular acidification rate (ECAR) determined in parallel with respiration shown in (b). (d) Bars show ATP synthesis rate in synaptosomes of 6-month-old WT rats assessed with luciferin-luciferase assay in digitonin-permeabilized samples in the presence of malate and glutamate (M + G) and after the addition of 1 µM oligomycin (O) or 5 µM antimycin (a). *p < 0.05 vs. M+G.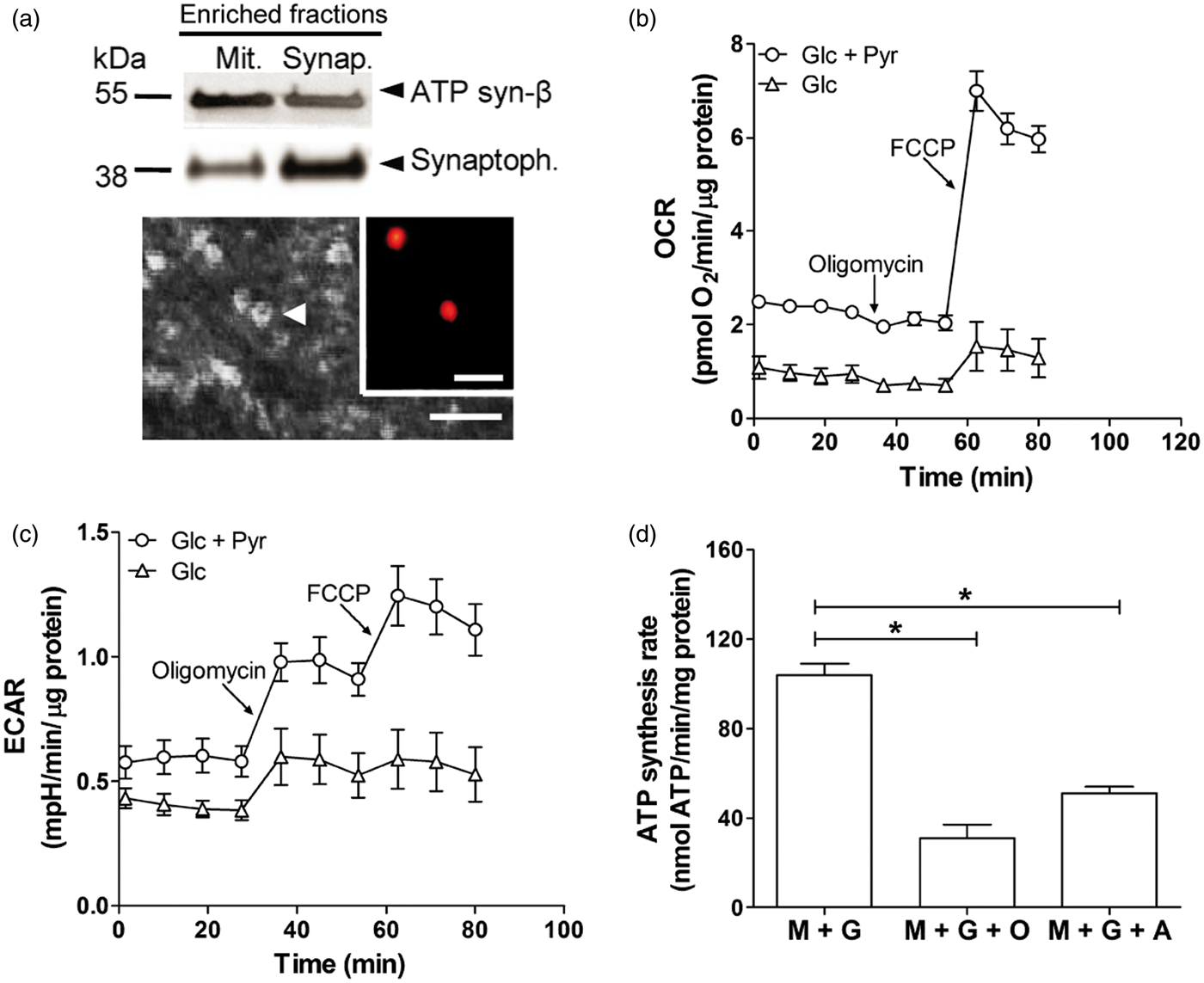
Since Aβ is known to localize to mitochondrial membranes, disrupt the electron transport chain, increase reactive oxygen species production, and cause mitochondrial damage,
25
we evaluated if these events could be responsible for mitochondrial dysfunction in our setting. In accordance with our previous observation in whole hippocampal homogenates,
15
we detected in synaptosomes of 6-month-old Tg(+/−) rats the presence of SDS-resistant Aβ oligomers (Figure 2a). Then, we compared the metabolic response of synaptosomes from Tg(+/−) and WT rats and showed that when maximal OCR was expressed as a fraction of WT basal value,
26
differences between WT and Tg(+/−) rats reached significance (271 ± 16 % vs. 190 ± 24 %; p < 0.05) (Figure 2b) concordant with the respiratory deficits detected in isolated hippocampal mitochondria. Additionally, decrements in the respiratory control rate (RCR) (2.6 ± 0.4 pmolO2/min/µg protein vs 3.1 ± 0.2 pmolO2/min/µg protein; p = 0.05) and in the spare respiratory capacity (2.6 ± 0.8 pmolO2/min/µg protein vs. 4 ± 0.3 pmolO2/min/µg protein; p < 0.05) were detected in synaptosomes from Tg(+/−) as compared to WT. Furthermore, the similar ECAR profile of WT and Tg(+/−) rats in basal conditions (0.6 ± 0.1 mpH/min/µg protein and 0.5 ± 0.2 mpH/min/µg protein, respectively) showed no significant differences after the addition of oligomycin (1 ± 0.1 mpH/min/µg protein vs 1 ± 0.2 mpH/min/µg protein; p > 0.05) or FCCP (1.2 ± 0.1 mpH/min/µg protein vs 1.1 ± 0.3 mpH/min/µg protein; p > 0.05).
Mitochondrial function of hippocampal synaptosomes from 6-month-old Tg(+/−) rats. (a) Representative Western blotting (upper panel) of pools corresponding to WT and Tg(+/−) synaptosomes showing Aβ oligomers (≈ 24 kDa) in Tg(+/−) in contrast to WT samples. Protein content of each lane is shown by anti-synaptophysin immunoreactivity (≈ 38 kDa) (lower panel). (b) Bars show the oxygen consumption rate (OCR) of synaptosomes of WT and Tg(+/−) rats expressed as % of WT basal value. *p < 0.05 vs. WT. (c) Representative Western blotting of synaptosomes of WT and Tg(+/−) rats immunoreacted with the synaptosomal (PSD-95) and mitochondrial ATP synthase β-subunit (ATP syn-β) protein markers (upper panel). Bars (lower panel) show the semi-quantification of 6–7 biological replicates of mitochondrial proteins normalized by synaptosomal content expressed in arbitrary units (A.U.) Data are means ± SEM. p = 0.9. (d) Bars show the citrate synthase activity of WT and Tg(+/−) synaptosomes expressed as nmol/min/mg protein. Data are means ± SEM. p = 0.6. (e) Bars show the specific activity of respiratory complex I, complex II and α-KGDHC of WT and Tg(+/−) synaptosomes expressed as nmol/min/mg protein. Data are means ± SEM. *p < 0.05 vs. WT. (f) The endogenous PGC-1α, NRF-1, and IDE-Met1 transcript levels were determined by RT-qPCR from hippocampal homogenates of WT and Tg(+/−) rats. Each point represents the mean value of at least three independent experiments performed by triplicate for each sample normalized by TATA-binding protein (TBP). The mean ± SEM in Tg(+/−) relative to WT are shown. Values between dashed lines (+1.5 and −1.5) were considered not different from WT=1. (g) Electron micrograph of a portion of the CA1 hippocampal region from WT and Tg(+/−) rats. Arrows, synapses; arrowheads, mitochondria. Scale bar = 2 µm. Inset, high magnification showing a representative mitochondria (M) and synapse (S). Scale bar = 500 nm. Bars (left panel) show the number of synapses/volume. Data are means ± SEM. p = 0.07.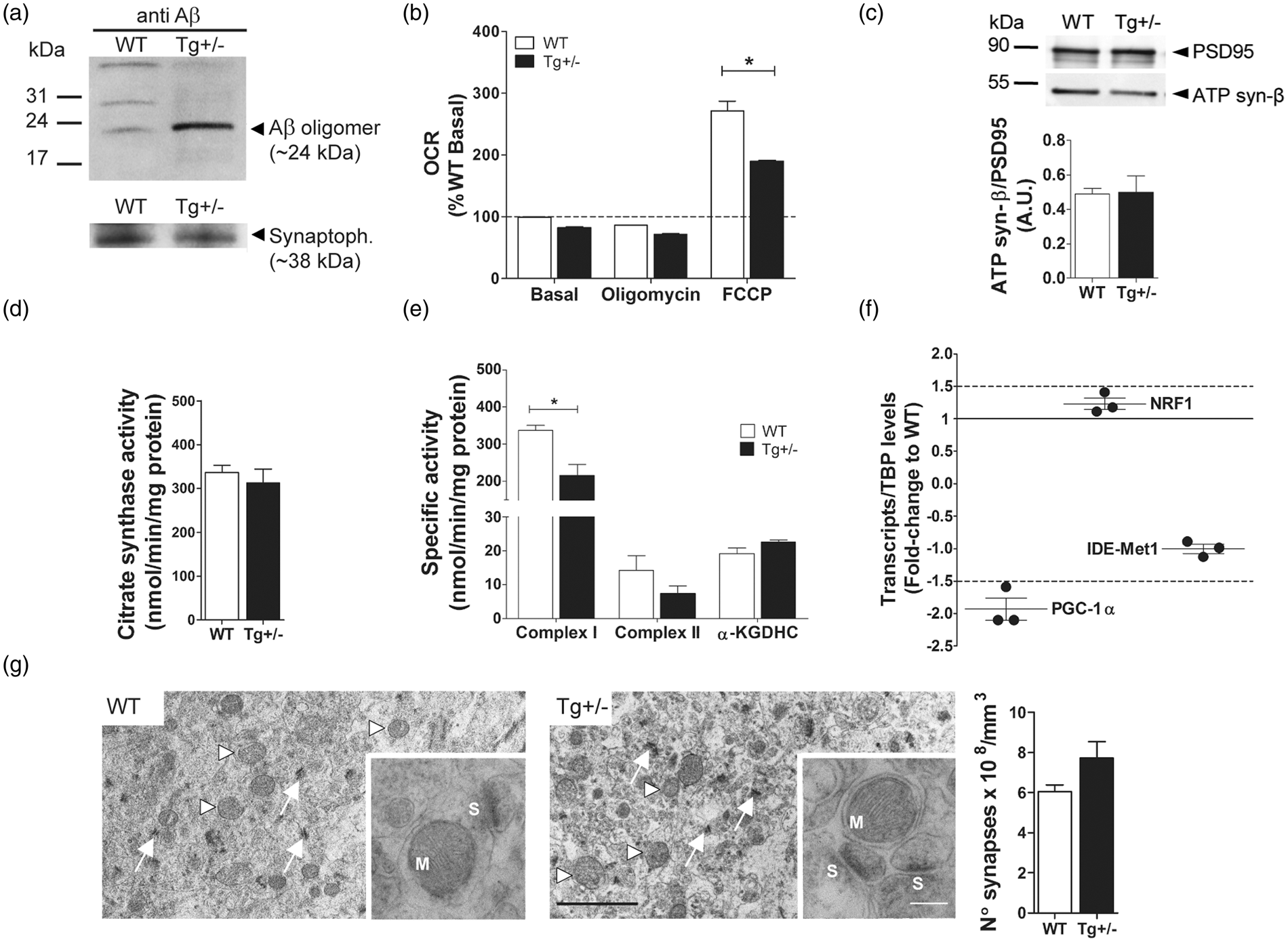
Synaptosomes of Tg(+/−) rats show decreased complex I enzymatic activity as compared to WT
We wondered whether the decrease in spare respiratory capacity and mitochondrial respiratory control ratio may be the consequence of impaired neuronal anterograde transport of mitochondria from the soma to the synapses. Thus, we determined by two different experimental approaches the number of mitochondria in synaptosomes of 6-month-old Tg(+/−) and WT rats. No significant differences were detected between WT and Tg(+/−) animals in the amount of mitochondrial protein (ATP synthase β subunit) as determined by Western blotting (Figure 2c) and expressed as arbitrary units (A.U.) (0.49 ± 0.1 vs. 0.5 ± 0.1), nor in the activity of citrate synthase enzyme (a stable mitochondrial matrix protein) (Figure 2d) (336.4 ± 16.5 vs. 312.5 ± 32.1 nmol/min/mg protein). To further analyze the respiratory chain mitochondrial complexes and the tricarboxylic acid (TCA) cycle functionality, we evaluated complex I, complex II and α-KGDHC activities at the synapses of WT and Tg(+/−) rats of 6 months of age. Our results showed (Figure 2e) significant decrements in complex I (338 ± 13 nmol/min/mg vs. 216 ± 29 nmol/min/mg; p < 0.05) but not in complex II (16 ± 5 nmol/min/mg vs. 7 ± 2 nmol/min/mg; p = 0.1) nor in α-KGDHC (19 ± 2 nmol/min/mg vs. 23 ± 1 nmol/min/mg; p = 0.09) enzymatic activities suggesting that bioenergetic impairments at the synapses may be due to a direct impact of mitAβ on complex I but not on α-KGDHC as shown in AD brains.27,28 To assess whether in Tg(+/−) rat brains there is a dysregulation of PGC-1α/NRF-1/IDE-Met1 pathways mediated by Aβ accumulation as reported in the AD brain and in vitro, 7 we determined the levels of these transcripts in iAβ positive cells isolated by LCM. Since the quality control of RNA (RIN<6) precluded its utilization for qRT-PCR and to partially answer this question, we used RNA isolated from hippocampal homogenates. In contrast to NRF-1 and IDE-Met1 genes that did not show variation, in 6-month-old Tg(+/−) rats transcript levels of PGC-1α gene decreased 1.9-fold, as compared to aged-matched controls (WT=1) (Figure 2f), suggesting that in Tg(+/−) rats the PGC-1α pathway is altered. In addition, we evaluated by transmission electron microscopy whether there were any synaptic changes at the CA1 region in Tg(+/−) as compared to WT rats. Stereological analysis of the images (Figure 2(g), left and middle panels) showed that the number of synapses/mm3 (Figure 2(g), right panel) and the morphology of mitochondria were similar between Tg(+/−) and WT rats (diameter: 0.4 ± 0.1 µm vs. 0.4 ± 0.1 µm; axial ratio: 1 ± 0.1 µm vs. 1 ± 0.2 µm; circularity ratio: 1.4 ± 0.4 µm vs. 1.3 ± 0.3 µm; p = n.s. for all cases). Western blotting of PSD-95, a marker of synaptic integrity29,30 did not show significant differences between Tg(+/−) and WT rats (Figure S4, Supplementary Information). Together, these results suggest that the metabolic changes in Tg(+/−) synaptosomes were not due to a reduced number of mitochondria or synaptic loss.
PQQ prevented cognitive impairment and synaptic mitochondrial dysfunction of Tg(+/−) rats
Assessment of spontaneous locomotion and anxiety-related behaviors.
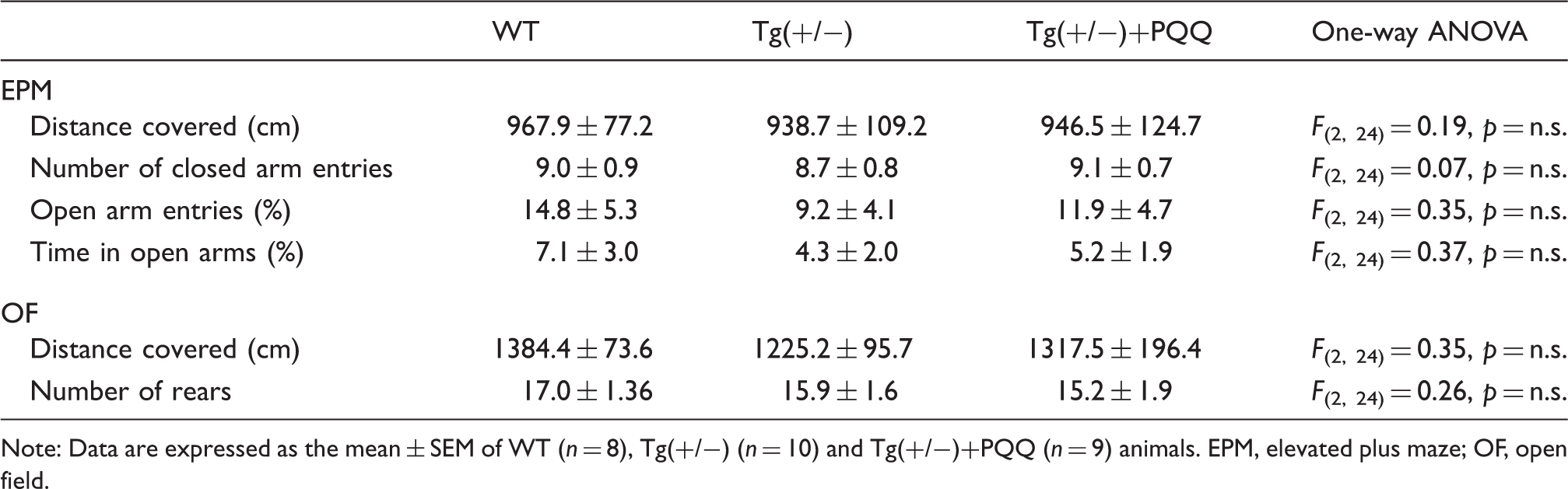
Note: Data are expressed as the mean ± SEM of WT (n = 8), Tg(+/−) (n = 10) and Tg(+/−)+PQQ (n = 9) animals. EPM, elevated plus maze; OF, open field.
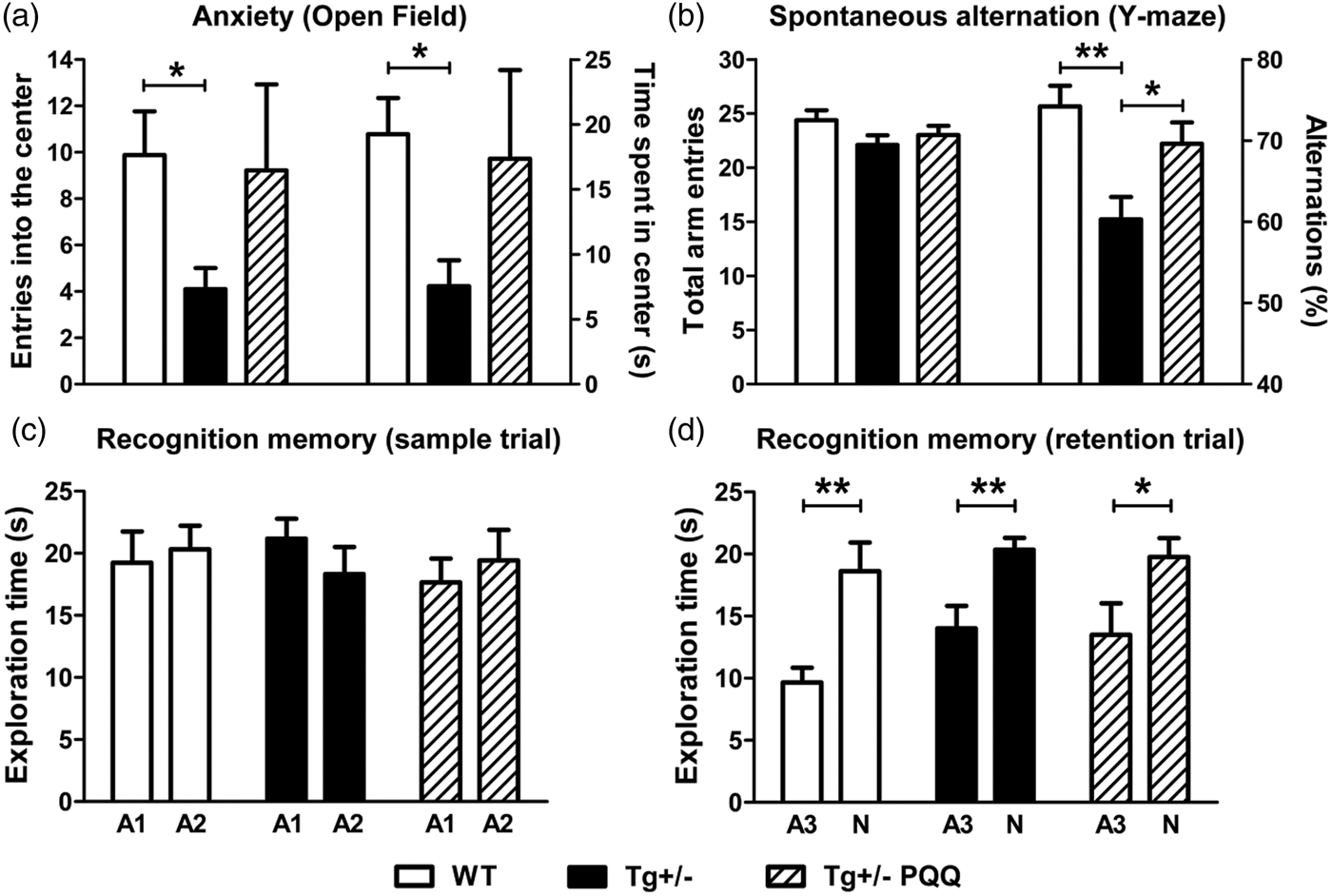
Effect of BioPQQ on anxiety, spatial working memory and recognition memory. Bars show in (a), entries into the center (left axis) and time spent in the center (right axis) of the OF; (b) total arm entries (left axis) and percentage of alternation (right axis) in the Y-maze; (c) exploration times for the two identical objects (A1 and A2) during the sample trial in the NORT; (d) exploration times for the familiar (A3, identical to A1 and A2) and novel (N) object. Data are means ± SEM of 8–10 rats per group. *p < 0.05, **p < 0.01.
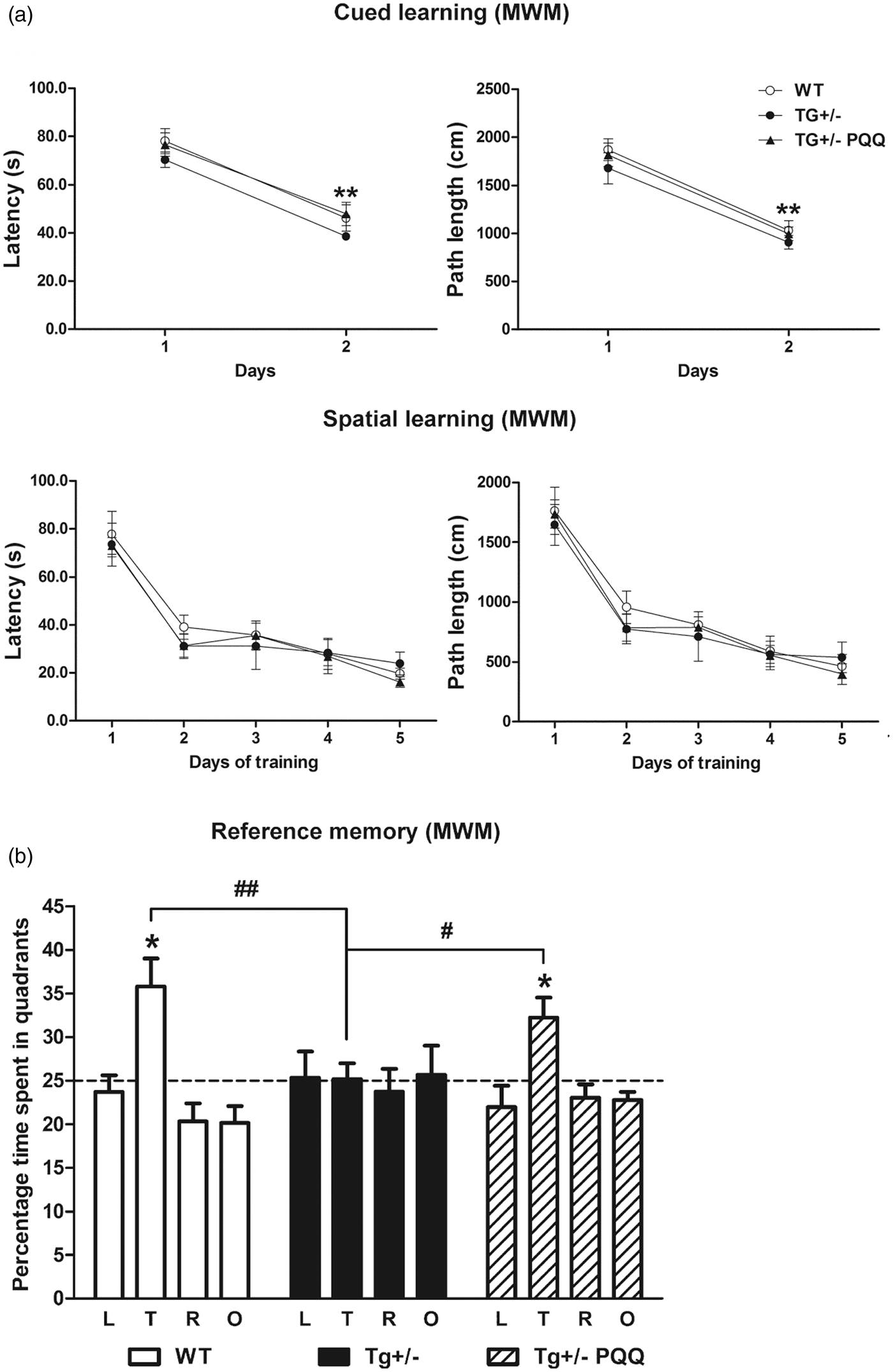
Treatment with BioPQQ prevented the spatial reference memory deficit exhibited by Tg(+/−) rats. (a) Graphs show latencies (upper-left panel) and paths length (upper-right panel) during the first and second day of the cued learning and latencies (lower-left panel) and paths length (lower-right panel) throughout the five days of spatial learning in the MWM test. (b) Bars show the percentage of time spent in quadrants (L, left; T, target; R, right; O, opposite). Data are means ± SEM of 8–10 per group. *p < 0.05 vs. percentage of time expected by chance (dashed line), #p < 0.05, ##p < 0.01.
To assess whether the cognitive improvement was related to a better metabolic response at the synapses, we determined the spare respiratory capacity of synaptosomes. When we expressed the spare respiratory capacity as a fraction of WT, differences between Tg(+/−) and Tg(+/−)+BioPQQ rats reached significance (69 ± 7% vs. 97 ± 3%; p < 0.05) (Figure 5a). This result suggests that cognitive improvements due to BioPQQ may be related to a better synaptic mitochondrial functionality. The improving effect of BioPQQ was not mediated by promotion of mitochondrial biogenesis or reduction of iAβ levels, since no significant differences were observed in the number of mitochondria as determined by qPCR from DNA of iAβ positive cells isolated by LCM from Tg(+/−) and Tg(+/−)+BioPQQ (Figure 5b) or in the levels of SDS-soluble Aβ determined by ELISA in hippocampal homogenates of Tg(+/−) and Tg(+/−)+BioPQQ rats (9.2 ± 1.7 pg Aβ/mg vs. 8.2 ± 2.8 pg Aβ/mg; p=0.75). Moreover, at the synaptic level, no significant differences were observed in the amount of a mitochondrial protein (ATP synthase β-subunit) determined by Western blotting (Figure 5c). Knowing that BioPQQ may exert an anti-oxidant effect,
31
we assessed TBARS in hippocampal mitochondria from Tg(+/−)+BioPQQ animals. Our results showed that BioPQQ treatment reversed the oxidative damage to phospholipids (Figure 5d) (3.4 ± 0.4 nmol/mg protein vs. 2 ± 0.1 nmol TBARS/mg protein; p < 0.05).
Biochemical impact of BioPQQ on synaptic mitochondrial functionality and brain oxidative stress in 6-month-old Tg(+/−) rats. (a) Bars show the spare respiratory capacity of synaptosomes from Tg(+/−) rats under control (black) or BioPQQ (dashed) supplemented diet expressed as % of WT. *p < 0.05 vs. Tg(+/−) + BioPQQ; (b) The gene copy number of endogenous ND1 was determined by qPCR from CA1 iAβ positive neurons microdissected by LCM from Tg(+/−) and Tg(+/−)+BioPQQ rats. Each point represents the mean value of at least three independent experiments performed by triplicate for each sample normalized by cystic fibrosis transmembrane conductance regulator (CFTR) (left) and ND4 (right) gene, respectively. The mean ± SEM in Tg(+/−)+BioPQQ relative to Tg(+/−) is shown. Values between dashed lines (1.5 and −1.5) were considered not different from Tg(+/−)=1. (c) Representative Western blotting of synaptosomes of WT, Tg(+/−) and Tg(+/−)+BioPQQ rats immunoreacted with the synaptosomal (PSD-95) and mitochondrial ATP synthase β-subunit (ATP syn-β) protein markers (upper panel). Bars show the semi-quantification of 6–7 biological replicates of mitochondrial protein ATP syn-β normalized by PSD-95 content expressed in arbitrary units (A.U.). F(2,13), p = 0.35. (d) Bars show TBARS accumulation expressed as nmol/mg protein in hippocampal mitochondria of Tg(+/−) rats exposed to control (black) or BioPQQ (dashed) diet. Data are means ± SEM. *p < 0.05.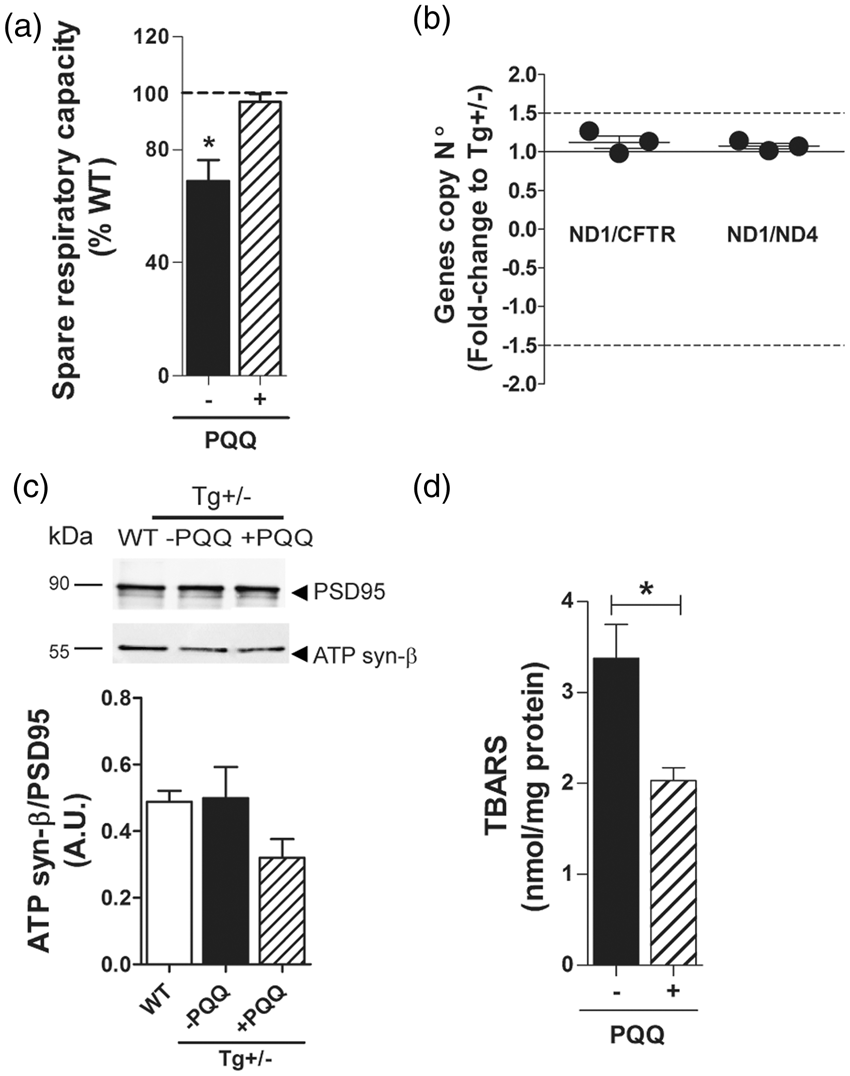
Discussion
The primary objective of the present study was to analyze early synaptic bioenergetics compromise in the progression of the AD-like amyloid pathology. To reach this goal, we resorted to use transgenic rats. Rats have advantage over mice because they are phylogenetically closer to humans, have postnatal CNS development, and show a more complex brain connectome and a richer behavioral display. 32 Different transgenic rat models of AD were developed in the last 10 years. 33 Among these, the McGill-R-Thy1-APP transgenic rat is of particular interest because AD-like pathology is achieved with a single transgene (human APP with 2 familial Alzheimer's disease mutations) and a single insertion site per allele making of it an AD transgenic model with a low transgene dosage. We opted to use hemizygous (+/−) adult animals (6-month-old) because they display iAβ accumulation and cognitive deficits similar to early AD. First, we showed that there are a number of significant biochemical abnormalities in mitochondria derived from bulk preparations of hippocampal tissue in Tg(+/−) rats. An effect of the expression of the human APP751 transgene with the Swedish and Indiana mutation and/or its proteolytic products including Aβ, on oxygen consumption is clearly observed in Tg(+/−) animals. Inhibition of active (state 3) and resting (state 4) respiration was detected in 6-month-old Tg(+/−) rats as compared to WT. These effects were evident when complex I substrates were used and may be indicative of complex I decreased activity, as described in AD brains. 34 Moreover, a significant decrement in mitochondrial ATP production rate was observed in Tg(+/−) hippocampi, suggesting the existence of a profound bioenergetic impairment in the brain of these animals. Different molecular mechanisms have been proposed for complex I inactivation including the synergistic inhibition produced by peroxynitrite-mediated reactions, reactions with free radical intermediates of lipid peroxidation and amine–aldehyde adduction reactions. More recently, alterations in complex I assembly intermediates precluding the proper formation of supercomplexes or respirasomes has been proposed. 35 The decrease in the oxygen consumption rate detected in 6-month-old Tg(+/−) rats may be due to an alteration in the redox equilibrium in the hippocampus in these animals. Mitochondria from 6-month-old Tg(+/−) rats produced more hydrogen peroxide and increased the accumulation of lipid oxidation products (TBARS). These results indicate that under basal metabolic conditions, Tg(+/−) brain is undergoing a sustained mitochondrial oxidative damage which may be responsible of the respiratory impairments with age.
Our results provide strong evidence that in Tg(+/−) rats there is an increasing mitochondrial vulnerability associated to the expression of the human transgene. However, we showed that 8-OHdG positive cells, described in AD brains, 36 are absent in the hippocampus of 6-month-old Tg(+/−) rats, but present in aged Tg2576 mice. Differences observed in oxidation of mtDNA mediated by ROS 37 in both AD animal models may be due to different expression levels of the human transgene (Tg2576 mice express mutated human APP at 7-fold while Tg(+/−) rats at 2-fold over endogenous APP) and/or the brain Aβ burden (Tg(+/−) rats show intracellular accumulation while Tg2576 mice extracellular deposits).
Isolated mitochondria from hippocampal homogenates possess a number of disadvantages to identify neuronal specific deficits and organella functionality in the cytoplasmic environment. To overcome this situation, we performed synaptosomal assays, as the synaptosome is by definition derived from neurons. Although isolated synaptosomes were not 100% pure, fluorescence microscopy indicated that broken synaptosomes were undetectable. Our results showed that maximum respiration, obtained in the presence of an uncoupler, was lower in equivalent amounts of synaptosomes (with equivalent mitochondrial content) isolated from Tg(+/−) rats than in those isolated from WT. In a broader sense, we compared the spare respiratory capacity of the synaptosomes and demonstrated the existence of mitochondrial dysfunction under conditions of energetic load. Spare respiratory capacity reflects the ability of mitochondria to meet increased energy demand with increased respiration and it is considered as a primary factor to define survival of neurons under stress. 38 Synaptosomes from Tg(+/−) rats also presented a decrease in the respiratory control ratio, and this parameter (sensitive to substrate oxidation and proton leak) also supports the existence of mitochondrial dysfunction, and has the virtue of internal normalization. 21 In addition, the examination of the ECAR profile of synaptosomes showed that the nerve terminals from Tg(+/−) rats are unable to compensate for the loss of mitochondrial respiration with glycolytic activity. A shift towards glycolytic energy production was suggested to be a protective “short-term” mechanism in neuronal tissue that eventually leads to a “long-term” bioenergetic deficit and cell death. 39 In this sense, our results ex vivo are opposite to previous findings showing that an increased flux through glycolysis “protects” primary neuronal cultures from death induced by Aβ. 40 Respiratory properties of synaptosomes from commonly used Tg mouse models of late-AD have been reported and showed no consistent bioenergetic defects as compared to WT mice. 13 By contrast, here we show that, similar to what was detected at early stages in AD brain, 41 synaptosomes isolated from Tg(+/−) rats show respiratory dysfunction suggesting that, in this model, interaction of oligomeric Aβ with mitochondria at the synaptic level may be responsible of the bioenergetic failure. In addition, synaptosomes of Tg(+/−) showed reduced respiratory chain complex I catalytic activity but not complex II or α-KGDHC as compared to WT rats. Together this profile suggests in Tg(+/−) rats a pattern of differential sensitivity of mitochondrial enzymes to oxidative damage similar to what has been found in AD brains at early stages of the disease. It was previously reported that when one of the components of α-KGDHC was knocked-down in a Tg mouse model of amyloid deposition, increments in ROS and protein tyrosine nitration were observed. 42 Our results suggest that iAβ accumulation generates ROS by a mechanism different from α-KGDHC inhibition. Experimental evidence suggest that PGC-1α is a powerful regulator of ROS removal by increasing the expression of numerous ROS-detoxifying enzymes with a critical impact in aging and AD. 43 In agreement with this concept, here we showed that in Tg(+/−) rats hippocampal level of PGC-1α is reduced, and the amount of H2O2 increased and mitochondrial metabolism impaired. Moreover, the identification that PGC-1α mRNA expression is reduced in Tg(+/−) brain as compared to WT, reinforces the concept that this pathway has a functional significance in mitochondrial activity and AD pathology. Synaptic loss correlates with cognitive impairment in demented patients. 44 However, we showed that the number of synapses per volume is not affected at CA1 in Tg(+/−) rats where iAβ accumulates. Since CA1 is a critical region of the cortico-hippocampal system for memory processing, our results suggest that functional rather than structural changes may account for the memory impairments in this animal model. It was recently reported that in a setting of energy failure due to mitochondrial dysfunction, neurons may attenuate neurotransmitters endocytosis to preserve ATP consumption. 45 In this regard, defects in vesicle recycling have been linked to short-term synaptic plasticity, memory, and cognitive impairment. 46 Alternatively, known mechanisms of Aβ toxicity include the disruption of intracellular Ca2+ homeostasis and over stimulation of synaptic and extra-synaptic glutamatergic receptors 47 with potential impact in behavior without gross synaptic integrity alterations. Recently, a longitudinal testing of hippocampal plasticity in freely behaving McGill-R-Thy1-APP homozygous animals revealed an age-dependent and persistent inhibition of long-term potentiation without a change in baseline synaptic transmission in the CA1 area suggesting that synaptic long-term potentiation is selectively disrupted in these animals prior to extracellular deposition of Aβ. 48 These in vivo experiments are in agreement with our in vitro biochemical tests and corroborate the impact of the metabolic impairments in cognition and behavior. Brain hypometabolic state occurs in patients with mild cognitive impairment and was also observed in Tg mouse models of AD with cerebral amyloid angiopathy (arcAβ mice) prior to the detection of Aβ plaque pathology. 49 This evidence suggests that neurotoxic soluble Aβ species may disrupt brain blood flow compromising the supply of oxygen and essential metabolic precursors. 50 It is important to note that Tg(+/−) rats do not show cerebral amyloid angiopathy detectable by immunohistochemistry. Further analysis should be performed in isolated microvessels in order to evaluate the existence of vascular Aβ in these animals and its potential impact on hippocampal mitochondrial deficits.
At the level of the synaptosome, our results showed that the restricted electron transport capacity, possibly due to complex I inactivation, results in a slow progressive bioenergetic failure of a sub-population of vulnerable terminals when challenged with an increased energy demand. This phenotype could be, in part, responsible for the poor performance in the hippocampal dependent tasks.
Interestingly, the treatment with BioPQQ prevented the cognitive impairment as shown by the performance in the Y-maze and in the probe trial of the MWM test. Our results are in accordance with previous reports showing that PQQ treatment improves learning abilities in the MWM and prevents reference memory deficits when rats were subjected to hyperoxia. 31 Moreover, PQQ blocks spatial learning and memory deficits associated with aging 51 in rats. Furthermore, administration of PQQ in combination with a low dose of lithium restored the spatial learning and memory impairments in the APP/PS1 transgenic mouse model of AD. 52 In the present study, we showed that the treatment with BioPQQ partially restored the bioenergetic synaptic deficits in Tg(+/−) rats by mechanisms beyond mitochondrial biogenesis suggesting that other reported neuroprotective effects of BioPQQ such as its anti-oxidant effect, 31 its interaction with the NMDA receptor redox modulatory site, 53 and/or the stimulation of nerve growth factor production could underlie the prevention of bioenergetics and cognitive deficits.
In sum, to our knowledge this is the first report showing bioenergetics deficits in synaptosomes from a rodent model of AD and we propose that this rat model could provide insight into the bioenergetic changes which occur in vivo at early-stages of the disease, reinforcing the utility of this model as a platform for understanding the earlier biological basis of this progressive neurodegenerative disease and its pharmacologic manipulation.
Footnotes
Funding
Grants from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET-PIP 378) (to LM), Alzheimer's Association (IIRG-11-205127) (to EMC), Agencia Nacional de Promoción Científica y Tecnológica (PICT 2013-318) (to EMC); Canadian Institute for Health and Research (MOP-97776) (to ACC), Comisión Sectorial de Investigación Científica (CSIC) (to CQ) and Agencia Nacional de Investigación e Innovación (ANII) (to CQ). PVMA and PG are supported by CONICET fellowships. SDC is holder of a Charles E Frosst-Merck post-doctoral fellowship. LM, EMC, and MCL are investigators of CONICET. ACC is holder of a McGill University Charles E Frosst-Merck Chair in Pharmacology.
Acknowledgements
We would like to thank Cecilia Rotondaro and Ágata Fernández Gamba from Fundación Instituto Leloir for their helpful assistance in histologic and imaging process techniques and to Juan Félix López Téllez from Andalusian Centre for Nanomedicine and Biotechnology (BIONAND) for the electron microscopy analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
PVMA, NM and PG performed research; PVMA, CQ and LM designed research; PVMA, CQ, NM, PG, PE, AC, MCL, EMC and LM analyzed data; PG performed statistical analysis; CQ, PE, SDC and ACC contributed with unpublished reagents/analytic tools; PVMA, PG and LM wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.