
Abstract
Gene therapy has become an important modality for a wide range of therapeutic indications with a rapid increase in the number of therapeutic candidates being developed in this field. Understanding the molecular biology underlying the gene therapy is often critical to develop appropriate safety assessment strategies. We aimed to discuss some of the commonly used gene therapy modalities and common preclinical toxicology testing considerations when developing gene therapies. Non-viral gene delivery methods such as electroporation, microinjection, peptide nanoparticles and lipid nanoparticles are deployed as innovative molecular molecular construct which are included in the design of novel gene therapies and the associated molecular biology mechanisms have become relevant knowledge to non-clinical toxicology. Viral gene delivery methodologies including Adenovirus vectors, Adeno-Associated virus vectors and Lentivirus gene therapy vectors have also advanced considerably across numerous therapeutic areas, raising unique non-clinical toxicology and immunological considerations. General toxicology, biodistribution and tumorigenicity are the pillars of non-clinical safety testing in gene therapies. Evaluating the tumorigenicity potential of a gene editing therapy often leverages molecular pathology while some translational challenges remain. Toxicology study design is entering a new era where science-driven customized approaches and program specific considerations have become the norm.
The Rise of Gene Editing Therapies
Gene therapy presents a compelling prospect in treatment of various indications such as cancer, cardiovascular and metabolic diseases and neurodegenerative disorders, some of which were previously thought to be incurable. 1 Progress with a range of gene editing methodologies have transformed the drug development industry and offer opportunities for innovative therapeutics in areas with important medical gaps. Some notable gene editing approaches include the use of Inlaid Base Editors (IBE), Clustered Regularly Interspersed Short Palindromic Repeats (Crispr-Cas9), Zinc Finger Nucleases (ZFN), and Transcription-Activator Like Effector Nucleases (TALEN). The Food and Drug Administration (FDA) has received more than 900 investigational new drug (IND) applications for ongoing clinical studies in the gene therapy arena. As of January 2020, it has approved 4 gene therapy products, and projects to approve 10-20 cell and gene therapy products annually by 2025.2,3 Of note, is the recently approved CAR-T cell therapy for Mantle Cell Lymphoma (MCL), called Tecartus. 4 MCL is a rare cancer of the B-cells, a type of non-Hodgkin’s lymphoma, that develops on the outer edge of the lymph nodes. Mature B cells possess a transmembrane glycoprotein called CD19 that serves as a biomarker for both normal and neoplastic B cells. 5 Tecartus involves genetically modifying the patient’s T cells using a replication incompetent retroviral vector to express a chimeric antigen receptor (CAR) encompassing a murine anti-CD19 linked to CD28 (CAR anchoring) and CD3 (T cell activation) domains.4,6 The anti-CD19 CAR T cells are replicated and infused as an autograft into the patient where they can identify and eradicate CD19 expressing B cells. 4
Gene therapy is a burgeoning contender in modern drug discovery and development. However, the extent of therapeutic effect of these methods is vastly influenced by the delivery mechanism or mode of administration used, which ultimately dictates the efficiency, safety, and subsequent clinical application of the therapy. 7 Gene delivery can be accomplished through various physical (electroporation, microinjection, lipid nanoparticles, peptides) or viral (adenovirus, adeno associated virus, lentivirus) methods which are discussed and elaborated below. 8
Non-Viral Gene Delivery Methods
Electroporation
Electroporation (EP) is perhaps the most widely used non-viral gene delivery technique attributing to precise dose control, versatility in cell types that can be used, and relatively high transfection efficiency and cell viability. 9 First described by Neumann et al. in 1982, it allows intracellular delivery of genetic material by exposing cells to an electrical impulse. Subjecting the cell membrane to an electrical impulse causes a potential difference across the membrane, leading to a lipid bilayer disruption that forms an opening called the aqueous pathway. 9
Traditionally, Bulk Electroporation (BEP) in cell suspension involved placing a cell suspension cuvette between an anode and cathode. This method, although simple, fast, and independent of cell type, lacked dose control and efficiency, due to uncontrollable membrane disruptions. 9 Other systems using flow electroporation transfection, nucleofection or germline genome editing have now been developed to refine methodologies and address these issues.
More recently, commonly used BEP systems such as MaxCyte, use flow EP to allow for scalable transfection. Flow EP involves displacing cells through 2 electrodes while electroporating them with a transient electric field. 10 The process is repeated until all cells in a lot are electroporated, thereby significantly increasing the volume of cell suspension transfection from what was previously possible. MaxCyte can transfect up to 20 billion cells at a time. 11 Frangoul et al. (funded by CRISPR Therapeutics and Vertex Pharmaceuticals) utilized MaxCyte GTx to electroporate healthy donor CD34+ hematopoietic stem and progenitor cells with CRISPR-Cas9 to modify the erythrocyte ɣ-globin and fetal hemoglobin repressor gene, BCL11 A (CTX001) to create an indel. Two patients with sickle cell disease and transfusion dependent β thalassemia (both of which are caused by BCL11 A activity) who received a single intravenous infusion of CTX001 exhibited improvement of disease manifestations. 12
Other instruments such as the Nucleofector 4d (Lonza), much like the Maxcyte, use cell-specific electrical parameters to transfect a gene of interest directly into the nucleus of the cell and can do so both in suspended and adherent cells. 13 The Neon cell transfection system (ThermoFisher) involves an electric field supplied within a pipette tip filled with the substrate and cells to be transfected. Once transfected, the cells can directly be transferred to the tissue culture dish via the pipette. 14
While both the Nucleofector 4d and Neon render remarkable performance for bulk mammalian cell transfection, they prove challenging to automate and integrate for scaled clinical production. The MaxCyte GTx by way of flow-based electroporation provides for an increased throughput in an integrated, automated workflow. 15
Apart from the instruments above, electroporation can also be performed in vivo through techniques such as Genome editing via Oviductal Nucleic Acid Delivery (GONAD). In 2015, Takahashi et al. first demonstrated GONAD as an approach for germline genome editing involving the direct injection of the Cas9/gRNA complex into the oviduct at the 2-cell embryo stage (1.5 days postcoitum) of a pregnant mouse using a glass capillary pipette, followed by in-situ electroporation of the oviducts with tweezer type electrodes (NEPA 21).16,17 This in-situ process eliminates zygote isolation, ex-vivo handling and subsequent implantation into the female thus reducing the margin of error, costly apparatus and need for a wide-ranging set of skilled technicians. 16
Despite its advantages, the GONAD method has a relatively low indel mutation efficiency (28-31%) in 2-cell embryos along with a high rate of mosaicism in the resulting offsprings, which prompted Takabayashi et al. to develop the i-GONAD, or improved GONAD method. The i-GONAD method employs the GONAD technique with ribonucleoprotein complexes (comprising of the Cas9 coupled with a synthetic gRNA) at the .7 days postcoitum zygote stage. This method resulted in genomic indels at higher efficiency (∼97%). 18
Nakano et al. employed i-GONAD to generate hemagglutinin (HA) tagged knock-in mice to study the Activating Transcription Factor 5 (ATF5) that is pivotal for murine olfactory sensory neuron survival and differentiation, due to restricted accessibility of immunoprecipitation-grade ATF5 antibodies. CRISPR/Cas9 was used to create the ATF5-HA tag knock-in mice followed by in situ electroporation in the oviducts. The ATF5-HA fusion protein was then successfully detected in the nuclei of the olfactory sensory neurons. Such applications of GONAD can prove instrumental in studying protein expression, localization and interaction. 19
Microinjection
Microinjection is the oldest employed technique in genetic manipulation, with the first use dating back to early 1900s when the pioneer, Dr. Barber used a minute, carefully crafted glass capillary tube to aspirate bacterial cells and inject them into plant cells to study the ensuing effects. 20 It has since found incredible application in biomedical and genetic engineering. Microinjection is a straight forward technique that utilizes a glass micro-diameter (.5-15 µm) micropipette to directly inject genetic material into the cytoplasm or nucleus of a cell. 20 Many widely available microinjectors use variable air pressure to selectively inject micro or nano volumes of fluid into cells, thus facilitating a convenient and precise mode of gene delivery. Fisher Scientific’s Eppendorf FemtoJet allows for microinjecting volumes as small as femtoliters, to both adherent and suspended cells. 21
Kurtz et al. in an attempt to characterize the porcine sex determining region SRY, used the FemtoJet to microinject 2 CRISPR/Cas ribonucleoproteins to create SRY knockout male pigs. The resulting genetically male pigs exhibited smaller but whole, internal and external female genitalia, thus unraveling the significance of the SRY gene in male porcine sex determination, and possibly humans, in extrapolation. 22 Although the use of microinjection of CRISPR/Cas RNP was successful, the SRY-KO males failed to develop as compared to wild type, the cause of which was unclear. The authors hypothesized the possibility that the ‘Y-chromosomal gene and hormone expression hampered the development of female genitalia in SRY-KO pigs’. 22
Regardless of commercially available instruments, microinjection requires extremely skilled technicians along with meticulous ex vivo handling including immense attention to detail in terms of zygote holding site, injection site, and injection volume. Eto et al. have developed the Integrated Automated Embryo Manipulation System (IAEMS), which is a fully automated electrical manipulation system for pronuclear microinjection designed to circumvent the human error and issues that may arise in current manually maneuvered microinjection devices. 23 The IAEMS software rotates the zygote and visualizes the pronucleus in 3D to establish the most optimal coordinates to allow for micropipette injection and then determines the ideal pressure conditions for solution administration based on survival rates of zygotes. 23 The team has successfully created transgenic and knock-in mice using this technology. 23
Peptide Nanoparticles
Peptides or short amino acid chains, are versatiles tools to facilitate gene delivery given their ability to traverse biological barriers (Cell Penetrating Peptides, Nuclear Localization Signals etc.), high stability, and low cytotoxicity and immunogenicity. 24 Human serum albumin (HSA), along with possessing the aforementioned merits of a peptide, is relatively pH and temperature stable, abundantly present in blood plasma, possesses a long half-life (19 days), and has several subdomains for ligand binding. 25 It is also easily metabolized, purified, and injected, making it an attractive candidate for gene delivery, especially in oncology. 26 In addition, tumor cells usually express high levels of albumin receptors thus assisting HSA transcytosis leading to HSA nanoparticles accumulating in tumor tissues. 27 However, limited encapsulation and protection of the genetic material present an obstacle to successful and efficient transfection using HSA. 26 Guan et al. developed a cationic HSA (cHSA) by surface modification using ethylene diamine. 26 The resulting cHSA was then electrostatically combined with a Simian Virus 40 T-antigen nuclear localization signal (NLS) to allow for nuclear pore delivery, and plasmid DNA (pDNA). This cHSA/NLS/pDNA complex proved to be a safe and efficient mode of gene delivery when tested for in vivo immunogenicity (blood IL-12 and IFN-α levels comparable to control) and anti-tumor effects (tumor inhibition rate of 57.3%) in BALB/c nude mice. 26
Further, Cheng et al. synthesized stearyl-polyethyenamine (stPEI) to allow for better binding of HSA to pDNA, to create a stPEI/HSA/pDNA nanoparticle. This nanoparticle was then non-covalently linked to CRISPR/Cas9 and small interfering (si)RNA to successfully knock out the immune checkpoint protein programmed cell death ligand 1 (PD-L1), which is involved in tumor evasion. 27
Lipid Nanoparticles
Liposomes were initially characterized for drug delivery in the 1980's because of their biocompatibility, nontoxicity and flexibility in size (.025-10 µm diameter), and composition. 28 Since then, multiple liposome based drugs have been approved for intravenous administration worldwide, which has laid the groundwork for the investigation and development of lipid nanoparticles (LNP) as gene delivery systems. 29 A typical LNP consists of an ionizable lipid, cholesterol to provide rigidity, polyethylene glycol to assist with bilayer formation, and a helper lipid to allow for ease of endocytosis and fusion. 30 LNPs are appealing due to their efficient encapsulation of cargo, durable and scalable manufacturing process, and long shelf life (∼1 yr at 4°C). 29 The advent of cationic lipids has resolved the issue of lipid-nucleic acid electrostatic instability that arose due to the inherent negative charge on both molecules. 29 More recently, ester groups comprising biodegradable ionizable lipids that retain positive charge due to protonation of the amine group, have been utilized for mRNA delivery to non-human primates (NHP). 31 The NHPs were subjected to weekly repeated dosing over 5 weeks up to 2 mg/kg, administered via intravenous infusion. The study reported uniform circulation of LNPs, efficient protein expression, quick release of lipid from tissues and no signs of toxicity or liver damage, believed to be associated with repeated dosing of LNPs. 32
The efficiency of liver targeted LNP based therapies can be attributed to the adsorption of Apolipoprotein E onto the LNP surface that aids in hepatocyte targeting, as can be seen in the newly developed RNAi delivery LNP, Onpattro (Alnylam Pharmaceuticals). 33 The formation of said 'biomolecular corona’ around the LNP due to interaction with biomolecules present in the blood, is the key to cell and tissue specific tropism. 33 The corona comprises of both, tightly bound high affinity surface proteins (hard corona), and a loosely bound dynamic protein layer that changes based on LNP-cell interactions (soft-corona). A major challenge with LNP’s is tissue specific targeting beyond the liver. 33 Understanding, and analysis of the corona can influence the development and success of LNP based gene therapy and tissue specific tropism. 33 Francia et al. have elaborated how ‘LNPs physicochemical characteristics, such as size and charge,’ may advance the targeting specificity and efficiency of LNP based gene delivery. For example, a dominantly cationic lipid composition has been shown to redirect biodistribution from the liver to the spleen and then to the lungs. 33 LNP specificity, however, remains a challenge to conquer given the influence of the surrounding biological environment on the ever-changing biomolecular corona.
Viral Gene Delivery Methods
Adenovirus (AdV) Gene Therapy Vectors
Most widely utilized as a viral vector, Adenoviruses are non-enveloped, non-integrating, episomal, double stranded DNA viruses with an insert packaging capacity of up to 35Kb. 34 Adenoviral [Coxsackievirus and adenovirus receptor (CAR)] and integrin receptors (αvβ3 and αvβ5) are naturally found on the surface of most human cells, which implies not only increased transduction efficiency but also increased levels of transgene expression.35,36 However, AdV are also the viral vectors with the highest number of identified human serotypes (∼57) thus facilitating a pre-existing immunity. 34 This inherent immunogenicity elicited by AdVs poses restrictions on repeat administration in a single patient. 34 That being said, certain rare serotypes like 2, 26 and 35 still present the prospect of being utilized for gene therapy.34,35 The high seroprevalence of AdV5 (ranging from 35% in the United States to 90% in Ivory Coast) presents a challenge in vector development and application. 37 Chimpanzee-derived AdV due to their low prevalence in human blood have been employed widely to overcome this issue. 37 The development of high capacity (HC), 'gutless', or 'helper dependent’ AdV, has allowed for increased packaging capacity as well as a means to reduce immunogenicity in the host. 35 HC-AdVs lack all viral coding genes [apart from the 5' and 3' inverted terminal repeats (ITR) that function as origin of replication] thereby increasing the viral cloning capacity, and decreasing immunogenicity. 35 Essential genes required for viral replication are supplied via trans-acting elements such as a helper virus (AdV or Herpes simplex virus) or plasmid DNA.35,38 Helper virus associated contamination can be mitigated with either modifying the helper virus sequence with ‘stuffer’ sequences that minimize contamination or employing negative selection against the helper virus using a recombinase mechanism (Cre/LoxP or FLP/frt system). 34
Jin et al. exploited the high genome packing capacity of HC-AdVs to pack an all-in-one Cas9-sgRNA-mCherry (fluorescent marker) vector in AdV type 5. 39 The all-in-one vector was created using the Gateway technology that employs bacteriophage Lambda site specific recombination, to target the circadian clock genes Bmal1 and Per1 in the human cell line U20S. 39 The group noted ∼100% AdV transduction efficiency alongside an effective gene knock-out, thereby eliminating any need for clonal selection. 39 Such approaches can prove instrumental in streamlining and accelerating the gene editing process. The first ever commercial adenoviral gene therapy was approved in China in October 2003. 40 Shenzhen SiBiono GeneTech’s Gendicine, a recombinant human p53 adenovirus (human wildtype p53 and Adv type 5) used to treat head and neck cancers, has been shown to have a total response rate of up to 96% (complete and partial) in greater than 30,000 patients over a span of 12 years. 40 Gendicine’s success has ushered a global, concerted effort toward gene therapy advancements.
Adeno-Associated Virus Gene Therapy Vectors
Perhaps the fastest growing gene delivery modality involves the use of Adeno-associated virus (AAV). AAV are non-enveloped, single stranded DNA viruses of the Parvovirus family with a genome size of ∼4.7Kb. 41 The AAV genome is relatively simple, comprising of the rep (non-structural protein coding) and cap (structural protein coding) genes flanked by T-shaped ITRs, that can be easily engineered to create recombinant AAV (rAAV). 41 Low pathogenicity and immunotoxicity make AAVs a desirable candidate as a gene delivery vector; however, since AAVs occur naturally in humans and NHPs, pre-existing antibodies reduced efficiency of vector delivery.41,42 This can be circumvented to a limited degree by plasmapheresis to remove all immunoglobulins, which has shown to be effective in patients with low neutralizing antibody (NAb) titer. 43 Another approach involves removing NAb’s in vitro by incubating patients’ serum with ‘AAV beads’ (i.e., beads with covalently bound AAV particles). 43 The most promising technique however, uses a streptococcal cystein protease, imlifidase (IdeS) that cleaves IgG into its component Fab and Fc fragments, thereby leading to a complete digestion of IgG. 43 Treatment of mice immunized with intravenous human IgG with IdeS resulted in AAV8 expression comparable to naïve mice. 43
Other AAV downsides include potential variability in the biodistribution in target tissues and lack of cell specific targeting, but viral capsid protein engineering presents an opportunity to enhance transduction efficiency and targeting. 41 Although not previously observed, Hanlon et al. reported full length and fragmented AAV genome integration (of up to 47%) at CRISPR mediated double strand break sites in the targeted cell cultures of murine neurons, muscle, and cochlea, suggesting that insertion site mutagenesis should be a safety factor to evaluate in AAV based CRISPR-Cas9 therapy. 44 The Central Nervous System (CNS) and eye have been the target organ for a number of rAAV gene therapies under development. While the eye is amenable to direct rAAV delivery, delivery to the brain can be direct (intraparenchymal injections), via cerebrospinal fluid (intrathecal administration) or intravenously (for certain serotypes like AAV9/AAVrh.10 that can cross the blood brain barrier). 42 These CNS routes of administration present associated toxicity, such as immune response due to off target gene expression or mild to moderate Dorsal Root Ganglion (DRG) inflammation/necrosis. Off target gene expression toxicity can be dealt with by confining gene expression to desired cell/tissue via specific promoters. Another strategy is to include miRNA binding regions in the 3’-UTR regions to de-target expression in those cells whose miRNA binds to the UTR region. 42
Regardless, AAVs continue to be a popular choice as gene delivery vectors, given their safety profile. As of 2019, there were 149 exclusive clinical trials for AAV based gene therapy, with AAV serotype 2 being the most abundantly applied. 45
Recently, AAV9 vector-based miRNA gene therapy was used to treat Huntington’s disease in transgenic sheep expressing the full disease-causing Human Huntingtin (HTT) gene. 46 The AAV9-miRNA targeted exon 48 of the HTT mRNA, thereby causing a 50-80% reduction in the HTT protein in the striatum, which was also discernable in the caudate nucleus and putamen. 46 The pre-clinical study concluded safe and effective miRNA-mediated silencing of HTT mRNA. 46
There remain a limited number of FDA approved AAV gene therapies on the market such as Glybera, the first European Medicine Agency (EMA) approved Lipoprotein Lipase Deficiency therapy (which was later withdrawn from the market as a result of commercial failure due to lack of demand), Spark Therapeutics' Luxterna and Novartis' Zolgensma.47,48
Lentivirus Gene Therapy Vectors
Lentiviruses are part of the retrovirus family characterized by single stranded, positive sense RNA genome. 49 The lentiviral genome comprises of the gag (structural protein coding), pol (reverse transcription/host integration) and env (viral capsid glycoprotein) genes flanked by 5' and 3' long terminal repeats (LTR). 49 Much like other viral vectors, endogenous genes required for replication and viral capsid are supplied in trans to allow for increased packaging capacity. 49 The removal of the vpr, vpu, and nef auxiliary genes (responsible for proliferation and infection) in the second generation lentiviruses aids in improving the safety profile, which is further improved by removal of the 3′-LTR (comprising of TATA box and transcription factor-binding sites) in the third generation lentiviral vector, thereby creating a ‘self-inactivating (SIN)’ system. 37
Lentiviral tropism can be altered by pseudotyping the envelope glycoproteins, for example. Pseudotyping with vesicular stomatitis virus glycoprotein (VSV-G) allows for lentiviral vector entry through the basolateral surface of the epithelium. 49 As with all retroviral vectors, the leading disadvantage for lentiviral vectors continues to be insertional mutagenesis. The risk is mitigated, but not eliminated even in third generation lentiviral systems, wherein strong promoter/enhancer sequences can instigate insertional mutagenesis. 37 The introduction of chromatin insulators (DNA regulatory sequences) that inhibit interactions between the vector and cell genome without compromising transgene expression or other packaging/replication elements can help improve the safety of lentiviral vectors. 50
As per the analysis conducted by Loza et al. in 2019, no evidence of insertional mutagenesis had been noted in the 1350 patients enrolled in clinical trials administering the third generation SIN lentiviral vectors. Regardless of the limited risk for insertional mutagenesis, it remains an important safety consideration during development. Kim et al. have developed an accurate and reproducible method termed CReVIS-seq to map lentiviral insertions sites. The approach involves 1) shearing/fragmenting genomic DNA 2) in vitro circularization and ligation of DNA 3) CRISPR/Cas 9 mediated selective linearization of viral LTR sequence 4) ligation of sequencing adaptors to linearized fragments and library preparation 5) high throughput sequencing, sequence alignment and analysis of data to identify integration sites. 51 This technique provides a platform to map genome wide lentiviral integration sites, without having to sequence the entire genome, making it a relatively inexpensive and time efficient process. 51 Nevertheless, lentiviral vectors remain attractive given their broad host range, efficient transduction of both proliferating and non-proliferating cells, and long term transgene expression.49,52 The first lentiviral vector gene therapy Kymriah was approved in 2017 for treatment of acute lymphoblastic leukemia, and many therapies continue to be in various developmental phases. 52
Viral vectors have been instrumental to the development of gene therapy with Adenoviruses dominating the clinical landscape (50% of 1140 viral vector mediated gene therapy clinical trials), followed by AAVs (28%) and Lentiviruses (22%) as of early 2021. 37
Safety Testing Considerations and Clinical Applications
Summary of Various Safety Parameters of Different Gene Editing Modalities.
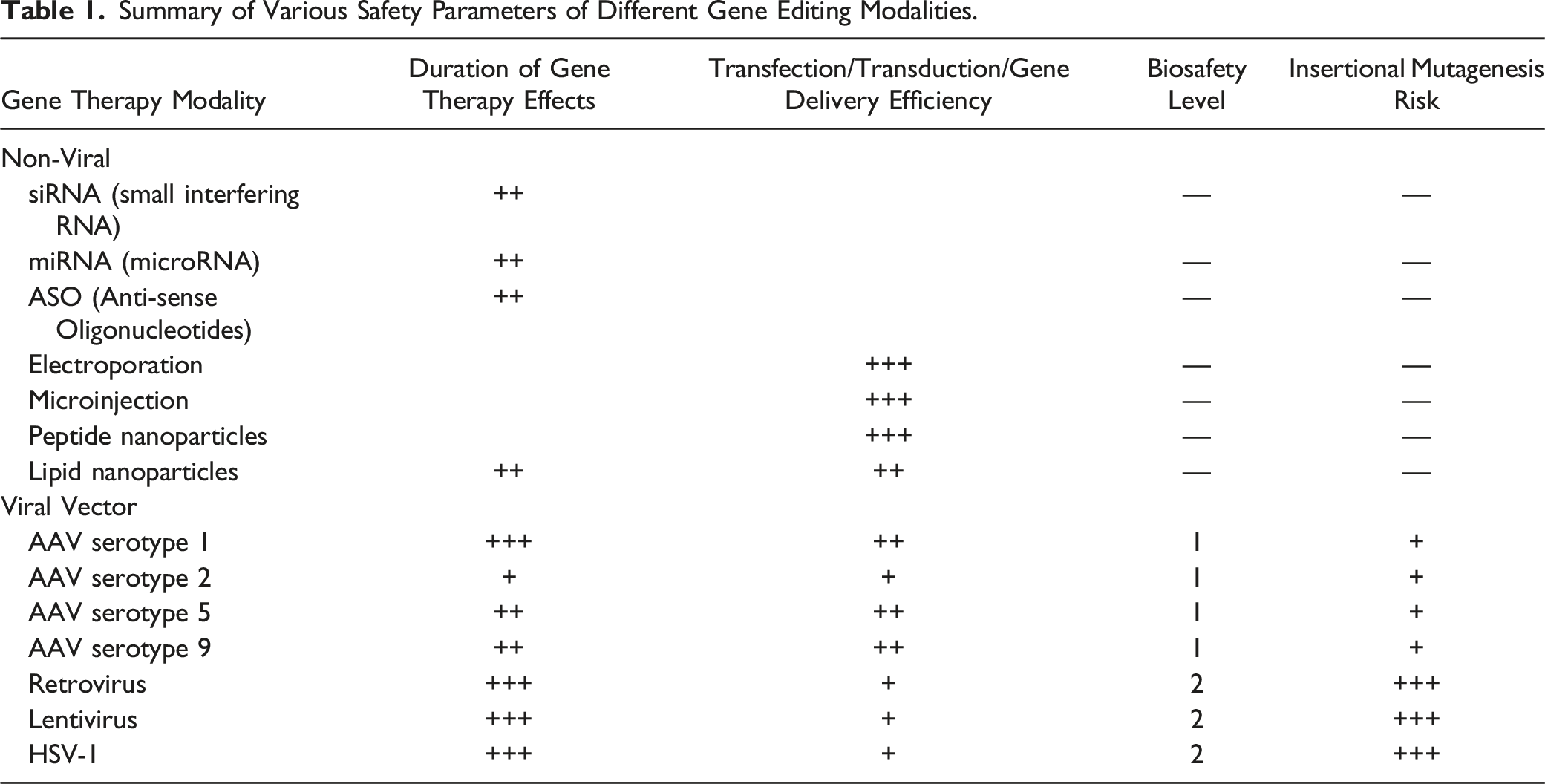
Similarly, an understanding of viral integration sites can help assess safety risks associated with insertional mutagenesis. A deep learning model called the DeepVISP (Deep learning for Viral Integration Site Prediction) developed by Xu et al. has been trained to predict viral integration sites in the human genome using the integration data available for the oncogenic viruses, hepatitis B virus (HBV), human herpesvirus (HPV), and Epstein-Barr virus (EBV). 55 With an area under curve value-receiver operating characteristic (AUC-ROC) of .8, the model is increasingly accurate at classifying viral genome from human genome, along with the ability to identify cis-acting elements involved in viral integration (AUC-ROC values signify the ability of a model to distinguish one class from another. A value of .5 indicates no discriminatory ability i.e., a prediction is as good as random chance. .8 is acceptable and .9 is excellent).55,56 Further training and testing of the DeepVISP model, in extrapolation, may become instrumental in detecting integration sites for episomal viruses. To further the comprehension of not only the insertion site, but also the associated viral sequence, Artesi et al. developed a sequencing technique called the PCIP-Seq (pooled CRISPR inverse PCR-sequencing), which much like the CReVIS-seq for lentivirus, employs the CRISPR/Cas gRNA system to cleave in vitro circularized host DNA at the site of viral insertion, and then utilizes PCR for elongation and sequencing. 57 The cleaved sequences are pooled, aligned and analyzed to create long reads of viral integration site sequences. 57 PCIP-seq has been shown to identify integration sites and single nucleotide polymorphisms (SNP) in human T cell leukemia virus-1 (HTLV-1), bovine leukemia virus (BLV), human immunodeficiency virus (HIV-1), and human papillomavirus (HPV), suggesting a wide range of applications for the technique. 57
General toxicology, biodistribution and tumorigenicity are the pillars of non-clinical safety testing in gene edited cell therapies. Therefore, there needs to be substantial investment in performing standardized safety assessment in animal models, and developments geared towards improving holistic measurement of animal physiological parameters (weight, temperature, blood pressure etc.), genetically diverse animal models, and multi-level computational models of whole genome networks that allow examination of gene expression and regulation may aid in further strengthening conventional pre-clinical safety assessments. 58 In 2002, the International Council of Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) established a gene therapy discussion group that has developed consideration documents addressing virus and vector shedding, oncolytic viruses and the risk of inadvertent germline integration of gene therapy vectors. 59 The considerations for virus and vector shedding provide recommendations for clinical/non-clinical study designs that encompass the properties of the virus, appropriate analytical assay to quantify viral shedding, animal species to be used, dose levels and route of administration, postdose sampling, analysis and interpretation as well as evaluation of third party transmission (ICH M6 Guidelines). 59 The considerations for oncolytic viruses provide recommendations for characterization of viruses based on tumor selectivity, viral variants, adventitious agent testing, selection of appropriate animal models as well as associated toxicity, pharmacological profile, biodistribution, immune response and biosafety (ICH S12 Guideline). 59 Lastly, the considerations for the risk of inadvertent germline integration of gene therapy vectors provide guidance on the risk factors such as vector type, dose, route, and site of administration that may cause germline integration as well as a need to establish a case-by-case approach for evaluation to minimize risk of viral integration. 59
Tumorigenicity is an evident part of safety assessment for gene edited cell therapy products. As previously described, AAV vectors have a lower rate of genome integration when compared to Lentivirus vectors, the rate of which is expected to correlate with the risk for tumorigenicity. The post-engraftment monitoring duration in non-clinical tumorigenicity studies needs to be carefully determined as spontaneous murine tumors can dramatically complicate study conduct since it becomes crucial to determine the origin of the tumors. Tumors from a murine origin are part of the test system characteristics and are not treatment related. The decision process to determine the tumor origin in the murine model can lead to extensive post-necropsy molecular pathology investigations (Figure 1), hence the imperative to select an optimal duration prior to the appearance of a large number of spontaneous murine tumors in the model. Flow chart addressing the procedure to evaluate tumorigenicity of a gene-edited cell therapy.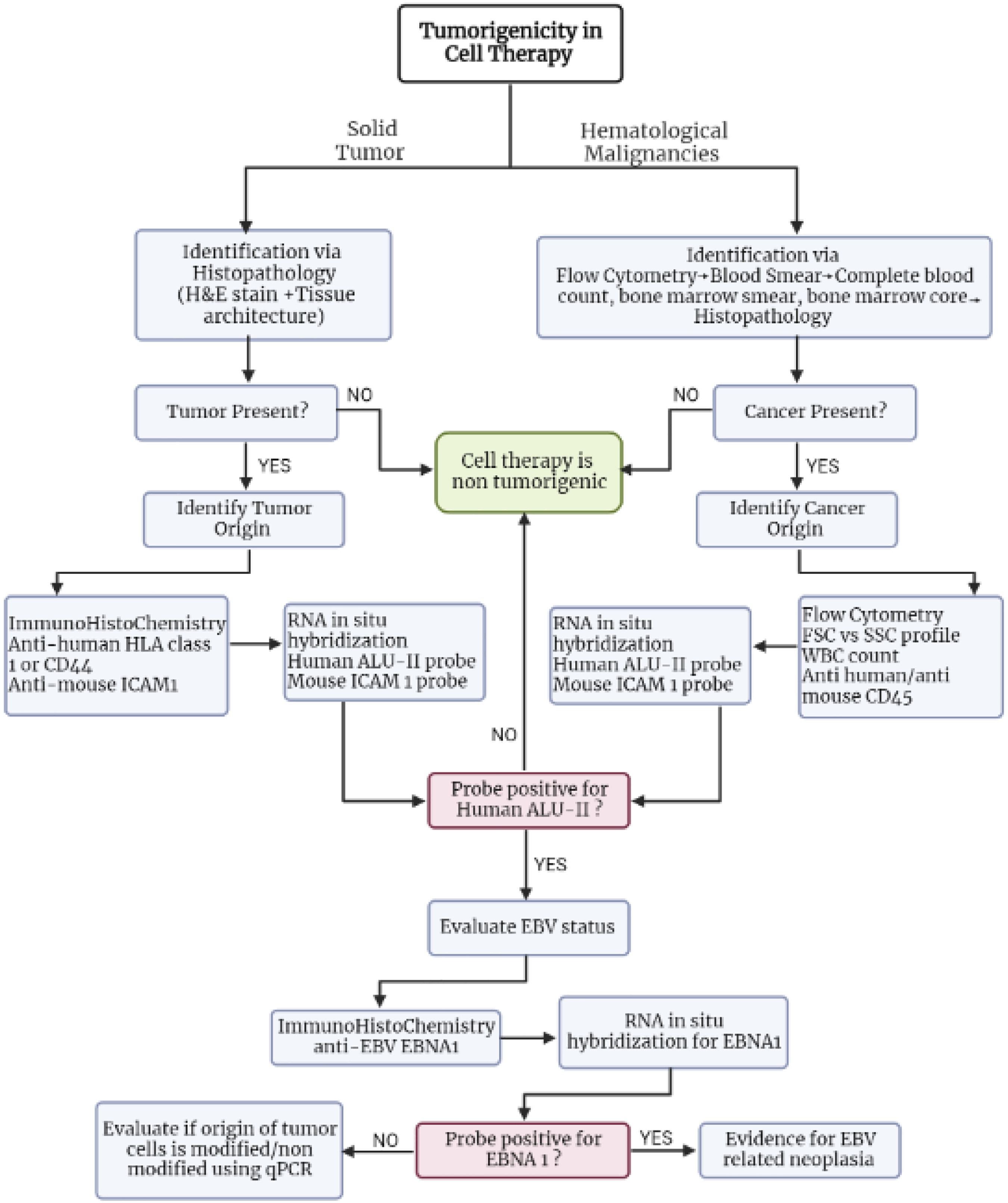
The source of the tumor cells (i.e., whether proliferating cells are of animal model such as mouse, or from the engrafted hHSPCs) in any potentially tumorigenic cell therapy product is essential in assessing the safety of the therapy. Therefore, a comprehensive approach is needed for the diagnoses and determination of origin of both solid tumors and hematological malignancies.
Solid Tumors: Presence of tumor mass in tissue of interest is identified by standard Hematoxylin and Eosin (H&E) staining and examining tissue architecture. Cells exhibiting tumorigenic characteristics are then stained with anti-mouse (ICAM1 or CD54) and anti-human (HLA class 1 or CD44) antibodies to determine the origin of tumor. Intercellular Adhesion Molecule 1 (ICAM1) is a surface glycoprotein, the expression of which is highly induced in the tumor microenvironment. 60 The human leukocyte antigen class 1 (HLA) are transmembrane proteins present on all nucleated cells. 61 Following antibody staining, an RNA in-situ hybridization assay, such as RNAScope is used to visualize binding of the Human ALU II and mouse ICAM1 probes to target sequence. The ALU II probe detects the human specific short interspersed ALU elements present in great abundance (.5-1million repeats) in human cells and is immensely advantageous in identifying human biomarkers. 62 If the resulting hybridization is positive for the human ALU II probe, implying the tumor is originating from engrafted human cells, then an immunohistochemistry test and RNA in situ hybridization is performed to evaluate the presence of Epstein Barr Virus (EBV) biomarker EBNA-1, to check for an underlying EBV infection. This is done as EBV infection of transplanted hHSPCs has been linked to lympho-proliferative disease and tumorigenicity in immunocompromised animals.63,64 A positive probe hybridization for EBNA-1 indicates EBV induced neoplasia, while a negative result solicits further investigation into the nature of origin of tumor cells, i.e., modified, or unmodified origin, via qPCR.
Hematological Malignancies: 60 Presence of hematological malignancies can be explored using flow cytometry to evaluate Forward Scatter (FSC- to distinguish based on immune cell size) and Side Scatter (SSC- to distinguish based on granularity) profiles, abnormal white blood cell (WBC) counts, blood and bone marrow smear to detect and quantify leukemic cells/lymphoma. Cells exhibiting malignant characteristics are then stained with anti-mouse and anti-human CD45 antibodies to determine the origin of cancer. RNA in situ hybridization with Human ALU II and mouse ICAM1 probes is employed to ascertain if the origin of cancer is mouse or human cells. The proceeding steps are similar to those applied in evaluating solid tumors, i.e., EBV infection status is assessed in case of a positive ALU II probe. Otherwise, cell therapy in question is deemed non-tumorigenic.
Although advances in gene therapy have opened opportunities for the treatment of rare diseases and disorders, many frontiers still remain unexplored and are stirring the curiosity of the scientific community. Allogeneic gene therapy has sparked an industry wide interest in investigating gene therapy as an off-the-shelf treatment. 65 It differs from autologous therapy in that the source of cells in allogeneic gene therapy is a healthy donor other than the patient. 65 The healthy donor cells can potentially be scaled up to create a master cell bank which can then be used to create working cell banks (differentiated cells) based on individual patient requirements. 65
Allogeneic therapy was first successfully performed in 1968 for a patient with Severe Combined Immunodeficiency Disorder (SCID). 66 Since then, allogeneic therapy has been advanced and implemented to cure not just SCID but other Primary Immunodeficiency Disorders (PID). Allogeneic hematopoietic stem cell transplantation continues to be the only compelling treatment option for certain disorders such as β-hemoglobinopathies. 67 Organogenesis' recently approved Gintuit, is the first commercially available allogeneic product. 68 It is a cellular sheet comprised of human keratinocytes and bovine collagen, intended to be used as a topical scaffold for the treatment of mucogingival conditions. 68
Allogeneic gene edited cell therapy is an attractive prospect given its cost effectiveness, scalability and manufacturing, quality of starting material and product (healthy donor cells), potential to rapidly treat a large patient population (off-the-shelf) and distribution, especially in countries lacking advanced infrastructure needed for autologous cell products.65,69 However, despite the apparent benefits, allogeneic therapy presents a major challenge with Graft vs Host Disease (GvHD), wherein the engrafted allogenic T-lymphocytes are activated by antigens from the recipient, largely due to the lack of thymic depletion of autoreactive lymphocytes which normally occurs during lymphocyte maturation processes. 69 GvHD can lead to diminished or absent therapeutic effect, and even mortality. 69 That said, there exist various methods to manage GvHD. Traditional management of GvHD involves prolonged immunosuppression using corticosteroids, which is typically associated with the expected adverse effects from this drug class and causing malignancy relapse. 70 More current treatment approaches include Ibrutinib, an irreversible Bruton’s Tyrosine Kinase (BTK) inhibitor. 70 BTK is a tyrosine kinase expressed on hematopoietic cells, the activity of which is essential for B-cell survival and proliferation. 70 Ibrutinib, therefore, leads to lack of circulating B cells and subsequently a lack of immune response in patients. 70 Other treatments, like Ruxolitinib, target the Janus Kinase Inhibitor (JAK 1/2), which leads to a reduction in CD4+ regulatory T cells. 71
The potential for GvHD is also an important part of the safety evaluations when developing gene edited allogenic lymphocytes. Gene edited lymphocyte products can be tested for their potential to induce GvHD non-clinically with severely immunodeficient mouse models, which allow for long term human cell engraftment. Low level total body irradiation (i.e. 2 Gy) can be used as immunoconditioning to increase the engraftment level in the mouse model. To demonstrate the sensitivity of the test system to identify GvHD, an experimental group (e.g. 15 males and 15 females) receiving non-gene edited human lymphocytes can be included (Figure 2). Signs of GvHD with non-gene edited lymphocytes can be observed starting within approximately 2 weeks in the mouse model. Monitoring of the murine model for 12 weeks post-lymphocyte engraftment is often considered adequate to assess the GvHD potential for allogenic gene edited lymphocytes. Kaplan Meier curve to illustrate GvHD survival outcome in irradiated murine models receiving unedited human T cells.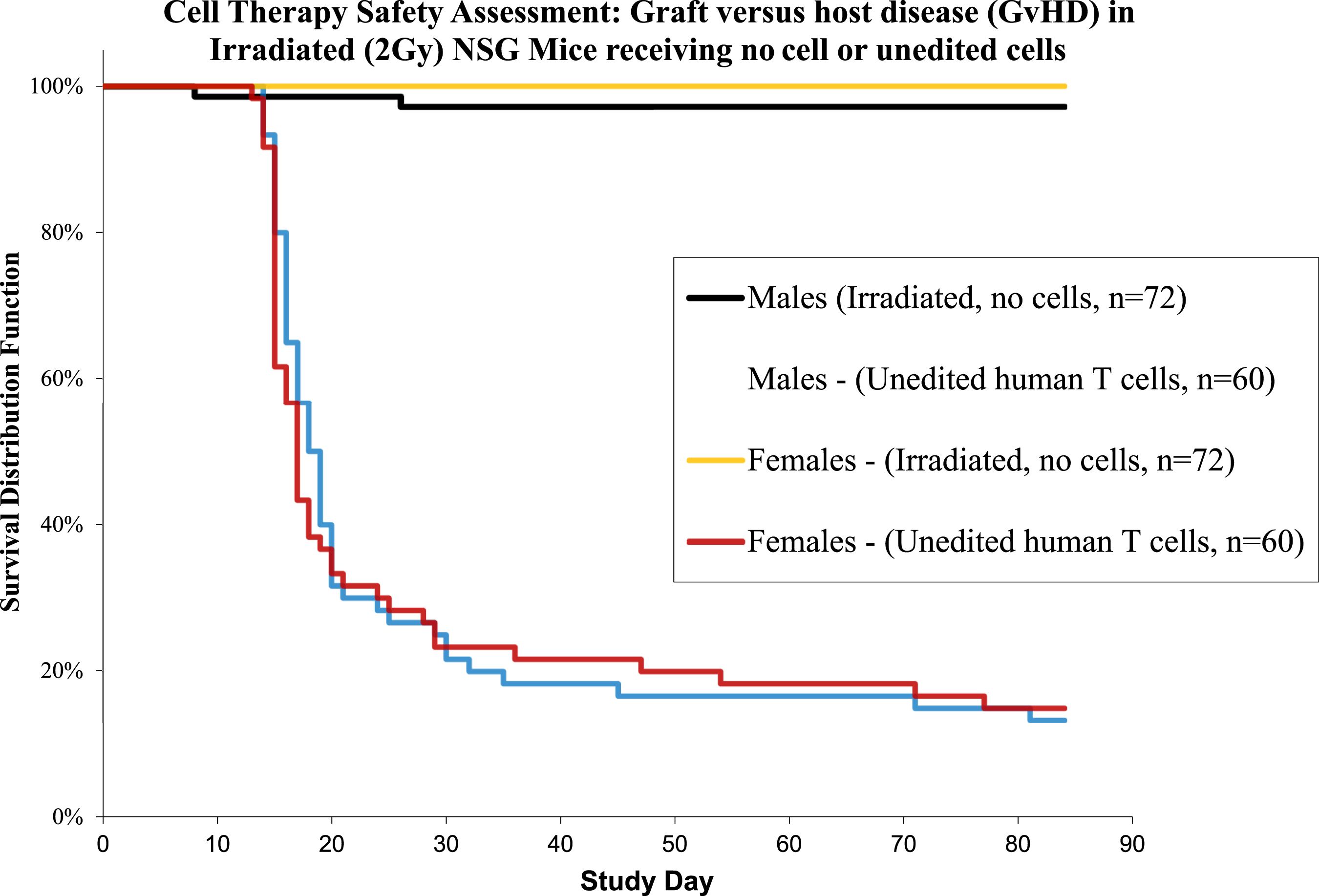
Various allogeneic CAR T (chimeric antigen receptor T cell) therapies are also being developed. T lymphocytes from a single healthy donor can be collected via leukapheresis, followed by viral vector mediated expression of CAR and subsequently amplified. 72 Post amplification, the cells expressing native TCR (surface T cell receptors) can be removed via filtration using anti-αβ TCR antibodies. 72 The resulting TCR negative allogeneic CAR T cells can be frozen and stored for shipment when needed. 72 Two major challenges that arise include immune response induced elimination of the CAR T cells by the host or allogeneic T cell induced GvHD. 72
Another promising candidate for allogeneic cell therapy is being developed to treat viral infections ensuing allogeneic hematopoietic stem cell transplant (HSCT). 73 Tzannou et al. generated virus specific T (VST) cells from third party donors for Epstein-Barr virus (EBV), adenovirus (AdV), cytomegalovirus (CMV), BK virus (BKV), and human herpesvirus 6 (HHV-6). 73 These VST cells were administered to 38 patients in a phase 2 clinical trial and were discovered to be safe and effective in the treatment of post allo-HSCT viral infections. Such a VST bank could provide a wide antiviral infection protection and be the first of its kind off the shelf product. 73
Autologous cell therapy is currently more widely implemented and persists longer in the patient, with no significant risk of immunogenicity, but it trails with its own drawbacks in that not all clinical centers have access or capital to perform required procedures, such a leukapheresis in the case of CAR T therapy.65,69 There are also commercial and manufacturing issues that may arise due to heterogeneity arising in each unique batch of cells from the patient. 65 Further, time is of the essence in autologous therapies as the process of isolation, modification and infusion of modified cells back into the patient may cause discomfort and delays. 65
Conclusion
Regardless of the nature of gene edited therapy, it can be said with fair conviction that it is indeed a revolutionary path to patient care, the advent of precision medicine. The accelerating scale at which gene editing modalities are onboarded in drug development is set to transform the fundamentals of non-clinical toxicology. The science supporting our discipline is expanding at an exponential pace, offering exceptional opportunities but also challenges to our toxicologist community.
Footnotes
Authorship Statement
Srishti Vats contributed to conception and design, contributed to analysis and interpretation, and drafted manuscript; Cristina Ballesteros contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Selly Hung contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Samantha Sparapani critically revised manuscript; Karen Wong contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Julius Haruna contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Christian Li contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Simon Authier contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.