
Abstract
There is emerging evidence suggesting that a cortical stroke can cause delayed and remote hippocampal dysregulation, leading to cognitive impairment. In this study, we aimed to investigate motor and cognitive outcomes after experimental stroke, and their association with secondary neurodegenerative processes. Specifically, we used a photothrombotic stroke model targeting the motor and somatosensory cortices of mice. Motor function was assessed using the cylinder and grid walk tasks. Changes in cognition were assessed using a mouse touchscreen platform. Neuronal loss, gliosis and amyloid-β accumulation were investigated in the peri-infarct and ipsilateral hippocampal regions at 7, 28 and 84 days post-stroke. Our findings showed persistent impairment in cognitive function post-stroke, whilst there was a modest spontaneous motor recovery over the investigated period of 84 days. In the peri-infarct region, we detected a reduction in neuronal loss and decreased neuroinflammation over time post-stroke, which potentially explains the spontaneous motor recovery. Conversely, we observed persistent neuronal loss together with concomitant increased neuroinflammation and amyloid-β accumulation in the hippocampus, which likely accounts for the persistent cognitive dysfunction. Our findings indicate that cortical stroke induces secondary neurodegenerative processes in the hippocampus, a region remote from the primary infarct, potentially contributing to the progression of post-stroke cognitive impairment.
Introduction
Stroke is the leading cause of acquired disability. 1 Of those who survive, over half remain physically dependent and approximately two-thirds have some form of neurological impairment in the years following stroke. While motor impairments are the most common and widely recognised post-stroke complications, 2 the neurological deficits caused by stroke go well beyond motor outcomes, with up to 80% of stroke survivors suffering from cognitive impairment.3–5 Such cognitive impairment may persist long-term after the ischemic injury, and affect different cognitive domains such as memory, learning and executive functions. 6 A previous history of stroke is known to be a major risk factor for development of dementia. 7 In a recent report from the Oxford Vascular Study, the one-year standardized morbidity ratio for incidence of dementia was found to be 47.3% in survivors of a severe stroke, 5.8% following a minor stroke and 3.5% in those with a transient ischaemic attack compared to age and sex matched population. 8 Given this, a better understanding of the neural mechanisms that underlie the development of persistent cognitive impairment following stroke is crucial for both early detection as well as development of targeted treatments.
It has been previously identified that stroke triggers a wave of secondary damage that causes the progressive and inexorable loss of brain tissue at sites connected to the area damaged by the initial infarction, a phenomenon known as secondary neurodegeneration (SND).9,10 SND develops within days after the primary infarction but may continue for some weeks, months and even years after the initial stroke event, 9 and has been observed in both rodents and humans.11–15 SND has recently been implicated as a potential modulator to a number of late phase functional disturbances.16,17 The key hallmarks of SND include neuronal loss, neuroinflammation and accumulation of neurotoxic proteins.9,10 Previous studies from our group and others have consistently reported the occurrence of these observations in the thalamus and substantia nigra.11–14,18–22 While hippocampal atrophy has been reported after ischemic stroke,15,23,24 little is known about the underlying molecular and cellular mechanisms. This is significant, as the hippocampus is known to be highly interconnected with both the cerebral cortex 25 and thalamus, 26 making it a potential key site for SND following stroke.
Recently, our group has identified that photothrombotic stroke within the motor and somatosensory cortices produces significant impairment in a hippocampal-dependent visual discrimination task using touchscreen based assessment at 14 days post-stroke. 27 Interestingly, although our cortical stroke model does not cause direct damage to the hippocampus, we identified neuronal loss, astrogliosis and increased accumulation of neurotoxic proteins within the hippocampus at 14 days post-stroke. These results clearly demonstrated that, despite the primary infarct being confined to the cortex, the disturbances extended well beyond this to include remote areas such as the hippocampus. While being an important landmark study to build an understanding of the mechanisms behind post-stroke cognitive impairment, this study left a number of important questions unanswered. Firstly, what is the timeline of progression of cognitive deficits following stroke? Secondly, what is the association between post-stroke functional outcomes and the key hallmarks of SND processes? In order to answer these questions, a longitudinal study encompassing both longer time-points and a battery of different cognitive tasks is necessary.
The primary aim of this study was to investigate the long-term impact of a cortical stroke at the motor and somatosensory cortices on motor and cognitive outcomes. Specifically, we assessed motor function using the cylinder and grid walk task, associative memory and learning using the paired associate learning task (PAL) and cognitive flexibility using visual discrimination reversal task (VDR). Our secondary aim was to explore the neuropathological changes for hypothesis generation of potential mechanisms behind these deficits, which could then be tested in subsequent studies. The progress of brain tissue loss was assessed throughout the rostrocaudal extent of the ischemic lesion. Furthermore, we investigated the pattern of spatiotemporal changes of neuronal loss, gliosis and accumulation of amyloid-beta (Aβ) within both the primary cortical infarction sites and ipsilateral hippocampal regions at 7, 28 and 84 days post-stroke.
Materials and methods
See Supplementary Materials and Methods for detailed protocols. The data that supports the findings for this study are available from the corresponding author upon reasonable request.
Experimental design
Animal research was undertaken in accordance with the Animal Research: Reporting of In Vivo Experiments guidelines. Experiments were approved by the University of Newcastle Animal Care and Ethics Committee (A-2013-340) and conducted in accordance with the New South Wales Animals Research Act and the Australian Code of Practice for the use of animals for scientific purposes. All the experimental groups were randomized, and all outcome analyses were performed in a blinded manner. A total of 85 mice (C57BL/6 male, 10 weeks old) were used in this study (Figure 1(a)). Details on animal numbers for each experiment and inclusion/exclusion criteria are provided (Supplementary Figure 1).
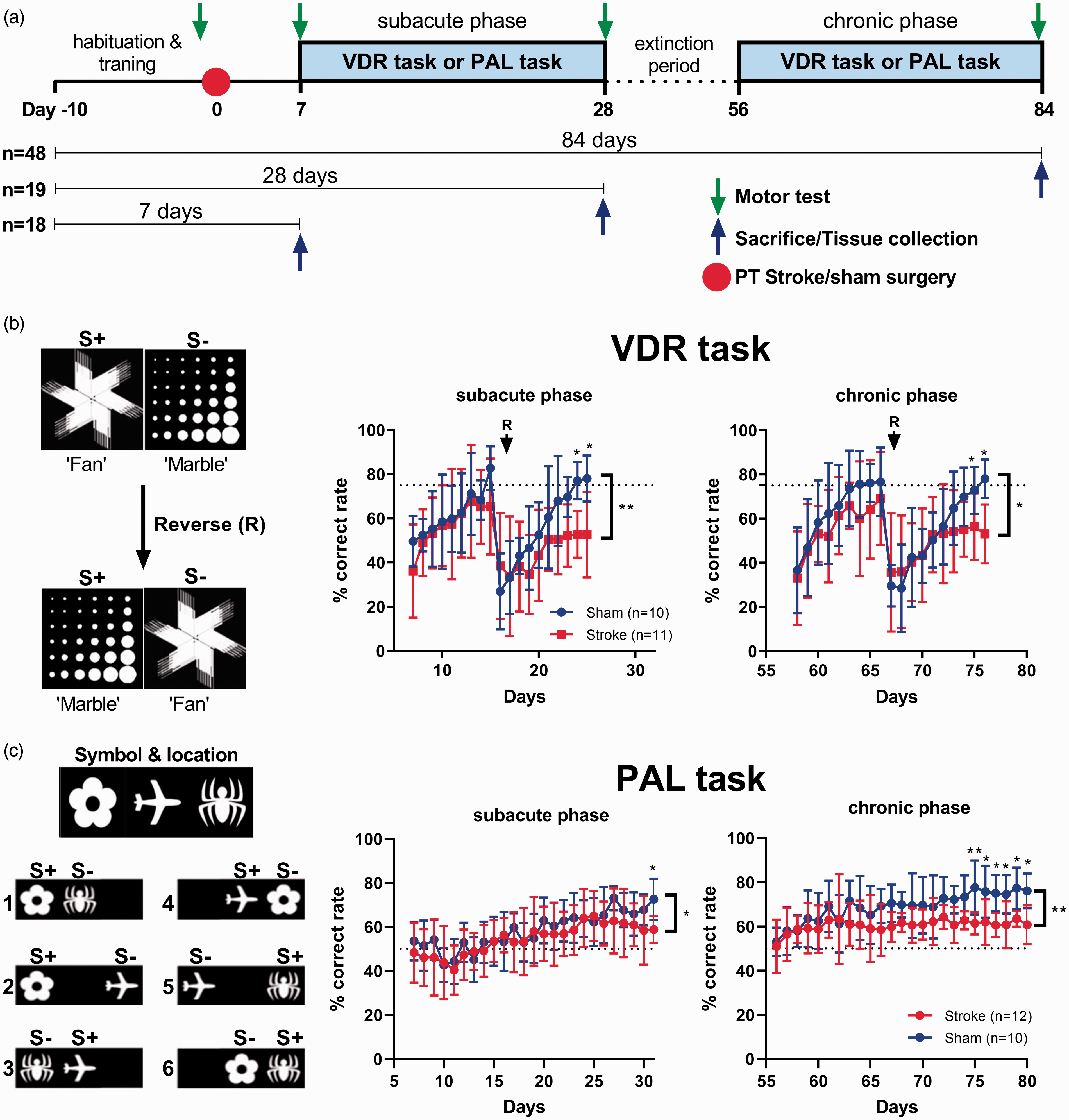
Cortical stroke impairs cognitive function. (a) Experimental design timeline. (b) First cohort of mice underwent assessment on a visual discrimination reversal (VDR) task during both the earlier (sub-acute phase) and later (chronic phase) stages post-stroke. We found a significant decrease in % correct responses in stroked mice compared to sham both in the subacute and chronic phase. (c) Second cohort of mice was assessed on the paired-associate learning (PAL) task in an earlier stage post-stroke (subacute phase) and at a later stage (chronic phase). We found a significant decrease in the % correct responses in stroked mice compared to shams, both in the sub-acute and chronic phases following stroke. Mean±SD (Two-way ANOVA and Sidak's multiple comparisons). *p < 0.05; **p < 0.01. PT: photothrombotic.
The first cohort (sham n = 10, stroke n = 14) were assessed on the VDR task and the brain tissue was used for protein analyses. The second cohort (sham n = 10, stroke n = 14) were assessed using a mouse touchscreen platform for the PAL task and underwent motor testing. The brain tissue from this cohort was used for histological analyses. For both cohorts, at day 0, mice were subjected to either photothrombotic occlusion or sham surgery. At day 7 post-stroke, mice were assessed on a mouse touchscreen platform on either the PAL task or VDR task. Upon completion of the task, mice were allowed to rest for one month (extinction period). At day 56 after stroke, the VDR task or PAL task was re-started. Motor tests were performed on second cohort one day before stroke/sham surgery and at days 7, 28 and 84 post-stroke. Finally, mice were sacrificed at day 84 post-stroke. A third cohort of mice were sacrificed at either 7 days (stroke n = 18) or 28 days (stroke n = 19) post-stroke, and the brains used for subsequent protein or histological analyses for the 1 week or 1 month time points. This cohort was not subjected to functional testing.
Sample size calculation
Sample size was estimated using G*Power 3.1 software. We required n = 10 per group for the first and second cohorts, with an estimated standard deviation, SD = 7 and an effect size of Cohen’s d = 1.7 allowing a type 1 error of 5% with the power of 80%.
Photothrombotic occlusion
Photothrombotic occlusion was performed as previously described. 28
Cognitive testing
Mouse touchscreen operant chambers (Campden Instruments Ltd., UK) were used in the cognitive testing as described with modifications,29,30 and testing was conducted in a blinded and randomised manner. Specifically, associative memory and cognitive flexibility was assessed using the PAL task and the VDR task respectively.
Motor test
Cylinder test and grid walk test were chosen as motor assessments. 31 Motor tests were performed one day before stroke surgery and at days 7, 28 and 84 after stroke.
Tissue processing
Mice were sacrificed at different time points after surgery: 7, 28 or 84 days. Brains were either paraformaldehyde fixed (histological analyses) or fresh frozen (western blotting) as previously described.27,32
Histological analysis
Cresyl Violet and Sudan black staining was performed as previously described.27,33 Free-floating fixed sections were used for immunofluorescence and were co-immunostained using standard protocols as previously described.19,20 The quantitative analysis was undertaken specifically in the peri-infarct territory (Bregma 0.0 mm) and ipsilateral hippocampus (Bregma −1.5 mm). Two areas (0.5 mm by 0.5 mm, corresponding to motor and somatosensory cortices), 0.01 mm from infarct, were selected for the analysis of peri-infarct territory (Figure 3(b)), and the values were pooled (Figures 3(b), 4(a) and 5(a)). Refer to Supplementary Table 1 for the analysis of individual peri-infarct regions. The CA1 and dentate gyrus (DG) sub-regions of ipsilateral hippocampus were selected for analysis (Figure 3(c)).
Protein extraction and Western blotting
Protein homogenates from the peri-infarct and the hippocampus samples were obtained and Western blotting performed as previously described.27,31
Statistics
All data were analysed using GraphPad Prism v7.02. The primary outcome measurement was differences between sham, 7 days, 28 days and 84 days post-stroke. All values reported are mean±SD. Data from tissue loss, western blotting and immunofluorescence labelling were analysed using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc comparison. Cognitive tests and motor tests were analysed using a 2-way ANOVA, followed by Sidak multiple comparisons. P < 0.05 was considered statistically significant.
Results
Cognitive flexibility is impaired long-term after photothrombotic stroke
Seven days after the induction of the stroke targeting the motor and somatosensory cortices, mice underwent assessment on a mouse touchscreen platform for VDR task to assess cognitive flexibility. The task was performed for 25 consecutive days (sub-acute phase, 7-28 day), followed by a one month rest period (extinction period). Then, the VDR task was re-initiated at 56 days post-stroke (chronic phase, 56-84 day) (Figure 1(a)).
During the sub-acute phase, we found a significant decrease in the percentage of correct trials in stroked mice compared to shams (F(1,19)=11.99, p = 0.0026), a significant time effect (F(21,399)=10.72, p < 0.0001) and no significant interaction effect (F(21,399)=1.367, p = 0.1300). In addition, post hoc analysis indicated a significant decrease in the percentage of correct trials in the last two days of the VDR task (p < 0.05, respectively) in stroked mice compared to shams. During the chronic phase, we also found a significant decrease in the percentage of correct trials in stroked mice compared to shams (F(1,19)=7.867, p = 0.0113), a significant time effect (F(18,342)=11.37, p < 0.0001), and no significant interaction effect (F(18,342)=1.083, p = 0.3673). Post hoc analysis indicated a significant decrease in the percentage of correct trials in the last two days of VDR task (p < 0.05, respectively) in stroked mice compared to shams (Figure 1(c)). A range of metrics from the VDR task were also collected for temporal analysis. There were no significant differences between groups either in time to complete 36 trials, the number of total correction trials completed, or the number of trials completed within 60 min (Supplementary Figure 2(c) and (d)).
Associative memory is impaired long-term after photothrombotic stroke
Associative memory was assessed using the PAL task at sub-acute (7-28 day) and chronic phases (56-84 day), as described above (Figure 1(a)). During the sub-acute phase, we found a significant decrease in the percentage of correct trials in stroked mice compared to sham (F(1,20)=4.855, p = 0.0394), a significant time effect (F(4,80)=42.31, p < 0.0001), and no significant interaction effect (F(4,80)=1.888, p = 0.1207). In addition, post hoc analysis indicated a significant decrease in the percentage of correct trials on the last day of the PAL task in stroked mice compared to shams (p < 0.05; Figure 1(b)). During the chronic phase, we also found a significant decrease in the percentage of correct trials in stroked mice compared to shams (F(1,20)=9.925, p = 0.0050), a significant time effect (F(4,80)=16.74, p < 0.0001), and significant interaction effect (F(4,80)=3.261, p = 0.0157). Post hoc analysis indicated a significant decrease in the percentage of correct trials at the last six days of the PAL task (p < 0.05, respectively) in stroked mice compared to shams (Figure 1(b)). A range of metrics from the PAL task were also collected for temporal analysis. There was no significant effect either in time to complete 36 trials or in the number of total correction trials completed. There was, however, a significant decrease in the number of trials completed within 60 minutes in the sub-acute phase in stroked mice compared to shams (Supplementary Figure 2(a) and (b)).
Motor deficits are persistent long-term after photothrombotic stroke, with only modest spontaneous recovery over time
The progression of motor deficits long-term after photothrombotic stroke was assessed using two motor tests: the cylinder task and the grid walk task. We performed these tests prior to stroke to establish a baseline, and at 7, 28 and 84 days post-stroke. Locomotor asymmetry was evaluated using a cylinder task to evaluate the paw preference mice exhibit for stabilising themselves whilst rearing within a cylinder. Data on asymmetry scores indicated that there were no significant differences in paw preference prior to stroke. However, at day 7, 28 and 84 post-stroke, the stroke group exhibited a significantly stronger preference for using their unaffected paw relative to sham (p < 0.001 at day 7, 28 and 84) (Figure 2(a)). Motor function was further assessed using the grid walk task, evaluating the ability of mice to effectively place their paws on an elevated grid during locomotion. There was no difference in the number of foot faults before stroke. At day 7, 28 and 84 post-stroke, the number of foot faults on the affected side (contralateral) was significantly higher in stroked mice compared to shams (p < 0.001 at day 7, 28 and 84) (Figure 2(b)).
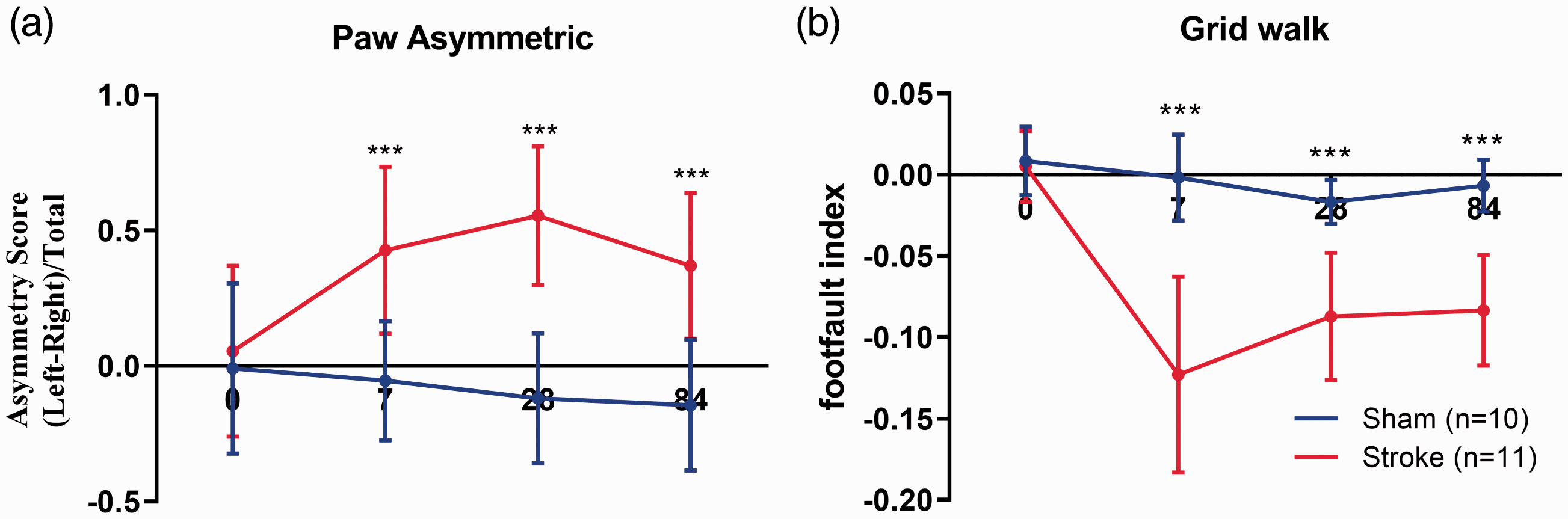
Cortical stroke impairs motor function. (a) Locomotor asymmetry was evaluated at baseline (one day before stroke induction) and at 7, 28 and 84 days post-stroke using the cylinder test. (b) Foot faults were also evaluated using the grid walk test. Stroked mice demonstrated significant deficits in both tests compared to shams, with a slight (non-significant) recovery over time. Mean±SD (Two-way ANOVA and Sidak's multiple comparisons). ***p < 0.001.
Photothrombotic stroke causes persistent brain tissue and neuronal loss
We used Cresyl Violet staining to estimate the tissue loss at Bregma 1.0 mm, 0.0 mm, −1.0 mm and −1.5 mm. Rostrocaudal analysis of tissue loss revealed a significant increase in tissue loss at Bregma 0.0 mm, 1.0 mm, −1.0 mm and −1.5 mm at all time points compared to sham (Figure 3(a)). Peak tissue loss was observed at 7 days post-stroke and then gradually reduced, suggesting some spontaneous recovery from ischemic brain injury. We also evaluated the effect of stroke on white matter tract loss using Sudan Black staining of myelin, given that stroke-induced SND is a disruption of connections between the cortex and hippocampus. Specifically, we focused on the corpus callosum between Bregma 1.0 mm and −1.5 mm, and assessed white matter tract loss as the difference between the contralateral (CL) and ipsilateral (IL) hemispheric area (mm2). We found a significant increase in white matter structural loss in the corpus callosum at Bregma 0.0 mm at 7, 28 and 84 days post-stroke compared to shams. Additionally, we observed white matter loss at Bregma −1.0 mm and −1.5 mm in stroked mice compared to shams at both 7 and 28 days, but not 84 days, post-stroke (Supplementary Figure 3).
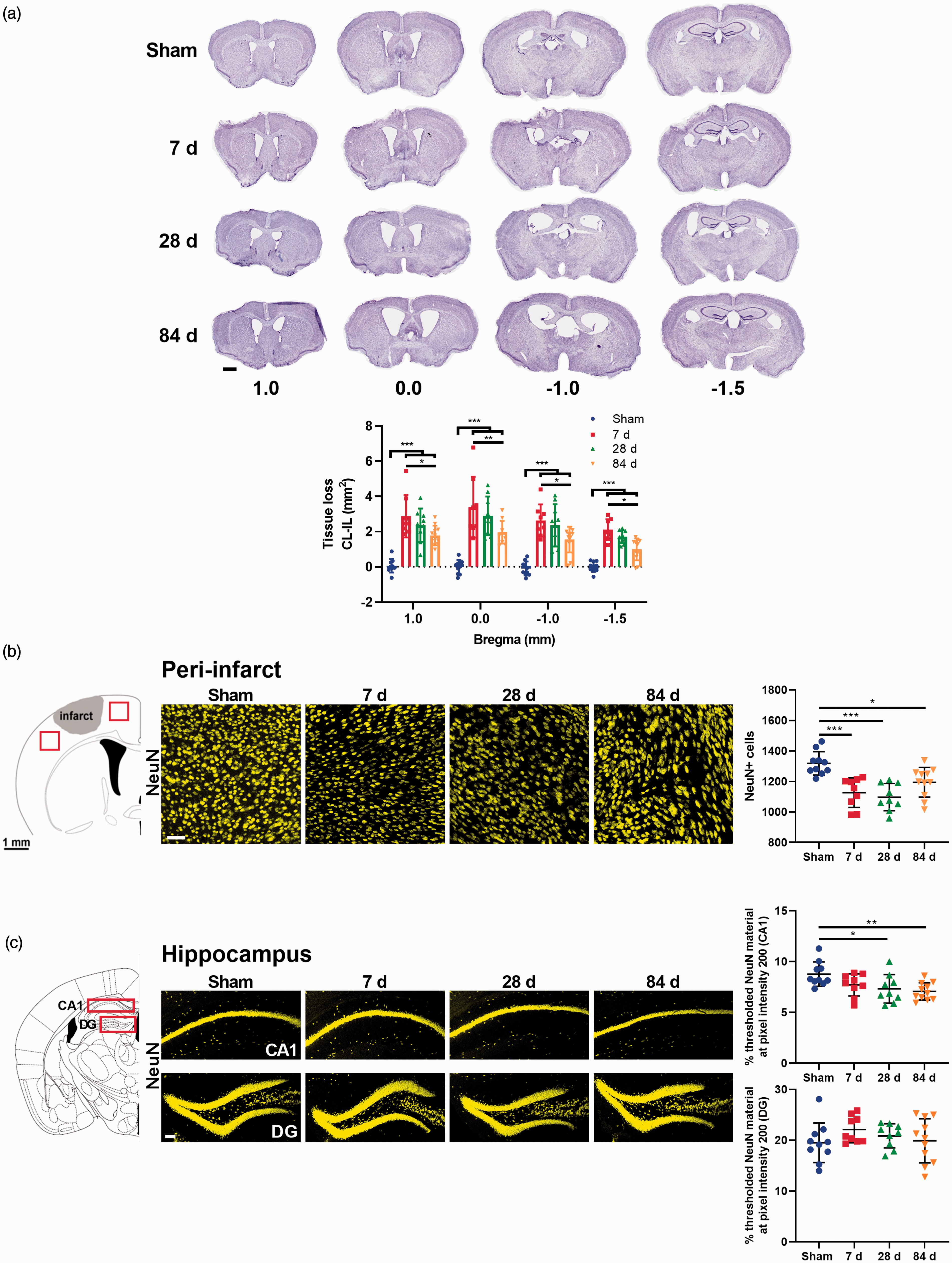
Cortical stroke induces neuronal loss within the peri-infarct area and hippocampus. (a) Representative images of Cresyl violet staining at Bregma 1.0, 0.0, −1.0, −1.5 mm. Tissue loss was calculated as contralateral (CL) hemisphere area−ipsilateral (IL) hemisphere area. Stroke significantly increased tissue loss at all Bregma levels (mm2). (b) Representative immunofluorescence labelling for NeuN in the peri-infarct. Left panel: Red squares represent the area of the peri-infarct region selected for immunofluorescence analyses. The presented values were pooled. Refer to Supplementary Table 1 for the analysis of individual peri-infarct regions. Right panels: higher magnification images (scale bar = 50 μm). (b) Representative immunofluorescence labelling for NeuN in the hippocampus. Left panel: red squares indicate the location of CA1 and dentate gyrus (DG) examined. Right panels: higher magnification images (scale bar = 100 μm). Mean±SD (One-way ANOVA and Turkey’s multiple comparison test). *p < 0.05; **p < 0.01; ***p < 0.001.
We performed immunofluorescence-based cell counting within the primary infarct area and hippocampus using the mature neuronal marker (NeuN) to investigate post-stroke neuronal loss. We performed automated NeuN+ cell count in peri-infarct. Data showed a significant decrease in NeuN+ cells in the peri-infarct of stroked mice at all time points compared to shams (p < 0.001 at 7 days, p < 0.001 at 28 days and p < 0.05 at 84 days) (Figure 3(b)). In the hippocampus cell bodies are very densely packed, making NeuN+ cell counting difficult. Therefore, optical density was assessed using thresholding analyses in both the CA1 and DG subregions of the ipsilateral hippocampus. We found a significant decrease in threshold material for NeuN in the CA1 subregion at 28 (p < 0.05) and 84 days (p < 0.01) in stroked mice compared with shams, however, there were no significant differences within the DG (Figure 3(c)).
Stroke induces transient astrogliosis in the peri-infarct and hippocampus
To study the dynamic change of reactive astrocytes, we immunostained brain samples corresponding to the peri-infarct region (Bregma 0.0 mm) and hippocampus (Bregma -1.5 mm) using the astrocytic marker GFAP. Under normal conditions, astrocytes in the mouse cortex usually express little GFAP. As expected, at 7 and 28 days post-stroke mice exhibited a significant increase in the GFAP-positive area within the peri-infarct region compared to shams (p < 0.001) (Figure 4(a)). However, there were no statistically significant differences between shams and stroke mice at 84 days post-stroke in the peri-infarct. It should be noted that the glial scar is observed in this region. In the CA1 subregion of the ipsilateral hippocampus, we observed a significant increase at days 7 (p < 0.001) and 28 (p < 0.05), but it was reduced to basal levels by day 84 (Figure 4(b)). In the DG, we only observed a significant increase in the % of area covered by GFAP at 7 days post-stroke (p < 0.01) (Supplementary Figure 4(a))
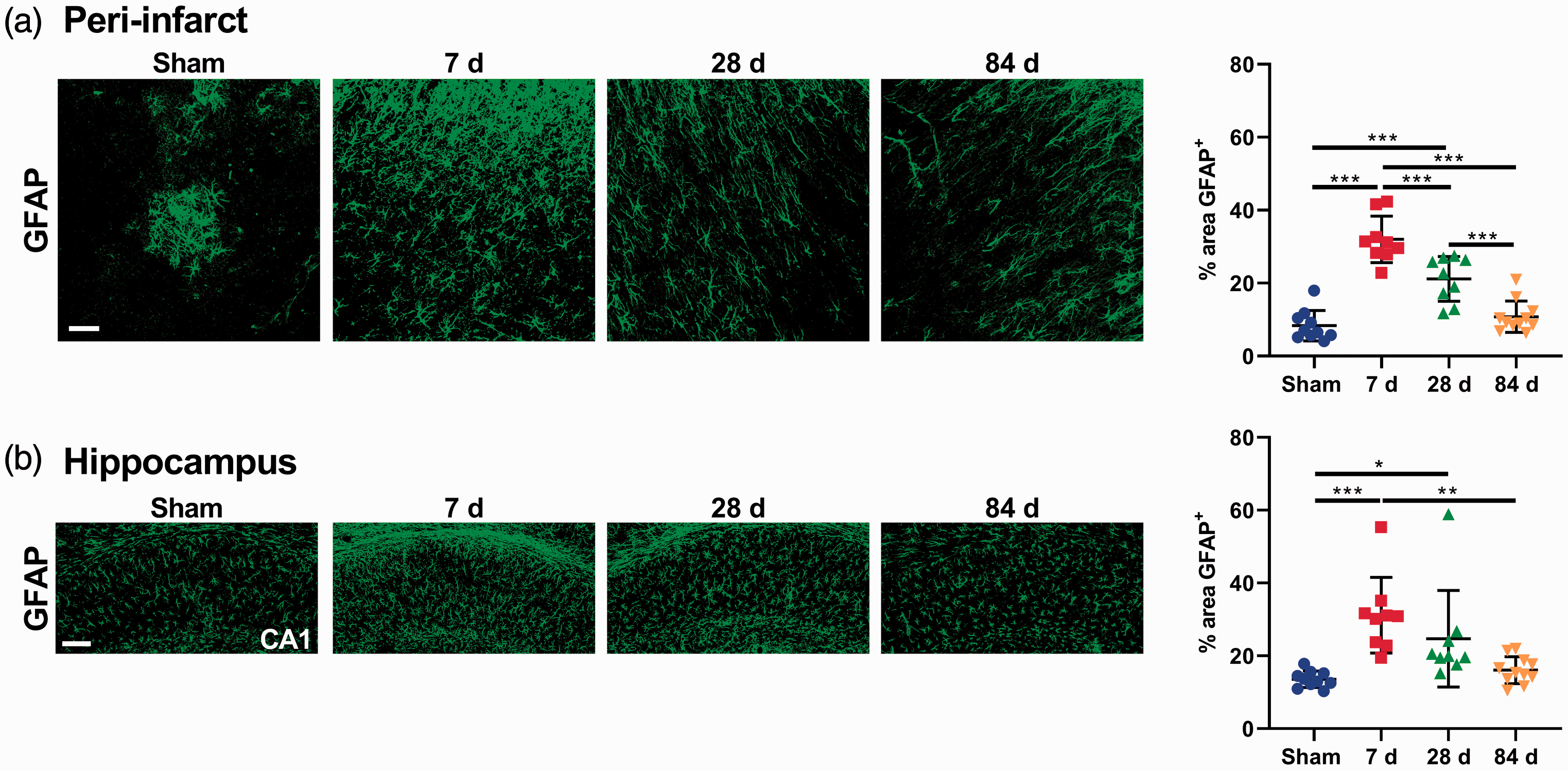
Cortical stroke promotes astrogliosis. (a) Representative immunofluorescence images of peri-infarct area labelled with GFAP (scale bar = 50 μm) and quantification of reactive astrogliosis (% area of GFAP+). (b) Representative immunofluorescence images of the CA1 region of the hippocampus labelled with GFAP (scale bar = 100 μm) and quantification of reactive astrogliosis. Reactive astrogliosis peaked at 7 days in both regions, which returned to basal levels by day 84. Mean±SD (One-way ANOVA and Turkey’s multiple comparison test). *p < 0.05; **p < 0.01; ***p < 0.001.
Persistent microglia activation long-term after stroke in peri-infarct and hippocampus
We performed immunofluorescence labelling of Iba-1, a widely used microglial/macrophage marker, to investigate the long-term changes in microglial activation. High-resolution confocal images were taken, and microglia morphological analyses were subsequently undertaken within the peri-infarct area and CA1 and DG subregions of the ipsilateral hippocampus. Further, we analysed the expression of a microglial transmembrane protein (CD11b) using western blotting.
In the peri-infarct area, morphological analysis indicated that the microglia had undergone a shift toward a classically activated phenotype. Parameters indicating morphological microglial activation (increase in cell number, increased soma area, decreased cell area, decreased cell radius, decreased number of primary branches, branch length, number of branch points and increased cell solidity) were strongest 7 days post-stroke (Figure 5(a)), with similar changes in microglial activation parameters also seen at 28 and 84 days post-stroke. At day 84, parameters including cell number and soma area were not significantly different to sham mice; however, there was a significant difference in all other parameters. These results indicate that there is persistent microglial activation up to 84 days post-stroke in the peri-infarct region. CD11b protein levels were significantly increased at 7 days post-stroke, but were reduced to basal levels by day 28, and further deceased at 84 days post-stroke (Supplementary Figure 6(a)).
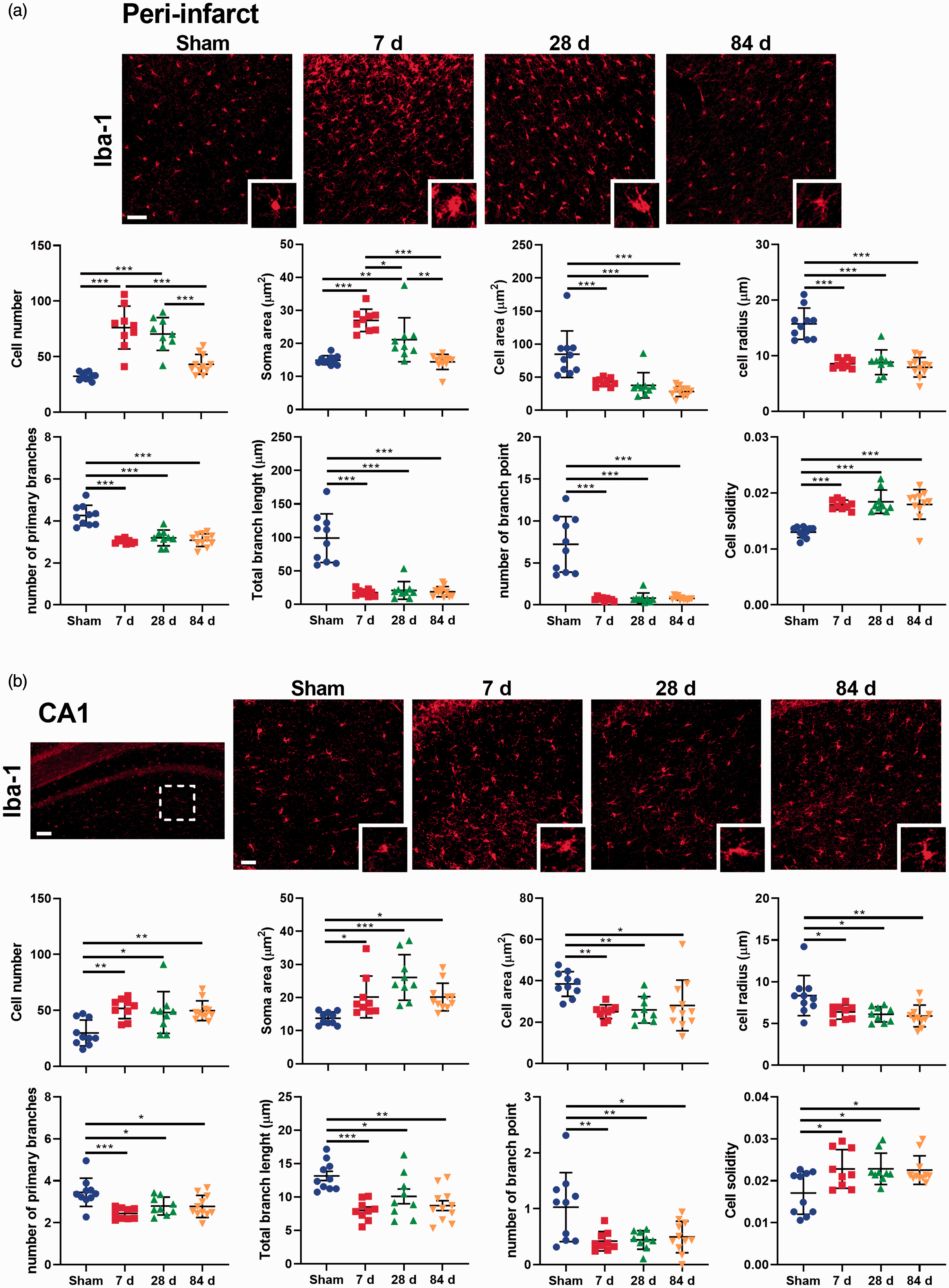
Cortical stroke induces microglial activation (a) Representative immunofluorescence images of the peri-infarct region and quantification of microglia morphology over time (scale bar = 50 μm). (b) Representative immunofluorescence images of the CA1 region of the hippocampus and quantification of microglia morphology over time (scale bar = 100 μm; scale bar of magnification = 50 μm). White dotted square represents the area of morphological analysis. Mean±SD (One-way ANOVA and Turkey’s multiple comparison test). *p < 0.05; **p < 0.01; ***p < 0.001.
Within the ipsilateral hippocampus, microglial morphology analysis also revealed activated microglia within this region. Relative to the sham group, following stroke the microglia within the CA1 area exhibited changes in all the parameters studied. Microglial activation (as indexed by an increase in cell number, increased soma area, decreased cell area, decreased cell radius, decreased number of primary branches, branch length, number of branch points and increased cell solidity) was observed at 7 days post-stroke, and persisted up to 84 days post-stroke (Figure 5(b)). Regarding the DG, we observed a significant increase in the number of cells but there were no significant changes in other parameters (Supplementary Figure 4(b)). Western blotting analysis indicated that CD11b protein levels were significantly increased at 7 and 28 days post-stroke compared with sham mice (Supplementary Figure 6(b)).
Photothrombotic stroke leads to oligomerisation and accumulation of amyloid-β both in the peri-infarct zone and in the hippocampus
We investigated Aβ oligomerisation status using protein homogenates taken from the peri-infarct and the ipsilateral hippocampus. Specifically, we quantitated the pentamer (25 kDa), intermediate size oligomers (30 kDa), decamer (50 kDa) and dodecamer (56 kDa) forms of Aβ. These Aβ oligomers are linked to cellular pathology and cognitive decline.34,35 In the peri-infarct area, we observed a significant increase in pentamer, decamer and dodecamer in stroked mice compared with shams at 7, 28 and 84 days (around 10-20 fold increase) (Figure 6(a)). In the ipsilateral hippocampus, we observed a significant increase in dodecamer at all time points in stroked mice compared to shams (around 2 fold increase) (Figure 6(b)).
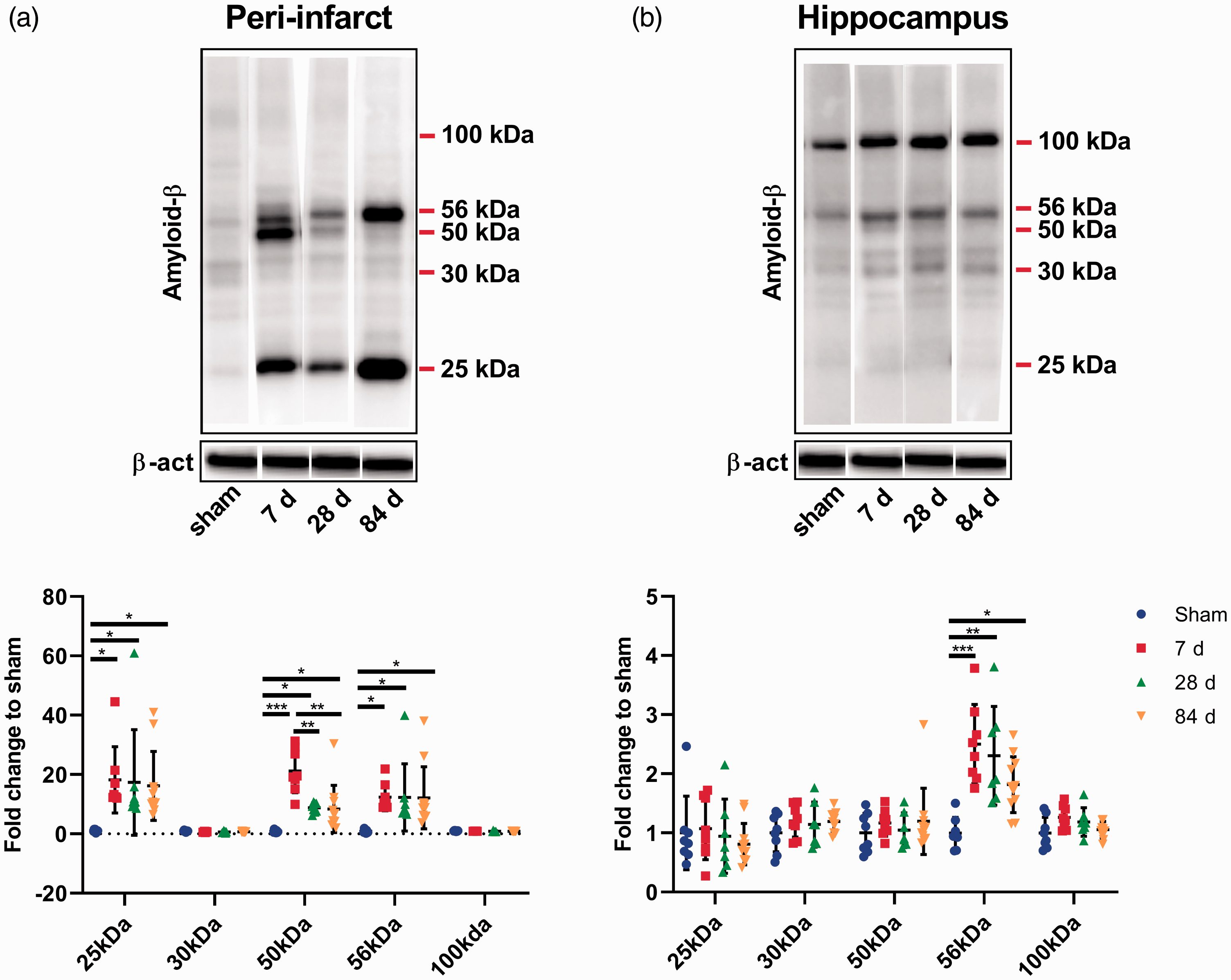
Stroke leads to amyloid-β (Aβ) oligomerization. Our analyses focused on different molecular weight oligomers (25 kDa, 30 kDa, 50 kDa, 56 kDa and 100 kDa). (a) Representative western blot and quantification of Aβ expression profile within the peri-infarct region. Stroke leads to significant increase in Aβ 25 kDa, 50 kDa and 56 kDa at 7, 28 and 84 days. (b) Representative western blot and quantification of Aβ expression profile within the hippocampus. Stroke leads to significant increase in only Aβ 56 kDa at 7, 28 and 84 days. Mean±SD (One-way ANOVA and Turkey’s multiple comparison test). *p < 0.05; **p < 0.01; ***p < 0.001.
To further assess the spatial distribution and accumulation of Aβ, we immunostained brain sections from different Bregma regions corresponding with the peri-infarct region and ipsilateral hippocampus using an antibody against Aβ. As expected, limited immunofluorescence signal was observed within the peri-infarct region and hippocampus in shams. We found a significant increase in Aβ immunoreactivity in the peri-infarct region of stroked mice at all time points compared to shams (p < 0.001 at 7 days, p < 0.001 at 28 days and p < 0.01 at 84 days, Supplementary Figure 5(b)). At day 84, we identified a change in the Aβ distribution pattern, from a scattered distribution to accumulation around the cerebral vessels (Figure 7(a)). In the CA1 subregion of the ipsilateral hippocampus, we detected a significant increase in Aβ immunoreactivity at all time points post-stroke (p < 0.001 at 7, 28 and 84 days, Supplementary Figure 5(b)). At day 84, Aβ was observed to accumulate around the cerebral vessels in the CA1, but to a lesser degree compared to the peri-infarct region (Figure 7(b)). We only observed a significant increase in Aβ immunoreactivity in the DG at 84 days in stroked mice compared with shams (p < 0.001, Supplementary Figure 5(b)).
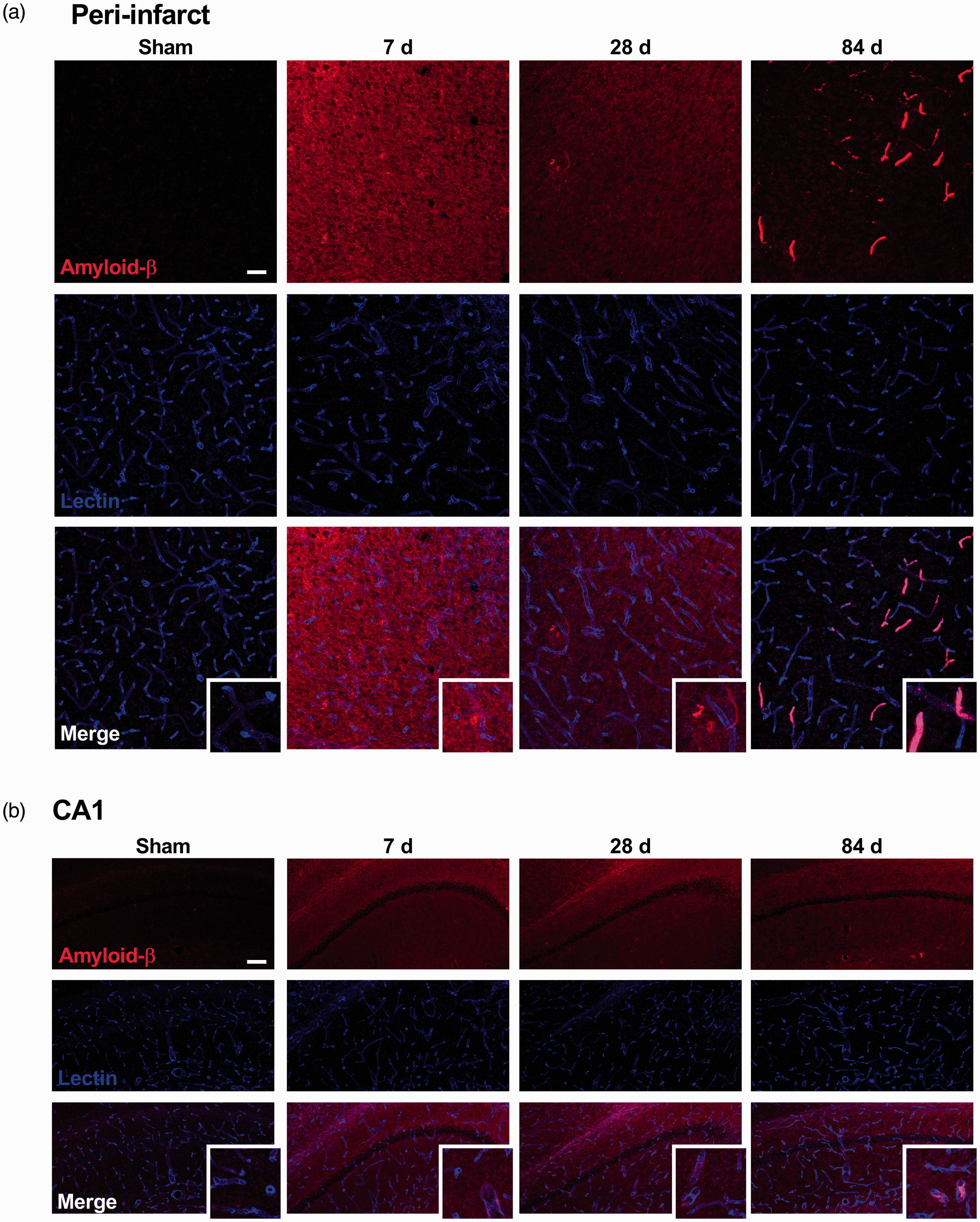
Stroke leads to amyloid-β (Aβ) aggregation which changes in distribution over time. (a and b) Representative immunofluorescence images of Aβ immunostaining (red) and Lectin (blue) and high magnification detail (peri-infarct scale bar = 50 μm, CA1 scale bar = 100 μm). Aβ expression switches from a scattered distribution in the parenchyma of the brain up to 28 days post-stroke to a localisation around the vessels at 84 days post-stroke in the peri-infarct region and in the CA1 sub-region of the hippocampus. At day 84, Aβ was observed to accumulate around the cerebral vessels in the peri-infarct region, and a lesser degree in the CA1 subregion of the hippocampus. For cumulative threshold analysis of Aβ immunostaining, see Supplementary Figure 5B.
Discussion
The main finding of the present study is that cortical photothrombotic stroke gives rise to a persistent impairment in associative memory, as well as in cognitive flexibility, up to 84 days after the initial stroke. Cognition, as assessed by a mouse touchscreen platform for the PAL and VDR tasks, showed significant and persistent deficits long-term following stroke. To the best of our knowledge, this is the first study to demonstrate persistent impairment in VDR task following a unilateral cortical photothrombotic stroke at the motor and somatosensory cortices. In contrast, motor impairment slightly improved over time after the cortical photothrombotic stroke. Secondly, we observed significant tissue and neuronal loss post-stroke that persisted for at least 84 days in both the peri-infarct region and in CA1 subregion of the ipsilateral hippocampus. We also investigated the involvement of neuroinflammation post-stroke and found that microglia activation persisted for up to 84 days, while astrogliosis peaked at 7 days and then decreased overtime. Lastly, we showed an increase in Aβ soluble oligomers in both regions following stroke, and a shift in the spatial distribution of these aggregates towards the cerebral vasculature. Collectively, these novel observations provide compelling evidence that stroke affects multiple cognitive domains and that these deficits are persistent, whereas motor function shows modest recovery overtime. Such cognitive deficits were associated with concomitant neuropathology in the ipsilateral hippocampus, including neuronal loss, activation of microglia and accumulation of Aβ, despite of this region being remote from the site of primary infarct.
We used a photothrombotic stroke model targeting the motor and somatosensory cortices of mice. As expected, cortical photothrombotic stroke resulted in a clear impairment of motor function at 7, 28 and 84 days post-stroke, which is consistent with previous studies.36,37 Following stroke, mice showed preferential use of the unaffected forelimb and an increase in foot faults when using the impaired limb, with the greatest motor deficits observed on post-stroke days 7 (grid walk task) and 28 (paw asymmetry). At post-stroke day 84, we observed a modest spontaneous recovery of motor function. Concomitantly, Cresyl Violet staining showed peak tissue loss at day 7 post-stroke, before being reduced thereafter. We also observed a gradual loss of neurons in the peri-infarct area up to day 28, and then a gradual increase at day 84. This is consistent with the functional remapping of the sensorimotor cortex that we hypothesise to be responsible for the slight recovery in motor function seen in the chronic phase post-stroke in our model. Such remapping within the peri-infarct region is driven both by axonal sprouting of new connections and by the birth of new neurons, which migrate from the subventricular zone to the cortex of the peri-infarct region. 38 Indeed, neurogenesis can be stimulated by cerebral ischemia, with previous work demonstrating that the number of neural progenitor cells is significantly increased in the peri-infarct region at 3–7 days post-stroke in the photothrombotic model.39,40 Further, at 56 days following photothrombotic stroke, mice showed a functional reorganization of the sensory cortical maps for both the forelimb and hindlimbs, as well as a modest but significant increase in motor neuron sprouting has been observed. 41 This was accompanied by the recovery of performance on a skilled reaching task, although interestingly, in contrast to the current study, deficits on both the cylinder and grid-walking task remained evident, suggesting that different types of sensorimotor function may recover at different rates depending on stroke size and location. 42 Promisingly, emerging studies have shown that the modulation of GABAergic signalling using pharmacological approaches37,43 or optogenetic stimulation 44 after stroke can promote motor recovery.
Neuroinflammation is an important process that occurs in the brain after cerebral ischemia, encompassing the activation of astrocytes and microglia. This response is at first beneficial and necessary to promote brain repair; however, it becomes detrimental when it is prolonged.45–47 In this study, we observed an intense astrogliosis response, as characterized by the excessive expression of GFAP, at 7 and 28 days post-injury in the peri-infarct region, which subsided by day 84. Indeed, astrogliosis is an early response in the days following stroke, which evolves to form the mature glial scar forms to wall off the infarcted tissue. 48 Next, we comprehensively assessed the alterations of microglial morphology using Iba-1 immunolabeling and the expression of microglial transmembrane protein, CD11b. Our in-depth analysis of the microglial response revealed that microglial activation status peaked at 7 days post-stroke in the peri-infarct region, before shifting to a less activated state by day 84, a similar pattern to the observed astrocytic response. We observed a concomitant loss of neurons in the peri-infarct area up to day 28, and then a gradual increase at day 84. The close temporal correlation between the activation of astrocytes and microglia and neuronal loss highlights the potential importance of astroglial- and microglial-mediated neuroinflammatory responses as a modulator of neuronal survival. These results suggest that reduced neuroinflammation as well as brain tissue and neuronal recovery in the peri-infarct region over time most certainty contribute to the spontaneous motor recovery.
Next, we examined the long-term impact of cortical stroke on cognition. In our previous study, we demonstrated that our stroke model induced impairment in the ability of mice to discriminate between two images with a high degree of similarity at 14 days post-stroke. 27 Further, this photothrombotic stroke model has been reported to elicit long-term deficits in learning and memory, as assessed by the Morris water maze. 49 Critically, given that stroke affects a range of cognitive domains, and these effects are known to be persistent long-term, we sought to further understand the devastating effects of stroke on cognition, by using a mouse touchscreen platform. 50 This mouse touchscreen platform is recognised to avoid some of the limitations associated with classical assessments. For instance, the “classical gold standard” Morris water maze is hampered by significant interpretational challenges, the aversive nature of the protocol, and shares limited resemblances with human cognitive assessments.51–53 Specifically, in the current study, we used the translationally relevant mouse touchscreen platform to assess different cognitive domains. The PAL task assesses associative memory domain. 54 This task has been extensively used in the clinical assessment of cognition, 55 and the task available for rodents is analogous to the one used in clinical research. 56 We have recently identified similar PAL task impairment in both humans and mice in chronic phase after stroke. 57 Further, we demonstrated that the PAL task is sensitive to detect the effects of pharmacological/therapeutic interventions on cognitive recovery in mice after cortical photothrombotic stroke.29,58 On the other hand, the VDR task includes both visual discrimination and reversal learning,30,59 with discrimination learning providing a measure of perceptual ability to discriminate between two stimuli, while reversal learning has been used to examine cognitive flexibility. 30 One of the critical variables that we were interested in evaluating in this study is whether the cognitive deficits observed at the subacute phase (7-28 day) would persist or further decline when mice were tested in the chronic phase (56-84 day). We observed a significant decrease in the percentage of correct trials in the subacute phase in stroked mice. Furthermore, when these mice were tested in the chronic phase months after stroke, we detected an even greater reduction in the percentage of correct trials in stroked mice compared to shams. While we observed modest spontaneous motor recovery after stroke in mice, a novel and important observation is that a stroke within the motor and somatosensory cortices resulted in persistent cognitive impairment (up to 84 days). Such progressive and persistent impairment in cognitive function mirrors that observed in stroke patients.3,7,60 For instance, Levine et al. demonstrated that stroke was associated with long-term cognitive dysfunction over 6 years after the event. 3
Previous research has shown that the PAL task is dependent on the dorsal hippocampus, 61 while the hippocampus is also known to play a role in the VDR task. 62 The hippocampus receives highly processed multi-modal sensory information from the cortex via its connections with the entorhinal cortex, making it a potential key site for the development of SND pathology. 25 Additionally, the hippocampus is also highly inter-connected with nuclei of the thalamus, such as the anterior and mediodorsal nuclei, which are known to play a critical role in cognitive function. 26 Given that the thalamus is known to be a key site for SND pathology, it is possible that Wallerian degeneration from the thalamus to the hippocampus may drive the development of SND in this area. Although, currently little is known about the importance of SND processes in the hippocampus long-term after stroke. Previous studies, including ours, have focused on other SND sites, such as the thalamus, or on earlier post-stroke time points.11,18,20,22,63 In our recent study, we documented a reduction in NeuN-positive cells in the CA1 region at 14 days following stroke. 27 Based on this result, we hypothesised that this reduction in neurons in the hippocampus might be an indicator of the persistent cognitive deficits observed post-stroke. Indeed, with the findings of this study we have confirmed this hypothesis, demonstrating persistent neuronal loss at 28 and 84 days post-stroke in the CA1 sub-region of the ipsilateral hippocampus. The persistent loss of neurons over time in the CA1 subregion is a critical finding, due to the integral role that this region plays in cognitive processes such as learning and memory.64,65 Furthermore, although most strokes do not directly affect the hippocampus, it is well established that the CA1 sub-region of the hippocampus is more sensitive to hypoxia-ischemia insults and neurodegeneration than other areas of the brain.23,24,66 Interestingly, we observed the DG sub-region of the hippocampus to be spared, which could be due to stimulation of neural cell proliferation within the DG after stroke. 67 Together these results support an important role for SND processes in the hippocampus and may contribute to the ongoing cognitive impairment observed after stroke.
There are several possible contributors to the observed persistent loss of CA1 neurons. We examined neuroinflammation in the ipsilateral hippocampus, observing a peak in reactive astrogliosis at 7 days post-stroke, which returned to basal levels by day 84 in the CA1 sub-region, similar pattern as observed in the peri-infarct region. These results suggest that astrocytes might play a role in neuroinflammation in the early stages post-stroke, but this effect is dampened in chronic phase following stroke. In contrast, microglial activation followed a different pattern, at least within the CA1 subregion of the hippocampus, with activated microglia observed at day 7, and persisted up to 84 days post-stroke. Together, these findings show that neuroinflammation driven by microglia lasts long-term after stroke, whereas the astrocytic response appears to resolve. While microglial activation was already detectable by day 7 post-stroke, we did not observe hippocampal neuronal loss at that time point. Therefore, microglial activation appears to precede neuronal death, suggesting that it may contribute to the delayed neuronal death within the hippocampus by creating a pro-inflammatory microenvironment. It is well described that brain injury and cerebral ischemia switch microglia into a prime state, where they have an increased pro-inflammatory response to secondary damage signals. 68 This is supported by previous experimental stroke studies demonstrating that chronic activation of microglia is detrimental to the survival of new hippocampal neurons and impairs cognition, whereas inhibition of microglial activation improves neurogenesis and cognitive performance. 69 Together, this evidence suggests that a potential exaggerated and prolonged microglial response might contribute to neuronal dysfunction and degeneration within the hippocampus, manifesting as post-stroke cognitive impairment. As microglia are a very dynamic cell population, capable of migrating and changing their morphology in order facilitate a variety of functions, the increased number of microglia could be due to migration from the cortex or in-site activation in the hippocampus. Nevertheless, there is regional heterogeneity in microglia populations that could make it possible to answer this question in future studies by doing single cell RNA sequencing.70,71
Taken together, we have identified important differences between the processes occurring in the primary injury site (peri-infarct) and SND sites (ipsilateral hippocampus). In the peri-infarct region, neuronal loss and neuroinflammation peaked early after stroke and spontaneously recovered at later time points, which could explain the observed motor improvements. Contrary to this, in the ipsilateral hippocampus, a region remote from the primary infarct, neuronal loss and microglial activation persisted out to 84 days post-stroke, supporting our findings of long-term cognitive deficits after stroke.
Furthermore, previous studies have linked Aβ accumulation with cognitive impairment and neurodegenerative conditions.34,35,72 Numerous clinical and experimental studies have now described that Aβ deposition occurs in the peri-infarct region 73 and the thalamus10,74 post-stroke, and that such accumulation of neurotoxic proteins is highly associated with cognitive impairment in stroke patients. 75 Indeed, we have recently reported increased levels of Aβ in the hippocampus at 14 days post-stroke. 27 In the current study we found significantly increased Aβ oligomerization in both the peri-infarct and hippocampal regions after stroke. Interestingly, we observed a consistent increase of the 56 kDa oligomer at all time points in both regions, in keeping with multiple studies which have suggested that these soluble Aβ oligomers, and not the plaques, are linked to cognitive decline in the context of Alzheimer’s disease.35,72 There is emerging evidence suggesting that Aβ oligomers are responsible for neuronal loss after experimental stroke via an uncontrolled feedforward neurodegeneration loop.10,76,77 Aβ oligomers released from ischemic cells can trigger pathological activation of microglia through various pathways, which in turn induce a pro-inflammatory and high oxidative stress microenvironment, both of which lead to neuronal death. 78 For instance, increased Aβ by chronic stress can exacerbate the loss of neurons in sites of SND, 77 whereas reduction of Aβ can alleviate secondary neuronal damage and cognitive function.10,33,58 It should be noted that Aβ oligomers can also evoke pericyte-mediated constriction of cerebral capillaries, and cause cerebrovascular dysregulation. 79 In addition to our protein analyses, we assessed the spatial distribution of these Aβ oligomers using immunostaining. Strikingly, we observed that the distribution of Aβ shifted over time, where at day 7 and 28 the Aβ adopted a scattered distribution in the brain parenchyma, whereas at 84 days post-stroke the Aβ was observed to accumulate around the cerebral vessels. Cerebral amyloid angiopathy or Aβ deposits around the cerebral vessels was observed in the peri-infarct region, and a lesser degree in the CA1 subregion of the hippocampus. Our results complement a previous study by Howe et al. which demonstrated that stroke induces increased perivascular deposition of Aβ, and this effect is aggravated in aged mice at 30 days post-injury. 80 Further, trans-synaptic spread of Aβ pathology from cortex to hippocampus has been reported in Alzheimer's disease models,81,82 and similar mechanism may occur in the context of stroke induced SND. Our findings suggest that Aβ oligomers are not merely a consequence of stroke, but also actively contributing to the death of neurons via various possible mechanisms, leading to post-stroke cognitive impairment.
In conclusion, here we demonstrated that a unilateral cortical photothrombotic stroke at the motor and somatosensory cortices causes impairments in associative memory and cognitive flexibility which persist for months after the initial cortical infarction. Additionally, our spatiotemporal analyses provide further critical insights regarding the cellular and molecular events leading to this impairment in cognition. We demonstrate that there is persistent neuronal loss in an area critical in cognition, the CA1 subregion of the ipsilateral hippocampus. Such neuronal loss and cognitive impairment was associated with persistent neuroinflammation and increased Aβ oligomerization that creates a hostile microenvironment for neuronal survival and proliferation, which come together to create a “perfect storm” of SND post-stroke. Our findings thus highlight the critical importance of microglia and Aβ as potential key mediators in the progression of secondary damage post-stroke. Additionally, we observed a modest spontaneous recovery of the motor function during the chronic phase, which was linked to brain recovery processes occurring in the peri-infarct area. This may be drive spontaneous functional remapping of the peri-infarct region following stroke. This spatiotemporal study has clear advantages by tracking translationally relevant functional outcomes and associated neuropathological changes long-term after experimental stroke. As previous studies have documented changes in the contralateral hemisphere after stroke,20,77,83 it would be of future interest to investigate both hemispheres of the brain. Further research is required to fully elucidate the brain mechanisms that underlie the persistent cognitive deficits seen post-stroke, including how these are related to the development of SND. As such, therapeutic strategies targeting neuroinflammation and neurotoxic protein accumulation may be promising approaches to promote brain recovery and enhance functional outcomes post-stroke.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X211005877 - Supplemental material for More than motor impairment: A spatiotemporal analysis of cognitive impairment and associated neuropathological changes following cortical photothrombotic stroke
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211005877 for More than motor impairment: A spatiotemporal analysis of cognitive impairment and associated neuropathological changes following cortical photothrombotic stroke by Sonia Sanchez-Bezanilla, Rebecca J Hood, Lyndsey E Collins-Praino, Renée J Turner, Frederick R Walker, Michael Nilsson and Lin Kooi Ong in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Acknowledgements
LKO, FRW and MN acknowledge ongoing support from NHMRC Centre for Research Excellence in Stroke Recovery and Rehabilitation. LKO and SSB acknowledge support from Research Advantage for ECR Higher Degree by Research (HDR) Scholarship and Greaves Family Postgraduate Scholarships in Medical Research (HMRI 1054). LKO acknowledge support from IBRO-APRC Travel & Short Stay Grant, International Society for Neurochemistry Career Development Grant and Monash University Malaysia. LECP and RJT acknowledge support from NeuroSurgical Research Foundation and Perpetual.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Hunter Medical Research Institute (HMRI 896), Faculty of Health and Medicine Pilot Grant, Priority Research Centre for Stroke and Brain Injury Research Support Grant, Mary Costello Alzheimer’s Pilot Grant and The University of Newcastle, Australia.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Sonia Sanchez-Bezanilla – involved in the concept and design of the study, performed the majority of the experiments, analysed and interpreted the data, prepared the first draft of the manuscript, revised and edited of subsequent drafts of the manuscript.
Rebecca J. Hood – involved in the experiments, analysed and interpreted the data, revised and edited of subsequent drafts of the manuscript.
Lyndsey E. Collins-Praino – contributed to interpretation of the results, revised and edited of subsequent drafts of the manuscript.
Renée J. Turner – contributed to interpretation of the results, revised and edited of subsequent drafts of the manuscript.
Frederick R. Walker – supervised the study, contributed to interpretation of the results, revised and edited of subsequent drafts of the manuscript, and obtained funding.
Michael Nilsson – supervised the study, contributed to interpretation of the results, revised and edited of subsequent drafts of the manuscript, and obtained funding.
Lin Kooi Ong – involved in the concept and design of the study, involved in the experiments, supervised the study, analysed and interpreted the data, prepared the first draft of the manuscript, revised and edited of subsequent drafts of the manuscript, and obtained funding.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.