
Abstract
The effectiveness of ketogenic diets and intermittent fasting against neurological disorders has brought interest to the effects of ketone bodies on brain cells. These compounds are known to modify the metabolism of neurons, but little is known about their effect on astrocytes, cells that control the supply of glucose to neurons and also modulate neuronal excitability through the glycolytic production of lactate. Here we have used genetically-encoded Förster Resonance Energy Transfer nanosensors for glucose, pyruvate and ATP to characterize astrocytic energy metabolism at cellular resolution. Our results show that the ketone body beta-hydroxybutyrate strongly inhibited astrocytic glucose consumption in mouse astrocytes in mixed cultures, in organotypic hippocampal slices and in acute hippocampal slices prepared from ketotic mice, while blunting the stimulation of glycolysis by physiological and pathophysiological stimuli. The inhibition of glycolysis was paralleled by an increased ability of astrocytic mitochondria to metabolize pyruvate. These results support the emerging notion that astrocytes contribute to the neuroprotective effect of ketone bodies.
Introduction
There is abundant evidence that ketone bodies are neuroprotective. For example, in GLUT1 deficiency, a pediatric condition characterized by impaired glucose delivery through the blood-brain barrier, 1 the treatment of choice is the ketogenic diet.2,3 Ketone bodies, the ketogenic diet and intermittent fasting have also been shown to be neuroprotective in cell models, animal models and clinical cases of epilepsy, Alzheimer’s, Parkinson’s and Huntington’s disease, neurodegenerative conditions characterized by early local deficits in brain glucose consumption. 4 Underscoring the role of glucose metabolism in neurodegeneration, it was recently reported that Alzheimer’s disease is exacerbated by GLUT1 deficiency. 5
The ketogenic diet and prolonged fasting induce the production of ketone bodies by the liver, mostly beta-hydroxybutyrate (BHB) and acetoacetate, which reach millimolar levels in blood and are oxidized in peripheral tissues for the production of metabolic energy. Under normal circumstances the brain is almost exclusively fueled by glucose, but if ketone bodies are available, they are efficiently utilized and a reduction of brain tissue glucose consumption is observed.6,7 The neuroprotective effect of ketone bodies has been linked to changes in glucose metabolism.8,9 While both neurons and astrocytes can efficiently oxidize ketone bodies, 10 their respective roles on the change in tissue glucose consumption provoked by ketone bodies is not well defined. Before reaching neurons most glucose passes through astrocytes 11 where a fraction is converted to lactate. Lactate may sustain neuronal firing and synaptic activity over short periods,12,13 but glucose is an absolute requirement for long-term neuronal function, antioxidation and inhibition of apoptosis.14–16
The question therefore arises as to whether ketone bodies may modulate the consumption of glucose by astrocytes, affecting the delivery of glucose and lactate to neurons. In the present study we have used Förster Resonance Energy Transfer (FRET) sensors for metabolites to investigate possible effects of chronic exposure to the ketone body BHB on the handling of glucose by astrocytes. Our results in vitro and in vivo show a strong inhibitory effect on astrocytic glucose usage, pointing to astrocytic metabolism as a possible target for neuroprotection.
Materials and methods
Standard reagents and inhibitors were acquired from Sigma or Merck. Plasmids encoding the FRET sensors FLII12Pglu700µΔ6, 17 ATeam 1.0318 and Pyronic 19 are available from Addgene (www.addgene.org). Viral vectors Ad FLII12Pglu700µΔ6, Ad ATeam and Ad Pyronic (all serotype 5) were custom made by Vector Biolabs. The adeno-associated virus (AAV9) expressing FLII12Pglu700µΔ6 under the control of the short gfaABC1D promoter was generated at the École Polytechnique Fédérale de Lausanne, Switzerland. Design, production and titration of the AAV9 vector for transgene expression in astrocytes have been described previously. 20 Magnesium Green acetoxymethyl ester (AM) and SNARF-5 F 5 -(and-6)-carboxylic acid AM were obtained from Molecular Probes.
Animals, cultures, stereotaxis and brain slices
Procedures involving animals were carried out according to the Guide for the Care and Use of Laboratory Animals, National Research Council, USA. Procedures were approved by the Centro de Estudios Científicos Animal Care and Use Committee or by the Landesuntersuchungsamt Rheinland-Pfalz, Koblenz (23 177–07). The reports of the procedures comply with the ARRIVE guidelines. Mixed neuronal glial primary cultures (days 8–16) were prepared from mixed F1 1 - to 3-day-old mice (C57BL/6 J x CBA/J), as described previously. 21 To estimate astrocytic glucose in acute hippocampal slices, 2-month-old female mice (C57BL/6 J × CBA/J) were anesthetized with an intraperitoneally injected mixture of ketamine (100 mg per kg bodyweight) and xylazine (10 mg per kg bodyweight). The head was fixed in a stereotactic apparatus and the eyes were kept wet with an ointment (vitamin A eye cream; Bausch & Lomb, Switzerland). A 1.4-mm diameter craniotomy was drilled using a dental drill and 1 µL of AAV9-GFAP- FLII12Pglu700µΔ6 (2.21 × 1014 vg/ml) was injected into the hippocampus. The open skin was treated with Lidocaine (4%) and sutured. After surgery the animals were kept warm, monitored until recovery from anesthesia and returned to their cages. The animals were maintained on a 12-h day/night cycle at constant room temperature with free access to water and standard mouse fodder in the animal facility of CECs. The concentration of ketone bodies in urine samples was determined with Ketone Body Assay Kit (EnzyChrom TM). After 6 weeks, animals were killed by cervical dislocation and 200 -µm-thick coronal hippocampal slices were prepared as described previously. 22 Organotypic hippocampal slice cultures were prepared according to literature 23 with some modifications. In brief, hippocampal slices (400 µm) were cut with a McIlwain tissue chopper (Mickle Laboratory Engineering Company, United Kingdom) from 5 - to 7-day-old C57BL/6 mice under sterile conditions. Slices were maintained on biopore membranes (Millicell standing inserts, Merck Millipore, Germany) in an interface between humidified normal atmosphere (5% CO2, 36.5℃) and culture medium, which consisted of 50% Minimal Essential Medium, 25% Hank’s balanced salt solution, 25% horse serum and 2 mM L-glutamine and 5 mM D-glucose at pH 7.4 in an incubator (Memmert, Germany). The culture medium (1 ml) was renewed every 3 days. After 7 days of culture, the slices were transduced by overnight incubation with 5 × 106 PFU of Ad FLII12Pglu700µΔ6 and imaged after another 4–8 days. To expose cultured cells and slices to ketone bodies, the culture medium was renewed with fresh culture medium containing 2 mM BHB. Fresh medium without BHB was used as control condition. After 3 days of exposure, cells and slices were transferred to a microscope rig and imaged as described below.
Fluorescence imaging
Detailed protocols for the use of the fluorescent sensors for glucose, lactate and pyruvate are available.24–26 Cultured cells and acute slices were imaged with an upright Olympus FV1000 confocal microscope and a 440-nm solid-state laser. Alternatively, cells were imaged with Olympus IX70 or BX51 microscopes equipped with Cairn Research monochromators and Optosplits and either a Hamamatsu Orca or Rollera camera. Cells were superfused at room temperature with a 95% air/5% CO2-gassed solution of the following composition (mM): 112 NaCl, 3 KCl, 1.25 CaCl2, 1.25 MgCl2, 2 glucose, 1 Na-lactate, 10 HEPES and 24 NaHCO3, pH 7.4 and tissue slices with a 95% O2/5% CO2-gassed solution of the following composition (mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1.25 CaCl2, 1.25 MgCl2, 10 glucose and 26 NaHCO3, at pH 7.4. The intact biopore membrane carrying organotypic hippocampal slices was inserted into the recording chamber of an upright microscope (BX50WI, Olympus) equipped with a monochromator (Polychrome IV, Till Photonics), Optosplit (Cairn Research, UK) and a cooled CCD camera (Till Photonics). The slices were superfused continuously with a CO2/HCO3- buffered saline containing (in mM): 136 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 24 NaHCO3, 1.25 NaH2PO4; 2 glucose, 1 lactate, pH 7.4 at room temperature (22–24℃).To measure the permeability mediated by monocarboxylate transporters (MCTs), mixed cultures were loaded with 18 µM SNARF-5 F 5 - (and -6)-carboxylic acid AM for 15 min at 25℃ and imaged with an LSM 700 Zeiss confocal microscope under continued superfusion with a buffer containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 0.5 NaH2PO4, 10 glucose, 10 HEPES, pH 7.4 at room temperature (22–24℃).
Data are presented as means ± SEM, except during exposure to experimental treatments where SEM are omitted for clarity. Metabolic fluxes are expressed as change in amount of substrate per liter of distribution volume per unit time (µM/s). Fluxes may be translated into more traditional units used for isotopic in vitro measurements by taking into account the astrocytic water distribution space (e.g. 6.4 µL/mg of protein 27 ). Comparison with data obtained in intact brain tissue, e.g. FDG-PET, may be achieved by considering the distribution space of glucose in brain tissue (0.77 mL/gram of tissue 28 ). For example, the rate of glucose consumption reported below in protoplasmic astrocytes (7 µM/s; see below) translates into 0.32 µmol/min per gram. As a reference, FDG-PET glucose consumption by mouse brain tissue has been measured at 0.61 µmol/min per gram. 29 Differences between experimental groups were assessed with the Student's t-test. p values < 0.05 were considered significant and are indicated with an asterisk (*).
Results
This work investigated the possible impact of a chronic exposure to ketone bodies on astrocytic energy metabolism. Glucose metabolism was characterized using FLII12Pglu700µΔ6,
17
a genetically-encoded FRET nanosensor for glucose that is insensitive to glucose-6-phosphate.
30
The method for the use of this sensor to quantify glucose consumption is available elsewhere.21,25 In short, the glucose sensor is expressed in cells (Figure 1a) and glucose consumption is measured by FRET microscopy as the rate of cytosolic glucose depletion after addition of cytochalasin B (Figure 1b), a non-competitive blocker of glucose uptake through GLUT1.
31
Chronic exposure of cultured astrocytes to 2 mM BHB, a concentration similar to that observed in plasma during a ketogenic diet, resulted in an 80% decrease of the rate of glucose consumption (Figure 1c).
Chronic BHB exposure inhibits glucose consumption by cultured astrocytes. (a) Expression of the glucose sensor FLII
12
Pglu700µΔ6 in astrocytes of mixed cortical cultures. Bar = 50 µm. (b) Rationale of the method to measure the rate of glucose consumption by monitoring cytosolic glucose in the presence of the GLUT blocker cytochalasin B (20 µM). (c) Measurement of glucose consumption rates of astrocytes exposed for 3 days to control conditions (gray symbols) or to 2 mM BHB (open symbols). Bars represent the average rates obtained in three separate experiments (20–30 cells per experiment).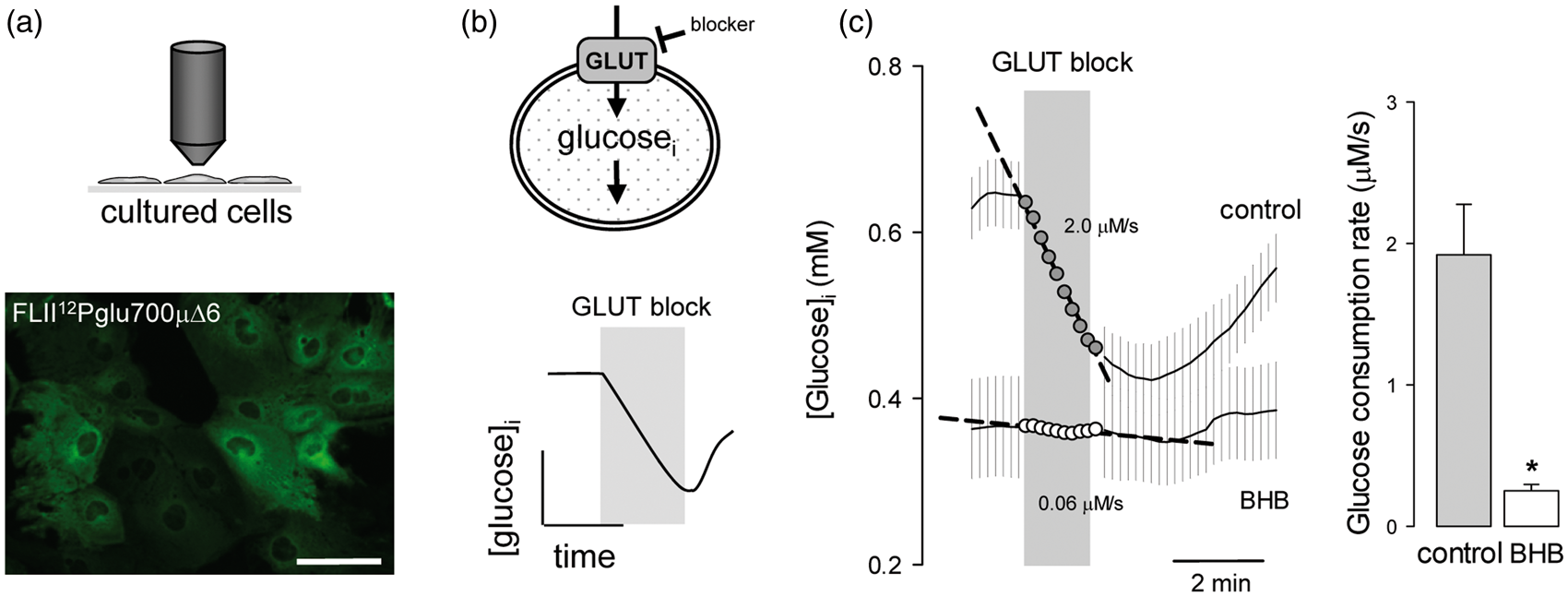
The consumption of glucose and production of lactate by astrocytes is acutely modulated by extracellular K+,32–34 the level of which is proportional to the intensity of excitatory synaptic activity and axonal firing.
35
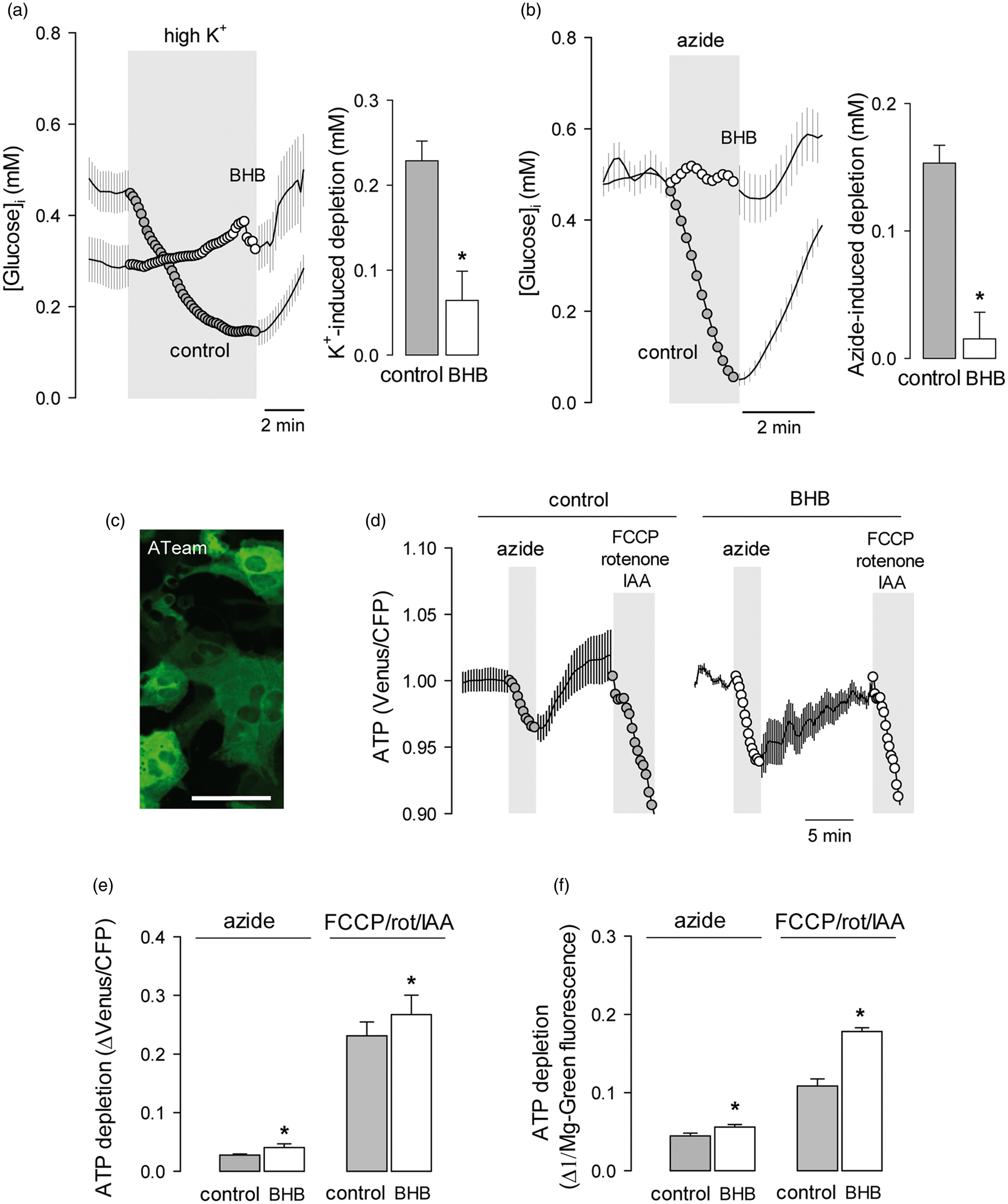
The activation of glycolysis by high [K+] in astrocytes is evidenced as a decrease in cytosolic glucose proportional to the degree of metabolic stimulation. As shown in Figure 2(a), after 3 days of exposure to BHB, astrocytes became less sensitive to high K+ (Figure 2a). The strong stimulation of astrocytic glycolysis that is normally observed in response to inhibition of oxidative phosphorylation (OXPHOS) was also blunted by BHB treatment (Figure 2b), despite significant ATP depletion, as detected independently with the FRET ATP nanosensor ATeam
18
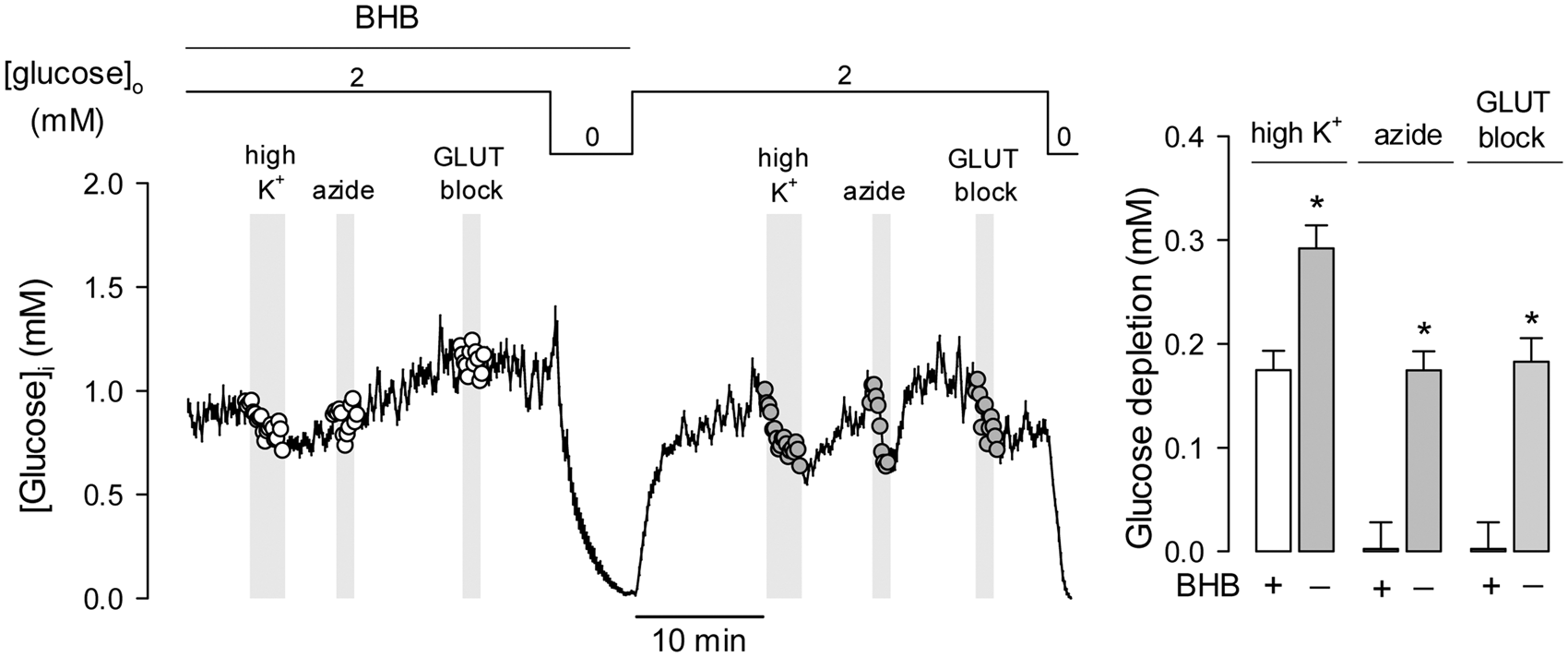
and the fluorescent probe Magnesium Green (Figures 2c–f). The effects of the ketone body were reversible. In the experiment illustrated in Figure 3 astrocytes that had been treated with BHB for 3 days displayed a small response to high K+ and negligible rates of glucose consumption and response to OXPHOS inhibition. Removal of BHB led to a significant recovery of all three parameters, indicating that the metabolic modulation requires the continued presence of BHB and that it involves signals acting in the order of minutes.
Chronic BHB exposure inhibits the stimulation of astrocytic glycolysis by K+ and OXPHOS inhibition. Effect of raising extracellular K+ from 3 to 12 mM (a) or inhibiting OXPHOS with 5 mM azide (b) on astrocytic glucose in control cultures (gray symbols) or in cultures exposed for 3 days to 2 mM BHB (open symbols). Bars represent the average extent of glucose depletion recorded after 5 min stimulation in three separate experiments (20–30 cells per experiment). (c) Expression of the FRET ATP sensor ATeam in astrocytes of mixed cortical cultures. Bar = 50 µm. (d) Effect of 5 mM azide or a cocktail composed of 2 µM FCCP, 2 µM rotenone and 500 µM ioadoacetic acid (IAA) on astrocytic ATP in control cultures (gray symbols) or in cultures exposed for 3 days to 2 mM BHB (open symbols). Data are normalized relative to the beginning of the experiments. (e) Average ATP depletion after 3-min exposure measured with ATeam in three separate experiments similar to those shown in (d) (6–8 cells per experiment). (f) Average ATP depletion after 3-min exposure, measured with the dye Magnesium Green in three separate experiments analogous to those shown in (d) (20–30 cells per experiment). The effects of chronic BHB exposure on astrocytic glycolysis are reversible. Cultures were pre-incubated with 2 mM BHB for 3 days and then sequentially exposed to 12 mM K+, 5 mM azide and the GLUT blocker cytochalasin B (20 µM). After transiently withdrawing glucose for calibration purposes, cultures were superfused with a buffer devoid of BHB, followed by reapplication of K+, azide and cytochalasin. Bars represent the average extent of glucose depletion measured at the end of the stimulations in three separate experiments (20–30 cells per experiment). Previous experiments showed that glucose metabolism in astrocytes is not affected by transient glucose depletion.
21
Ketone bodies are oxidized in mitochondria. To test for possible effects of BHB on mitochondrial metabolism, we measured the capacity of astrocytic mitochondria to metabolize pyruvate using the genetically-encoded FRET nanosensor Pyronic according to a protocol described previously.
19
In brief, cells expressing the sensor (Figure 4a) were first incubated in a buffer containing pyruvate as sole energy substrate, and then exposed to a cocktail of inhibitors of pyruvate transport at the plasma membrane. We have observed that in astrocytes in culture the uptake of pyruvate is partly mediated by an AR-C155858-sensitive MCT
36
and partly by a pathway sensitive to the non-specific inhibitor probenecid (Garrido-Gerter, P. and Barros, L.F., unpublished data). The rate of decrease in the concentration of pyruvate after inhibition of both pathways is therefore equivalent to the rate of pyruvate consumption by mitochondria (Figure 4b and literature
19
). As shown in Figure 4(c), a 3-day exposure of astrocytes to BHB caused a small but significant increase in mitochondrial pyruvate consumption. This enhancement of the ability of BHB-treated astrocytes to metabolize pyruvate suggests that the reduction of glycolysis is not explained by a decrease in pyruvate uptake by mitochondria but by an inhibition of glycolysis, possibly by enhanced mitochondrial ATP production from BHB. The steady-state cytosolic concentration of pyruvate in pyruvate-bathed cells was higher in BHB-treated cells than in control cells (103 ± 3 µM and 81 ± 2 µM, respectively, n = 6 experiments) despite their higher consumption, pointing to parallel upregulation of pyruvate transport at the cell surface. Confirming this notion, BHB treatment significantly increased MCT activity (Figure 4d and e), computed as the rate of intracellular acidification in response to either lactate or BHB.
37
This increase in MCT activity may be interpreted as an adaptive response to the presence of BHB.
Chronic BHB exposure stimulates mitochondrial pyruvate consumption and surface MCT activity. (a) Expression of the pyruvate sensor Pyronic in astrocytes of mixed cortical cultures. Bar = 50 µm. (b) Rationale of the method to measure the rate of mitochondrial pyruvate consumption by monitoring cytosolic pyruvate in the presence of the pyruvate transport blockers AR-C155858 (1 µM) and probenecid (1 mM). (c) Measurement of pyruvate consumption rates of astrocytes exposed for 3 days to control conditions (gray symbols) or to 2 mM BHB (open symbols). Bars represent the average rates obtained in three separate experiments (20–30 cells per experiment). Monocarboxylate transporter activity was measured by monitoring the rate of intracellular acidification in the presence of 10 mM lactate (d) or 10 mM BHB (e) in control cells or in cell treated for 3 days with 2 mM BHB. Bars represent the average rates obtained in six separate experiments (10–20 cells per experiment).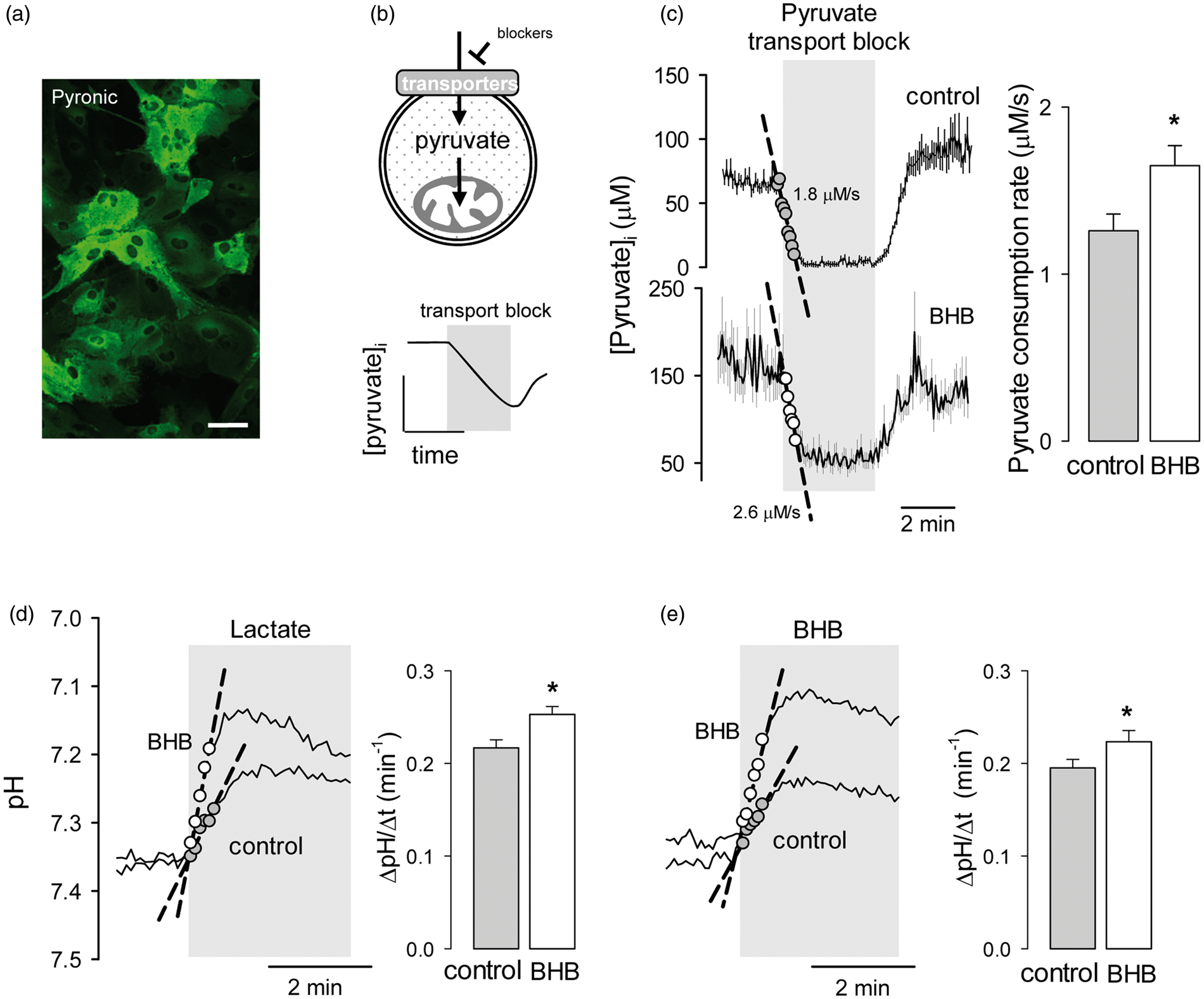
Next, the glucose sensor was expressed in protoplasmic astrocytes in organotypic hippocampal slices (Figure 5a), prepared according to a revised protocol that optimizes the functional and metabolic properties of the tissue.
38
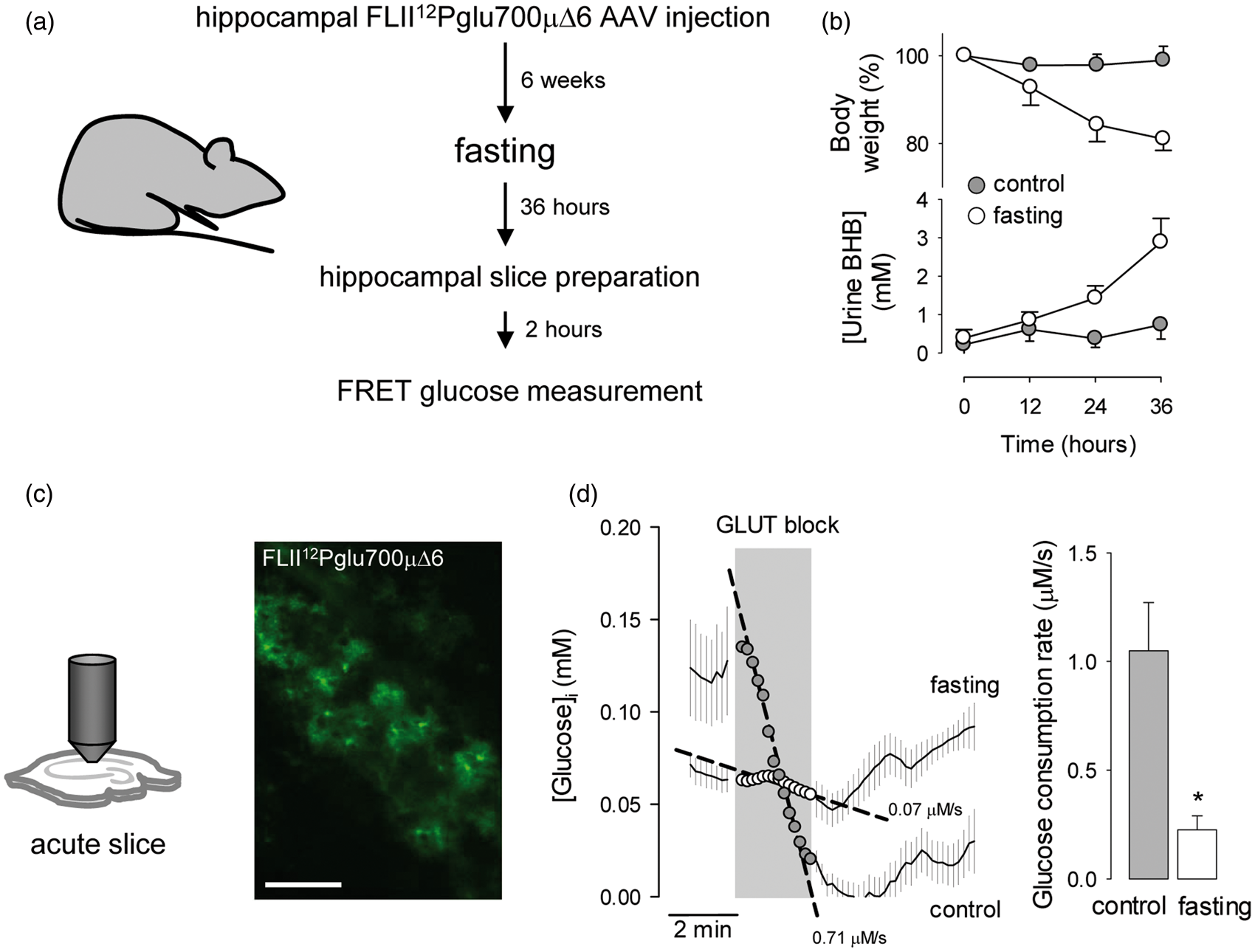
As with cultured cells, a 3-day treatment of tissue slices with BHB provoked a marked inhibition of astrocytic glucose consumption (Figure 5b). To approach the effect of ketone bodies in vivo, the rate of astrocytic glucose consumption was measured by FRET in acute hippocampal slices prepared from ketotic animals (Figure 6a). Firstly, a recombinant adeno-associated virus coding for FLII12Pglu700µΔ6 under the control of the short gfaABC1D promoter was stereotactically injected into the hippocampus of 3-month-old mice. After an expression time of 6 weeks, the animals were food-deprived for 36 h to induce ketosis,
39
which was confirmed by a marked increase in ketone body urine concentration (Figure 6b). At the end of the starvation period, the animals were euthanized and hippocampal slices were prepared,
22
followed by estimation of glucose consumption by FRET microscopy. To prevent loss of ketone body effect (Figure 3), the brain slices of food-deprived animals were prepared in the presence of 2 mM BHB. Food-deprivation had no apparent effect on the general level of activity of the animals. Imaged in acute hippocampal slices, protoplasmic astrocytes showed their typical size and morphology, with small somata and abundant branching processes (Figure 6c). Application of the GLUT block protocol showed a significant decrease in the rate of astrocytic glucose consumption in slices prepared from ketotic animals (Figure 6c).
Chronic BHB exposure inhibits astrocytic glycolysis in organotypic hippocampal slices. (a) Expression of the glucose sensor FLII12Pglu700µΔ6 in astrocytes in organotypic hippocampal slices. Bar = 100 µm. (b) Measurement of astroytic glucose consumption rate with 20 µM cytochalasin B in organotypic slices exposed for 3 days to control conditions (gray symbols) or to 2 mM BHB (open symbols). Bars represent the average rates obtained in three separate experiments (8–10 cells per experiment). Inhibition of astrocytic glycolysis in ketotic mice. (a) Protocol for the estimation of astrocytic glucose consumption in ketotic mice. (b) Effect of food deprivation on body weight and ketone body concentration in urine. (c) Expression of the glucose sensor FLII12Pglu700µΔ6 in astrocytes in acute hippocampal slices. Bar = 100 µm. (d) Measurement of astrocytic glucose consumption rate with 20 µM cytochalasin B in acute hippocampal slices of control (gray symbols) and 36-h fasting mice (open symbols). Bars represent the average rates obtained in three separate experiments (5 –8 cells per experiment).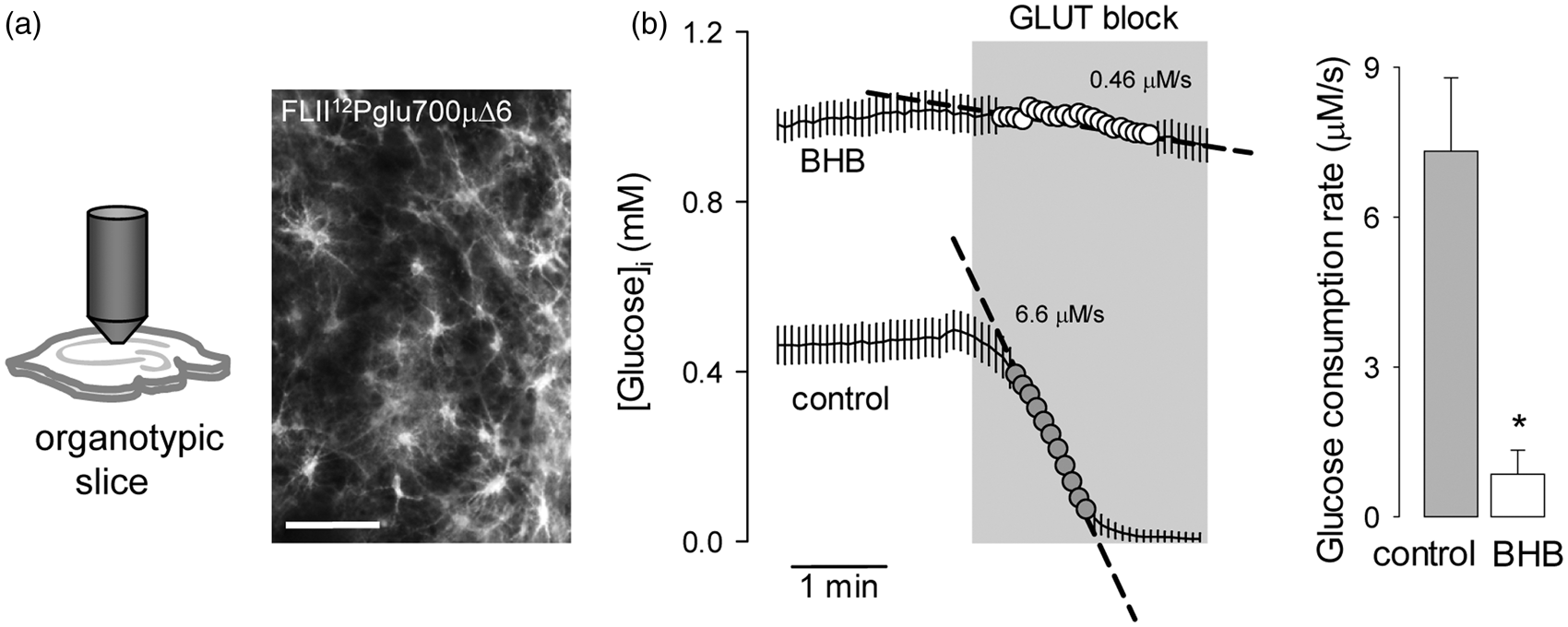
Discussion
We report a strong inhibitory effect of chronic exposure to the ketone body BHB on the consumption of glucose by astrocytes and on the capacity of astrocytic glycolysis to respond to physiological and pathological stimuli. However, the ability of astrocytes to metabolize pyruvate was unaffected by BHB. These findings suggest that the reduction of whole brain glucose consumption observed during ketosis involves astrocytes. Considering the importance of astrocytic glucose and lactate for neuronal function and survival, we propose that the modulation of astrocytic glucose metabolism by ketone bodies contributes to neuroprotection.
The reduction of astrocytic glucose consumption in response to BHB was a robust phenomenon, observed in cultured cells and in protoplasmic astrocytes in organotypic hippocampal slices. Lower astrocytic glucose consumption was also found in acute hippocampal slices prepared from animals that had been made ketotic by food deprivation. Furthermore, BHB severely impaired the ability of astrocytes to upregulate their glycolysis in response to K+, a mechanism whereby active neurons acutely stimulate astrocytic glycolysis and lactate release.32–34 BHB also blunted the response of astrocytic glycolysis to chemical anoxia, despite ATP depletion of similar extent to that observed in control cells. This suppression of the Pasteur Effect suggests that ketone bodies interfere with the regulatory interaction between mitochondria and the glycolytic machinery. As astrocytes consume glucose and are interposed between neurons and the vasculature,
11
a weaker glycolysis in the presence of BHB is predicted to increase the availability of glucose for neuronal usage (Figure 7). A reduced glycolysis may also impair astrocytic lactate export, a process thought to be involved in intercellular signaling and fueling, whose extent is a matter of current debate and investigation. Underscoring the role of glucose metabolism in epilepsy, pharmacological inhibition of lactate dehydrogenase (LDH) decreased neuronal excitability and protected against seizures.
40
Modification of astrocytic glucose dynamics by BHB. Under normal conditions glucose enters astrocytes where a fraction is metabolized to lactate. During ketosis, BHB is efficiently metabolized by mitochondria and astrocytic glycolysis becomes inhibited. This increases the availability of glucose to neurons and reduces their exposure to lactate.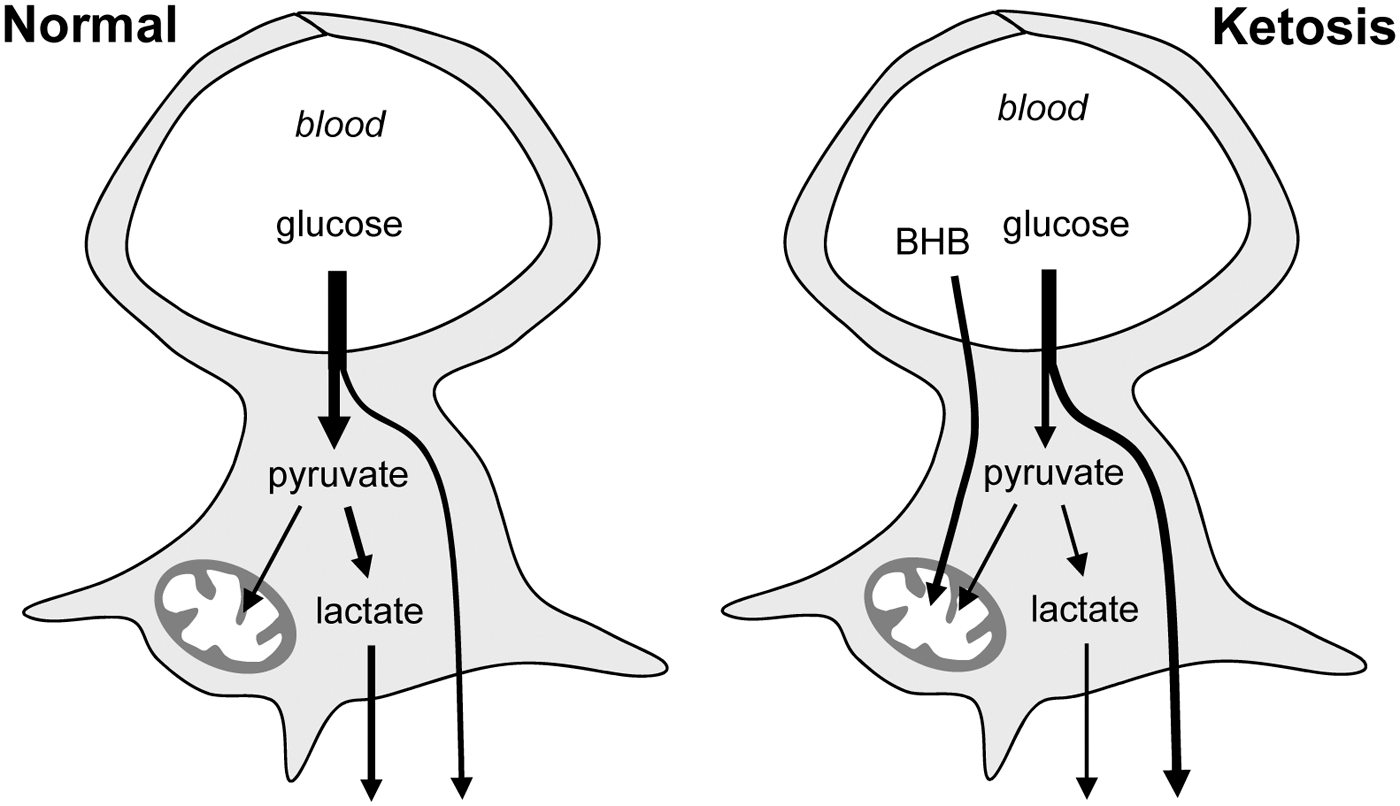
Multiple mechanisms contribute to the neuroprotective effect of ketone bodies. Neurons can effectively utilize ketone bodies,7,10,41,42 leading to the activation of the neuronal potassium channel KATP. 43 Consistently, reduced glucose oxidation in Bcl-2-associated death (BAD) knockout mice conferred resistance to seizures, an effect shown to be mediated by KATP channels. 8 In addition, ketone bodies have been shown to compete with the chloride binding site at synaptic glutamate vesicles, thereby reducing excitatory neurotransmission. 44 The importance of appropriate glucose delivery to neurons is highlighted by the observation that GLUT1 reductions in the blood-brain barrier – but not in astrocytes – exacerbate neurological abnormalities in a mouse model of Alzheimer's disease. 5 In this context, astrocytic glycolysis and astrocytic lactate release and the mechanisms that modulate them in response to neuronal activity may be considered therapeutic targets.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was partly supported by a joint grant from the Comisión Nacional de Investigacion Científica y Tecnológica (CONICYT)-Chile and the Deutsche Forschungsgemeinschaft (DFG) to LFB and JWD (DFG-12, DE 231/25-1) and by Fondecyt 1130095 to LFB. The Centro de Estudios Científicos (CECs) is funded by the Chilean Government through the Centers of Excellence Basal Financing Program of CONICYT.
Acknowledgements
We thank Karen Everett for critical reading of the manuscript and Dr Bruno Weber for kindly providing the FLII12Pglu700µΔ6 AAV vector.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Designed research: IR, HMB, JDW, LFB; Performed research: RV, IR, PG, IF, LF, KA; Analyzed data: RV, IR, PG, IF, LF, HMB, JWD; Wrote the paper: LFB.