
Abstract
Stroke is the second leading cause of death and the most frequent cause of adult disability. Neuronal Per-Arnt-Sim domain protein 4 (Npas4) is an activity-dependent transcription factor whose expression is induced in various brain insults, including cerebral ischaemia. Although previous studies have demonstrated that Npas4 plays a critical role in protecting neurons against neurodegenerative insults, the neuroprotective effect of Npas4 in response to ischaemic brain injury remains unknown. In this study, we used a loss-of-function approach to examine the neuroprotective potential of Npas4 in the context of ischaemic damage. Using oxygen and glucose deprivation, we demonstrated that the knockdown of Npas4 in mouse cortical neurons resulted in increased susceptibility to cell death. The protective effect of Npas4 was further investigated in vivo using a photochemically-induced stroke model in mice. We found a significantly larger lesion size and increased neurodegeneration in Npas4 knockout mice as compared to wild-type mice. Moreover, we also showed that ablation of Npas4 caused an increase in activated astrocytes and microglia, pro-inflammatory cytokines interleukin-6 and tumour necrosis factor alpha levels and a switch from apoptotic to necrotic cell death. Taken together, these data suggest that Npas4 plays a neuroprotective role in ischaemic stroke by limiting progressive neurodegeneration and neuroinflammation.
Introduction
Stroke is the second leading cause of death and the most frequent cause of disability in adults worldwide. 1 Ischaemic stroke is more common than that of haemorrhagic stroke, accounting for an estimated 87% of all strokes. 2 Cerebral ischaemia, which occurs as a result of sudden impairment of blood circulation to an area of the brain, initiates a complex and highly interconnected cascade of cellular and molecular events leading to cellular damage and loss of neurological function. 3 Immediate early genes (IEGs) are induced in response to ischaemic injury and as a member of IEGs, 4 Npas4 has also been shown to be upregulated following focal cerebral ischaemia.5,6
Npas4 belongs to the basic Helix-Loop-Helix (bHLH)–PAS family of transcription factors that have important roles in many essential developmental, physiological and pathological processes. 7 It has been reported that Npas4 controls the development of γ-aminobutyric acid (GABA)ergic inhibitory synapses by regulating the expression of activity-dependent genes and therefore, may play a role in maintaining the balance between excitatory and inhibitory neuronal activities in the brain. 8 Subsequent studies have revealed a functional role for Npas4 in learning and memory formation.9,10 Due to its central role in neuronal connectivity, Npas4 has been linked to autism, depression and cognitive disorders.11,12
Upregulated Npas4 expression has been documented following cerebral ischaemia, seizure and kainate-induced hippocampal injury.5,6,13–15 While the role of Npas4 in these conditions is not fully understood, it has been suggested that induction of Npas4 in response to various brain insults may play an important neuroprotective role in the brain. Although not in the context of ischaemic damage, there is evidence that Npas4 has a neuroprotective function. In vitro studies have shown that Npas4 expression induced by preconditioning with 50 mM potassium chloride for 90 min conferred neuroprotection in F-11 neuronal cells after potassium cyanide treatment and that this protective effect was abolished following knockdown of Npas4 by RNA interference. 14 A similar result was also observed in primary hippocampal neurons preconditioned for 16 h with 50 µM bicuculline (a GABA receptor antagonist that causes bursts of action potentials) to induce Npas4 expression. Stimulation of Npas4 expression was found to be neuroprotective when cells were later challenged with either growth factor withdrawal or staurosporine treatment (a classical inducer of apoptosis). 16
The neuroprotective property of Npas4 in response to acute neurological injury has also been demonstrated in vivo using various animal models. In a rat model of seizure, dorsal hippocampal neurons transduced with adeno-associated virus to overexpress Npas4 were protected against kainic acid-induced cell death with a 92% inhibition of cell death relative to the uninjected contralateral hemisphere. 16 Conversely, mice lacking Npas4 were shown to be more susceptible to neuronal damage in the hippocampus when challenged with kainate. The treatment, which was not fatal for the wild-type littermates, resulted in death in more than one third of Npas4 knockout (KO) mice and furthermore, the remaining mice exhibited severe brain damage with strong glial fibrillary acidic protein (GFAP) immunoreactivity. 15
These in vitro and in vivo findings imply that Npas4 confers neuronal protection against several neurodegenerative stimuli. However, little is known about the neuroprotective role of Npas4 in ischaemic stroke. In this study, we used a loss-of-function approach to examine the neuroprotective potential of Npas4 in the context of ischaemic injury both in vitro and in vivo. Firstly, primary mouse cortical neurons were subjected to oxygen and glucose deprivation (OGD), an in vitro model of ischaemia, to assess the protective effect of Npas4 in cultured cells. Next, we subjected both wild-type and Npas4 KO mice to photochemical stroke and compared the extent of brain injury in each genotype using various histological and immunohistological methods.
Materials and methods
Mice
Primary cultures of mouse cortical neurons were prepared from embryonic day 21 or postnatal day 0 wild-type C57BL/6 mice (Laboratory Animal Services, The University of Adelaide, SA, Australia). The transgenic mouse Npas4tm1Meg (MGI:3828099), established on a C57BL/6J background was a gift from Prof. Yingxi Lin. All experimental mice were maintained on a 12 h light/dark cycle with ad libitum access to food and water. Experimental procedures complied with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and were approved by The University of Adelaide Animal Ethics Committee (Animal Ethics #S-2009-159 and #M-2011-058). All sections of this report adhere to the ARRIVE Guidelines for reporting animal research.
Primary neuronal cultures
The frontal–lateral cortical lobes were dissected from embryos/pups, stripped of meninges, mechanically chopped, and cells were then dissociated with 0.25% trypsin/ethylenediaminetetraacetic (EDTA) (Gibco, Carlsbad, CA, USA) and DNase I (Sigma, St. Louis, MO, USA) with gentle trituration, and subsequently filtered through a 40 µm cell sieve (BD Biosciences, Franklin Lakes, NJ, USA). Cells were resuspended in Neurobasal medium (Gibco) supplemented with B27 (Gibco), 10% foetal bovine serum (FBS; Gibco), 2 mM GlutaMAX (Gibco), and 100 U/mL penicillin and 100 µg/mL streptomycin (Sigma), and plated at a high density of 200,000 cells/well in 48-well plates (for biochemical assays) or at a low density of 100,000 cells/coverslip in 24-well plates (for immunofluorescence analysis) precoated with 0.1% poly-
Cells were maintained for five to six days in vitro (DIV) in serum-free culture medium at 37℃ in a humidified 5% CO2 atmosphere before conducting experiments (time line shown in Figure 1(a)). Additionally, 5 μM cytosine β- Experimental design and Npas4 expression following OGD. Mouse cortical neurons grown for five to six DIV were treated with Endo-Porter along with or without MO for 48 h to allow uptake before being subjected to 2.5 h of OGD or normoxia (as control parallel cultures). Following incubation for additional 24 h under normoxia (recovery), cultures were assessed for changes in neuronal death and apoptotic activity (a). qRT-PCR analysis of Npas4 mRNA expression (b) in cortical neurons subjected to 2.5 h of OGD and 24 h of recovery under normoxia as compared to normoxia-treated cultures. Values are means ± SD (n = 5). **P = 0.0079 vs no OGD control. Representative Western blot analysis of Npas4 (c) in OGD-exposed cortical neurons following treatment with or without Npas4 antisense MO. β-actin was used as a loading control.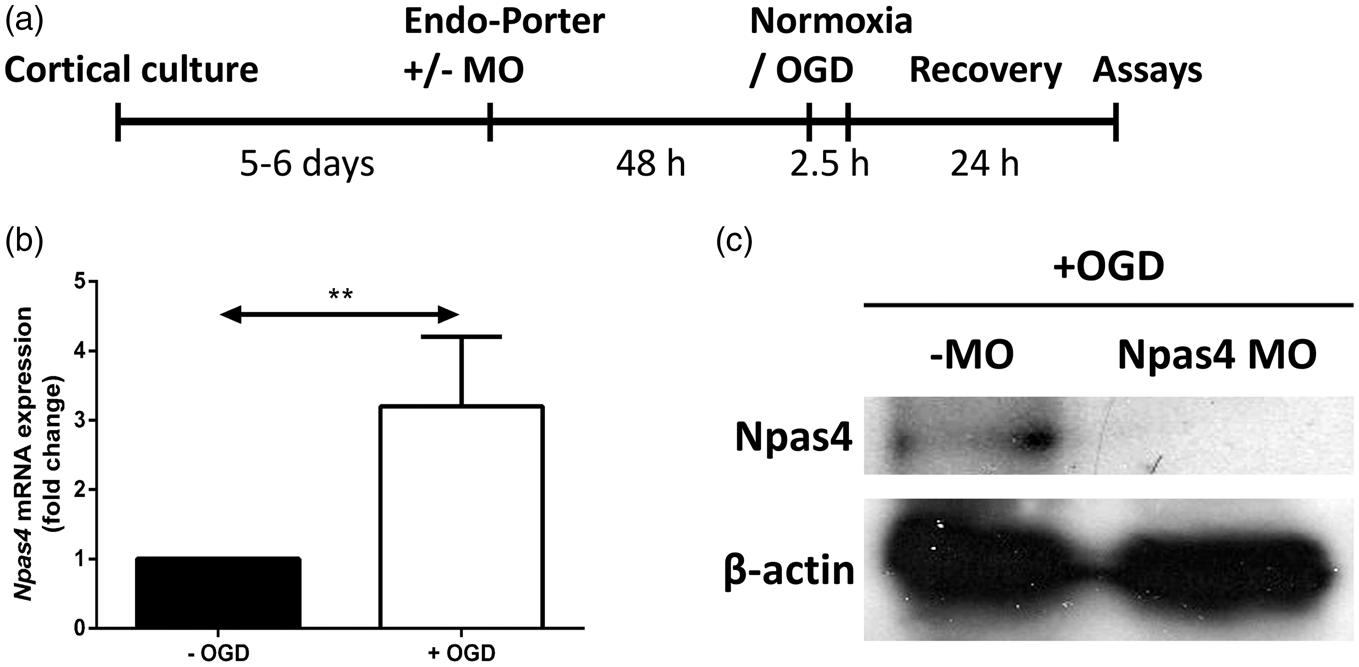
Knockdown of Npas4 in cortical neurons by translation blocking
Transient knockdown of Npas4 protein expression in mouse cortical neurons was achieved by translation inhibition using 10 μM mouse Npas4 antisense morpholino oligonucleotide (MO) (5′-GCCCTTGGTGGATCGGTACATGACT-3′; Gene Tools, Philomath, OR, USA) along with 5 μM of the delivery vehicle Endo-Porter (Gene Tools) according to the manufacturer’s instructions. A total of 10 μM scrambled MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′; Gene Tools) was used as a negative control – this MO has not been reported to have other targets or generate any phenotypes in any known test system except human beta-thalassemic haematopoietic cells (Gene Tools product information).
Cortical cultures were separated into six treatment groups: Endo-Porter alone with or without OGD (no MO control group), Endo-Porter + standard control MO with or without OGD (standard negative control MO group) and Endo-Porter + mouse Npas4 antisense MO with or without OGD (Npas4 knockdown MO group).
Oxygen and glucose deprivation
To model cerebral ischaemia-like conditions in vitro, cortical cultures were subjected to a short phase of OGD. Briefly, OGD was initiated by replacing culture medium with glucose-free Dulbecco’s modified Eagle medium (DMEM; Gibco) and incubating cells at 37℃ in a 3.5 L airtight chamber with an AnaeroGen sachet (Oxoid, Hampshire, UK), which reduces oxygen levels to <1% within 30 min (Oxoid product information). Cells were returned to glucose-containing culture medium 2.5 h after OGD and incubated under normoxia for an additional 24 h to simulate in vivo reperfusion. Control cultures were incubated in high-glucose DMEM (Gibco) for the same periods of time under normoxic (95% air/5% CO2) conditions.
Quantitative real-time PCR
Total RNA was isolated using the High Pure RNA Isolation Kit (Roche, Mannheim, Germany) following the manufacturer’s instructions. First-strand cDNA synthesis was performed using Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA) primed with 10 mM oligo(dT) and random primers (Promega, Madison, WI, USA). Quantitative real-time PCR (qRT-PCR) was performed in triplicate using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) with 50 ng of template cDNA for relative quantification of gene expression.
Npas4 and β-actin (internal control) primers with the following sequences were used: Npas4, forward 5′-AGCATTCCAGGCTCATCTGAA-3′, reverse 5′-GGCGAAGTAAGTCTTGGTAGGATT-3′; and β-actin, forward 5′-ACGGCCAGGTCATCACTATTG-3′, reverse 5′-CCAAGAAGGAAGGCTGGAAAA-3′. ABI Prism 7000 (Applied Biosystems) was used for all qRT-PCR experiments and data were analysed with DataAssist software v3.0 (Applied Biosystems) using the comparative Ct (ΔΔCt) method. Npas4 expression was normalised to β-actin expression and fold change was relative to no OGD control.
Western blot analysis
Knockdown of Npas4 protein expression in mouse cortical neurons using MO was assessed using Western blot analysis. Cells were lysed using lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing protease inhibitor cocktail (Roche). Following sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred onto 0.45 µm polyvinyl difluoridine (PVDF) membranes (Millipore, Billerica, MA, USA) and after blocking, the membranes were incubated overnight at 4℃ with one of the following primary antibodies diluted in 0.1% phosphate-buffered saline with Tween 20 (PBST): rabbit anti-β-actin (1/5,000; Sigma) or rabbit anti-Npas4 (1/15,000; a kind gift from Prof. Yingxi Lin). After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:50,000; Rockland, Gilbertsville, PA, USA) for 2 h at room temperature. Proteins were detected using enhanced chemiluminescence (ECL) substrate (Millipore) according to the manufacturer’s instructions.
Propidium iodide exclusion assay
Neuronal cell death was quantified by propidium iodide (PI) exclusion. PI (Sigma) was added to the culture medium at a final concentration of 2.5 µg/mL and incubated at 37℃ for 15 min, after which excess PI was washed off with Hank’s buffered saline solution (HBSS; Gibco) and 4′,6-diamidino-2-phenylindole (DAPI; Sigma) was added. At least 10 random fields were imaged using an AxioPlan2 microscope (Zeiss, Thornwood, NY, USA) and processed with the AxioVision 4.8 software (Zeiss) for each treatment group. Cell death was quantified by calculating the number of PI-labelled cells as a percentage of the total number of DAPI-labelled nuclei.
Measurement of caspase-3 activity
Early apoptotic activity was assessed using the Caspase-3 Colorimetric Assay Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Caspase-3 enzymatic activity was determined by absorbance readings at 405 nm with a microplate reader. Caspase-3 activity in the no MO control group without OGD was the baseline reference group.
Photochemical induction of focal cortical ischaemia
A total of 17 male mice (wild-type, n = 9; Npas4 KO, n = 8) aged 14–15 weeks were used for the study. Briefly, a light-sensitive dye (Rose Bengal; Sigma) was prepared fresh by dissolving in saline (10 mg/mL), filtering through a 0.45 µm filter (Minisart, Sartorius, France) and kept protected from light until use. Mice were intraperitoneally injected with Rose Bengal dye (100 mg/kg) prior to be being anaesthetised with inhaled isoflurane (Attane, Bomac, NSW, Australia). The mice were then secured in a stereotaxic frame in which isoflurane was continuously delivered via a nose cone. An incision was made in the scalp and the skull exposed to reveal Bregma. The stereotaxic frame was then used to locate the target area, i.e. the region of the skull overlying the right forelimb motor cortex (1.5 mm lateral to Bregma, left hemisphere). A flexible light guide (Zeiss) attached to a light-emitting diode cold light source CL 6000 (Zeiss) was then positioned above the target area. The light source was fitted with a green light filter (Zeiss) to restrict the wavelength of emitted light to 500–570 nm and an aperture of diameter 2.5 mm was placed over the tip of the light guide to focus the light beam onto the skull. To induce focal cerebral ischaemia, the target area was illuminated for 10 min with a luminous flux of 240 lumens after which the wound was sutured. Following the procedure, animals received a subcutaneous injection of 50 µL of 0.5% bupivacaine hydrochloride (Lyppard, Townsville, QLD, Australia) under the scalp and a 2% mupirocin ointment (Bactroban, Brentford, UK) was applied to the surgical site.
Histology and lesion size determination
At 3.5 h and 96 h after the induction of photochemical ischaemia, mice were killed by cervical dislocation, their brains removed, washed briefly in PBS and then fixed in 4% paraformaldehyde for 48 h at 4℃. Brains were then washed in PBS, embedded in paraffin wax and sectioned (5 µm sections). Beginning from just prior to the appearance of the lesion, every 20th section was transferred to glass slides until the end of the lesion was reached giving a total of 22–30 sections per brain with a spacing of 100 µm between consecutive sections (i.e. covering a total distance of 2.2–3.0 mm). Sections were stained with haematoxylin and eosin (H&E) according to the standard protocol 17 and imaged using a NanoZoomer apparatus (Hamamatsu, Hamamatsu City, Japan). For each section, the area of the lesion and the area of the ispilateral hemisphere were determined by manually tracing their borders using NDP.view2 software (Hamamatsu) and the size of the lesion was expressed as a percentage of the ipsilateral hemisphere as previously published. 18 The mean value across all sections was calculated to give the average lesion size, which was compared between the two genotypes.
Fluoro-Jade C staining
Fluoro-Jade C (FJC; Millipore) staining was used to investigate neurodegeneration according to the manufacturer’s instructions with minor modifications. Briefly, brain sections were first deparaffinised through two 10 min changes of xylene, followed by rehydration through a graduated ethanol series to deionised water and subsequently, incubated in 0.06% potassium permanganate (Sigma) for 15 min. The sections were then transferred to a 0.1% acetic acid solution containing 0.0001% FJC and 0.0001% DAPI (Sigma) for 30 min. After the processes of washing and drying, the sections were cleared in xylene and coverslipped with DPX (Sigma). All sections were scanned using a Pannoramic Scan digital slide scanner (3DHistech, Budapest, Hungary). For quantification of degenerating neurons, at least three random non-overlapping fields of view were selected from the lesion core of each stroke brain at 40× objective magnification using the Pannoramic Viewer software package (3DHistech). The entire quantifying procedure was performed blinded.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling
For terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labelling (TUNEL) staining, the DeadEnd Colorimetric TUNEL System (Promega) was used following the manufacturer’s instructions. Briefly, biotinylated nucleotides were incorporated at the 3′-OH DNA ends using the enzyme TdT. HRP-labelled streptavidin was then bound to these biotinylated nucleotides. HRP was then detected using diaminobenzidine. The apoptotic nuclei were stained dark brown using this procedure and all sections were scanned using a Pannoramic Scan digital slide scanner (3DHistech). For quantification of apoptotic cells, at least three random non-overlapping fields of view were selected from the lesion core of each stroke brain at 20× objective magnification using the Pannoramic Viewer software package (3DHistech). The entire quantifying procedure was performed blinded.
Immunohistochemistry
Brain sections were deparaffinised, rehydrated and microwaved in sodium citrate buffer (pH 6.0, 5–20 min) or Tris-EDTA buffer (pH 9.0, 5–20 min) for antigen retrieval. Sections were blocked with 10% normal donkey serum (Jackson ImmunoResearch, West Grove, PA, USA) in PBS/0.3% Triton X-100 (Sigma) at room temperature for 1 h and incubated overnight at 4℃ with one of the following primary antibodies diluted in blocking buffer: rabbit anti-apoptosis-inducing factor (AIF, 1:500; Abcam, Cambridge, UK), rabbit anti-GFAP (1:250; Dako, Glostrup, Denmark), rabbit anti-interleukin 6 (IL-6, 1:400; Abcam), rabbit anti-ionised calcium binding adaptor molecule 1 (Iba-1, 1:500; Wako, Osaka, Japan), rabbit anti-nitrotyrosine (NT, 1:100; Millipore), rabbit anti-Npas4 (1:2000) or rabbit anti-tumour necrosis factor alpha (TNF-α, 1:100; Abcam). Negative controls were treated similarly but were incubated in the absence of the primary antibodies. Following washes in 0.1% PBST, sections were incubated with Cy3-conjugated anti-rabbit IgG (1:200; Jackson ImmunoResearch) and counterstained with DAPI (Sigma). All sections were scanned using a Pannoramic Scan digital slide scanner (3DHistech). For quantification of AIF-positive, GFAP-positive and Iba-1-positive cells, at least three random non-overlapping fields of view were selected from the penumbra of each stroke brain at 20× or 40× objective magnification using the Pannoramic Viewer software package (3DHistech). For quantification of IL-6-positive and TNF-α-positive cells and nitrotyrosine score, at least three random non-overlapping fields of view were selected from the lesion core of each stroke brain at 40× objective magnification. All quantifications were performed by a blinded researcher.
Statistical analysis
Results are expressed as means ± standard deviation (SD). The fold change in Npas4 expression and the quantification of FJC staining, TUNEL assay and immunohistochemistry were analysed using Student’s t-test. The statistical significance between different MO treatment groups subjected to OGD or normoxia was assessed with one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test. P-value < 0.05 was considered to be statistically significant. All analyses were performed using Prism 6.0 (GraphPad Software, San Diego, CA, USA).
Results
Knockdown of Npas4 led to increased OGD-induced neuronal death
In this study, we used OGD as an in vitro model of cerebral ischaemia to examine the neuroprotective effect of Npas4 (Figure 1(a)). We found that Npas4 transcript levels were significantly upregulated in primary mouse cortical neurons subjected to 2.5 h of OGD (with OGD: mean 3.2, SD ± 1.0 fold, P = 0.0079) (Figure 1(b)). After verifying that treatment with Npas4 antisense MO effectively reduced Npas4 protein expression under OGD conditions (Figure 1(c)), we assessed the effect of Npas4 knockdown on neuronal cell viability following OGD.
Using PI staining, we found that cortical neurons treated with Npas4 MO displayed a significant increase in cell death (mean 53.2, SD ± 5.3%) in comparison to cells treated with vehicle alone (Endo-Porter) (mean 36.5, SD ± 2.5%, P = 0.0003) or standard negative control MO (mean 37.4, SD ± 1.5%, P = 0.0002) (Figure 2(a) and (b)). OGD-induced neuronal death did not differ between the no MO control and scrambled MO control groups. Next, we examined the effect of knocking down endogenous Npas4 expression on early neuronal apoptosis after OGD. By measuring the levels of activated caspase-3, we found no significant difference in the levels of apoptotic activity between each MO treatment group following OGD exposure (Figure 2(c)). There were no statistically significant differences between the three treatment groups incubated under normoxic condition.
Cell death determination (PI exclusion assay) and apoptotic activity of cortical neuronal cultures treated with or without MO after OGD. Representative images and quantification of cell death by PI assay (a and b) in cortical neurons subjected to OGD or normoxia for 2.5 h followed by 24 h recovery at normoxia. Values are means ± SD (n = 4). ***P = 0.0003 vs no MO control or ***P = 0.0002 vs scrambled MO control. Quantification of neuronal early apoptotic activity by caspase-3 activity (c) in cortical cultures at 24 h after OGD or normoxia. Values are means ± SD (n = 5). KD: Npas4 antisense knockdown; PI: propidium iodide; Scr: scrambled MO; MO: morpholino oligonucleotide; OGD: oxygen and glucose deprivation. Scale bars = 50 µm.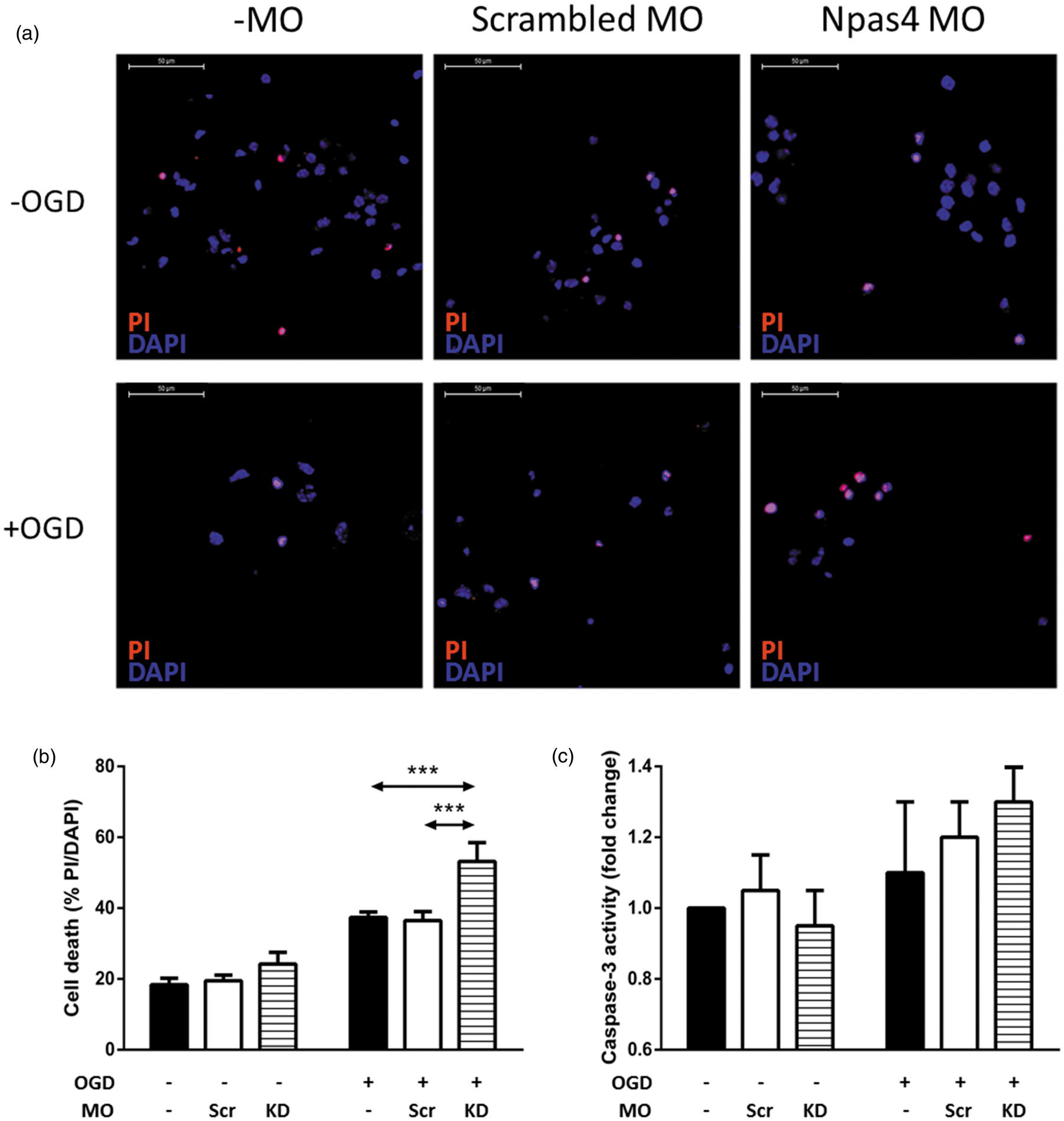
Post-stroke cortical lesions were larger in Npas4 KO mice
Following our in vitro experiments that demonstrated a neuroprotective effect for Npas4 under OGD conditions, we next assessed the influence of Npas4 on neuroprotection in an in vivo context using a photochemical model of ischaemic stroke (Figure 3(a)). First, we used immunostaining to examine the pattern of Npas4 protein expression in the brain at 3.5 h following the induction of photochemical stroke. In line with our previous study,
5
strong upregulation of Npas4 protein expression was observed throughout the ipsilateral cortex with no upregulation seen in the contralateral hemisphere (Figure 3(b)). Next, we investigated the effect of Npas4 ablation on lesion size in the photochemical stroke model and found that the mean brain lesion area for Npas4 KO mice determined at 96 h post-ischaemia was significantly increased when compared with wild-type mice (wild-type: mean 14.2, SD ± 4.7 mm2; Npas4 KO: mean 20.9, SD ± 4.4 mm2, P = 0.0079) (Figure 3(c) and (d)).
Experimental time line and photochemical stroke in wild-type and Npas4 knockout mice. Mice were intraperitoneally injected with Rose Bengal dye. To induce focal ischaemia, the target area was illuminated for 10 min with a cold light source. Brains were processed for immunohistochemistry at 96 h post-stroke (a). Representative Npas4 immunostained coronal brain sections (b) from wild-type mice at 3.5 h following the induction of stroke. Representative H&E stained brain sections (c) from wild-type and Npas4 knockout mice. Average brain lesion size (% area of ipsilateral hemisphere) (d) for wild-type and Npas4 knockout mice when assessed at 96 h following cerebral ischaemia. Values are means ± SD (wild-type: n = 9; Npas4 knockout: n = 8). **P = 0.0079 vs wild-type mice. Scale bars = 2000 µm.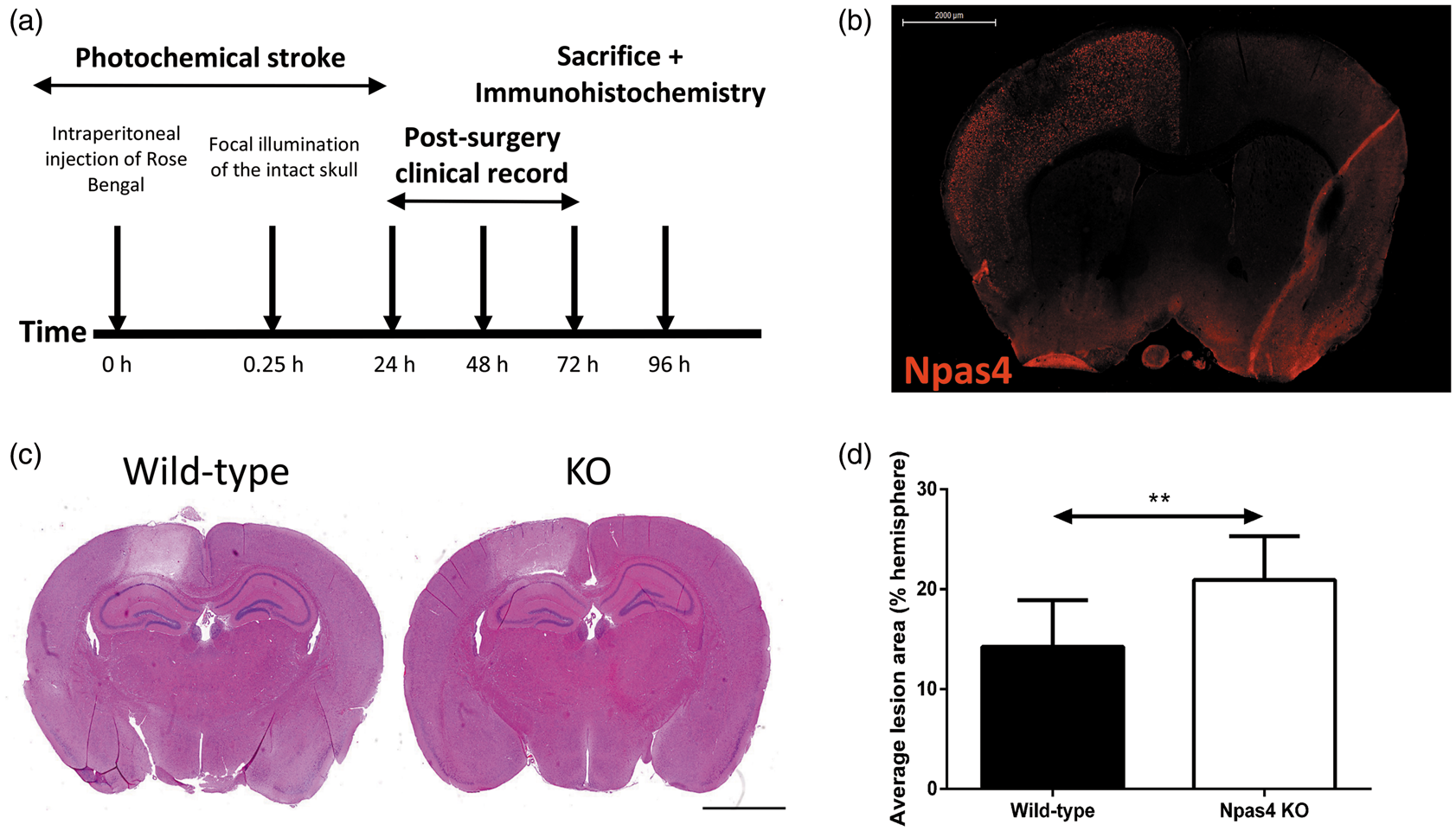
Lesions of Npas4 KOs showed increased neurodegeneration with reduced apoptosis
To assess the effect of Npas4 on neuronal protection, FJC staining was performed to evaluate neurodegeneration in mice subjected to photochemically-induced cerebral ischaemia. The number of FJC-positive degenerating neurons observed in the ischaemic core at 96 h post-ischaemia was significantly higher in Npas4 KO mice as compared to wild-type mice (wild-type: mean 19.8, SD ± 2.8; Npas4 KO: mean 32.9, SD ± 4.2, P = <0.0001) (Figure 4(a) and (c)).
Neuronal degeneration evaluation (FJC staining) and apoptosis detection (TUNEL assay) in the lesion core 96 h after brain ischaemia. Representative images and quantification of FJC (a and c) and TUNEL (b and d) stained brain sections from wild-type and Npas4 knockout mice subjected to focal ischaemic stroke. Values are means ± SD (wild-type: n = 9; Npas4 knockout: n = 8). ****P = <0.0001 and **P = 0.0053 vs wild-type mice for FJC staining and TUNEL assay, respectively. Scale bars = 50 µm (a) and 100 µm (b).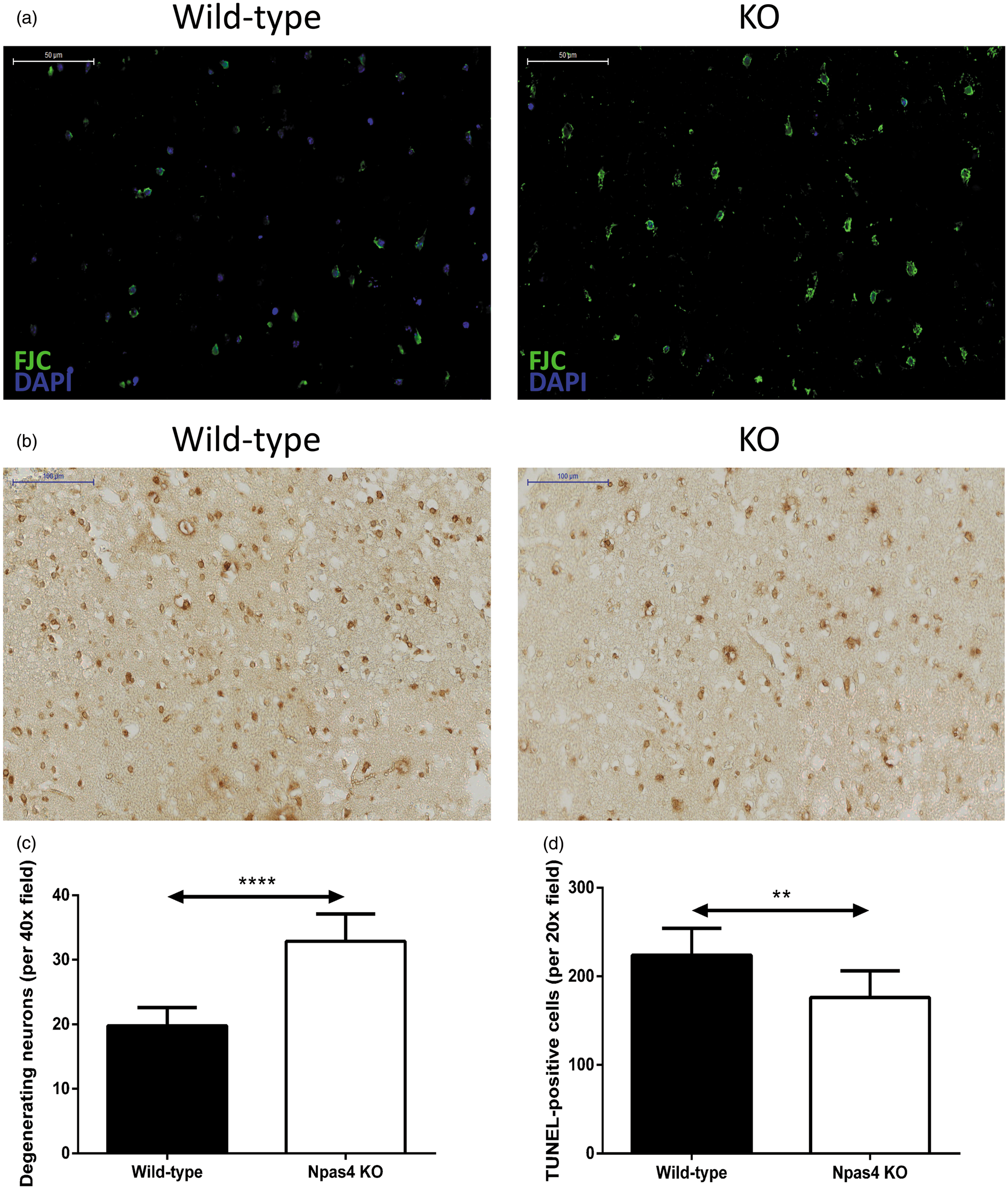
To understand the mechanism underlying the increased ischaemia-induced brain injury in Npas4 KO mice, we evaluated apoptosis in and around the lesion region using TUNEL assay. Interestingly, at 96 h post-stroke, brain sections from Npas4 KO mice showed a significant decrease in TUNEL-positive cells in the lesion core when compared with wild-type mouse sections (wild-type: mean 224.0, SD ± 30.2; Npas4 KO: mean 176.3, SD ± 30.0, P = 0.0053) (Figure 4(b) and (d)).
Increased AIF expression in the lesion periphery of wild-type mice without evidence of oxidative stress
To determine whether Npas4 induction leads to caspase-independent apoptosis, we analysed the expression of AIF (a protein involved in the caspase-independent pathway) and found a significantly higher number of AIF-positive cells in the penumbra of the ischaemic brain from wild-type mice as compared to Npas4 KO mice (wild-type: mean 70.3, SD ± 3.3; Npas4 KO: mean 27.4, SD ± 2.6, P = <0.0001) (Figure 5(a) and (c)).
Expression of AIF and nitrotyrosine in the lesion periphery and core, respectively, at 96 h following ischaemia. Representative images and quantification of AIF (a and c) and nitrotyrosine (b and d) stained brain sections from wild-type and Npas4 knockout mice subjected to photochemical stroke. Values are means ± SD (wild-type: n = 9; Npas4 knockout: n = 8). ****P = <0.0001 vs wild-type mice for AIF staining. Scale bars = 50 µm.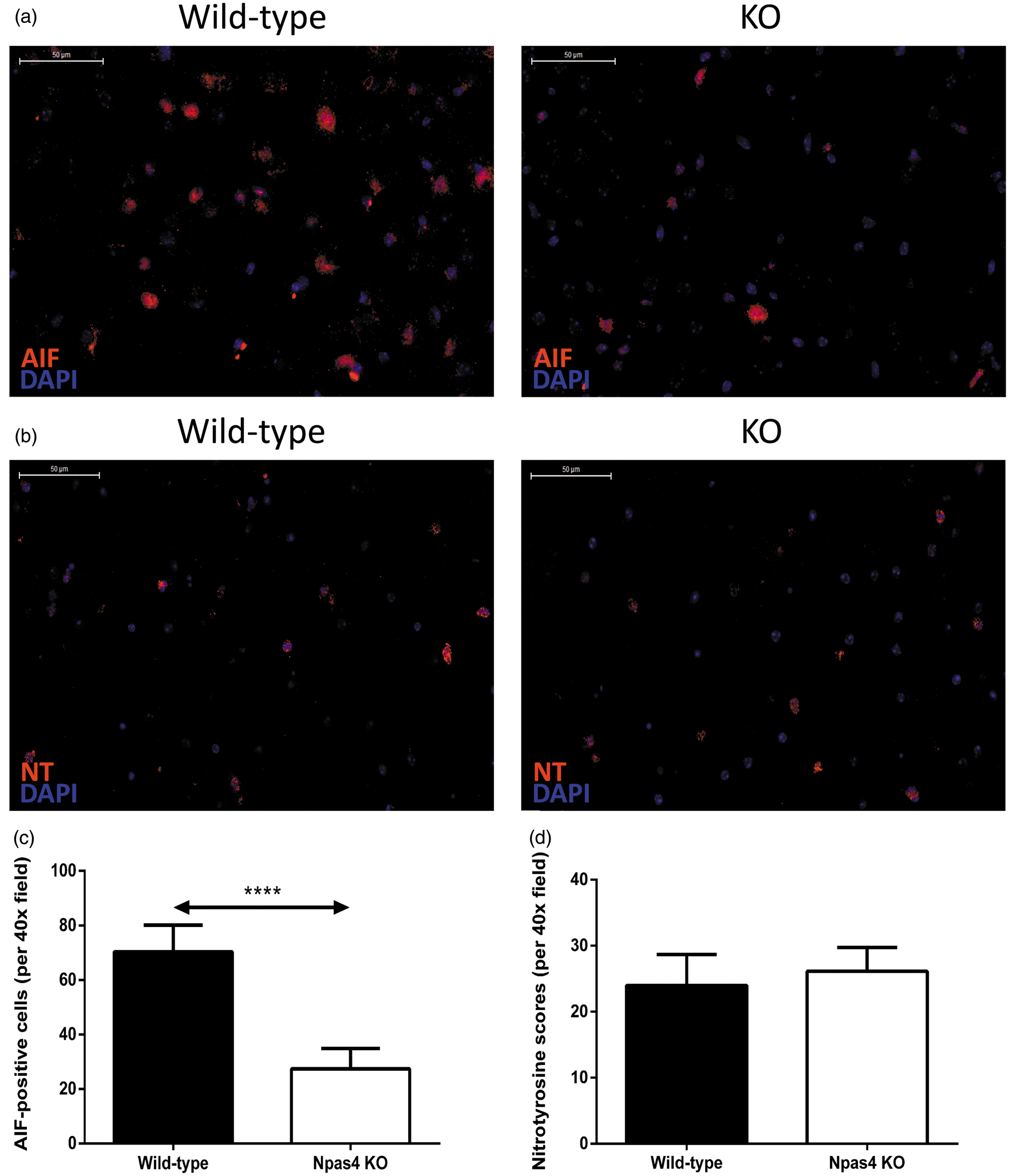
Ischaemia is known to induce the formation of reactive oxygen and nitrogen species, leading to oxidative stress that contributes to the pathogenesis of stroke. 19 Nitric oxide and superoxide interact to form peroxynitrite, a potent oxidant and nitrating species, which reacts with tyrosine in proteins to form nitrotyrosine, a stable end product of oxidation. 20 We next investigated whether oxidative stress contributes to the larger lesion size as observed in Npas4 KO mice subjected to focal cerebral ischaemia. Interestingly, there was no significant difference between nitrotyrosine levels of the wild-type and Npas4 KO mice at 96 h post-stroke (Figure 5(b) and (d)).
GFAP and Iba-1 expression were increased in the penumbra when Npas4 was absent
Given that inflammation may contribute to differences in the extent of post-ischaemic brain damage,
21
we compared the inflammatory cellular responses to focal ischaemia in wild-type and Npas4 KO mice. At 96 h after ischaemic stroke, the number of activated astrocytes (detected with GFAP antibody; Figure 6(a) and (c)) and microglia (detected with Iba-1 antibody; Figure 6(b) and (d)) were significantly higher in the penumbra of the ischaemic lesion from Npas4 KO mice when compared with wild-type mice (GFAP – wild-type: mean 22.8, SD ± 6.6; Npas4 KO: mean 59.00, SD ± 15.8, P = 0.0002. Iba-1 – wild-type: mean 48.1, SD ± 5.4; Npas4 KO: mean 83.9, SD ± 17.6, P = 0.0005).
GFAP and Iba-1 expression in the penumbral region at 96 h post-stroke. Representative images and quantification of GFAP (a and c) and Iba-1 (b and d) stained brain sections from wild-type and Npas4 knockout mice subjected to photochemical ischaemia. Values are means ± SD (wild-type: n = 9; Npas4 knockout: n = 8). ***P = 0.0002 and ***P = 0.0005 vs wild-type mice for GFAP and Iba-1 staining, respectively. Scale bars = 100 µm and 2000 µm (a) and 50 µm (b).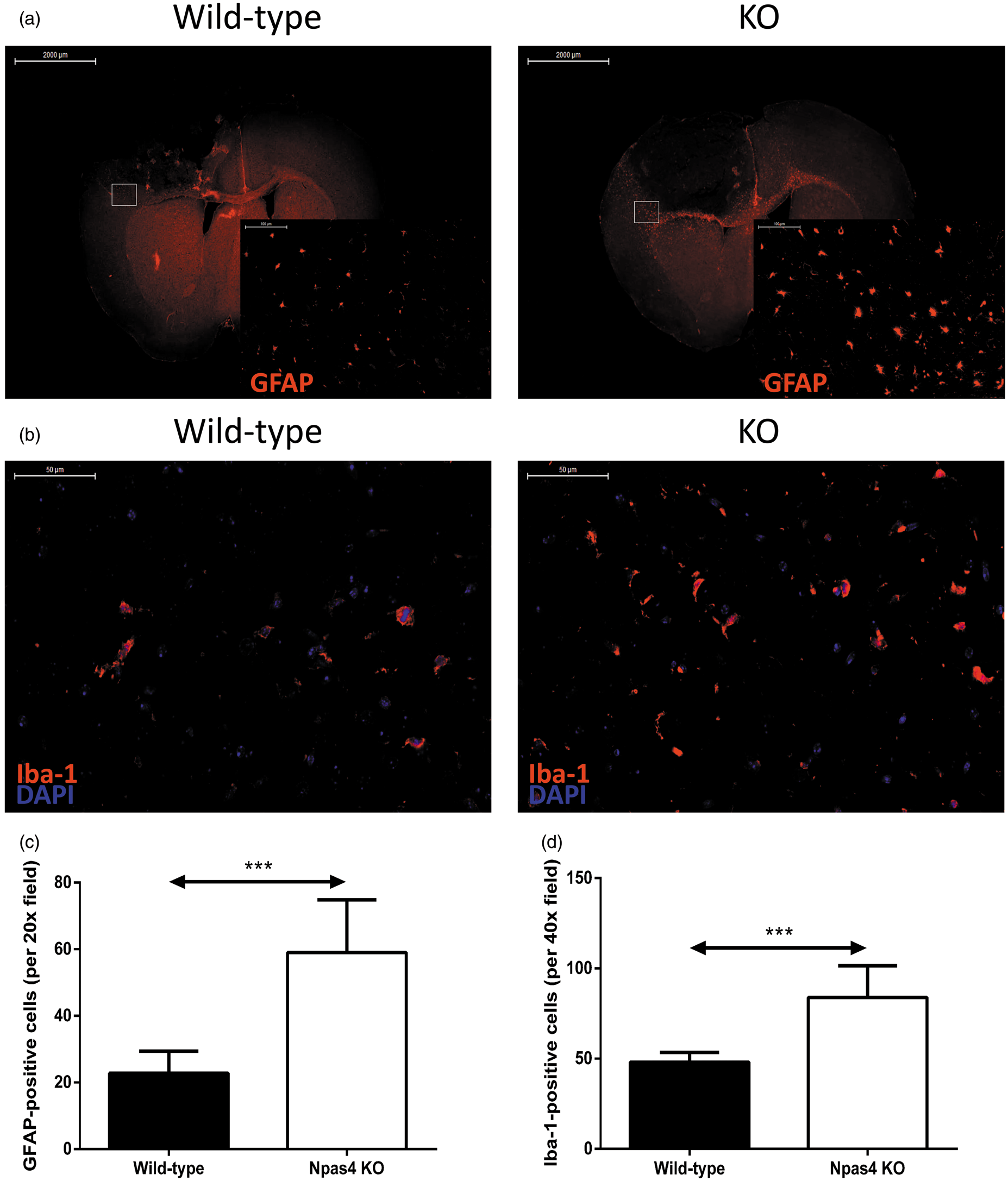
Elevated IL-6 and TNF-α in the lesion core of Npas4 null mice
Numerous studies have reported the crucial role of cytokines such as IL-6 and TNF-α, which are markedly increased in the acute phase after stroke onset, in the initiation and promotion of inflammation and the development of secondary brain injury.
22
We therefore investigated whether ablation of Npas4 also influences both IL-6 (Figure 7(a) and (c)) and TNF-α (Figure 7(b) and (d)) expression following cerebral ischaemia and found that significantly more cells positively stained for IL-6 and TNF-α were detected in the lesion core of Npas4 KO mice than in wild-type mice at 96 h post-stroke (IL-6 – wild-type: mean 12.7, SD ± 3.2; Npas4 KO: mean 34.9, SD ± 5.2, P = <0.0001. TNF-α – wild-type: mean 10.2, SD ± 0.5; Npas4 KO: mean 22.6, SD ± 1.1, P = <0.0001).
Expression of IL-6 and TNF-α in the ischaemic core at 96 h post-ischaemia. Representative IL-6 (a and c) and TNF-α (b and d) stained brain sections and quantification of staining from wild-type and Npas4 knockout mice subjected to photochemically-induced cerebral ischaemia. Values are means ± SD (wild-type: n = 9; Npas4 knockout: n = 8). ****P = <0.0001 vs wild-type mice for both IL-6 and TNF-α staining. Scale bars = 50 µm.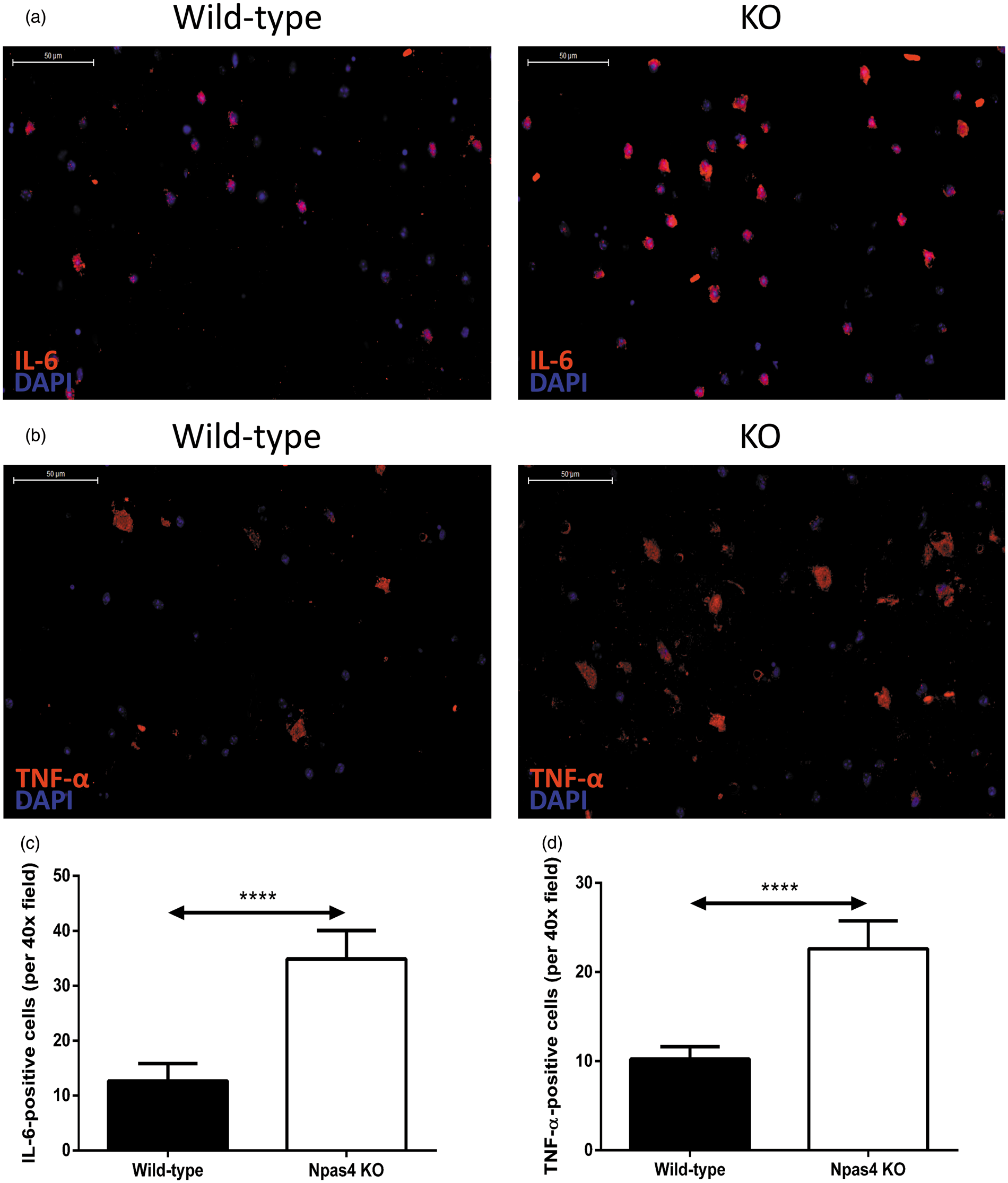
Discussion
Npas4 mediates a challenge-induced cellular neuroprotective response
Npas4 is an activity-dependent bHLH-PAS transcription factor whose expression is induced in various brain insults, including cerebral ischaemia.5,6,13–15 Although previous studies have demonstrated a role for Npas4 in the protection of neurons against neurodegenerative insults,14–16 the neuroprotective effect of Npas4 in response to ischaemic brain injury has yet to be elucidated. In this study, we used OGD as an in vitro model to examine the effect of Npas4 on neuronal survival following ischaemia. OGD treatment induced an upregulation of Npas4 in mouse cortical neurons. This is consistent with in vivo studies that showed that Npas4 expression is upregulated following experimental stroke in rodents.5,6 Using Npas4 antisense MO, we showed that the knockdown of Npas4 in cortical cultures resulted in increased neuronal susceptibility to OGD-induced cell death as demonstrated by increased PI membrane permeability. This indicates that Npas4 protects cultured neurons against OGD-induced neurotoxicity. Interestingly, we did not observe any change in the viability of Npas4 MO-treated cultures under baseline conditions (i.e. without OGD challenge). In the literature, there are differing reports on the role of Npas4 in neuronal survival under normal culture conditions. While one study reported that the knockdown of Npas4 gave rise to a slight increase in the basal level of cell death in cultured hippocampal neurons, another study found no effect on the overall health of the neurons with reduced Npas4 expression.8,16 Our data support the latter, which may indicate that the neuroprotective effect of Npas4 is only triggered in response to a biological challenge that threatens cell survival.
Modulation of apoptosis by Npas4 in cerebral ischaemia
OGD elicits a complex cascade of detrimental events leading to cell death via two separate processes: apoptosis (also known as programmed cell death, PCD) or necrosis. Two distinct apoptotic pathways have been demonstrated for their involvement in ischaemic injury. Classical apoptosis, i.e. caspase-dependent PCD, involves the activation of caspase-3 which cleaves numerous substrate proteins, including poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP). 23 Inactivation of PARP after cleavage by caspase-3 leads to DNA damage and apoptosis. 23 In addition to caspase-3-mediated apoptosis, emerging evidence implicates a significant role for caspase-independent pathway in PCD. 24 Such a mechanism involves the release of mitochondrial AIF, which translocates to the nucleus where it causes large-scale DNA fragmentation and cell death in a caspase-independent manner. 25 In this study, we assessed OGD-induced neuronal death using the fluorescent dye PI, which labels cells with loss of membrane integrity. Using this assay, we observed an increase in cell death in neuronal cultures subjected to OGD and Npas4 antisense MO treatment. However, PI does not distinguish between necrotic and apoptotic cells. Therefore, to determine which processes Npas4 is acting upon, we performed caspase-3 activity assay to quantify the number of cells undergoing apoptosis after OGD. The lack of substantial effects of Npas4 knockdown on neuronal caspase-3 activity suggests that Npas4 may preferentially mediate caspase-independent apoptotic and/or necrotic cell death rather than conventional caspase-dependent apoptotic events following OGD.
The protective effect of Npas4 was also investigated in vivo using a photochemically-induced brain ischaemia model in wild-type and Npas4 KO mice. We found a significantly larger lesion size in Npas4 KO mice as compared to wild-type mice at 96 h post-stroke, a time point chosen to avoid the period of maximum brain swelling (due to disruption of the blood–brain barrier) which could unevenly affect lesion size calculation. 26 FJC staining has a high affinity for degenerating neurons. Here, we performed FJC staining to further evaluate the neuronal protective potential of Npas4. In accordance with H&E staining, the number of degenerating neurons in the lesion core after ischaemic insult was significantly higher in Npas4 KO mice than in wild-type mice. These results confirm that Npas4 has a neuroprotective role in brain ischaemia.
Accumulating evidence has demonstrated that PCD contributes to the loss of neuronal cells caused by ischaemic injury. 27 Interestingly, using the TUNEL assay (which labels fragmented DNA in apoptotic cells), Npas4 KO mice subjected to focal cerebral ischaemia showed a significant decrease in the number of apoptotic cells in the lesion core when compared with wild-type mice. This suggests that the induction of necrosis, rather than apoptosis, may be responsible for the aggravated brain injury in mice lacking Npas4. Replacement of cell death types has been demonstrated in various studies. For example, depending on the cell types and stimuli, inhibition of caspase-8 can interrupt death signalling and favour a switch from apoptotic to necrotic cell death. 28 In mouse embryonic fibroblasts deficient for pro-apoptotic proteins Bax and Bak, alkylating DNA damage induced a necrotic type of cell death that was dependent on adenosine triphosphate (ATP) depletion, a condition also observed after ischaemic stroke.3,29 Interestingly, the possible role of Npas4 in modulating apoptosis has also been highlighted by a study which demonstrated that Npas4 could directly upregulate the expression of Bax. 14 Moreover, to elucidate if Npas4 is indeed involved in caspase-independent apoptosis, we performed immunostaining using the anti-AIF antibody and found that there were significantly more cells positive for AIF in wild-type mice than in Npas4 KO mice. Therefore, it is possible that a reduction in Bax induction in Npas4 KO mice caused neuronal cells to undergo necrotic cell death following cerebral ischaemia instead of activating the apoptotic pathway. Together with our in vitro studies, these data demonstrate that Npas4 plays a role in caspase-independent apoptotic events and that the lack of Npas4 can induce a switch from ischaemia-induced apoptosis to necrosis.
Inflammation following ischaemic stroke
In stroke, there is an increasing amount of evidence demonstrating the relationship between inflammation and neuronal cell damage. 30 Neuroinflammation is one of the key pathophysiological mechanisms contributing to the progression of damage caused by ischaemia and is characterised by the activation of astrocytes and microglia. 31 In this study, we found that the number of activated astrocytes and microglia in the lesion periphery had significantly increased in Npas4 KO mice compared to wild-type mice 96 h after ischaemia. These findings indicate that knocking out Npas4 leads to excessive inflammation which is known to be a major contributor to the secondary brain damage following ischaemic insult. 32
Cytokines are upregulated in the brain after a variety of insults, including ischaemic stroke. It has been shown that cytokines are expressed not only in cells of the immune system but are also produced by resident brain cells, including neurons and glial cells. 3 In this study, we evaluated the effect of Npas4 KO on IL-6 and TNF-α expression following cerebral ischaemia as it has been demonstrated previously that, in stroke patients, the serum levels of both IL-6 and TNF-α correlate with brain lesion size and stroke severity.33,34 We observed that Npas4 KO mice had both increased IL-6 and TNF-α expression levels and larger lesions which are in accordance with previous reports.
In ischaemic brain injury, IL-6 and TNF-α are largely thought to act as pro-inflammatory cytokines, which exacerbate brain damage and participate in the initiation of early inflammation. 22 Hence, the data presented suggest that the increased cerebral damage in Npas4 KO mice following focal ischaemia may be attributed to an excessive inflammatory response induced by IL-6 and TNF-α overexpression. Interestingly, it has been shown that IL-6 also has an anti-apoptotic effect after ischaemic stroke. 35 In line with this study, we have demonstrated an inverse relationship between the number of cells that underwent apoptosis and IL-6 expression levels whereby a significant drop in apoptotic cell death was accompanied by an increased number of IL-6-positive cells in the lesion core of Npas4 KO mice when compared with wild-type mice. In addition, data from other studies have demonstrated that TNF-α can induce necrosis in cells that failed to activate the pro-survival NF-κB signalling cascade and apoptosis. 36 These results further support the hypothesis that the induction of necrosis as a result of apoptosis inhibition is responsible for the aggravated brain injury observed in Npas4 KO mice following cerebral ischaemia. Furthermore, IL-6 expression has been demonstrated to be associated with necrosis but not apoptosis. 37
An increasing number of studies now recognise that activated microglia are highly plastic cells that can exist in two different states: classically activated (M1) phenotype that is characterised by the release of pro-inflammatory mediators or alternatively activated (M2) phenotype that possesses neuroprotective properties. 38 M1 and M2 microglia can be converted into each other in response to their microenvironment. 38 In this study, we noted that TNF-α expression was upregulated in the lesion core of Npas4 KO mice. Given the fact that TNF-α has been shown to be able to polarise microglia towards the M1 phenotype, 39 microglia could be primed towards the M1 state in Npas4-deficient mice which may help explain the increased stroke-induced brain injury and inflammation observed in these animals.
Previously, it was reported that increased amounts of nitrotyrosine were detected in the brains of aged Npas4 KO mice, contributing to the neurodegenerative phenotype observed in these animals. 15 However, in this study, we demonstrated that the nitrotyrosine levels between wild-type and Npas4 KO mice subjected to photochemical stroke were not significantly different, indicating that oxidative stress does not contribute to the expansion of brain injury in Npas4 KO mice post-stroke.
Concluding remarks
Taken together, we conclude that Npas4, whose expression is robustly and transiently upregulated in the brain during the acute phase of ischaemia, plays a neuroprotective role because Npas4 deficiency increased the susceptibility of cultured neurons to cell death by OGD and exacerbated the severity of brain injury after photochemically-induced stroke in mice. The mechanism underlying the in vivo protective effect of Npas4 after ischaemic insult could be due to its capacity to regulate inflammatory response. The present study demonstrated for the first time that ablation of Npas4 increased activated astrocyte and microglial cell numbers, pro-inflammatory cytokines IL-6 and TNF-α levels and caused a switch from apoptotic to necrotic cell death, which can contribute to the excessive inflammatory reaction due to leakage of cellular contents, 40 following focal cerebral ischaemia. Thus, we hypothesise that in wild-type mice, Npas4 limits progressive neurodegeneration and neuroinflammation after ischaemia, thereby decreasing brain damage. This is highly valuable given that understanding how the volume of ischaemic stroke may be reduced through Npas4-dependent apoptotic and inflammatory pathways could lead to the development of new stroke therapies.
Footnotes
Funding
FCC was supported by the Adelaide Graduate Research Scholarship from the University of Adelaide.
Acknowledgements
The authors would like to thank Prof. Yingxi Lin for her generous gift of the anti-Npas4 antibody.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FCC conceived and designed research, acquired data, analysed and interpreted data and results, performed statistical analysis and drafted the manuscript. TSK conceived and designed research, acquired data, analysed and interpreted data and results, performed statistical analysis and was involved in drafting the manuscript. WKL acquired data, analysed and interpreted data and results, performed statistical analysis and was involved in drafting the manuscript. SAK conceived and designed research, contributed to analysis and interpretation of data and was involved in drafting the manuscript and providing critical review. MDL conceived and designed research, acquired data, contributed to analysis and interpretation of data and was involved in drafting the manuscript and providing critical review. All authors critically reviewed and approved the final manuscript. FCC and TSK are joint first authors. SAK and MDL are joint senior authors.