
Abstract
Replication-competent oncolytic virus (OV) therapies are a promising new modality for cancer treatment. However, they pose unique challenges for preclinical assessment, due in part to their tumor specificity and ability to self-replicate in vivo. Understanding biodistribution, immune cell responses, and potential effects of intratumoral replication on these outcomes are important aspects of the nonclinical profile for OVs. Herein, a single intravenous dose of vesicular stomatitis virus pseudotyped with the glycoprotein of lymphocytic choriomeningitis virus (VSV-GP), or a cargo-expressing variant (VSV-GP-[cargo]), was examined in both tumor-free and CT26.CL25.IFNAR-/- syngeneic tumor-bearing mouse models. Biodistribution and immune cell responses were characterized using different molecular pathology methods, including a strand-specific in situ hybridization method to differentiate administered viral genomes from replicated or transcribed viral anti-genome RNA. We identified distinct patterns of viral biodistribution and replication across tumor and nontumor sites but no major differences in biodistribution, off-tumor cell tropism, or immune cell responses between tumor-free and tumor-bearing mouse models. Our findings characterize key cellular changes following systemic exposure to VSV-GP, provide a better understanding of a nonclinical permissive tumor model for OV assessment, and demonstrate how current molecular pathology methods can provide a bridge between traditional biodistribution and pathology readouts.
Keywords
Introduction
Oncolytic viruses (OVs) are unique among cancer therapies in that they self-replicate.8,24 The general therapeutic concept is that OVs will efficiently replicate in cancer cells and trigger a targeted immune response against tumor antigens released by lysed cancer cells, while sparing healthy cells.6,13 This property of self-replication introduces a range of unique challenges to nonclinical OV development, as it may impact not just efficacy but also biodistribution (BD) and safety.3,28,32 Importantly, tumor-based replication may result in viral effects on nontumor sites. 3 Both tumor-bearing and tumor-free models have been used to support nonclinical development of OVs.7,17,33,39 To date, however, there have been no published studies directly investigating the effects of a tumor on OV BD, replication, and immune responses in mice. This information is important for understanding OV tumor selectivity (ie, potential for unwanted cytotoxicity in nontumor cells) and immunogenicity (ie, potential for excessive anti-viral immune responses).
In this report, we characterize early viral BD, replication, and immune cell responses associated with a single intravenous (IV) dose of vesicular stomatitis virus pseudotyped with the glycoprotein of lymphocytic choriomeningitis virus (VSV-GP) OV and a variant encoding a transgene cargo (VSV-GP-[cargo]). 26 The primary goal of these studies was to evaluate tumor effects on nontumor BD and replication and temporal/spatial dynamics underlying immune cell responses. In our previous work, 7 we described a central role for CD169+ macrophages in the early distribution, transcription, and replication of VSV-GP in the liver and spleen of a healthy (tumor-free) mouse model. Here, we compare the distribution, transcription, and replication dynamics in tumor-free and tumor-bearing mouse models, with a focus on spleen and liver, which were the sites with the greatest level of viral BD. The tumor model used a syngeneic CT26-CL25.IFNAR-/- tumor type, selected as it lacks the interferon-alpha (IFN α) receptor, 31 creating the deficient innate environment desired for OV therapies. We describe here differences between tumor and nontumor sites with respect to initial viral genome distribution and timing of peak replication. Furthermore, we establish that despite a high prevalence of CD169+ macrophages in tumors and initial capture of input viral genomes by these cells, the viral replication occurs mainly in neoplastic cells. Finally, by directly comparing signal in tumor-bearing and tumor-free mice, we were able to determine that the presence of a CT26.CL25.IFNAR-/- tumor did not impact the magnitude or timing of replication in nontumor sites.
Methods
In Vivo Studies
All animal experiments were performed in accordance with protocols reviewed and approved by the Institutional Animal Care and Use Committee of Boehringer Ingelheim Pharmaceuticals, Inc (Ridgefield, Connecticut). Female BALB/cByJ mice were purchased from Jackson Laboratories (Bar Harbor, Maine) at 7 to 8 weeks of age. The vendor facility has a comprehensive pathogen surveillance program that includes monitoring for 18 different mouse viruses. The animals were group-housed and acclimated for one week prior to the start of experiments.
On the day of tumor cell implantation, animals intended for tumor-bearing groups were briefly anesthetized with isoflurane and 100 µl of 1 × 106 CT26.CL25.IFNAR-/- tumor cells in phosphate-buffered saline were implanted via subcutaneous injection into the right flank of each mouse. The injection site was monitored daily until a palpable tumor formed. Tumors were then measured using digital calipers at least three times per week. Tumor volume was calculated using the equation for an elliptical rotational solid: π*(width)2*(length)/6, where length is the longer dimension. When a sufficient number of animals reached the target tumor volume range of 50 to 300 mm3, animals were randomized into groups and sampling time cohorts (n = 3 per time point) and re-housed with their cohort on the day prior to dosing.
VSV-GP, VSV-GP-[cargo], or vehicle buffer were administered to each animal via a single bolus IV injection at 109 TCID50 (50% Tissue Culture Infectious Dose) in 100 to 200 µl into the lateral tail vein. At predetermined time points (1, 6, 24, 72, or 168 hours), the animals were humanely euthanized by CO2 asphyxiation followed by exsanguination. Tissues (spleen, liver, heart, kidney, lung, and tumor) were then collected using aseptic necropsy techniques and halved. One half was flash frozen in liquid nitrogen for quantitative real-time polymerase chain reaction (RT-qPCR), and the other half was fixed in 10% neutral buffered formalin for 18 to 24 hours and then transferred to 70% ethanol for histology.
Tissue Fixation and Block Preparation
Fixed samples were trimmed, processed using standard histologic procedures, and embedded in paraffin block. Chilled tissue blocks were sectioned at 4 µm at full face onto positively charged slides. Formalin-fixed paraffin-embedded (FFPE) sections were then dried and stored at room temperature until use for immunohistochemistry (IHC) or in situ hybridization (ISH) methods.
In Situ Hybridization
Single-plex ISH was performed on a Leica BOND RX autostainer. The FFPE sections were baked in an incubator at 60°C for 1 hour and then added to the instrument, where they were deparaffinized. Endogenous peroxidase activity was quenched, and target retrieval was performed according to standard RNAscope manufacturer protocols (Advanced Cell Diagnostics, Newark, CA). VSV-GP is a negative-strand RNA virus; therefore, a custom-designed RNAscope probe for V-VSV-sense strand was used to detect the VSV-GP genomic RNA (negative-strand) in the M-P-N sequence region of the VSV genome (#858988, 40ZZ; Advanced Cell Diagnostics, Newark, CA). The commercially available RNAscope V-VSV-N probe (#453008, 20ZZ; Advanced Cell Diagnostics, Newark, CA) was used to detect both replicated VSV-GP anti-genomic (positive-strand) and transcribed mRNA in the N sequence region, as the mRNA and anti-genome sequences are indistinguishable with the RNAscope probe. Probes were detected using the RNAscope LSx Prefilled Brown detection kit (#322700, Advanced Cell Diagnostics, Newark, CA). Following protocol completion, slides were then transferred to an automated cover-slipper (Leica XL Stainer) for mounting and allowed to dry prior to imaging.
Immunohistochemistry
All steps for single-plex staining were performed on the Leica BOND RX instrument (Leica biosystems Deer Park, IL). To stain for CD169 by IHC, sections of FFPE tissues (spleen, liver, and tumor) were deparaffinized and run through heat-induced target retrieval (HIER) with Tris-EDTA pH 9.0 (ER2, #AR9640 Leica biosystems Deer Park, IL) for 20 minutes. Endogenous peroxidase activity was quenched with H2O2* for 15 minutes, and nonspecific binding was blocked using Protein Block (#X0909 Dako-Agilent, Stanta Clara, CA) with 10% normal donkey serum (#D9663 Sigma-Aldrich, St. Louis, MO) for 30 minutes. The anti-CD169 rabbit polyclonal antibody (#NBP2-30903 Novus Biologicals, Centennial, CO ) was diluted in diluent (#S3022-82-2 Dako-Agilent, Stanta Clara, CA) to 1 µg/ml and incubated for 30 minutes, followed by the EnVision Detection System with anti-rabbit horseradish peroxidase (HRP) for 30 minutes (#K411 Dako-Agilent, Stanta Clara, CA), color development with DAB (3,3’-diaminobenzidine) for 10 minutes, and hematoxylin* for 5 minutes (*components of the BOND polymer refine detection kit, #DS9800 Leica biosystems Deer Park, IL).
To stain for germinal centers (GCs), sections of FFPE spleens were deparaffinized and run through (HIER) with Tris-EDTA pH 9.0 (ER2, #AR9640 Leica biosystems Deer Park, IL) for 20 minutes. Endogenous peroxidase activity was quenched with H2O2* for 15 minutes, and nonspecific binding was blocked using Protein Block (#X0909 Dako-Agilent, Stanta Clara, CA) with 10% normal donkey serum (#D9663 Sigma-Aldrich, St. Louis, MO) for 1 hour. The anti-BCL6 rabbit monoclonal antibody (#ab172610 Abcam Waltham, MA) was diluted in diluent (Dako-Agilent, above) to 1 µg/mL and incubated for 1 hour, followed by polymer* secondary detection for 15 minutes, DAB* color development for 10 minutes, and hematoxylin* for 5 minutes (*components of the BOND polymer refine detection kit, #DS9800 Leica biosystems Deer Park, IL).
Upon run completion, slides were removed from the instrument and dehydrated through a series of increasing alcohol percentages, followed by xylene clearing. Slides were then transferred to an automated cover-slipper (Leica XL Stainer) for mounting and allowed to dry prior to imaging.
In Situ Hybridization + Immunohistochemistry Staining
Duplex ISH+ IHC was performed on the Roche Discovery Ultra instrument (Roche Diagnostics, Indianapolis, IN). The FFPE tissue sections were incubated at 65°C for 1 hour, deparaffinized (Xylene 5 minutes 2×, 100% ethanol 2 minutes 3×), then air dried flat at room temperature for 10 minutes. Offline antigen retrieval was performed using Target Retrieval Buffer (#322000) Advanced Cell Diagnostics, Newark, CA) according to the manufacturer’s protocol. Briefly, 1X target retrieval buffer was prepared fresh, and retrieval was performed in a pressure cooker (#DC2012 Biocare Medical, Pacheco, CA) at 95°C for 5 minutes. Slides were removed from pressure cooker and placed in waiting container of deionized water until added to Discovery Ultra instrument using the wet load option.
The duplex ISH-ISH method followed standard incubation and temperature recommendations for RNAscope, VS Universal HRP Detection Kit (#323210 Advanced Cell Diagnostics, Newark, CA) and RNAscope 2.5 VS probes V-VSV-sense probe (#858989, 40ZZ; Advanced Cell Diagnostics, Newark, CA), V-VSV-N probe (#453009 Advanced Cell Diagnostics, Newark, CA) or Mm-Ifnb1-No-XHs-C1 (#1045159-C1, 13ZZ; Advanced Cell Diagnostics, Newark, CA), Mm-TNFa (#311089 Advanced Cell Diagnostics, Newark, CA), and Mm-ifng-No-XHs-C1 (#1045169-C1 Advanced Cell Diagnostics, Newark, CA). The ISH probes were detected using the mRNA Teal chromogen (#760-256 Roche Diagnostics, Indianapolis, IN).
For duplex ISH staining with CD169 IHC, dual sequence was selected in the Discovery Ultra software. Following completion of the ISH sequence as described above, dual sequence was selected, and slides were blocked for 1 hour using Goat Ig (#76-6008 Roche Diagnostics, Indianapolis, IN), followed by Discovery inhibitor (#760-4840 Roche Diagnostics, Indianapolis, IN) and the neutralization option. The CD169 primary antibody (#NBP2-30903 Novus Biologicals, Centennial, CO) was diluted in Dako-Agilent diluent (#S3022-82-2 Dako-Agilent, Stanta Clara, CA) to 2 µg/ml and incubated for 1 hour. The secondary antibody UMap-Rb-AP (#760-4313 Roche Diagnostics, Indianapolis, IN) was applied for 2 minutes, followed by the Discovery Yellow chromogen (#760-239 Roche Diagnostics, Indianapolis, IN) for 48 minutes. Nuclei were detected using Hematoxylin II (#790-2208 Roche Diagnostics, Indianapolis, IN) for 4 minutes and Bluing Reagent (#760-2037 Roche Diagnostics, Indianapolis, IN) for 4 minutes.
For duplex ISH staining with CD3 IHC, dual sequence was selected in the Discovery Ultra software. Following completion of the ISH sequence as described above, slides were blocked for 1 hour using Goat Ig, followed by Discovery inhibitor and the neutralization option. The CD3-epsilon primary antibody (#85061S, clone D7A6E Cell Signaling, Danvers, MA) was diluted in Dako-Agilent diluent to 0.5 µg/ml and incubated for 1 hour. The secondary antibody UMap-Rb-AP was applied for 20 minutes, followed by the Discovery Yellow chromogen for 48 minutes. Nuclei were detected using Hematoxylin II for 4 minutes and Bluing Reagent for 4 minutes.
For multiplex staining, ISH was combined with CD169 and CD45R IHC primary antibodies using the triple sequence selection on the Discovery Ultra software. Following the ISH sequence as described above, slides were blocked with Goat Ig for 1 hour, followed by Discovery Inhibitor and neutralization. In the first IHC sequence, anti-CD169 primary antibody was diluted in Dako-Agilent diluent to 1 µg/ml and incubated for 1 hour, followed by detection with the secondary antibody UMap anti-Rb-AP, and Discovery Yellow chromogen for 48 minutes. In the second IHC sequence, slides were blocked with Goat Ig for 28 minutes, followed by Discovery Inhibitor and neutralization. The anti-CD45R (B220) primary antibody (#550286 BD Pharmingen, San Diego, CA) was diluted in Dako-Agilent diluent to 10 µg/ml and incubated for 1 hour, followed by secondary antibody OMap anti-Rat-HRP secondary (#760-4457 Roche Diagnostics, Indianapolis, IN), and Discovery Purple chromogen (#760-229 Roche Diagnostics, Indianapolis, IN) for 28 minutes. Nuclei were detected using Hematoxylin II for 4 minutes and Bluing Reagent for 4 minutes.
Following completion of all duplex and multiplex staining protocols, slides were washed and then transferred to an automated cover-slipper (Leica XL Stainer) for mounting and drying prior to imaging. Stained tissue sections were scanned using an Zeiss Axioscan KMAT microscope with PApo 20×/0.8 M27 objective (Carl Zeiss, White Plains, NY).
Digital Image Analysis
Digital image analysis algorithms were developed in Visiopharm software (version 2022.04 and 2023.09, Visiopharm, Denmark). The workflow involved two separate modules. The first module defined the tissue region of interest (ROI) from the background was done using standard thresholding techniques. Afterwards, a deep learning classifier trained to identify tissue folds was run to exclude any tissue folds from the ROI and then isolate the signal at 20× magnification. For chromogenic IHC images, nuclei and the chromogenic signals were initially detected inside the ROI. The nucleus detection was based on a deep learning–based nuclei detection classifier, whereas the detection of the chromogenic signal was based on the DAB deconvolution bands. For duplex stain signals, chromaticity deconvolution bands were used to identify the ISH and IHC marker signals. The output variables were positive marker area and tissue area. For duplex stained slides, the output variables included the CD169+ area alone, VSV ISH area alone, and the colocalized area of the two markers. The total marker area was defined by the sum of the marker area alone and the colocalized area. The algorithm results for each marker were normalized for each specimen by dividing the marker area by the total tissue area analyzed. The final normalized marker was reported and graphed as the percent of total tissue area.
RNA Extraction From Tissues and Real-Time Quantitative Polymerase Chain Reaction
Frozen spleen and liver tissues from tumor-free and tumor-bearing mice treated with VSV-GP were homogenized in TRIzol (ThermoFisher, Waltham, Massachusetts) with a Qiagen TissueLyser followed by chloroform extraction. Total RNA was purified from tissue lysates using the MagMax mirVana Total Isolation Kit on the KingFisher Flex Purification platform (ThermoFisher), according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using SuperScript VILO Master Mix (ThermoFisher, USA) following the manufacturer’s instructions. The RT-qPCR was performed with TaqMan Fast Advanced Master Mix and TaqMan Gene Expression Assays on the QuantStudio 12K Flex Real-Time PCR System (ThermoFisher). Target gene mRNA levels were normalized to the levels of murine endogenous control gene beta-2-microglobulin (B2m) and reported as relative expression. Real-time qPCR assays for target and housekeeper genes assays were purchased for interferon beta-1 (Ifnb1, #Mm00439552_s1), interferon gamma (Ifng, #Mm01168134_m1), tumor necrosis factor alpha (Tnfa, #Mm00443258_m1), CD169/Siglec1 (Siglec1, #Mm00488332_m1), caspase-3 (Casp3, #Mm01195085_m1), gasdermin D (Gsdmd, #Mm00509954_m1), and B2m (#Mm00437762_m1) (ThermoFisher, Waltham, Massachusetts).
Statistical Analysis
Statistical analysis was performed by comparing group means at each time point using unpaired, Mann-Whitney U-test. Statistical analyses were performed in Prism version 10.1.2 (GraphPad Software, La Jolla, California).
Results
Initial Biodistribution of Viral Genomes
To characterize the initial BD of viral genomes, we first administered a single IV dose of VSV-GP at 109 TCID50 to either tumor-free or CT26.CL25.IFNAR-/- tumor-bearing female BALB/c mice. Spleen, liver, heart, kidney, lung, and tumor tissues were collected at five time points: 1, 6, 24, 72, and 168 hours. Early viral BD was then characterized using an ISH method specific for VSV-GP genomic RNA BD. At 1 hour post-infection (hpi) in both tumor-free and tumor-bearing mice, genomic RNA hybridization puncta were observed primarily in cells of the sinusoidal/perisinusoidal region in liver (Figure 1A) and the marginal zone (MZ) in spleen surrounding white pulp regions (Figure 1B). In the heart, kidney, and lung, rare hybridization punctae were observed within scattered interstitial cells (Supplementary Figure S1A-C, respectively). In tumor, sparse hybridization punctae were also found in a low number of stromal cells (Figure 1C, arrow).
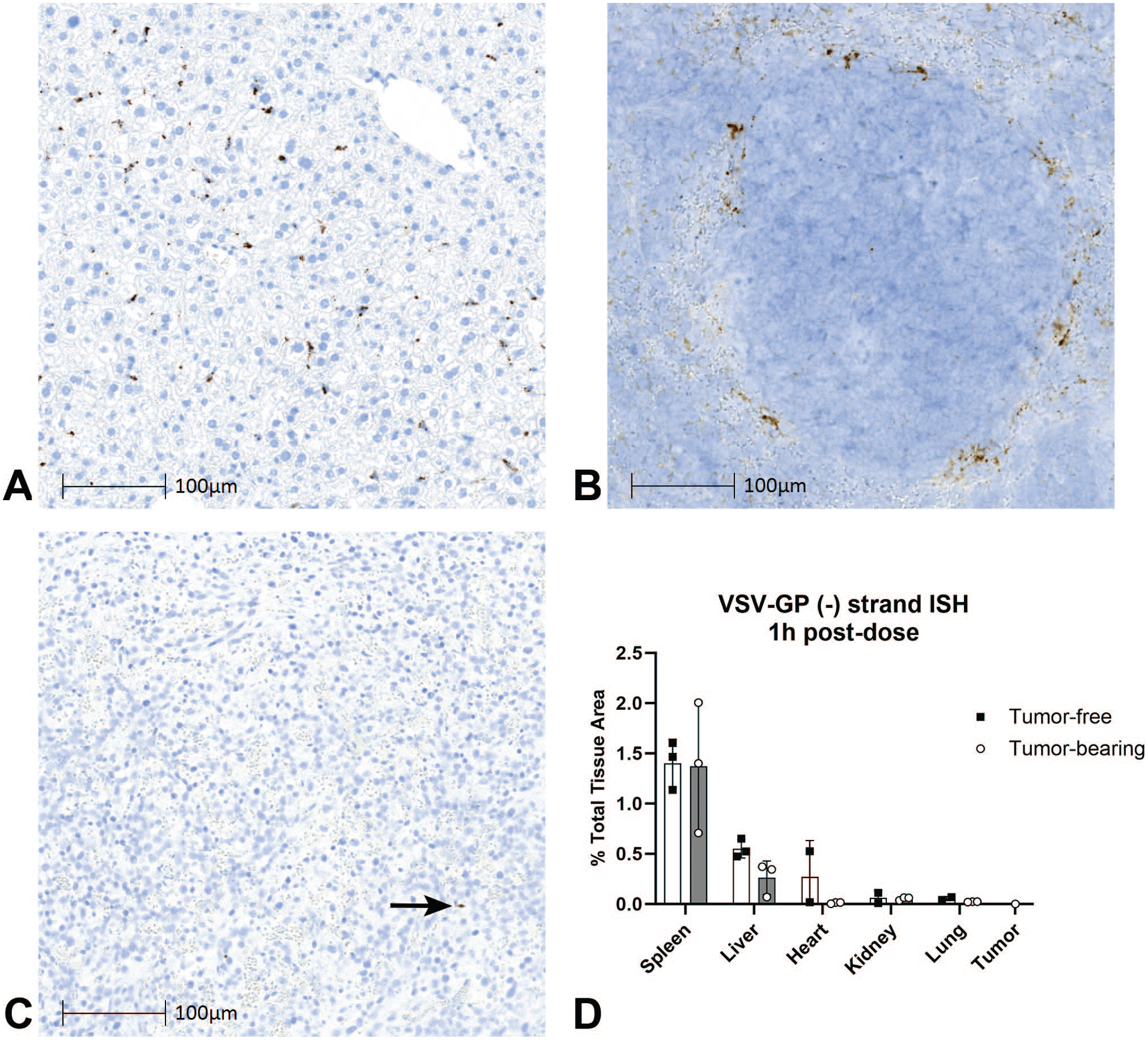
Initial biodistribution of viral genomes. VSV-GP genomic RNA ISH (DAB, brown stain) at 1 hpi in tumor-bearing mice administered a single IV dose of VSV-GP at 109 TCID50 in (A) liver, (B) spleen, and (C) tumor (arrow). (D) Quantification of the area of genomic RNA ISH as a percent of total tissue area. For each group, bars represent the mean values and error bars represent the standard deviation.
Next, to identify the primary tissues of initial VSV-GP BD and determine whether there were tumor effects, the area of genomic RNA was quantified using image analysis and reported as a percent of total tissue area analyzed. At 1 hpi, the greatest amount of genomic RNA was found in the spleen and liver of both tumor-free and tumor-bearing mice (Figure 1D). Across the five nontumor tissues examined, the tumor-free and tumor-bearing mouse models had no significant difference in genomic RNA, indicating that tumor presence did not impact the initial viral BD.
Next, we wanted to look specifically at the role for CD169+ macrophages in early BD across tissues. These cells have been shown to play an important role in viral surveillance and immune response,14,16,18,23,36 and we have previously shown that tissue-resident CD169+ macrophages of the spleen and liver were the primary site for initial distribution and subsequent transcription/replication in tumor-free mice. 7 Here, we developed a duplex ISH/IHC method to evaluate and quantify the colocalization of viral genomic RNA with CD169 (Siglec1) at 1 hpi. In all tissues examined from both tumor-free and tumor-bearing mice, the genomic RNA colocalized with CD169+ cells (Figure 2A liver, Figure 2B spleen, and Figure 2C tumor). The percent colocalization was greater than 94% across all tested tissues in both tumor-free and tumor-bearing models (Table 1), confirming that CD169+ macrophages are the primary site of initial VSV-GP BD after IV administration.
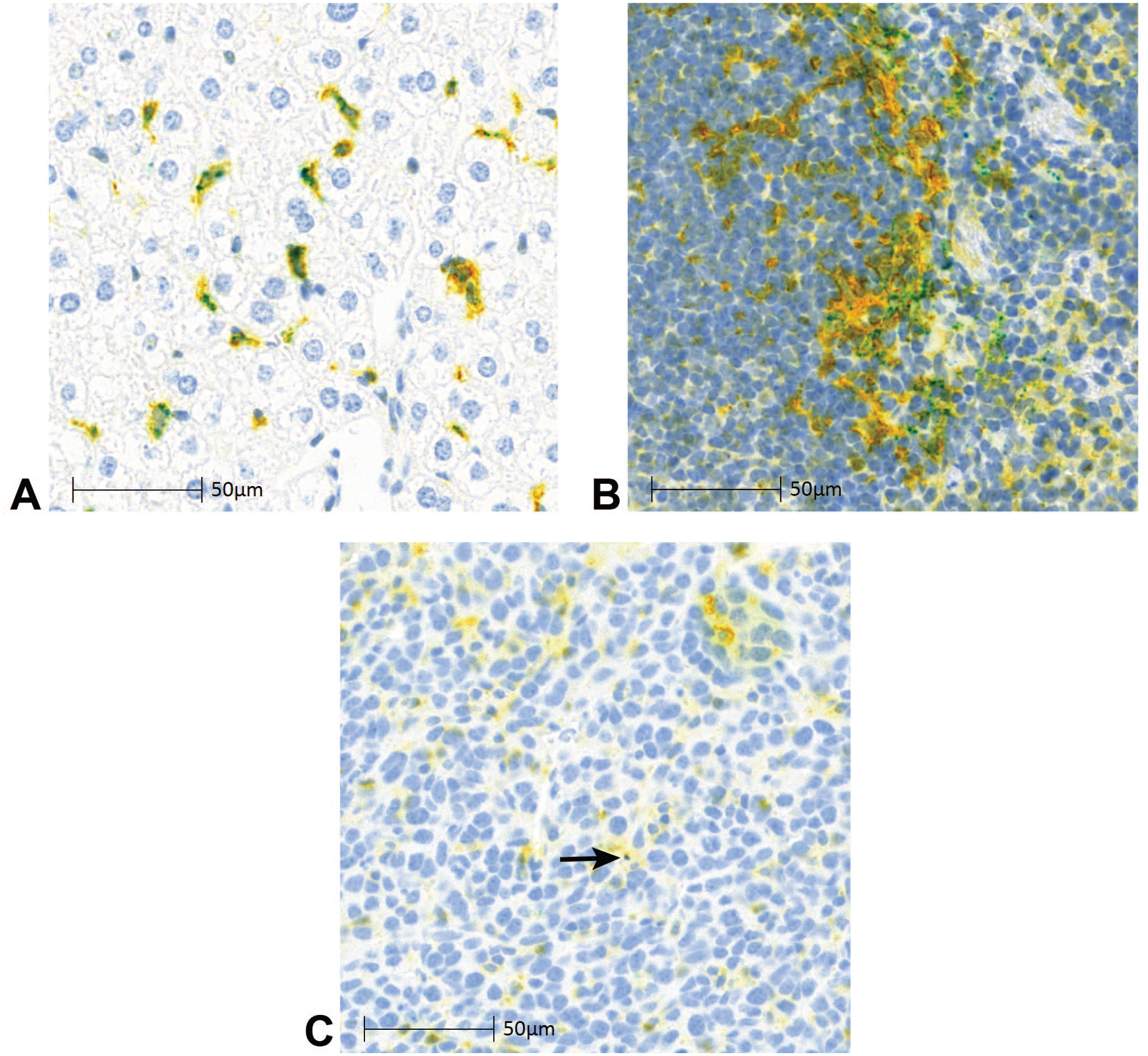
VSV-GP genomic RNA colocalizes to CD169+ macrophages. VSV-GP genomic RNA ISH (teal), CD169 IHC (yellow), and colocalized signal (green) at 1 hpi in tumor-bearing mice administered a single IV dose of VSV-GP at 109 TCID50 in (A) liver, (B) spleen, and (C) tumor (arrow).
Initial VSV-GP capture by CD169+ macrophages.
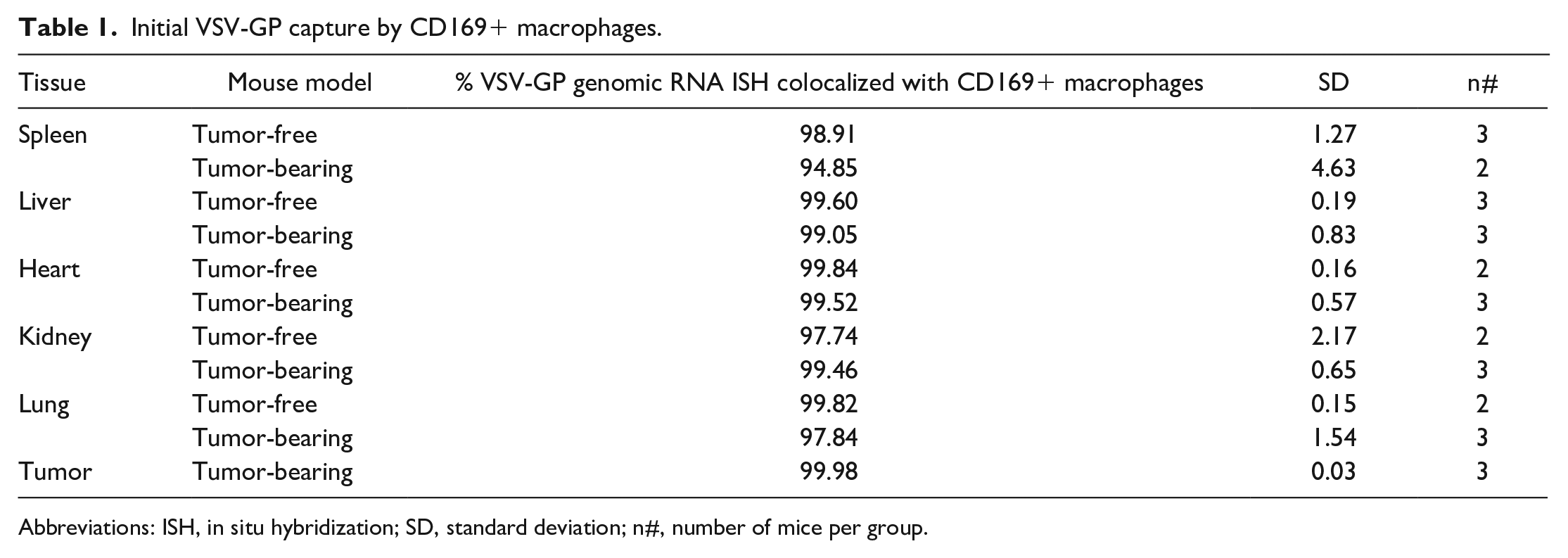
Abbreviations: ISH, in situ hybridization; SD, standard deviation; n#, number of mice per group.
Transcription/Replication Dynamics of Vesicular Stomatitis Virus Pseudotyped With the Glycoprotein of Lymphocytic Choriomeningitis Virus
We next investigated VSV-GP transcription/replication using an ISH probe specific for the VSV-GP anti-genomic RNA. Notably, the combined term “transcription/replication” is used hereafter to characterize the signal of this probe, given that it detects both transcribed VSV-N mRNA and replicated anti-genomes. With this method, we evaluated the colocalization of anti-genomic RNA hybridization punctae and CD169 IHC in liver, spleen, and tumor over time (1, 6, 24, 72, and 168 hpi) in both tumor-free and tumor-bearing models.
In the liver, anti-genomic RNA was detected starting at 1 hpi and visually colocalized predominantly with CD169+ macrophages (Figure 3A). As described by Gordon et al, 9 these cells likely represent a subset of Kupffer cells. The overall amount of anti-genomic RNA decreased slightly from 1 to 6 hpi (as the number of CD169+ macrophages decreased), whereas stronger anti-genomic RNA hybridization punctae were observed in the remaining CD169+ cells (Figure 3B). At later time points (24, 72, and 168 hpi), anti-genomic RNA was rarely detected (Figures 3C-E). The number of CD169+ cells also decreased in the liver at 6, 24, and 72 hpi compared to 1 hpi but then showed a marked increase in observed CD169+ cells between 72 and 168 hpi (Figure 3E).
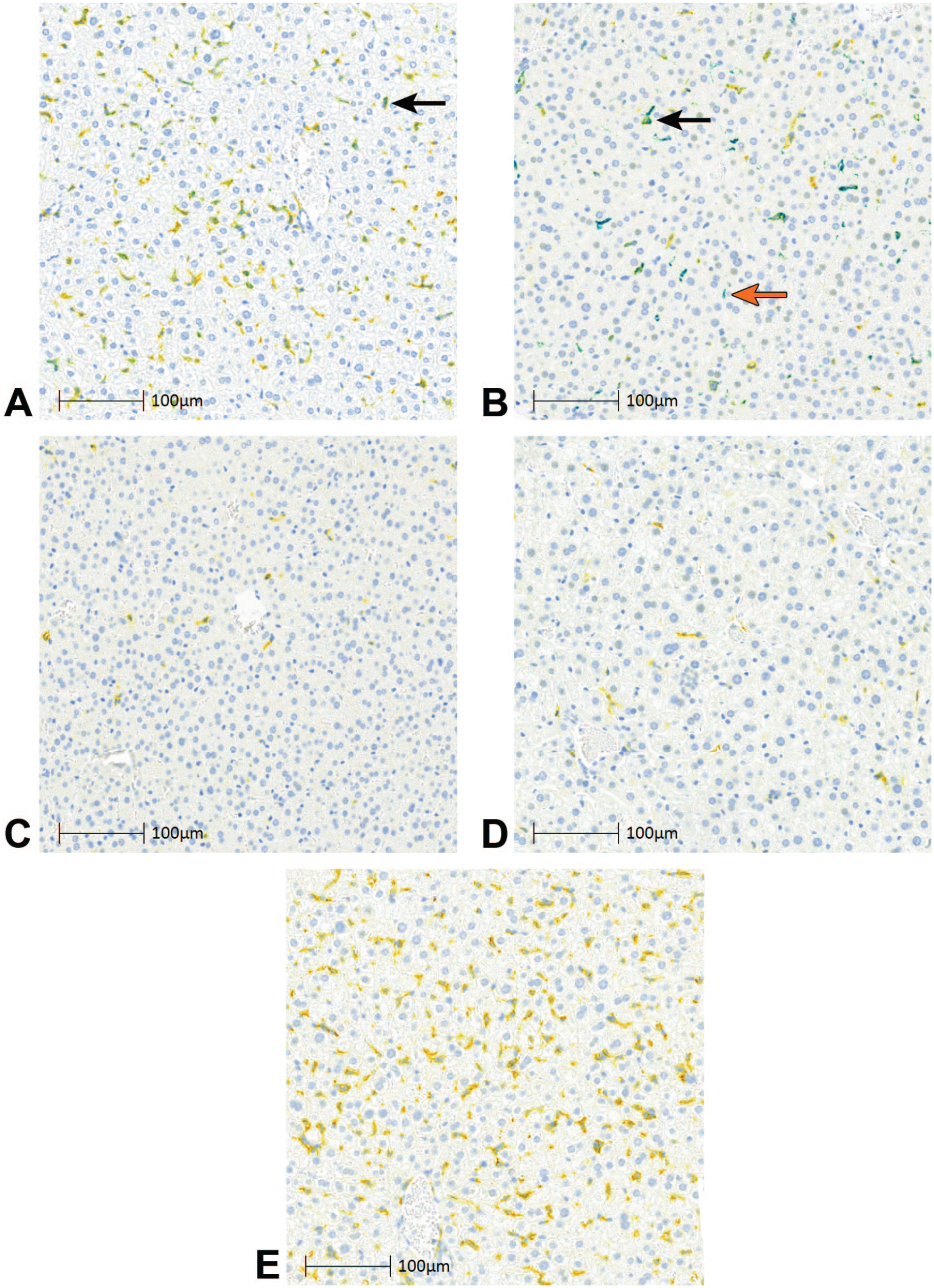
Transcription/replication dynamics of VSV-GP in liver. VSV-GP anti-genomic RNA ISH (teal), CD169 IHC (yellow), and colocalized signal (green) in liver of tumor-bearing mice administered a single IV dose of VSV-GP at 109 TCID50 at (A) 1 hpi, (B) 6 hpi, (C) 24 hpi, (D) 72 hpi, and (E) 168 hpi. Arrows indicate example teal signal (orange arrow) and green colocalized signal (black arrows).
In the spleen, there was abundant anti-genomic RNA at 1 hpi, which largely colocalized visually with CD169+ macrophages of the MZ (Figure 4A). In contrast to the liver, an increase in anti-genomic RNA was detected at 6 hpi within CD169+ macrophages. This signal was observed both in the MZ and scattered within the interior white pulp (Figure 4B). At 24 hpi, there was a marked reduction in both CD169+ macrophages and anti-genomic RNA (Figure 4C). In association with these changes, MZ regions showed decreased cellularity and increased cell debris indicative of cell lysis. At 72 hpi, there were rare CD169+ macrophages and rare if any anti-genomic RNA observed in the MZ or white pulp regions (Figure 4D). At 168 hpi, anti-genomic RNA remained scant and the CD169+ macrophages partially repopulated the MZ (Figure 4E and Supplementary Figure S2B).
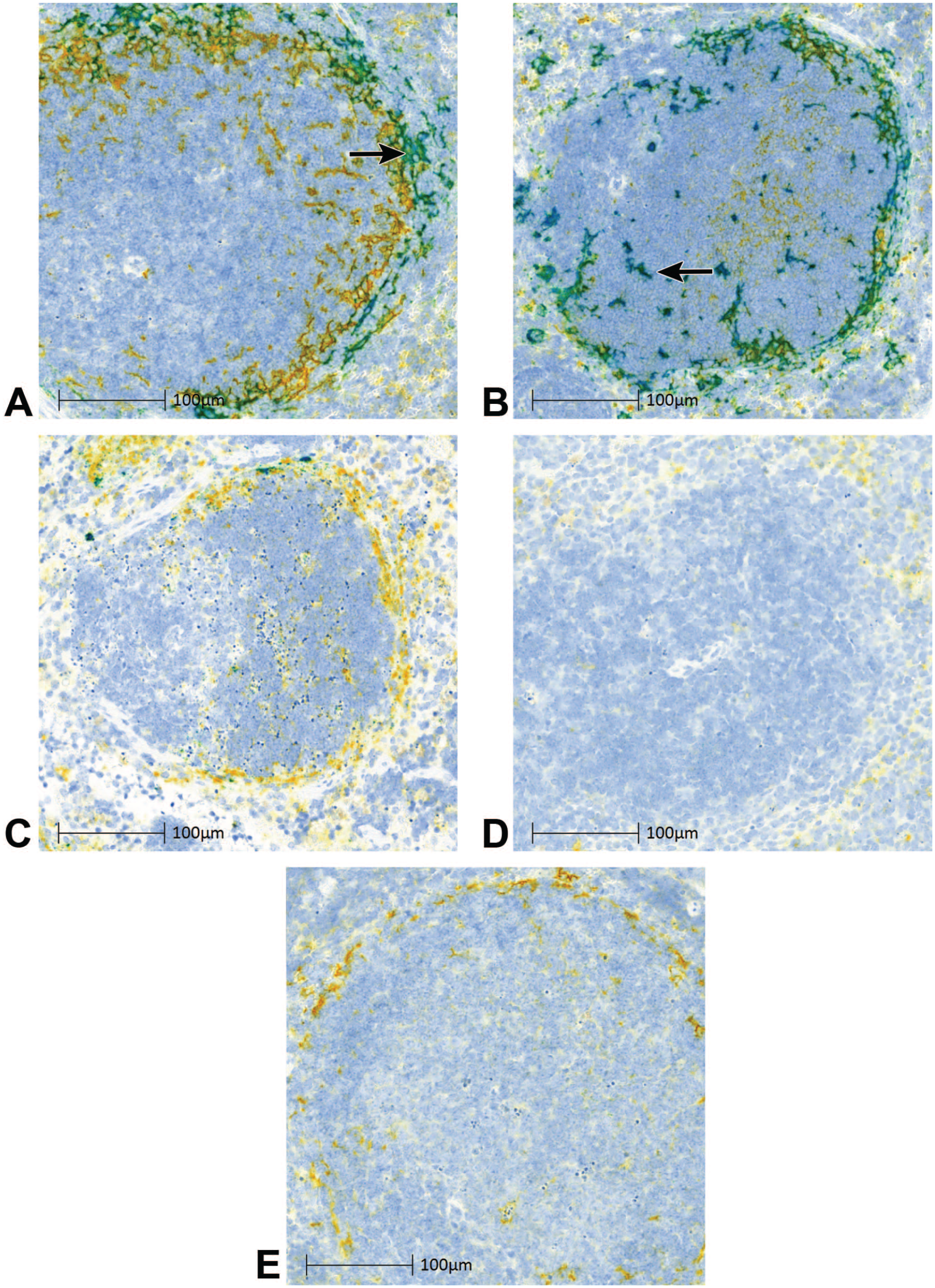
Transcription/replication dynamics of VSV-GP in spleen. VSV-GP anti-genomic ISH (teal), CD169 IHC (yellow), and colocalized signal (green) in the spleen of tumor-bearing mice administered a single IV dose of VSV-GP at 109 TCID50 at (A) 1 hpi, (B) 6 hpi, (C) 24 hpi, (D) 72 hpi, and (E) 168 hpi. Arrows indicate example colocalized signal (arrow 4A) teal signal (arrow 4B).
In tumor, rare anti-genomic RNA was detected at the 1 and 6 hpi time points (Figure 5A and B, black arrows). When present, these hybridization punctae visually colocalized primarily with CD169+ macrophages, which were strikingly abundant throughout the tumor samples. In contrast, at 24 hpi, and to a greater extent at 72 hpi, there were larger foci of anti-genomic RNA throughout the tumors (Figure 5C and D, orange arrows). At 168 hpi, there were still prominent foci of anti-genomic RNA, but they appeared smaller and more scattered compared with 72 hpi (Figure 5E). In contrast to the spleen and liver, the robust transcription/replication seen in the tumor starting at 24 hpi and persisting through 168 hpi often did not colocalize with CD169+ tumor-associated macrophages (TAMs) but instead occurred predominantly in the neoplastic cells.
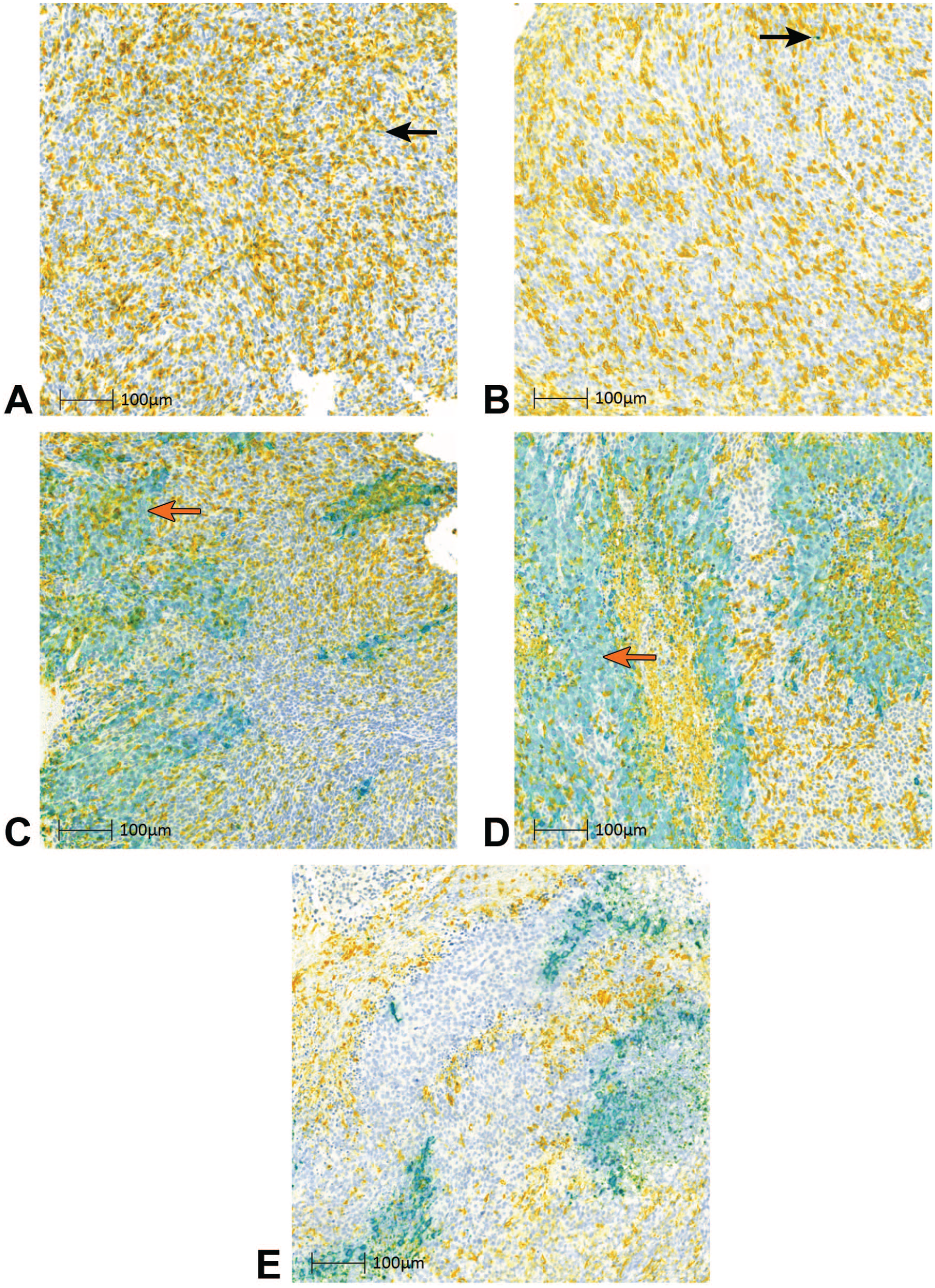
Transcription/replication dynamics of VSV-GP in tumor. VSV-GP anti-genomic RNA ISH (teal), CD169 IHC (yellow), and colocalized signal (green) in tumor of tumor-bearing mice administered a single IV dose of VSV-GP at 109 TCID50 at (A) 1 hpi, (B) 6 hpi, (C) 24 hpi, (D) 72 hpi, and (E) 168 hpi. Arrows indicate example teal signal (orange arrows) and green colocalized signal (black arrows).
Tumor Effects on Nontumor Tissue Transcription/Replication
To determine whether the presence of a tumor altered the magnitude or time course dynamics of transcription/replication at nontumor sites, the total area of anti-genomic RNA was quantified as a percent of the total tissue area analyzed. Tumor showed a distinct transcription/replication profile compared with liver and spleen, with a peak signal area (15%) at 72 hpi (Figure 6A). In the spleen and liver, the maximum signal area occurred at 6 hpi (5%) and 1 hpi (<1%), respectively. Notably, the burst of transcription/replication in tumor at 72 hpi did not result in a secondary peak for RNA in liver or spleen at a later time point, and no differences were found in anti-genomic RNA at any time point between tumor-free and tumor-bearing mice.
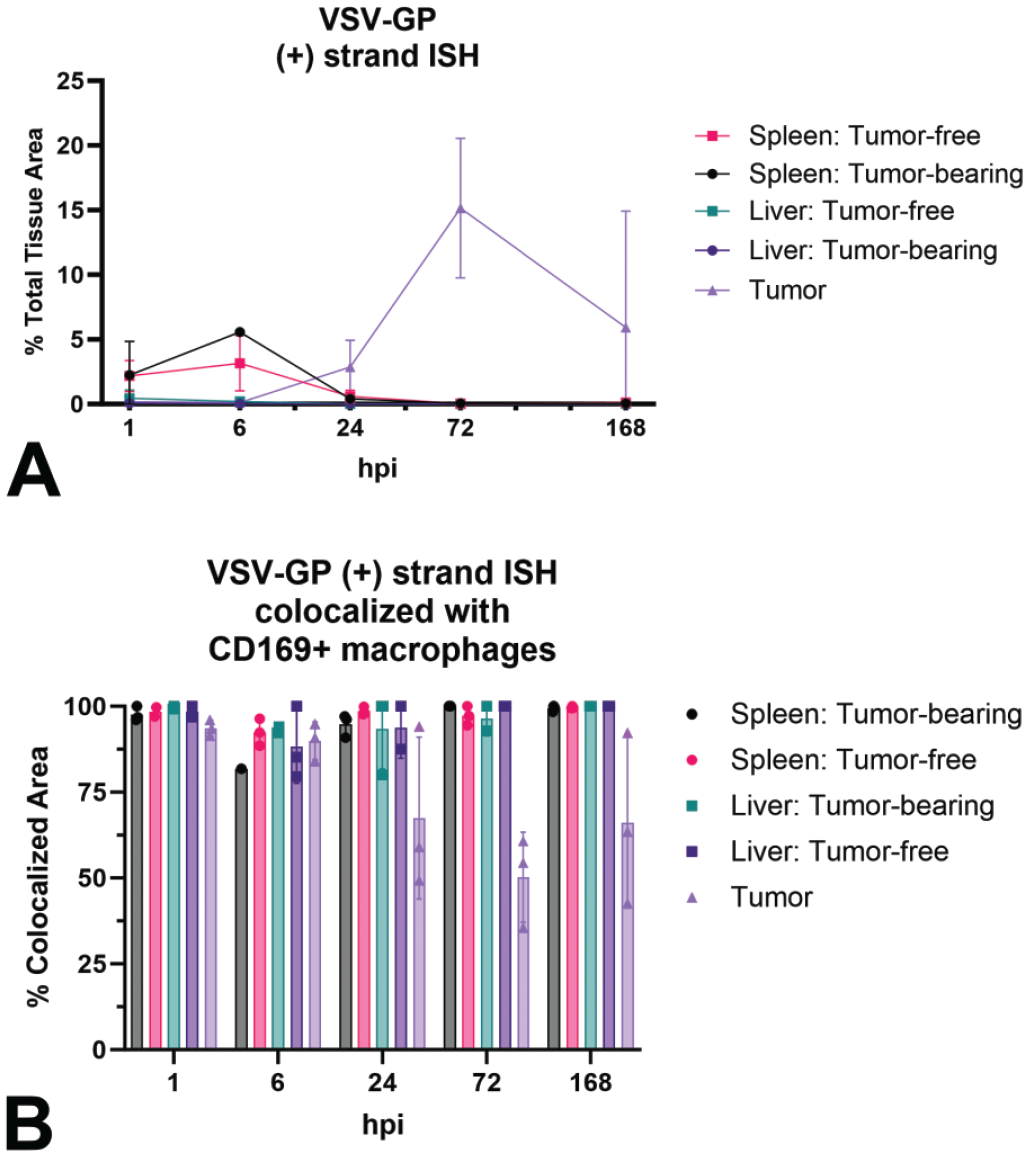
Quantification of total and macrophage-colocalized VSV-GP anti-genomic RNA over time. Quantification of anti-genomic RNA ISH signal in liver, spleen, and tumor of tumor-free and tumor-bearing mice administered a single IV dose of VSV-GP at 109 TCID50: (A) time course and (B) percent colocalized with CD169+ macrophages.
We next examined the extent to which the transcription/replication occurred within CD169+ macrophages, and if there were differences in temporal/spatial pattern or magnitude of transcription/replication between tumor-free and tumor-bearing mice. To do so, the percent of anti-genomic hybridization punctae colocalized with CD169+ macrophages was measured in spleen and liver. Across all time points, between 81% and 100% of anti-genomic RNA colocalized with CD169, confirming our previous descriptive findings that this macrophage subtype is the primary cell supporting transcription/replication in nontumor sites (Figure 6B). In tumors, colocalization with CD169 was also high (88%-90%) at 1 and 6 hpi but then decreased to 50% to 67% at 24, 72, and 168 hpi, consistent with the observation that at later timepoints, the signal was observed in primarily neoplastic cells in addition to TAMs.
Characterization of CD169+ Macrophages After Vesicular Stomatitis Virus Pseudotyped With the Glycoprotein of Lymphocytic Choriomeningitis Virus Administration
To further characterize the response of CD169+ macrophages to VSV-GP, we next wanted to determine whether the reduction in CD169+ macrophages observed by IHC in the tissue sections could be substantiated by the CD169 mRNA levels in matched snap-frozen tissue. To do so, we isolated total RNA from spleen and liver, then determined the quantity of CD169 (Siglec1) mRNA present relative to B2m across the time course. In treated mice, liver CD169 mRNA expression decreased from 1 hpi through 72 hpi before increasing at 168 hpi (Figure 7A). The CD169 mRNA expression in the spleens of treated animals decreased between 1 and 6 hpi, remained low through 72 hpi, and then increased by 168 hpi (Figure 7B). There were no statistical differences found between tumor-free and tumor-bearing mice.
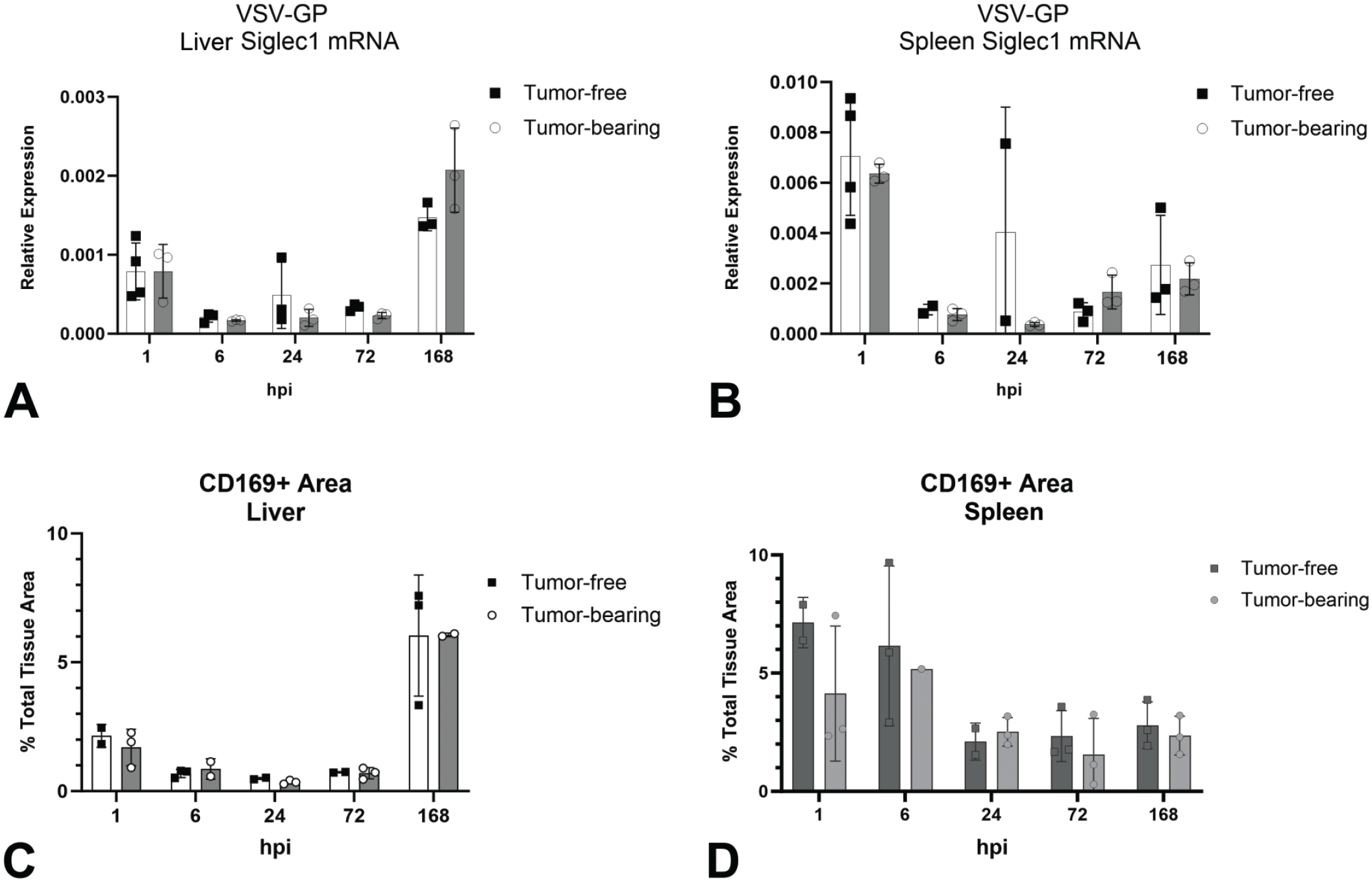
Characterization of CD169+ macrophages after VSV-GP administration. Quantification of CD169 IHC area in (A) liver and (B) spleen and relative expression of Siglec1 (Cd169) mRNA in (C) liver and (D) spleen of tumor-free and tumor-bearing mice following administration of a single IV dose of VSV-GP at 109 TCID50.
To compare mRNA and IHC protein results, we quantified the total CD169+ area in the liver and spleen based on the duplex positive-strand ISH/CD169 IHC method. In the liver, the percent CD169+ area decreased from 2% to <0.5% between 1 and 6 hpi and remained low through 72 hpi. The subsequent increase to 6% of CD169+ macrophage area at 168 hpi corresponded to the observed repopulation within the sinusoidal/perisinusoidal space (Figure 7C). In spleen, the percent CD169+ area decreased from 7% at 1 hpi to 2% at 24 and 72 hpi (Figure 7D). The MZ depletion of CD169+ cells observed on visual evaluation at 24 hpi, and the subsequent repopulation at 168 hpi were likely obscured in the whole section analysis.
In comparing tumor-free and tumor-bearing mice, no difference was found in CD169+ macrophage area at any time point for spleen and liver, indicating the presence of the tumor did not impact the response of these cells to VSV-GP. Overall, the observed decrease in CD169+ staining in spleen and liver was captured by the quantification using both qPCR for CD169 (Siglec1) mRNA and CD169+ IHC area. However, only analysis of bulk mRNA captured the repopulation in the spleen at 168 hpi.
Finally, we sought to explore the mechanism of cell death responsible for the decrease in CD169+ macrophages in the liver and spleen at 24 and 72 hpi. In our previous study, 7 in which similar depletion of CD169+ macrophages in spleen and liver were observed, IHC staining for cleaved caspase 3 showed minimal to no positive staining in the liver sinusoids or the splenic MZ at 24, 48, or 72 hpi (data not published). Here, we used qPCR to measure relative expression of Casp3 mRNA in spleen and liver and found no clear temporal pattern between 1 and 72 hpi in either liver (Figure 8A) or spleen (Figure 8B). We also tested the samples for Gasdermin D mRNA, a marker of the pyroptosis cell death pathway implicated in viral-mediated cell death.4,20 In the liver, there was a trend toward increased expression at 24 hpi; however, this higher level of Gsdmd mRNA was not significantly different compared with the other timepoints (Figure 8C). In the spleen, there was no clear temporal pattern between 1 and 72 hpi (Figure 8D).
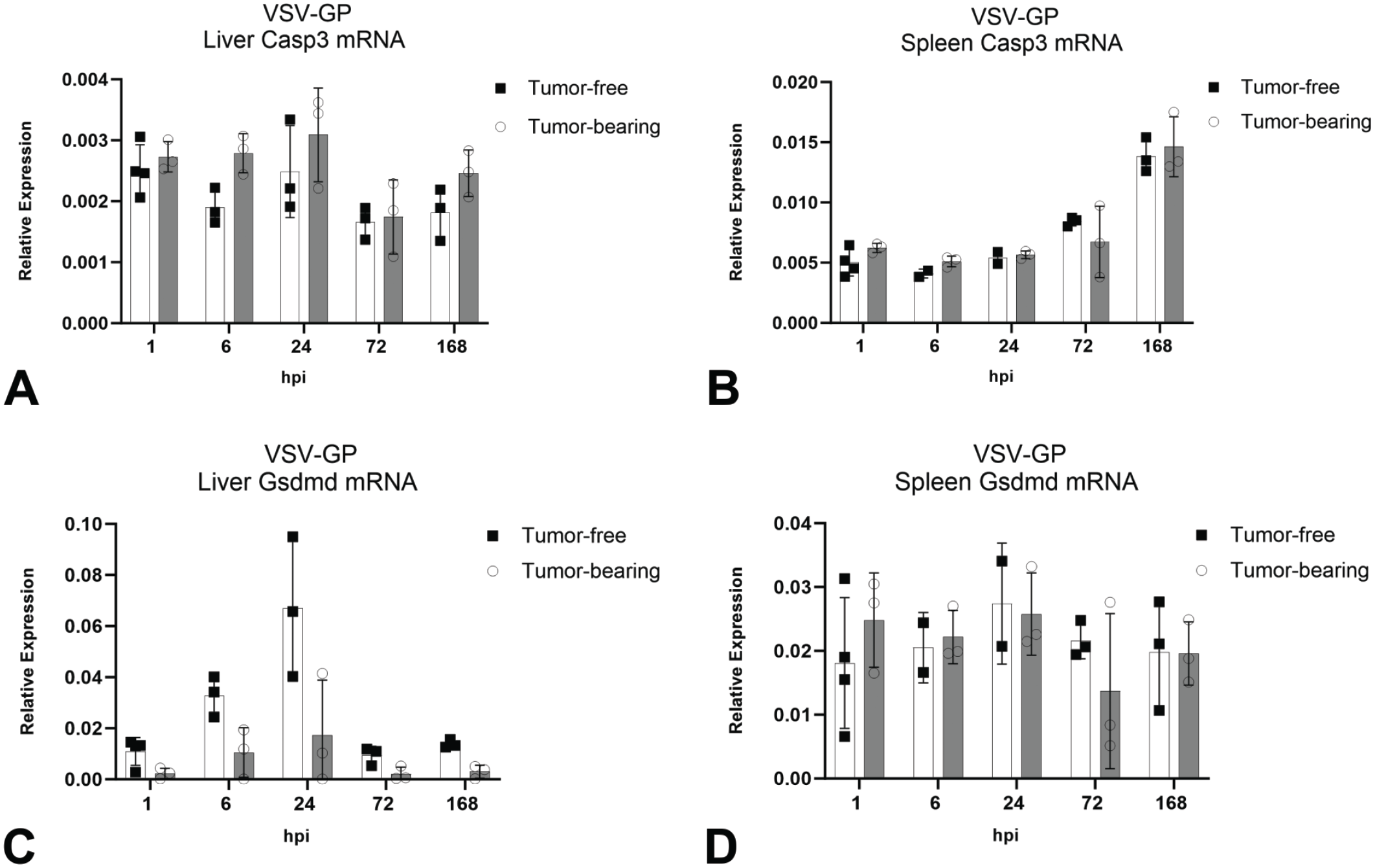
Relative quantification of cell death markers in liver and spleen. Relative expression of Casp3 mRNA in (A) liver and (B) spleen and Gasdermin D mRNA in (C) liver and (D) spleen of tumor-free and tumor-bearing mice following administration of a single IV dose of VSV-GP at 109 TCID50. For each group, bars represent the mean values and error bars represent the standard deviation.
Immune Cell Responses to Vesicular Stomatitis Virus Pseudotyped With the Glycoprotein of Lymphocytic Choriomeningitis Virus
Viral entry is known to induce a type-I IFN response, along with the induction of proinflammatory cytokines such as TNF.5,10,19,34,35,38 Previously, in a tumor-free mouse model, we found that the CD169+ macrophages of the spleen and liver were the main sites of Ifnb1 expression in response to VSV-GP, and that Ifnb1 peaked at 6 hpi in spleen and 1 hpi in liver. 7 Here, we compared expression of Ifnb1, Ifng, and Tnfa in spleen, liver, and tumor. We first tested bulk tissue from spleen for Ifnb1 mRNA expression and found that Ifnb1 expression peaked at 6 hpi (Supplementary Figure S3A). To compare the temporal/spatial expression of Ifnb1, spleen samples at 6 hpi were assessed for Ifnb1 expression using ISH duplexed with CD169 IHC. The main cell type expressing Ifnb1 was found to be CD169+ macrophages of the MZ in both the tumor-free and tumor-bearing mice (Supplementary Figure S3C and D). By contrast, the vehicle animals had little to no Ifnb1 ISH detected (Supplementary Figure S3E). Importantly, no significant differences were found in Ifnb1 magnitude of expression or temporal/spatial location between tumor-free and tumor-bearing animals, indicating the tumor did not impact splenic cytokines at these early time points.
We then tested bulk spleen for Tnfa and Ifng mRNA using qPCR. The Tnfa expression was consistent across time (Supplementary Figure S4A). Whereas Ifng expression increased from 1 to 24 hpi (Supplementary Figure S4B). Neither of these markers showed a significant difference between tumor-free and tumor-bearing mice.
We next tested bulk liver tissue for Ifnb1, Tnfa, and Ifng mRNA by qPCR. The baseline expression of Ifnb1 and Ifng at 6 hpi was similar in the liver and spleen, whereas Tnfa expression was nearly 18-fold lower in the liver than in the spleen. The Ifnb mRNA peaked at 6 hpi and tended to be higher in tumor-bearing mice, although no statistically significant differences were observed (Supplementary Figure S3B). Liver Tnfa mRNA peaked at 6 hpi before returning to baseline (Supplementary Figure S4C), and Ifng expression did not show any clear temporal patterns. Neither Tnfa nor Ifng mRNA exhibited a statistical difference between tumor-free and tumor-bearing animals (Supplementary Figure S4D). Overall, these results indicate that viral replication in the tumor did not impact cytokine mRNA expression in the liver.
Finally, we examined the Ifnb, Tnfa, and Ifng responses in the CT26.CL25.IFNAR-/- tumors. These tumor cells can produce IFN but lack the IFN-α receptor, 31 thereby blunting downstream type-I IFN signaling. First, using both Ifnb1 ISH and bulk Ifnb1 mRNA analysis, we determined the temporal/spatial pattern and quantified relative expression. Rare Ifnb1 hybridization punctae were detected at 1 and 6 hpi (Figure 9A and B, respectively). At 24 hpi, and to a greater extent at 72 hpi, foci of Ifnb1 hybridization punctae were detected (Figure 9C and D, respectively). Rare to no Ifnb1 hybridization punctae were detected at 168 hpi (Figure 9E). Similarly, the relative expression of Ifnb1 in the tumor was the highest at 72 hpi before returning to baseline at 168 hpi (Figure 9F). Like Ifnb1, the Tnfa mRNA expression in the tumor peaked at 72 hpi before returning to baseline (Supplementary Figure S4E). The Ifng remained unchanged throughout the time course in the tumor (Supplementary Figure S4F).
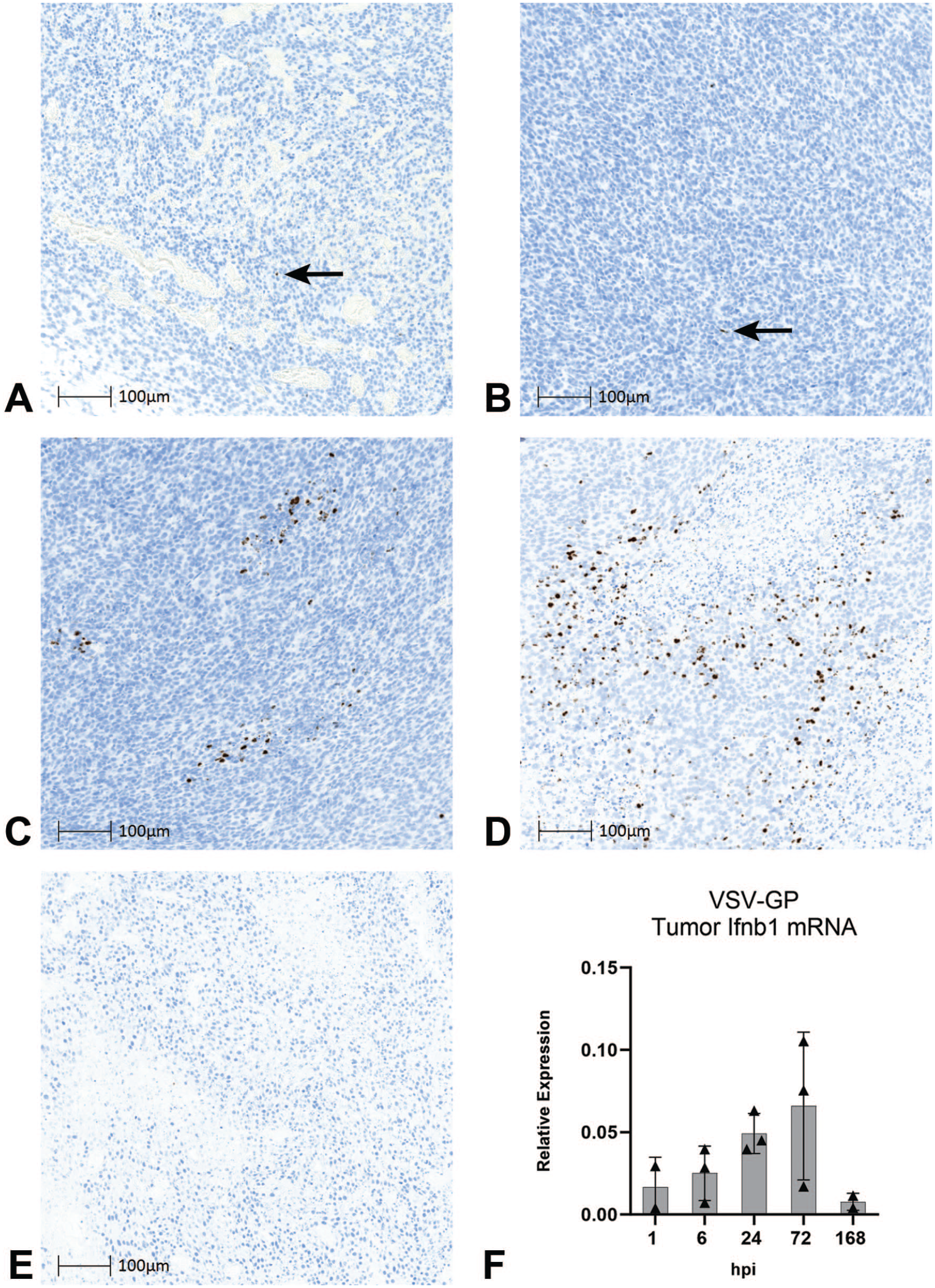
Interferon dynamics in tumor. Detection of Ifnb1 by ISH (DAB) in tumors from tumor-bearing mice following administration of a single IV dose of VSV-GP at 109 TCID50 at (A) 1 hpi, (B) 6 hpi, (C) 24 hpi, (D) 72 hpi, and (E) 168 hpi. Arrows indicate DAB signal (black arrows). (F) Relative quantification of Ifnb1 mRNA in tumors from tumor-bearing mice following administration of a single IV dose of VSV-GP at 109 TCID50. Bars represent the mean values and error bars represent the standard deviation.
Role of viral replication in immune responses
We next used a VSV-GP virus inactivated using ultraviolet (UV) light to investigate whether the observed immune cell responses in the spleen were dependent on viral replication. 30 We administered a single IV dose of either live VSV-GP-[cargo] at 108 TCID50 (1010 genome copies) or UV-inactivated (replication-incompetent) VSV-GP-[cargo] at 1010 genome copies to tumor-bearing mice and collected spleens at 1, 6, 24, 72, and 168 hpi. To demonstrate cellular localization of Tnfa and Ifng and determine the impact of replication competence on their expression, we used ISH for Tnfa mRNA duplexed with IHC for CD169. The Tnfa hybridization punctae localized primarily to CD169+ MZ macrophages in both groups; however, the magnitude of expression was greatly reduced in the UV-inactivated virus group (Figure 10A and B). No Tnfa hybridization punctae were observed in vehicle-treated mice at 6 hpi (Supplementary Figure S5A).
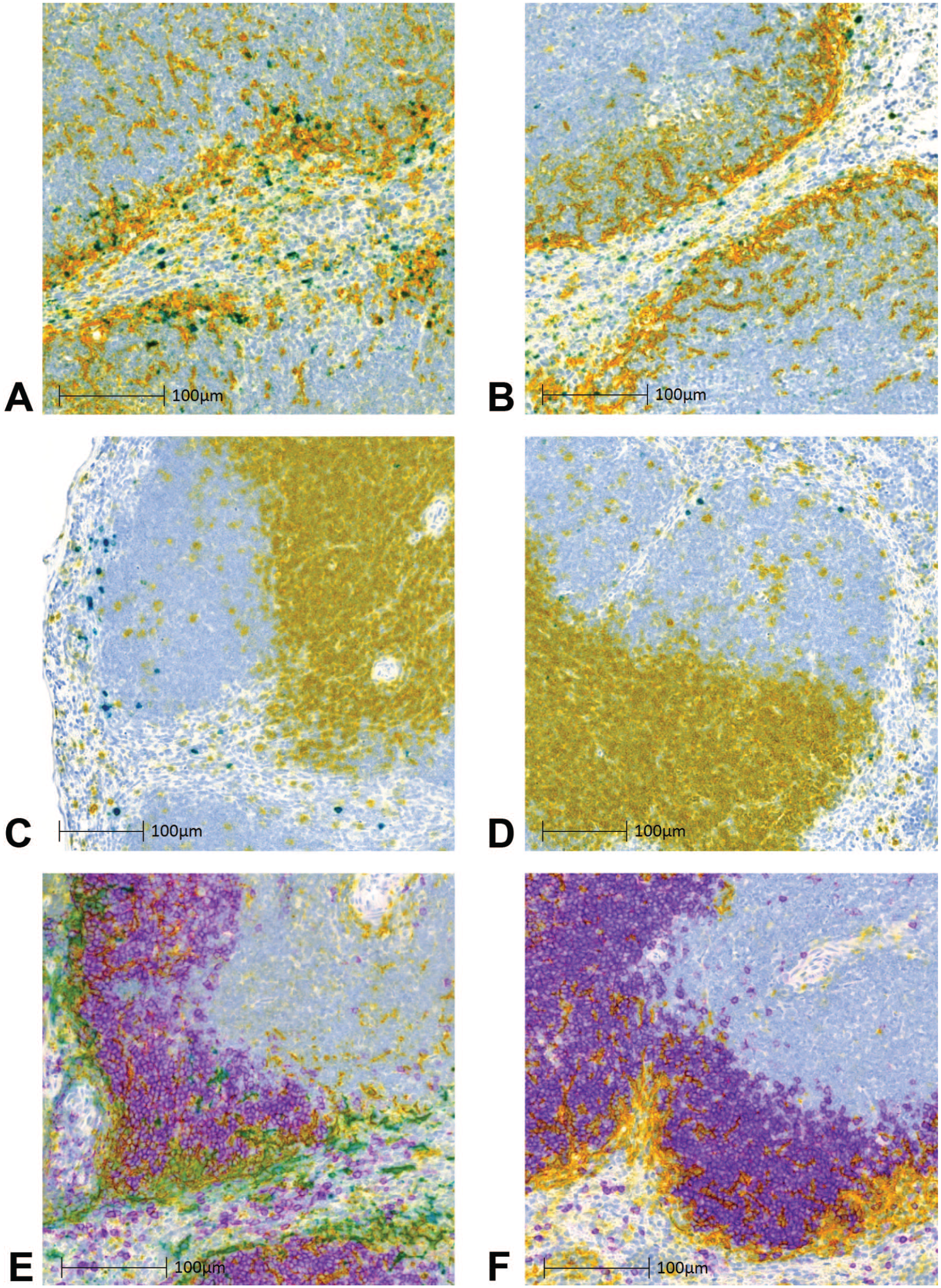
Role of viral replication in immune responses. Multiplex ISH and IHC staining of spleens from tumor-bearing mice at 6 hpi following a single IV dose of live or UV-inactivated (replication-incompetent) VSV-GP-[cargo] at 1010 genome copies (equivalent to live VSV-GP-[cargo] 108 TCID50). Tnfa mRNA ISH (teal), CD169 IHC (yellow), and colocalized signal (green) in mice treated with (A) live or (B) UV-inactivated virus. Ifng mRNA ISH (teal), CD3 IHC (yellow), and colocalized signal (green) in mice treated with (C) live or (D) UV-inactivated VSV-GP-[cargo] virus. VSV-GP anti-genomic RNA ISH (teal), CD169 IHC (yellow), CD45R (B220) IHC (purple), and colocalized VSV-GP anti-genomic RNA + CD169 (green) in mice treated with (E) live or (F) UV-inactivated virus.
Using Ifng mRNA ISH duplexed with CD3 IHC, we localized Ifng mRNA to the MZ/B-cell zone border in both treatment groups (Figure 10C and D). However, the magnitude of Ifng expression was reduced in the UV-inactivated group when compared to the live virus group. No Ifng hybridization punctae were observed in vehicle-treated group at 6 hpi (Supplementary Figure S5B).
Next, we wanted to assess the impact of viral transcription/replication on the location and interaction of MZ macrophages, B-cells, and T-cells in response to virus. To do so, we compared the results of multiplex staining using anti-genomic ISH, CD169 IHC, and CD45R (B220) IHC (as a marker of B-cells) in spleens from the live and UV-inactivated virus groups at 6 hpi. The localization of CD169+ MZ macrophages adjacent to B-cell zones, and their trafficking within these regions, has been described by others.12,25,29 Here, in the live virus-treated group, CD169+ macrophages containing anti-genomic RNA were found in the MZ along the outer border of white pulp B-cell zones. The interior B-cell zones also contained CD169+ macrophages both with and without anti-genomic RNA, although in lower numbers than the MZ (Figure 10E). In the UV-inactivated virus group, the CD169+ macrophages were in a similar distribution but with no anti-genomic RNA (Figure 10F). Here, we did not find evidence that the location or trafficking of CD169+ MZ macrophages in and around B-cell zones was impacted by viral replication.
Finally, to determine whether viral replication was necessary for splenic GC formation, we stained for BCL6 by IHC at 1 and 168 hpi. The BCL6 protein is expressed specifically on GC B-cells and thus used as a marker of B-cell activation in secondary lymphoid organs. 2 The live virus-treated group, in contrast to the UV-inactivated-treated group, showed an increase from 1 to 168 hpi both in the size and number of GCs (Figure 11A-D). The GCs from mice treated with the UV-inactivated virus were found to be more like vehicle-treated mice in both size and number at 168 hpi (Figure 11E). These results indicate that VSV-GP replication (and associated responses such as cell lysis) induces a more robust GC response than UV-inactivated virus.
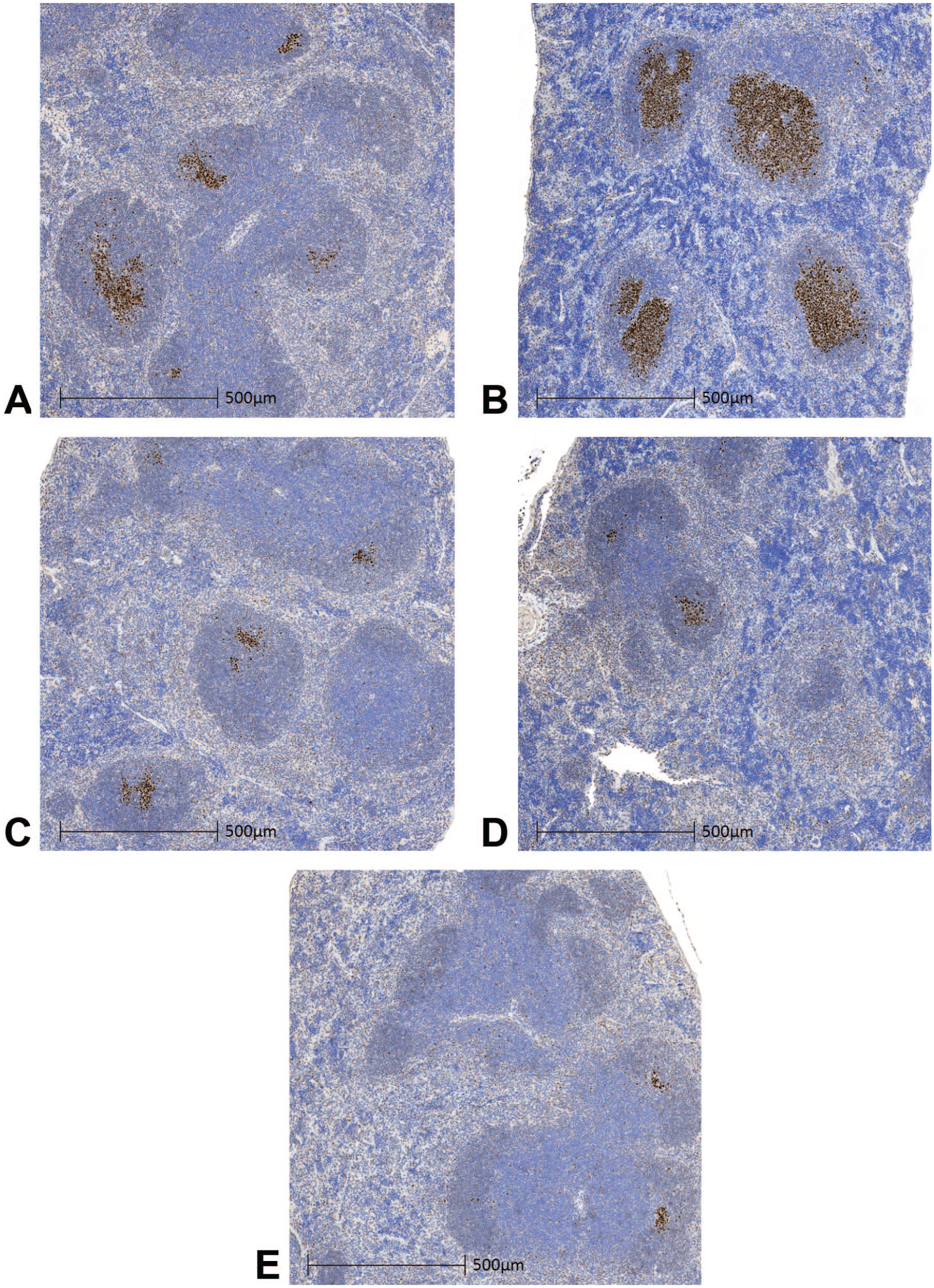
Role of viral replication in germinal center formation. Immunohistochemistry staining for BCL6 (DAB) on spleens from tumor-bearing mice following administration of a single IV dose of live or UV-inactivated (replication-incompetent) VSV-GP at 1010 genome copies (equivalent to live VSV-GP 108 TCID50). Representative images from mice treated with live virus at (A) 1 hpi and (B) 168 hpi, UV-inactivated virus at (C) 1 hpi and (D) 168 hpi, and vehicle control at (E) 168 hpi.
Discussion
Self-replication of OVs presents unique challenges to nonclinical development, as it may impact BD and safety and require a disease model to fully evaluate viral effects on nontumor sites. In this study, we applied different molecular pathology methods to better understand BD, replication dynamics, and immune cell responses of a replicating OV in tumor-free and tumor-bearing mouse models. We show that CD169+ macrophages play a central role in the initial BD of VSV-GP across key nontumor tissues. We also demonstrate that these cells are a primary site of both the transcription/replication and initial induction of cytokines that restrict replication of VSV-GP in liver and spleen. Furthermore, our results indicate that replication within CD169+ cells is a determinant of B-cell development within splenic GCs. No clear differences in VSV-GP BD, replication, or immune responses were observed between tumor-free and tumor-bearing mice. These findings support the hypothesis that the early burst of VSV-GP transcription/replication in CD169+ macrophages serves as a key priming event for downstream immune responses.11,15,21
Our results extend previous findings by Dambra et al by describing VSV-GP effects in a tumor-bearing model. Notably, the CT26.CL25.IFNAR-/- tumors exhibited several marked differences in viral response compared with nontumor sites. Peak transcription/replication occurred later, at 72 hpi in tumors vs 6 hpi in spleen and 1 hpi in liver. Cellular expression of key cytokines (Infb1, Ifng, and Tnfa) followed a similar pattern. During the peak window of tumor replication (24-168 hpi) tumor cells were a major site of VSV-GP transcription/replication, in contrast to nontumor sites where transcription/replication occurred almost exclusively within CD169+ macrophages. Importantly, the presence of a tumor did not impact early BD of viral genomes or the magnitude or timing of transcription/replication in nontumor tissues. Moreover, we found no evidence that viral replication in the tumor, after a single IV dose of VSV-GP, induces secondary infection in key nontumor sites out to 168 hpi.
Macrophages are a major component of early innate immune responses to viral infection.1,12,22,27 In organs such as the spleen, liver, and lymph node, they line endothelial channels and actively capture and clear viral particles from the blood. 22 This phagocytic uptake also drives an IFN response that inhibits viral replication and limits systemic infection.12,22 A third antiviral mechanism is described specifically for CD169+ macrophages, which have been shown to allow transient viral replication7,15 by decreasing IFN signaling.7,15 This “enforced” replication may in turn provide viral antigen for priming of the adaptive immune system. 37 Our findings collectively support this model. In key nontumor sites, we observed rapid uptake of viral genomes from the blood by CD169+ macrophages (at 1 hpi, in multiple tissues), followed by viral transcription/replication and interferon expression that was highly specific to this cell type. In the spleen, where CD169+ MZ macrophages surround lymphocyte-rich white pulp regions, blocking viral replication by UV treatment effectively inhibited activation of GC B-cells, suggesting that replication in MZ macrophages is a key step in the initiation of downstream adaptive responses.
In addition to the initial burst of VSV-GP replication in CD169+ macrophages, we document a subsequent decrease in these cells starting at 6 to 24 hpi in the liver and spleen. The mechanism of cell death for this effect is currently unknown. Here, we measured markers of both classical apoptosis (Caspase 3) and pyroptosis (Gasdermin D) but did not see a pattern that is consistent with induction of either pathway. It is also unclear how the transient loss of these macrophages following initial infection may impact BD and immune responses upon repeat dosing with VSV-GP.
In this work, we applied several different tissue-based methods to investigate the early dynamics of VSV-GP infection in tumor-free and tumor-bearing mouse models. Methods described here, in particular ISH assays for decoupling genomic and anti-genomic RNAs, can be applied to the nonclinical assessment of other OVs. Overall, our findings highlight tissue-resident CD169+ macrophages as a major determinant of initial viral BD, replication, and cytokine induction. Future directions of this work include further characterization of viral immune cell interactions and investigation of the impacts of route of administration (including IV vs intratumoral) and repeat-dose regimens on the viral BD, safety, and pharmacodynamics.
Supplemental Material
sj-pdf-1-tpx-10.1177_01926233241303904 – Supplemental material for Molecular Pathology Methods to Characterize Biodistribution and Pharmacodynamics of the Oncolytic Virus VSV-GP in a Nonclinical Tumor Model
Supplemental material, sj-pdf-1-tpx-10.1177_01926233241303904 for Molecular Pathology Methods to Characterize Biodistribution and Pharmacodynamics of the Oncolytic Virus VSV-GP in a Nonclinical Tumor Model by Andrea Matter, Karol Budzik, Saurin Mehta, Kathleen Hoyt, Richard Dambra, Adam Vigil, Joseph Ashour, Ernest Raymond, Elizabeth Clark and Charles Wood in Toxicologic Pathology
Footnotes
Acknowledgements
The authors acknowledge Cedric Cheminay, Kari Neier, and Michael Franti for helpful discussions and feedback. In addition, the authors thank Corey Cirillo, Richard Ruiz, Michelle Maugiri, Kaitlynn Graca, Ashlee Bell-Cohn, Saeed Akhand, Kathleen Bunosso, for excellent technical assistance, Audrey Brenot and Amy Mikolaichik for support with the mouse tumor-model, and Boehringer Ingelheim Animal Resources and their veterinary staff for professional animal husbandry and performing additional health checks to ensure the welfare of the research animals.
Author Contributions
Conceived and designed the overall research (AM, KB, RD, JA, AV, CW); supervised the research and provided feedback (EC, ER, JA, AV, CW); wrote the original manuscript and made figures (AM); edited the manuscript (AM, SM, KH, RD, JA, CW); designed in vivo studies (RD, JA, AM, CW); coordinated in vivo studies (RD); designed, analyzed, and interpreted molecular pathology assays (AM, KB, SM, CW); performed molecular pathology experiments (AM, KB, SM); designed and interpreted molecular biology assays (KH, ER, AM, CW); performed and analyzed molecular biology experiments (KH); performed statistical analyses(AM, KH); interpreted the overall results of the studies (AM, CW); secured necessary support for this work (JA, AV, CW).
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are/were employed at Boehringer Ingelheim Pharmaceuticals, Inc (BIPI) in Ridgefield, Connecticut, at the time of the experiments.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded entirely by BIPI.
Data Availability
Raw data, materials, and protocols are available from the corresponding author, AM, upon reasonable request. Some information specific to VSV-GP may be limited due to restrictions that could compromise proprietary information. The VSV-GP is part of an ongoing clinical development program, thus neither samples of the vector itself nor its full genetic sequence can be shared at this time.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.