
Abstract
Replication-incompetent adeno-associated virus (AAV)-based vectors are nonpathogenic viral particles used to deliver therapeutic genes to treat multiple monogenic disorders. AAVs can elicit immune responses; thus, one challenge in AAV-based gene therapy is the presence of neutralizing antibodies against vector capsids that may prevent transduction of target cells or elicit adverse findings. We present safety findings from two 12-week studies in nonhuman primates (NHPs) with pre-existing or treatment-emergent antibodies. In the first study, NHPs with varying levels of naturally acquired anti-AAV5 antibodies were dosed with an AAV5-based vector encoding human factor VIII (hFVIII). In the second study, NHPs with no pre-existing anti-AAV antibodies were dosed with an AAV5-based vector carrying the beta subunit of choriogonadotropic hormone (bCG); this led to the induction of high-titer antibodies against the AAV5 capsid. Four weeks later, the same NHPs received an equivalent dose of an AAV5-based vector carrying human factor IX (hFIX). In both of these studies, the administration of vectors carrying hFVIII, bCG, and hFIX was well-tolerated in NHPs with no adverse clinical pathology or microscopic findings. These two studies demonstrate the safety of AAV-based vector administration in NHPs with either low-titer pre-existing anti-AAV5 antibodies or re-administration, even in the presence of high-titer antibodies.
Keywords
Graphical abstract
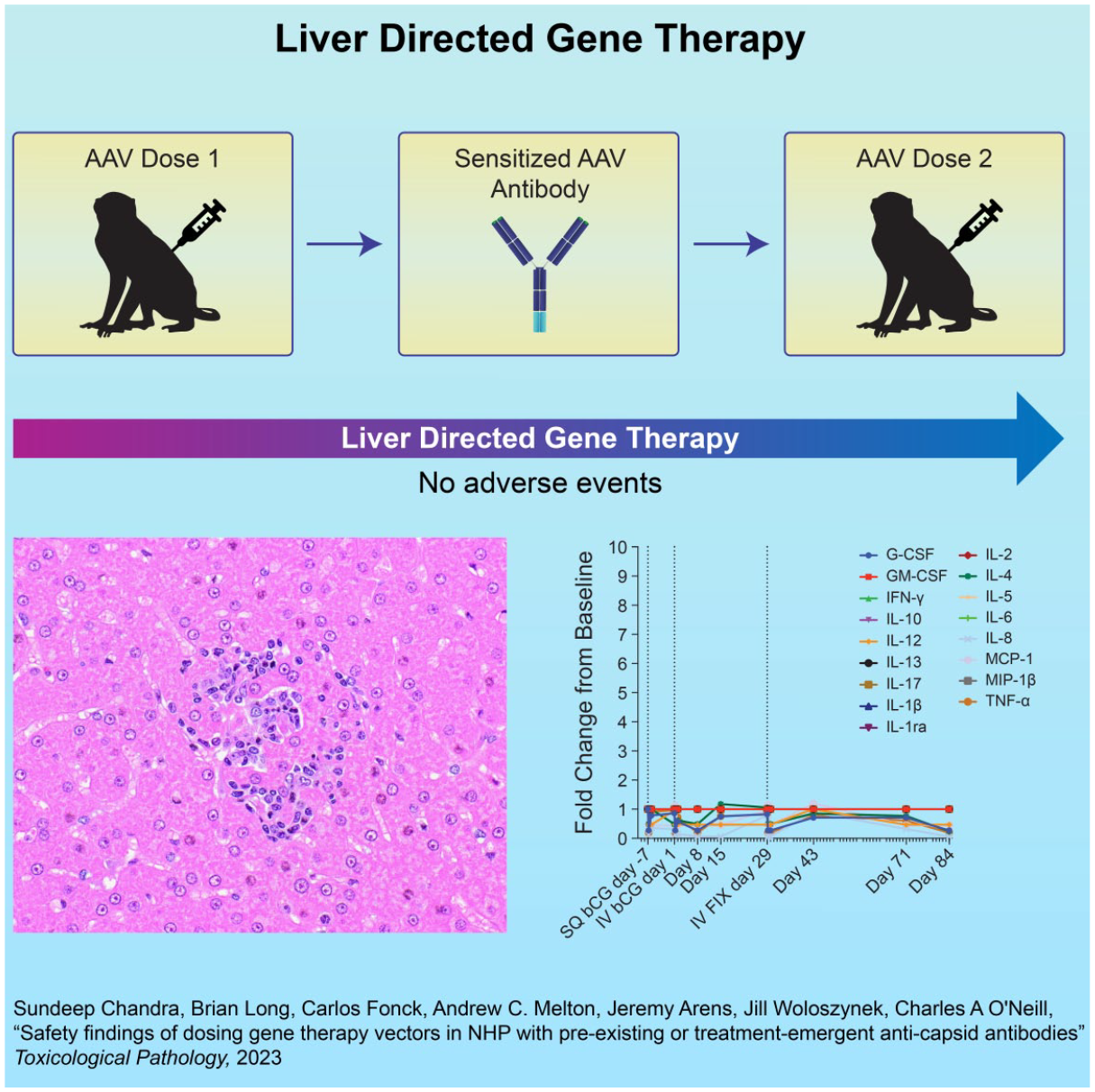
Introduction
Adeno-associated virus (AAV)-based vectors hold promise for gene therapy applications, in part because they are nonreplicating and nonpathogenic. AAV-based vector genomes generally persist in an epi-chromosomal state and drive long-term transgene expression with relatively low integration into the host genome. Though immunity against AAV is widely disseminated within the human population 12 with reported global seroprevalence estimates ranging from 35% to 90% for various AAV serotypes,5,17,20,21,24 wildtype (WT) AAV5 infection has not been definitively proven as the cause of any known human pathologies or diseases. 2 Notably, while pre-existing immunity to AAV varies by serotype and geographic region, AAV5 has lower seroprevalence compared to other serotypes. 17
Recent clinical studies have challenged the nonpathogenic profile of AAV infection. Following the initial discovery of an association of AAV integrations into oncogenes within a small subset of hepatocellular carcinomas (HCCs), 18 two additional clinical investigations reported findings of AAV2 infection in the livers of children with acute severe hepatitis, raising the possibility of a role of AAV in hepatitis in the presence of a helper virus.8,13,16 Nevertheless, causality between AAVs and acute severe hepatitis has not been thoroughly established and requires further investigation.
Although AAVs are not strongly immunogenic, they can nonetheless give rise to both humoral and cellular immune responses. As AAV-based vectors are administered directly to patients, pre-existing or recall immune responses to the viral vector capsid or the transgene product can interfere with the efficacy of AAV gene therapies. 24 Seroprevalence studies indicate that initial exposure to WT AAVs often occurs during childhood or adolescence4,24 when humoral and cellular immune responses directed against the AAV capsid might be mounted, although immune responses against AAVs may also be mounted in adults.1,30 As such, AAV-specific memory T-cells and B-cells might have lifelong persistence and be recalled upon recombinant AAV (rAAV)-mediated gene transfer, with the vector initiating a memory response at the time of infusion.
The humoral response results in the generation of anti-AAV neutralizing antibodies (NAbs), pre-existing due to WT AAV exposure or generated following therapeutic dose administration of rAAVs. Whereas pre-existing anti-AAV antibodies might prevent the use of AAV-based gene therapy in a particular individual altogether, administration of rAAV often gives rise to quantitatively higher antibody responses even in immunologically naïve patients, thereby precluding repeat dose administration. Iterative administration of AAV vectors of the same or antigenically similar serotype, which would be beneficial in certain instances, may only be possible with therapeutic interventions that either prevent the formation of the antibody response or subvert existing antibody response by other means. In addition to the humoral response, a modest cellular immune response can result in the targeted ablation of transduced cells and failure to establish long-term transgene expression in some instances. 27
We searched for but did not find toxicology reports of AAV-based vectors administered to NHPs in the presence of pre-existing antibodies that follow the natural course of exposure, nor have effects been reported following repeat dose administration in the presence of high-titer, capsid-specific antibodies that emerge following the initial dose. Since humoral and cellular immune responses are critical parameters for evaluating the safety and efficacy of AAV-based gene therapy, we conducted two separate NHP studies to evaluate the impact of both pre-existing and treatment-emergent antibodies following dose administration of an AAV5-based vector. The objective of Study 1 was to determine the safety and comparative pharmacodynamics of gene delivery and human FVIII-SQ (hFVIII-SQ) expression following a single bolus injection of BMN 270, an investigational AAV5-based gene therapy vector encoding B domain-deleted hFVIII-SQ for the treatment of hemophilia A, to NHPs with varying baseline pre-existing levels of anti-AAV5 total binding antibodies (TAb) and antibody or non-antibody transduction inhibitors (TI). The objective of Study 2 was to evaluate the impact of re-administration of an AAV5-based vector to previously dosed NHPs with high levels of treatment-emergent antibodies resulting from intentional exposure to the AAV5-based vector. Results detailing the safety and transduction efficiency of the AAV-based vector in NHPs with either pre-existing or treatment-emergent antibodies are discussed in this manuscript.
Materials and Methods
Study 1
Study design
The objective of this non-clinical study was to determine the pharmacodynamics of gene delivery and hFVIII-SQ expression following a single infusion of BMN 270 into cynomolgus monkeys with varying pre-existing levels of neutralizing AAV5 antibodies (AAV5 TAb assay) and non-antibody transduction inhibitors (AAV5 TI assay). To examine the impact of pre-existing AAV5 immunity on BMN 270-mediated gene transfer, this study was conducted in cynomolgus monkeys (Macaca fascicularis) that met pre-defined criteria for AAV5 TAb/TI status (TAb−/TI−, TAb-/TI+, or TAb+/TI+ as described below). For Study 1, 60 male monkeys were screened during the pre-treatment phase; of these, 15 were subsequently assigned to one of four study groups (Table 1) at Charles River Laboratories, Inc. (Reno, NV, USA). The animals were between 2.5 to 4.4 years of age and weighed between 2.4 and 3.3 kg prior to treatment. Group 1 (N = 3) included animals without anti-AAV5 TAb or detectable TI (TAb−/TI−; control group). Groups 2 (N = 4) and 3 (N = 3) included animals without detectable anti-AAV5 TAb but with detectable TI (TAb−/TI+; non-antibody inhibitors); lower TI titers (2-4) were enrolled in Group 2, while moderate TI titers (5-10) were enrolled in Group 3. Group 4 (N = 5) included animals with both anti-AAV5 TAb and detectable TI (TAb+/TI+). Dosing was initiated on June 8, 2016, designated as study Day 1, with the in-life study phase completion on August 2, 2016. BMN 270 was administered as a single, slow intravenous bolus injection on study Day 1, at a dose level of 6E13 vg/kg for all animals in each group.
Experimental study design for Study 1: male cynomolgus monkeys with varying levels of pre-existing TAb/TI titers were administered BMN 270, an AAV5-based vector encoding B domain-deleted human factor VIII (hFVIII-SQ).

Abbreviations: TAb = total antibody; TI = transduction inhibitor (titers at Screening and Baseline sample timepoints may differ); AAV = adeno-associated virus.
Test article
Test article BMN 270 was provided by BioMarin Pharmaceutical Inc. and stored at −80°C until use. BMN 270 was equilibrated at room temperature for approximately 1 hour on the day of use, prepared for administration under a laminar flow hood, and administered within 5 hours after preparation. The experimental design is presented in Table 1.
Assays and sample collection
Results describing specific details of the AAV5 TAb assay, AAV5 TI assay, FVIII-SQ protein assay, and FVIII TAb assay have been published previously.10,22 Briefly, in the AAV5 TAb assay, AAV5 antibodies were detected in cynomolgus monkey plasma with a sandwich electrochemiluminescence (ECL) assay on the Meso Scale Discovery (MSD) platform. Plasma samples were analyzed to determine the presence or absence of AAV5 antibodies; no titrations were performed to produce antibody titer values. Notably, the clinical AAV5 TAb assay differs from the non-clinical AAV5 TAb assay, as it uses a bridging ECL immunoassay and has a different screening cutpoint (SCP). Therefore, a direct comparison of results between them cannot be determined. 22
The cell-based AAV5 TI assay used HEK293T/17 cells and an AAV5-luciferase reporter vector. Plasma samples were first analyzed in a screening assay to identify the presence or absence of AAV5 neutralizing activity; samples with a normalized transduction signal below the SCP of 44.1% transduction were considered positive. Positive samples were further analyzed with the titer method to determine the relative magnitude of AAV5 TI (TI titer).
In the hFVIII-SQ protein assay, a sandwich ECL was used to measure the concentration of hFVIII, the transgene being expressed, in cynomolgus monkey plasma. 22 Standard curve regression analysis was used to report concentrations of study samples; the lower limit of quantitation (LLOQ) for the assay was 2.35 ng/mL hFVIII-SQ.
Finally, the hFVIII TAb assay measured hFVIII antibodies with a bridging ECL assay on the QuickPlex Imager. Plasma samples were analyzed to identify the presence or absence of hFVIII antibodies; samples with a normalized signal greater than or equal to the plate-specific SCP were considered positive. The limit of detection (LOD) was 1.93 ng/mL of the positive control hFVIII antibody.
Blood was obtained by venipuncture for routine hematology throughout the study at designated time points (Week −1 and Days 29 and 56). Coagulation parameters (activated partial thromboplastin time, fibrinogen, and prothrombin time) were assessed at Week −1 and Days 29 and 56; fasting clinical chemistry studies were performed at Week −1 and on Days 15, 29, 43, and 56. NHPs were euthanized by exsanguination on study Day 56. At necropsy, a complete set of tissues was collected and processed for routine light microscopic evaluation.
Blood was obtained by venipuncture for all assays. Time points included pre-treatment for all assays, as well as additional Days 29 and 56 for the AAV5 TAb and hFVIII TAb assays and Days 8, 15, 22, 29, 36, 43, 50, and 56 for the hFVIII-SQ protein assay. Plasma samples were shipped on dry ice within 7 business days following Day 29 and termination collection to Precision for Medicine (PFM, Redwood City, CA) for the AAV5 TAb assay, or BioMarin Pharmaceutical Inc. (San Rafael, CA) for the hFVIII-SQ protein and hFVIII TAb assays. Samples collected at protocol-specific time points were stored at −60° to −80° C and analyzed for anti-AAV5 TAb/TI, hFVIII-SQ protein, and hFVIII TAb.
Statistical analysis
Animal Groups 2, 3, and 4 were compared to Group 1, the control group (TAb−/TI−). Analysis of variance (ANOVA) was used to evaluate significant intergroup differences, followed by a multiple comparisons test. Assumptions allowing the use of a parametric ANOVA were verified with the Shapiro-Wilks test for normality and Levene’s test for homogeneity, with P ≤ 0.001 significance required to reject assumptions. A single-factor ANOVA was applied using animal grouping as the factor, with the P ≤ 0.05 level of significance. If parametric assumptions were not satisfied, then the Kruskal-Wallis non-parametric ANOVA was used to evaluate intergroup differences at P ≤ 0.05; Dunn’s multiple comparison tests were applied if the ANOVA was significant (P ≤ 0.05). A summary statistical report was generated by SAS®.
Study 2
Study design
Sixty male cynomolgus monkeys were screened for the presence of antibodies against the AAV5 capsid, as described in the screening phase for Study 1 and enrolled in one of two study groups (Table 2) at Charles River Laboratories, Inc. (Reno, NV, USA). The animals were between 2.5 and 3.0 years of age, weighed between 2.1 and 2.3 kg, and were screened negative for anti-AAV5 TAb prior to treatment.
Experimental study design for Study 2: male cynomolgus monkeys were administered an AAV5-based vector encoding hFIX or an AAV5-based vector encoding the bCG.

Abbreviations: AAV = adeno-associated virus; hFIX = human factor IX; bCG = beta subunit of choriogonadotropic hormone; IV = intravenous; NA = not applicable.
The NHPs in Group 1 (N = 3) served as positive controls and received one gene therapy dose at 3E13 vg/kg of an AAV5-based vector expressing human FIX (AAV5-hFIX) on Day 29 via intravenous (IV) injection. Following dose administration, Group 1 was expected to achieve maximal expression of the hFIX transgene (hFIX plasma protein concentration) since these NHPs pre-screened negative for anti-AAV5 TAb and did not receive gene therapy dose administrations prior to Day 29.
The NHPs in Group 2 (N = 3), which also pre-screened negative for anti-AAV5 TAb, received three total gene therapy doses. Animals received an initial dose of AAV5-bCG (5E12 vg) on study Day −7 via subcutaneous injection, and a second dose of AAV5-bCG at a dose level of 3E13 vg/kg on study Day 1 via IV injection. Group 2 animals were expected to mount an anti-AAV5 TAb response following the second administration of AAV5-bCG on study Day 1. Finally, animals in Group 2 were administered a dose of AAV5-hFIX at a dose level of 3E13 vg/kg on Day 29 via IV injection to evaluate the safety and pharmacodynamic effect of gene therapy administration in the presence of a high-titer anti-AAV5 TAb. Group 2 received the AAV5-hFIX vector on the same day as Group 1 animals to match the timing for antibody response.
Test article
Frozen test articles (AAV5-bCG and AAV5-hFIX) were stored at −80°C until use and thawed on the day of dose administration. Dose formulations were equilibrated at room temperature for approximately 0.5 hours prior to dosing and administered within 4 hours after thawing. The experimental design is presented in Table 2.
Assays and sample collection
A detailed description of the anti-AAV5 TAb assay method has been published previously and provided in the description for Study 1. 22 Meanwhile, anti-hFIX antibodies were assessed with a bridging ECL immunoassay and analyzed as the ratio of sample signal normalized to a pooled cynomolgus plasma control; plasma hFIX protein levels were assessed by a fluorescence ELISA assay.
Blood was collected for hematology, clinical chemistry, coagulation, and anti-AAV5 capsid antibodies by venipuncture on Days −14, 1 (predose), 15, 29 (predose), 43, 57, 71, and 84. Plasma levels of the hFIX transgene product were measured as well. NHPs were euthanized by exsanguination on study Day 85. At necropsy, a complete set of tissues was collected for histopathology and for DNA/RNA-qPCR analysis. A standard list of protocol-required tissues was processed for routine light microscopic evaluation. Anti-AAV5 TAb blood samples were collected by venipuncture on Days −14, 1 (predose), 15, 29 (predose), 43, 57, 71, and 84 and processed to plasma. Anti-hFIX TAb, hFIX, and bCG blood samples were collected by venipuncture on Days −14, 1 (predose), 29 (predose), 57, and 84 and processed to plasma.
Blood was collected for immunophenotyping by venipuncture on Days −14, 15, 43, 71, and 84. Whole blood samples were checked for clots and transferred to the appropriate laboratory; upon receipt, samples were stored at ambient temperature. Absolute cell counts were determined using BD TruCount™ tubes per standard operating procedure. Calculation of absolute counts for each cell population was performed using the relative percentages of each cell population and the corresponding lymphocyte or lymphocyte/monocyte/granulocyte count obtained using BD TruCount™ tubes with CD45 (SSC/CD45+). The cellular antigens and cell populations identified were quantified according to testing facility-validated methods. Longitudinal plots of immunophenotyping data were generated using GraphPad Prism™ software version 8.4.2.
Blood was collected for the AAV5 capsid IFN-γ ELISpot assay by venipuncture on Days −10, 15, 43, 71, and 84. Samples were mixed gently and kept at room temperature. Peripheral blood mononuclear cell (PBMC) samples were transferred at ambient condition for subsequent processing and frozen at 5-10 × 106 cells/mL/cryovial. Short-term (1-3 days) storage of PBMCs was in a −60° to −80° C freezer, while long-term storage (>3 days) was in a liquid nitrogen tank. PBMCs were assessed for ex-vivo cytokine release in response to Staphylococcal enterotoxin B (SEB), anti-CD3 (positive controls), dimethyl sulfoxide (DMSO; negative control), or two sponsor-derived AAV5 capsid peptide pools. 33 Several acceptance criteria were applied. Upon thawing of PBMCs, samples were required to be ≥70% viable. Samples were rejected if the positive control stimulation with PHA did not yield a positive response at or above the LOD (10 SFU/well). For test sample results with ≥ 30 SFU/well, the intra-triplicate %CV was required to be ≤ 30%. For samples with %CV > 30%, a Dixon’s Q test (P = 0.05) was performed for outlier removal. If no outlier was determined or the %CV > 30% even after outlier removal, then the sample was rejected or retested using a backup aliquot, if available. For the sample where the SFU > LOD, but < 30 SFU/well, results were not required to meet this %CV criterion, and results were reported with a comment that the result is > LOD but ≤ LLOQ.
Blood was collected for cytokine analysis by venipuncture on Days −7, 1, 8, 15, 29, 43, 71, and 84; the resultant plasma was divided into two tubes and frozen at −60° to −80° C until transferred to the laboratory. Samples were analyzed using a validated Luminex method for detection of IL-1β, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12/23 (p40), IL-13, IL-17A, MCP-1, MIP-1β, IFN-γ, TNF-α, G-CSF, and GM-CSF.
Statistical analysis
Statistical analysis was performed by the testing facility on all numerical data, with the exception of flow cytometry and cytokine data. For data collected in Provantis™, Levene’s test was used to assess the homogeneity of group variances parametric assumption at P ≤ 0.05. Datasets with ≥3 groups were compared using an overall one-way ANOVA F-test or Kruskal-Wallis test at P ≤ 0.05. These pairwise comparisons were conducted with a two-sided Dunnett’s or Dunn’s test, respectively, if the overall test was significant. All results were calculated using non-rounded values as per the raw data rounding procedure and may not have been exactly reproduced from individual data presented.
Ethical Treatment of Animals
These studies complied with all applicable sections of the Final Rules of the Animal Welfare Act regulations (CFR, Title 9), the Public Health Service Policy on Humane Care and Use of Laboratory Animals from the Office of Laboratory Animal Welfare, and the Guide for the Care and Use of Laboratory Animals from the National Research Council. The protocol and any amendments or procedures involving the care or use of animals in these studies were reviewed and approved by the Charles River Laboratories, Inc. Institutional Animal Care and Use Committee before the initiation of such procedures. The two NHP studies reported here were conducted between 2016 and 2019. The cynomolgus monkey was chosen for these two studies as it is accepted by regulatory agencies as a non-rodent species for use in preclinical toxicity testing. The number of animals and groups was considered the minimum needed to sufficiently characterize the effects of the test article without using an unnecessary number of animals. Dosage for these studies was based on previous unpublished data from BioMarin Pharmaceutical Inc.
Results
Study 1
In this study, we evaluated the safety and pharmacodynamics of gene delivery and hFVIII-SQ expression in cynomolgus monkeys with varying baseline Tab and TI titer status following a single bolus injection of BMN 270. At baseline (Day −7), hFVIII-SQ levels in all animals were below the LLOQ, confirming that the assay has no cross-reactivity for monkey FVIII. After administering BMN 270, all animals showed hFVIII-SQ levels above the LLOQ, with the exception of two of the five animals in Group 4 (Tab+/TI+; neutralizing AAV5 antibodies with screening TI titers of 6-59). Peak levels (Cmax) in all animals with quantifiable hFVIII-SQ fell between Days 22 and 36. Even though animals in all groups received the same dose level, the mean hFVIII-SQ concentration in Group 4 was lower than that in Group 1, which included anti-AAV5 Tab-/TI− animals (control group) with no evidence of pre-existing AAV5 immunity. The lower levels of hFVIII-SQ in Group 4 were observed across all three time points (Days 22, 29, and 36), suggesting that anti-AAV5 Tab+/TI+ positivity diminished the pharmacodynamic effectiveness of gene therapy administration. Levels of hFVIII-SQ in cynomolgus monkey plasma mean data across three different time points are presented in Table 3.
Study 1 mean (SD) hFVIII concentration (ng/mL) at three different time points in male cynomolgus monkeys administered BMN 270 without Tab/TI titers (Group 1) or with various levels of Tab/TI titers (Groups 2, 3, and 4).
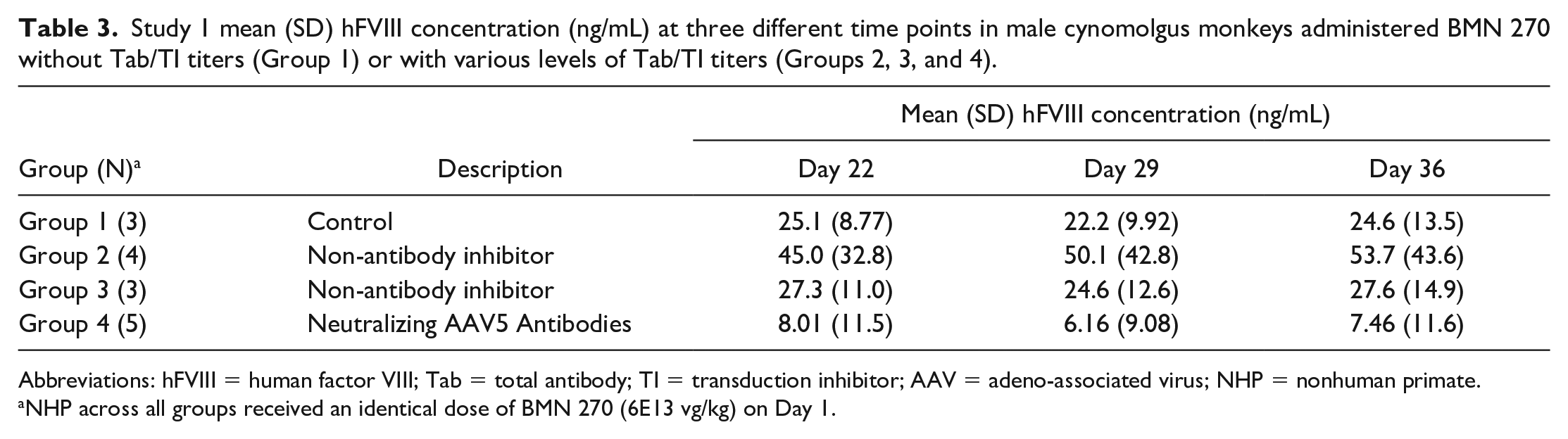
Abbreviations: hFVIII = human factor VIII; Tab = total antibody; TI = transduction inhibitor; AAV = adeno-associated virus; NHP = nonhuman primate.
NHP across all groups received an identical dose of BMN 270 (6E13 vg/kg) on Day 1.
Plasma samples from all animals were tested with the AAV5 Tab assay at baseline and post-treatment on Days 8, 15, 22, 29, and 56. By Day 8, all dosed animals were AAV5 antibody-positive, indicating that the onset of the AAV5 antibody response occurred rapidly after BMN 270 injection and with similar kinetics across all study groups. Plasma from all dosed animals remained positive for anti-AAV5 antibodies throughout the study until Day 56, without any apparent effect of pre-existing AAV5 immunity. 22
There were no clinical observations or body weight changes associated with the administration of BMN 270 to NHPs either with or without pre-existing Tab or TI titers. NHPs in Group 4 (Tab+/TI+ titers of 6-100) administered BMN 270 had a statistically significant increase (1.5X) in lymphocyte counts on Day 56 compared to Group 1 (Tab-/TI-). There were no changes in coagulation parameters or clinical chemistry including serum transaminases (alanine aminotransferase [ALT] and aspartate aminotransferase (AST); Table 4).
Group mean serum transaminases (ALT and AST; U/L)a in NHPs with various baseline titers from Study 1.
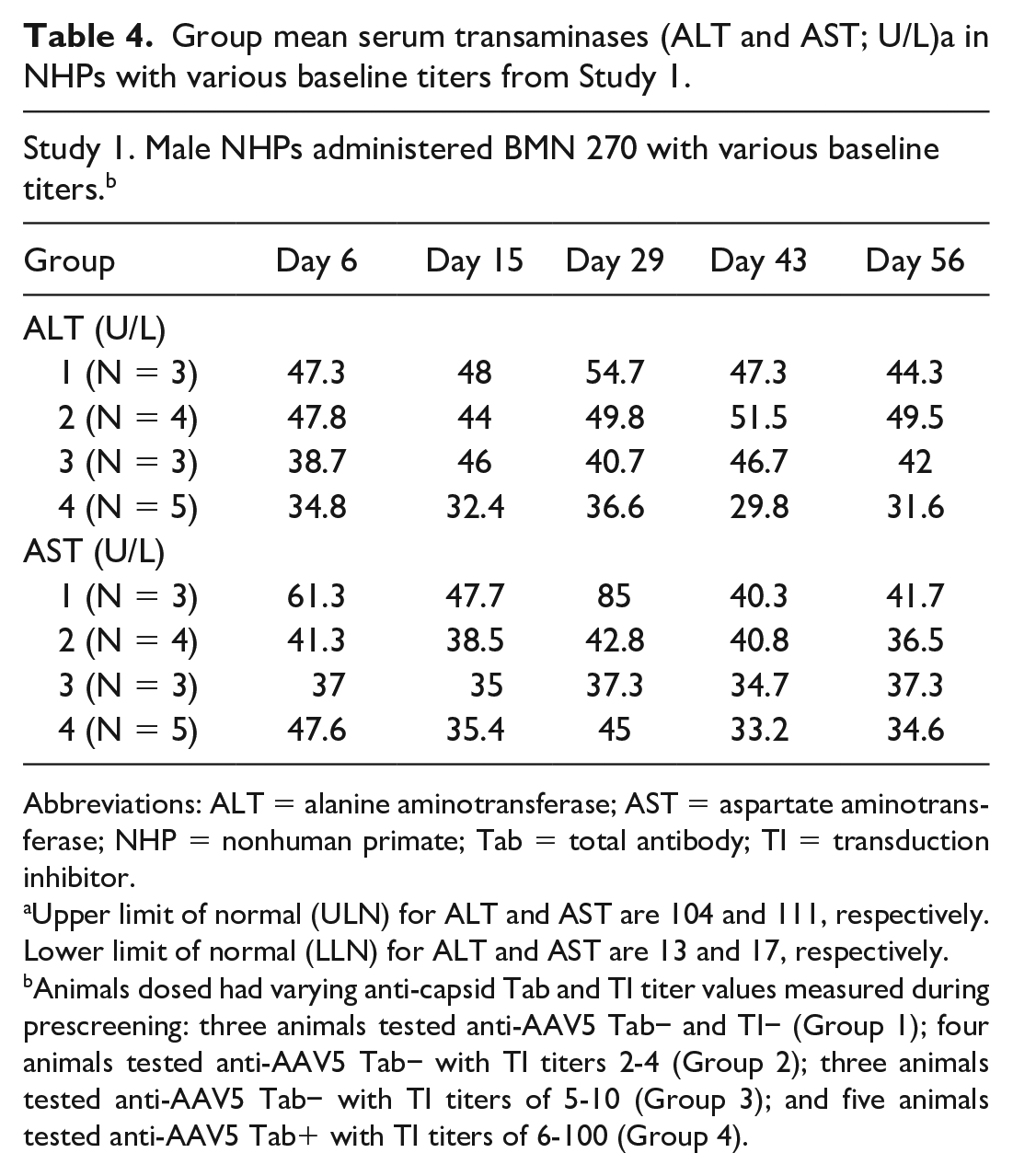
Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase; NHP = nonhuman primate; Tab = total antibody; TI = transduction inhibitor.
Upper limit of normal (ULN) for ALT and AST are 104 and 111, respectively. Lower limit of normal (LLN) for ALT and AST are 13 and 17, respectively.
Animals dosed had varying anti-capsid Tab and TI titer values measured during prescreening: three animals tested anti-AAV5 Tab− and TI− (Group 1); four animals tested anti-AAV5 Tab− with TI titers 2-4 (Group 2); three animals tested anti-AAV5 Tab− with TI titers of 5-10 (Group 3); and five animals tested anti-AAV5 Tab+ with TI titers of 6-100 (Group 4).
Group mean splenic weights were slightly higher (not statistically significant) in Group 4 animals compared to Group 1 (data not shown). However, in the absence of a true naïve/untreated control group, the splenic weight change was considered incidental with no relationship to Tab/TI status or to the administration of the test articles. Notably, spleen weights in cynomolgus monkeys, absolute and relative to body or brain weights, are highly variable (up to 5.11-fold) within a control group on an individual study. 9 Histopathologic examination of livers including major parenchymal organs from NHPs in all groups revealed no test article-related or adverse findings. Furthermore, a few scattered foci of mixed inflammatory cell infiltrates (Figure 1) observed across all treatment groups were considered incidental and normal background findings of cynomolgus monkeys.7,28 Overall, there were no microscopic changes in the spleen.
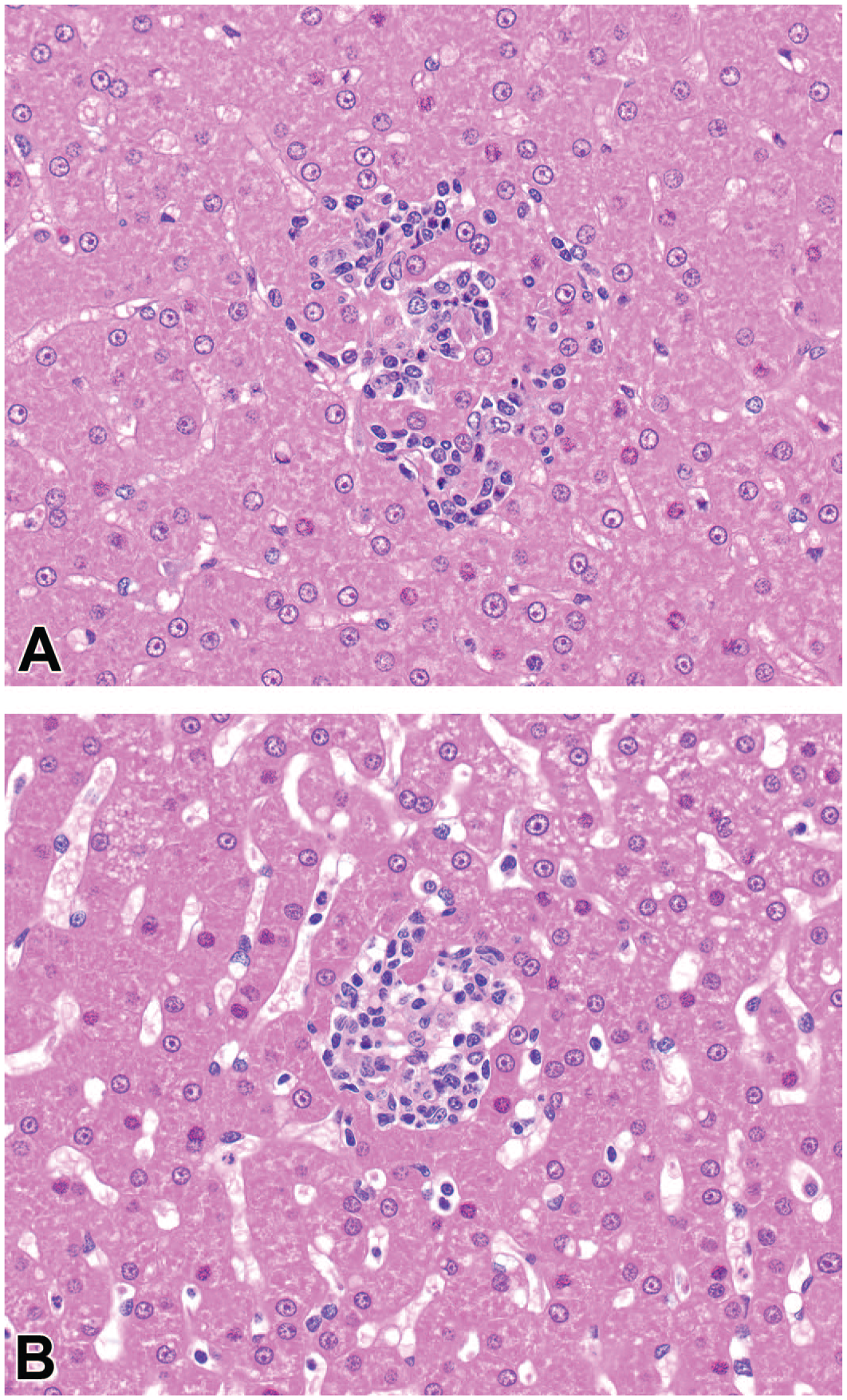
Livers from NHP administered an AAV5 vector either with pre-existing (A) or treatment-emergent anti-capsid antibodies (B) for liver-directed gene therapy. AAV indicates adeno-associated virus; NHP, nonhuman primate.
Study 2
In this study, we assessed the safety and pharmacodynamic effects of AAV5-based vector re-administration in previously dosed NHPs with treatment-emergent antibodies using a challenge dose of AAV5 encoding hFIX. All animals developed anti-AAV5 Tab following dosing with AAV5-hFIX (Group 1) and AAV5-bCG (Group 2) (Table 5). Group 1 animals, dosed with AAV5-hFIX on Day 29, had detectable titers on Day 43. Group 2 animals, receiving a subcutaneous dose of 5E12 vg AAV5-bCG on Day −7 and IV doses of 3E13 vg/kg AAV5-bCG on Day 1 and 3E13 vg/kg AAV5-hFIX on Day 29, had very low titers detectable in some animals on Day 1 (one week following the initial injection of AAV5-bCG on Day −7), with a robust titer response in all animals by Day 43. Notably, the purpose of the subcutaneous injection was to target the vector capsids to the draining lymph nodes in an attempt to elicit a more localized immune response, which would not have been achieved as efficiently with an IV or systemic administration.
Study 2: Group mean anti-AAV5 antibody levels and group mean hFIX levels in NHP.
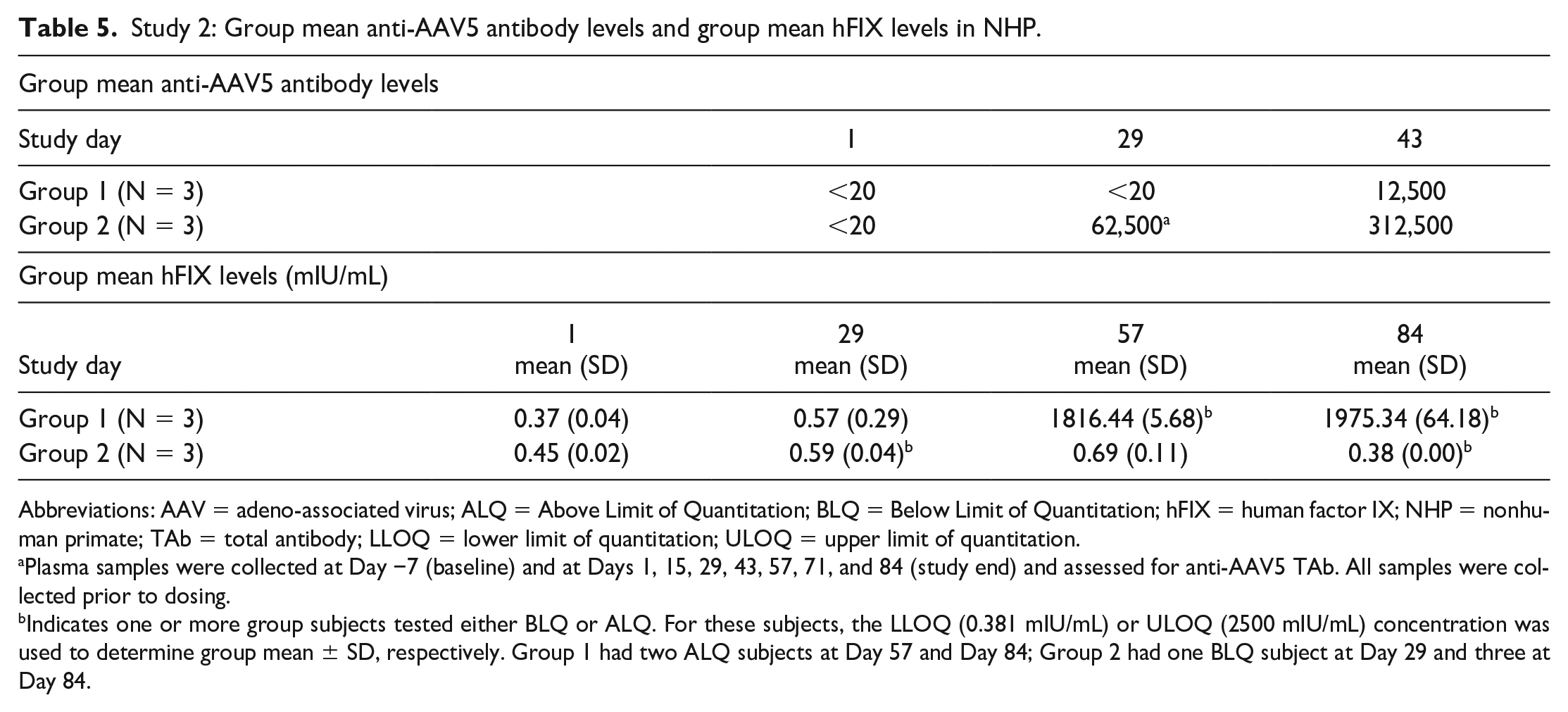
Abbreviations: AAV = adeno-associated virus; ALQ = Above Limit of Quantitation; BLQ = Below Limit of Quantitation; hFIX = human factor IX; NHP = nonhuman primate; TAb = total antibody; LLOQ = lower limit of quantitation; ULOQ = upper limit of quantitation.
Plasma samples were collected at Day −7 (baseline) and at Days 1, 15, 29, 43, 57, 71, and 84 (study end) and assessed for anti-AAV5 TAb. All samples were collected prior to dosing.
Indicates one or more group subjects tested either BLQ or ALQ. For these subjects, the LLOQ (0.381 mIU/mL) or ULOQ (2500 mIU/mL) concentration was used to determine group mean ± SD, respectively. Group 1 had two ALQ subjects at Day 57 and Day 84; Group 2 had one BLQ subject at Day 29 and three at Day 84.
Human FIX was observed only in AAV5-hFIX animals (Group 1; Table 5) that were not previously sensitized to AAV5 prior to challenge dose administration; hFIX plasma protein concentrations ranged from 5636.6 to 11,355.5 mIU/mL on Days 57 and 84. Human FIX plasma protein expression was not detected in the animals previously administered AAV5-bCG prior to AAV5-hFIX (Group 2), suggesting the anti-AAV5 capsid TAb response following the prior sensitizing dose administration was capable of neutralizing transduction by the second dose (data not shown).
In both treatment groups, there were no clinical observations, body weight changes, or food consumption changes among the animals during the course of the study. There were no changes in coagulation parameters or clinical chemistry, including serum transaminases (ALT and AST; data not shown). Similar to Study 1, histopathologic examination of major parenchymal organs from NHPs in all groups revealed no adverse findings. A few scattered foci of mixed inflammatory cell infiltrates observed across all treatment groups were considered incidental and normal background findings of cynomolgus monkeys.7,28
The percentage change in absolute leukocyte counts over baseline is shown in Figure 2. There were few notable differences in percentage change from baseline measures between Groups 1 and 2; however, AAV5-bCG administration on Day −7 and Day 1 in Group 2 resulted in a mean 2-fold increase (220% increase over baseline) in circulating CD3-/CD14+ monocytes on Day 15. The mean at Day 15 was increased by a single animal (2003), with a 301% increase over baseline. A less robust increase in monocytes (approximately 1.5-fold) was also observed on Days 71 (144% over baseline) and 84 (154% over baseline) in the same group (Group 2). By comparison, Group 1 mean percentage changes from baseline were 118% at Days 15 and 71, and 99% at Day 84.
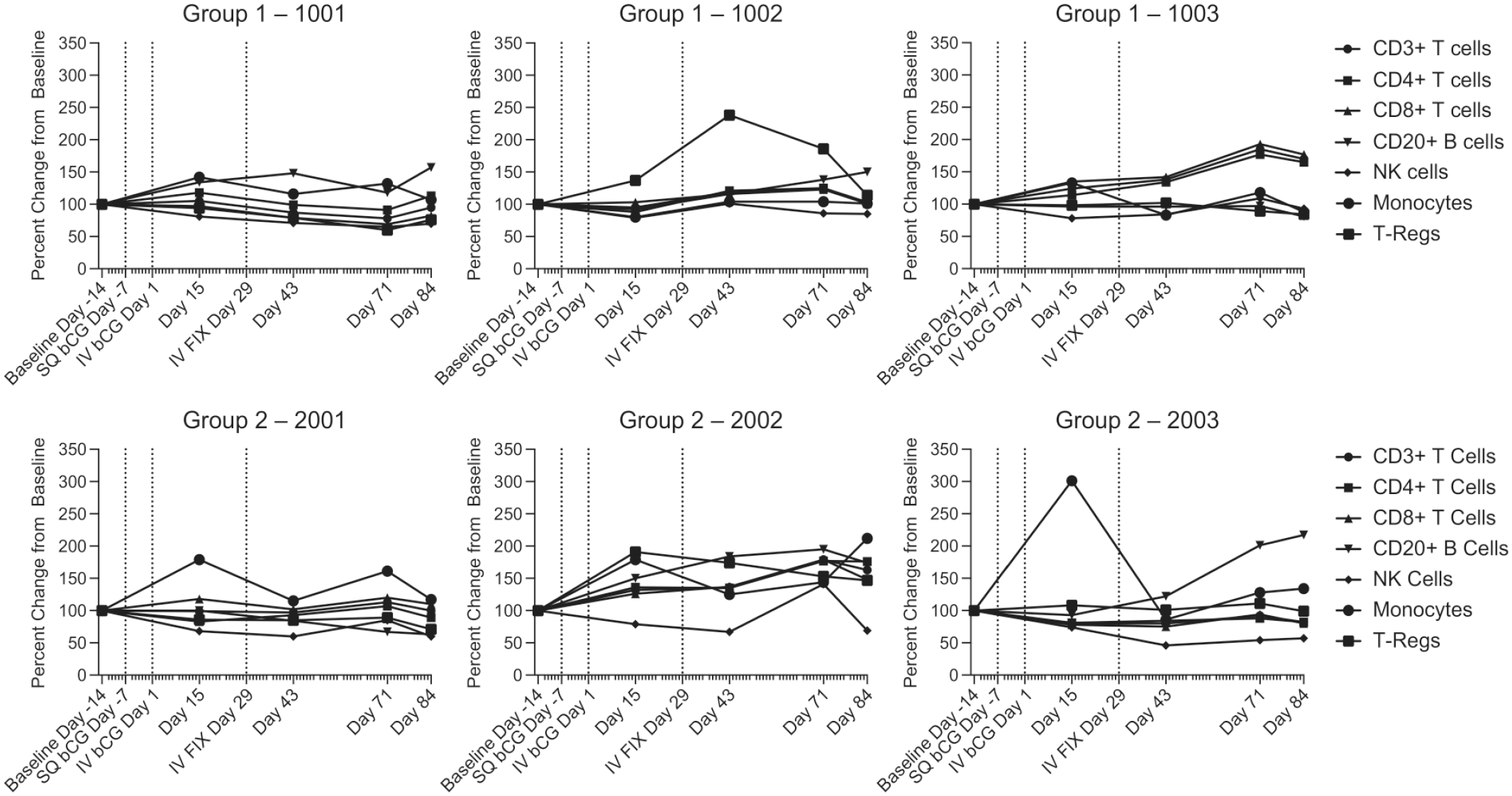
Whole blood collected by venipuncture on Days −14, 15, 43, 71, and 84 were immunophenotyped by flow cytometry and assessed for the percent change from baseline in CD3+ T cell, CD4+ T cells, CD8+ T cells, CD20+ B cells, NK Cells (CD3− CD16+), monocytes (CD3− CD14+), and regulatory T cells (T-Regs: CD4+/CD25+/FoxP3+). Vertical dotted lines indicate timepoints for gene therapy administration. SQ—subcutaneous at Day −7. IV—intravenous at Days 1 and 29. bCG indicates beta subunit of choriogonadotropic hormone; NK, Natural Killer; FIX, factor IX; IV, intravenous.
Consistent with the elevated whole blood monocyte counts, an increase in plasma monocyte chemoattractant protein-1 (MCP-1) concentration was present in all Group 2 animals (3.18, 7.01, and 6.56-fold relative to prestudy, respectively) on Day 1, at 6 hours postdose (Figure 3). One control animal in Group 1 yielded detectable MCP-1 values, but as this value was only 1.133-fold above baseline, it was considered within the range of normal variability observed at the testing facility. No further increases in this analyte were detectable following the Day 29 challenge dose. Plots of fold-change over baseline in individual animal cytokine data are shown in Figure 3. No test article-related changes were observed for interleukin-1 receptor antagonist (IL-1RA), IL-4, IL-6, IL-8, IL-12/23(p40), granulocyte-colony-stimulating factor (G-CSF), or tumor necrosis factor-alpha (TNF-α). While detectable levels were observed for each of these analytes, changes were not considered to be test article-related, as they were not observed in all group members and were generally within the range of prestudy responses. No test article-related changes were observed for granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-gamma (IFN-γ), IL-10, IL-13, IL-17A, IL-1β, IL-2, IL-5, or macrophage inflammatory protein-1β (MIP-1β), as all samples were below the LLOQ.
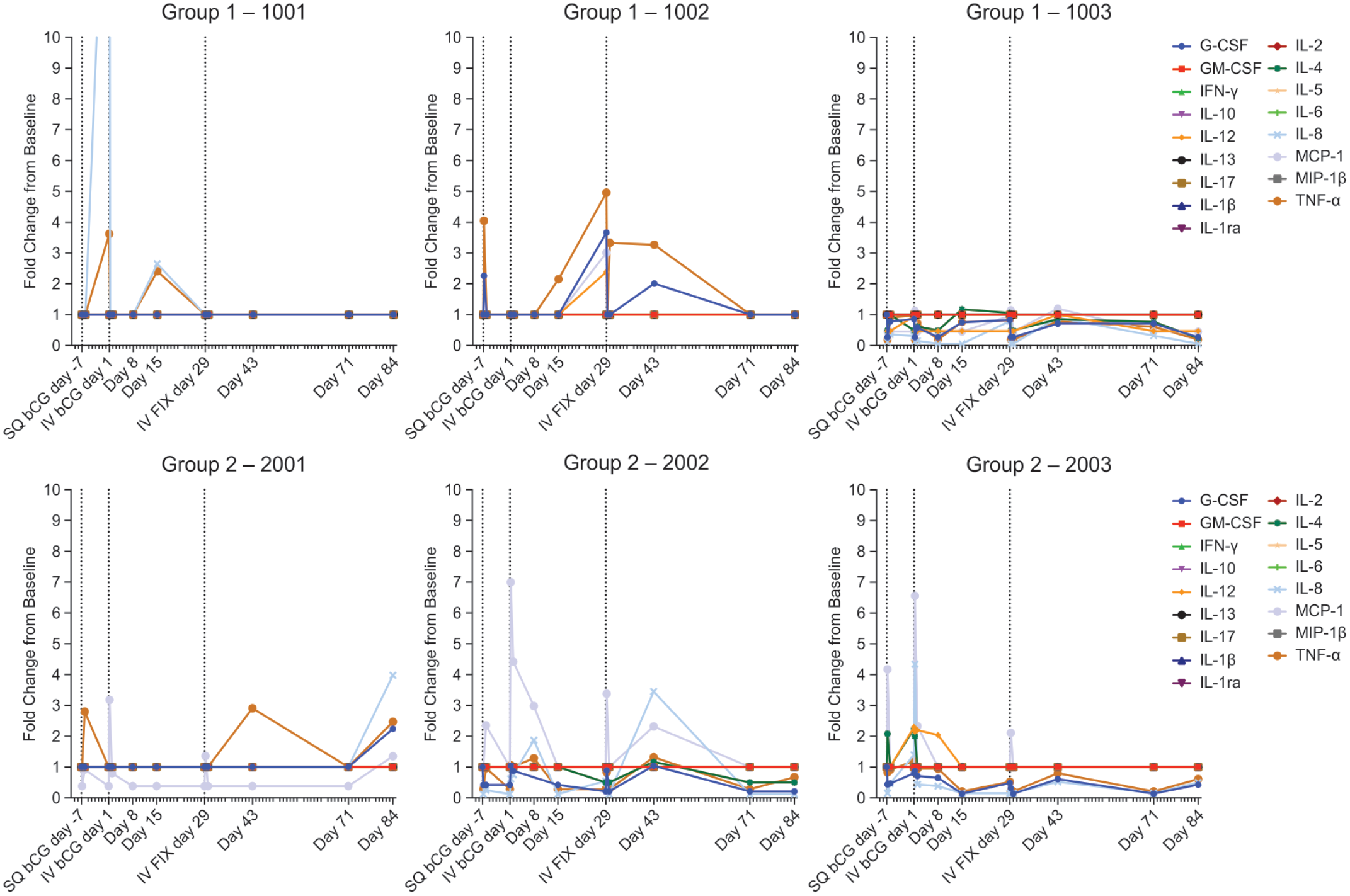
Plasma samples from the indicated time points were analyzed by bead-based multiplex sandwich immunoassay for circulating levels of IL-1β, IL-1RA, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12/23 (p40), IL-13, IL-17A, G-CSF, GM-CSF, IFN-γ, TNF-α, MCP-1, and MIP-1β. Individual animal data were compared to their respective predose results and expressed as fold-change from baseline for any observed trends (time- or dose-related changes). bCG indicates beta subunit of choriogonadotropic hormone; FIX, factor IX; G-CSF, granulocyte-colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon gamma; IL, interleukin; IV, intravenous; MCP-1, monocyte chemoattractant protein-1; MIP-1β, macrophage inflammatory protein-1β; TNF-α, tumor necrosis factor-alpha.
AAV5 capsid-specific cellular immune responses were measured by ELISpot assay for secreted IFN-γ from cryopreserved cynomolgus monkey PBMCs collected at Days −10, 15, 43, 71, and 85 and stimulated with AAV5 peptide pools. Based on the PBMC responses to control stimulations, the method was performed within acceptable parameters. No IFN-γ secretion in response to stimulation with AAV5 peptide pools was measured above the LOD for any animal at any time point. While all PBMC samples passed formal sample acceptance criteria, several samples (N = 4) showed a decreased response to stimulation with the positive control, phytohaemagglutinin lectin (PHA-L), indicating that the PBMC sample collection or immune responses in these samples may not have been optimal (data not shown). Overall, expression of hFIX was observed only in AAV5-hFIX animals (Group 1) that were not previously sensitized to AAV5 prior to the challenge dose administration.
Discussion
AAV-based vectors have rapidly become a vehicle of choice for therapeutic gene transfer. However, one potential challenge to the success of AAV-based gene therapy is the presence of circulating NAbs against AAV vector capsids that may prevent the successful transduction of target cells. Antibodies can be present in the blood of a patient prior to AAV-based treatment due to naturally acquired infections or as the result of prior treatment with an AAV-based vector.
Study 1, as reported here, was designed to examine the safety of administering an AAV5-based vector in the presence of known anti-AAV5 titers; transduction results from Study 1 have been previously published. 22 In some animals with pre-existing anti-AAV5 antibodies, injection of BMN 270 at 6E13 vg/kg resulted in lower levels of maximal hFVIII-SQ plasma concentration (Cmax) and area under the drug concentration-time curve (AUC) of 74.8% and 66.9%, respectively, compared with control animals without pre-existing antibodies. In contrast, animals with only non-antibody TI showed hFVIII-SQ plasma concentrations and liver vector copies comparable with those of controls. These findings suggest an association between pre-existing anti-AAV5 antibodies and reduced hFVIII-SQ levels. There were no adverse effects of dosing NHPs with known pre-existing AAV5 antibodies at the levels detected. As expected, by Day 8, all dosed animals were AAV5 antibody-positive, indicating that the onset of the AAV5 antibody response occurred rapidly after BMN 270 administration and with similar kinetics across all study groups, regardless of the presence of pre-existing AAV5 immunity. Plasma from all dosed animals remained positive for AAV5 antibodies throughout the study until Day 56, without any apparent effect of pre-existing AAV5 immunity. 22
Study 2 focused on re-dosing NHPs with the AAV5 capsid amid higher levels of induced AAV5 antibody. Prior to dosing, all animals in Groups 1 and 2 were negative for NAbs (titer <20), confirming that they were not previously exposed to AAV5. Antibody levels in Group 2 animals prior to re-dosing (Day 29) were rather high by normalized titer values (~62,500) and continued to increase following the second administration, with peak titers around 312,500 on Day 43. NHPs tolerated re-dosing of the AAV5-hFIX vector, showing no adverse clinical signs. Additionally, there were no clinical chemistry changes or histopathologic changes in the tissues at the end of the study. Expression of hFIX was observed only in AAV5-hFIX animals (Group 1) that were not previously sensitized to AAV5. Human FIX plasma protein expression was not detected in Group 2 animals: as expected, the anti-AAV5 NAbs elicited after the first vector administration prevented the successful transduction of the second vector, AAV5-hFIX.
Previous findings have described the impact of pre-existing antibodies or those elicited after the administration of AAV-based vectors on transduction efficiency.11,22,25,29,32,34 Notably, AAV is the cause of natural infections in many species, potentially inducing B-cell responses to the virus, although these are primarily infections without further sequelae. 6 In addition to humoral immunity characterized by NAbs or neutralizing titers, two other components of the immune system, namely the innate immune response and cellular response, are also critical from a safety perspective. A detailed understanding of the interaction of AAV-based vectors with the immune system is of great importance for the efficacy and safety of gene therapy applications.
The findings reported here demonstrate that AAV-based gene therapy is well-tolerated in NHPs, with consideration of carefully monitoring for toxicity with high doses of AAV-based vectors in preclinical studies. For example, a first-generation adenovirus-mediated FIX gene transfer to rhesus monkeys (N = 3) resulted in substantial, dose-dependent, dose-limiting liver toxicity characterized by elevated serum transaminase levels, hyperbilirubinemia, hypoalbuminemia, and prolonged clotting times. 23 In two of the animals, evidence of liver toxicity resolved; however, the highest dose recipient (1E11 vg/kg) demonstrated persistent hypofibrinogenemia, suggesting potentially permanent liver damage.
Elsewhere, another study reported severe toxicity after high vector doses (2E14 vg/kg) of an AAV9 variant (AAVhu68) were administered to juvenile NHPs (age 14 months; N = 3) and young piglets (age 7-30 days; N = 3). All three NHPs demonstrated transaminase elevations; these resolved without clinical sequelae in two of three. The other NHP developed acute liver failure and shock and was euthanized four days after vector injection. The piglets demonstrated no evidence of hepatic toxicity, but all three demonstrated proprioceptive deficits and ataxia within 14 days of vector injection, necessitating euthanasia. 14 The acute toxicity in this study was likely due to an innate immune response rather than a humoral response to the viral capsid; systemic toxicity may be a general property of high-dose IV delivery of AAV-based vectors in piglets regardless of the transgene or serotype used.
In addition to humoral antibody response and innate immune response, administration of AAV vectors can induce a cytotoxic T-cell response directed against the AAV capsid as well as the transgene product, particularly at high doses.1,26,27 A cytotoxic T-cell response, either against the capsid proteins or the transgene itself, is likely to be manifested by hepatic inflammation or hepatic injury, with a concomitant increase in serum transaminases. Elevated transaminases (ALT or AST) were not observed in either study at any time point and with no histopathologic changes at terminal necropsy. No IFN-γ secretion from stimulated PBMCs was detected by ELISpot assay in Study 2 (data not shown).
Despite the lack of adverse safety findings in these studies, multiple lines of evidence and the results described here demonstrate that both pre-existing and treatment-induced antibody responses significantly diminish the efficacy of gene transfer in AAV-based gene therapy. 22 Neutralizing antibodies can block interaction with AAV receptors to prevent vector uptake and transduction, while binding antibodies can cause the vector to be taken up by cells of the reticuloendothelial system (mostly in the spleen) via Fc receptor (FcR) and complement receptor-mediated phagocytosis.15,31 These research findings serve as an impetus for investigating interventions that can contend with AAV-specific antibodies; as such, recent studies suggest it may be possible to reduce pre-existing antibody titers to levels compatible with gene therapy dose administration.3,19
The results reported here indicate that re-dosing or inadvertent dosing of an AAV5-based capsid vector at the dose levels administered in these NHPs with varying pre-existing antibody levels is safe, without any adverse consequences. However, if AAV doses are increased when all else stays the same, adverse events related to activated immune system-associated microscopic changes cannot be ruled out. Extrapolation of safety findings based on AAV5 vectors to other serotypes should be conducted judiciously, as a variety of AAV serotypes with differing biodistribution properties are currently being evaluated for use in gene therapy.
Footnotes
Acknowledgements
The authors thank the members of the Translational Sciences, Pharmacological Sciences, and Study Execution Teams at BioMarin Pharmaceutical Inc. for their dedication and contribution to this manuscript. All authors critically reviewed the manuscript and provided substantive input during the drafting stages. Project management support was provided by Micah Robinson, PhD, Jonathan Morton, PhD, and Stephanie MacLeod, MS of BioMarin Pharmaceutical Inc.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Sundeep Chandra, Ph.D., has no conflicts of interest within the past 36 months to report. Brian Long, PhD, is currently a paid employee of BioMarin Pharmaceutical Inc. and holds stock ownership with BioMarin Pharmaceutical Inc. Carlos Fonck, PhD, was a paid employee of BioMarin Pharmaceutical Inc. from February 2017 to April 2020 and holds stock ownership with BioMarin Pharmaceutical Inc. Andrew Melton, PhD, is currently a paid employee of BioMarin Pharmaceutical Inc. and holds stock ownership with BioMarin Pharmaceutical Inc. Jeremy Arens, MS, is currently a paid employee of BioMarin Pharmaceutical Inc. and holds stock ownership with BioMarin Pharmaceutical Inc. Jill Woloszynek, BA, is currently a paid employee with Astellas Gene Therapies and held stock ownership with BioMarin Pharmaceutical Inc. from 2014-2022. Charles A. O’Neill, PhD, is currently a paid employee of BioMarin Pharmaceutical Inc. and holds stock ownership with BioMarin Pharmaceutical Inc. In addition, Dr. O’Neill has planned, pending, or issued patents.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this study was provided by BioMarin Pharmaceutical Inc.
Data and Materials Availability
Materials and protocols will be distributed to qualified scientific researchers for non-commercial, academic purposes. The AAV5-hFVIII-SQ vector and sequence are part of an ongoing development program, and they will not be shared.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.