
Abstract
Sequencing of the human genome and numerous advances in molecular techniques have launched the era of genetic medicine. Increasingly precise technologies for genetic modification, manufacturing, and administration of pharmaceutical-grade biologics have proved the viability of in vivo gene therapy (GTx) as a therapeutic modality as shown in several thousand clinical trials and recent approval of several GTx products for treating rare diseases and cancers. In recognition of the rapidly advancing knowledge in this field, the regulatory landscape has evolved considerably to maintain appropriate monitoring of safety concerns associated with this modality. Nonetheless, GTx safety assessment remains complex and is designed on a case-by-case basis that is determined by the disease indication and product attributes. This article describes our current understanding of fundamental biological principles and possible procedures (emphasizing those related to toxicology and toxicologic pathology) needed to support research and development of in vivo GTx products. This article is not intended to provide comprehensive guidance on all GTx modalities but instead provides an overview relevant to in vivo GTx generally by utilizing recombinant adeno-associated virus-based GTx—the most common in vivo GTx platform—to exemplify the main points to be considered in nonclinical research and development of GTx products.
Introduction
Ongoing advances in the understanding of gene modification techniques and molecular mechanisms of disease are fostering an explosion in innovative gene therapies designed to treat the specific gene defects in living cells that are responsible for many severe, debilitating, and lethal diseases in humans. Recent approvals in the United States of gene therapy (GTx)-based products, including Kymriah (tisagenlecleucel), Tecartus (brexucabtagene autoleucel), and Yescarta (axicabtagene ciloleucel) to combat B-cell acute lymphocytic tumors; Luxturna (voretigene neroparvovec-rzyl) to ameliorate RPE65-mediated inherited retinal disease; and Zolgensma (onasemnogene abeparvovec-xioi) for spinal muscular atrophy (SMA) represent the merest tip of the iceberg with respect to the numbers of GTx treatments projected to become available in the next several decades. A review of the scientific literature and web-based databases indicates that nearly 3000 clinical trials have been approved around the globe in the past 3 decades to develop novel gene therapies, including over 200 in each of the past several years. 1,2
Numerous gene delivery platforms are currently under development to facilitate gene modification. Such gene delivery platforms include viral vectors (eg, adenovirus [AdV], adeno-associated virus [AAV], herpesvirus, lentivirus, and other retroviruses), nonviral vectors (eg, naked DNA/plasmid DNA delivered via liposomes, nanoparticles, or exosomes), and various hybrid systems (eg., clustered regularly interspaced short palindromic repeats/CRISPR-associated protein [CRISPR/Cas] genome editing, virosomes). The choice of which GTx platform to use depends on several factors (eg, persistence and biodistribution patterns of transgene expression, host immune response) and may involve a distinct set of considerations relative to the platform used. Several brief examples will illustrate some of the complexities in selecting a GTx approach.
Primary points to consider in choosing a system for GTx are the attributes of the platform and the procedure for exposure. In terms of platform characteristics, viral vectors typically are preferred due to their superior ability to introduce (“transduce”) foreign DNA into a host cell (transduction efficiency); nonviral vectors are of interest due to their reduced cytotoxicity and immunogenicity relative to viral vectors; and hybrid systems are being pursued due to their manufacturing simplicity and versatility. 3 –5 Genome-integrating viral vectors (eg, lentivirus and other retroviruses) provide long-term transgene expression, but their tendency for random incorporation within an existing gene (ie, insertional mutagenesis) is a potential concern with respect to a test article-related tumorigenic response later in life. 6 –12 In contrast, vectors that do not need to integrate to express their transgene (eg, AAV, AdV) generally remain episomal and therefore have a lower likelihood of insertional mutagenesis. 13 –15
The means of host cell exposure to GTx may be either ex vivo or in vivo. In ex vivo GTx, cells are isolated from a patient and undergo gene modification outside the patient’s body, typically with a lentivirus vector (eg, Kymriah and Yescarta, where cultured T-cells are modified ex vivo to express a chimeric B-cell/T-cell receptor to boost their homing specificity and cytotoxic efficacy on neoplastic B-cells). For in vivo GTx, the vector is delivered locally or systemically into the living host (eg, Luxturna and Zolgensma, where a recombinant AAV [rAAV] vector carrying a transgene expression cassette to replace the defective gene is injected locally into the retina or systemically in the intravascular space, respectively). Ex vivo GTx permits higher control of gene modification in the exposed target cells prior to their infusion back into the patient, while in vivo GTx is injected directly and irretrievably into the patient, leading to both a higher likelihood of systemic exposure and a greater potential for modification of nontarget cells. When all these parameters are considered together, rAAV vectors are the leading platform worldwide for in vivo GTx due to their balance of excellent transduction efficiency, high level of transgene expression, and relatively limited toxicity. 16
Due to the novelty and lack of sufficient platform knowledge on the efficacy and safety assessment of GTx products, the regulation of this therapeutic modality is continuously evolving. Nonclinical safety assessments for GTx products require the design of nonclinical programs and specific nonclinical toxicity studies with a focus on individual test article attributes. Frequently, the study protocol is defined on a case-by-case basis that is determined by disease indication and product attributes, since no standard approach of necessary studies (like those recommended for small molecule test articles 17,18 ) has been devised. This flexible approach to nonclinical safety evaluation of GTx products is the same approach that was advanced when biological drugs were initially being developed in the early 1990s. 19 The approach has also been endorsed in the current regulatory guidance documents that have been issued for GTx-based products. 20,21 Accordingly, the Scientific and Regulatory Policy Committee of the Society of Toxicologic Pathology created a Working Group to frame “points to consider” to assess current practices and advise on approaches for the nonclinical research and development, including safety assessment, of GTx products. Given the fundamental differences between ex vivo and in vivo GTx and the much larger share of rAAV-based gene therapy (rAAV-GTx) currently in development or on the market, the Working Group focused the effort on in vivo rAAV-GTx.
Several attributes of rAAV vectors contribute to the popularity of their use for in vivo GTx. 22 First, many serotypes of wild-type AAV (wtAAV) are present naturally, each with differing selective cellular tropism and gene expression efficiency that are further refined and differentiated in the engineering of rAAV vectors. 23,24 Second, rAAV vectors are considered to generally have low pathogenicity and immunogenicity in mammalian cells and tissues. 25,26 Third, rAAV vector delivery directs persistent episomal transgene expression, not requiring integration into the genome to be biologically active and thus lowering the likelihood of insertional mutagenesis and late-onset disease (eg, cancer). 27,28
Although the focus of this document is on rAAV-GTx, the information presented here for planning and conducting rAAV-GTx safety assessments can be used for nonclinical development of other viral and nonviral in vivo GTx platforms. The safety concerns for other viral vectors are conceptually similar to those for rAAV-GTx, so many of the considerations raised in this manuscript regarding rAAV vectors will need to be addressed during nonclinical development of non-rAAV viral vectors. In like manner, nonviral GTx vectors will need to assess many of the major points raised for rAAV vectors in this article as the nonclinical safety testing for nonviral vectors will be influenced both by the genetic payload and the carrier/vehicle used for delivery. Therefore, the following points to consider in the nonclinical research and development of rAAV-GTx represent general principles that frequently apply to other modes of in vivo GTx.
General Considerations for Safety Assessment of In Vivo GTx Products
Safety assessment of in vivo GTx products occurs throughout the product research and development program. Similar to other biological drugs, the nonclinical program for an rAAV-GTx product has the following overall goals: (1) confirm the desired pharmacological effect and identify a biologically active dose range; (2) evaluate the safety of the test article using the proposed clinical route of administration (ROA), including the delivery device and method, in a relevant test species; and (3) develop a clinical safety monitoring and dose escalation plan based on the nonclinical safety profile and therapeutic index. 19 –21 However, due to the variety of gene delivery platforms and the complexity of the products, GTx safety assessment is approached on a case-by-case basis that is determined by disease indication and product attributes such as mode of gene modification, mechanism of action, and intended ROA. 29 Nonetheless, the essence of the case-by-case approach is that the principles of nonclinical safety evaluation are the same as for other modalities even though the practices for GTx products are different. 30 In other words, certain themes are common to all nonclinical development programs, including in vivo GTx products. 31 For example, assessing the potential toxicity of a GTx product at peak and persistent exposure (eg, 4-week and 13-week general toxicity studies) is an expectation, while other questions such as immunotoxicity, genotoxicity, reproductive toxicity, and carcinogenicity studies are only considered on a case-by-case basis depending on the unmet medical need and risk/benefit considerations. In particular, standard genotoxicity and carcinogenicity studies generally are not performed for GTx vectors like rAAV.
Initial in vitro and in vivo nonclinical proof-of-concept (discovery research) studies should have justification in a solid scientific rationale and be designed to demonstrate the biological effect of the vector distribution and transgene expression. The toxicologic pathologist should be acquainted with and whenever possible consulted on the general design considerations for such studies, although the pathologist’s main contributions will come in overseeing the collection and analysis of tissues and biofluids (eg, serum, urine, and cerebrospinal fluid [CSF]). Ideally, proof-of-concept studies in animals should mimic the planned clinical protocol with regard to ROA, dosing schedule, the use of novel dosing devices, and the measurements of biological markers of efficacy and safety. They may be performed under non-Good Laboratory Practice (GLP) conditions as long as there is adherence to good documentation practices. Each lot of an investigational GTx product used for in vitro and in vivo studies should be characterized, and a representative sample from each lot needs to be retained for potential future interlot comparison. The transgene expression cassette delivered by the vector and intended for use in humans needs to be tested in nonclinical animal species whenever possible. However, in some instances, the use of an animal species-specific orthologous transgene may be required if the human transgene is not biologically active in animals or if it may induce an immune response due to variations in amino acid sequences between the human transgene protein and the orthologous protein of the nonclinical animal species. Data acquired during proof-of-concept studies—including information on the level of transgene expression and the degree of correction of the defective phenotype, the optimal ROA and dosing regimen, the minimally and maximally effective doses, the biodistribution of the vector, the feasibility and safety of novel administration devices, and the identification of a susceptible and pharmacologically relevant animal species, strain, and/or model of disease for nonclinical testing—will ultimately inform the design of pivotal toxicity studies. 20,21
The goals of pivotal nonclinical toxicity studies are to identify local and systemic toxicities associated with the vector or transgene product and any adverse effects associated with the delivery method at the intended or multiples of efficacious dose levels. The toxicologic pathologist again will contribute chiefly by involvement in the study design and by overseeing collection and analysis of tissues and biofluids using conventional endpoints (eg, macroscopic findings, organ weights, clinical pathology values, histopathologic evaluation of hematoxylin and eosin [H&E]-stained tissues). However, pivotal toxicity studies for GTx products typically include a number of molecular pathology endpoints to evaluate the cellular location of the vector DNA, transcription, and translation. Pivotal toxicity studies are performed under GLP conditions when feasible, should employ a study design that mimics the planned clinical protocol in humans, and should use the investigational GTx product intended for use in clinical trials. Dose levels that bracket the maximally effective clinical dose, as determined during proof-of-concept studies, should be incorporated in the study design. Exposure, referred to as biodistribution in GTx studies, is typically included in pivotal toxicity studies and includes an assessment of vector DNA and transgene expression. Methods used to evaluate biodistribution typically include quantitative polymerase chain reaction (qPCR) for measuring vector DNA and reverse transcription polymerase chain reaction (RT-PCR) for measuring transgene messenger RNA (mRNA). 32 Evaluating transgene protein expression, when relevant, in tissues and/or biofluids may also be included. For novel viral serotypes and to aid in nonclinical species selection, preliminary biodistribution studies should be considered in order to broadly assess tissue distribution and species differences in tissue tropism. Additional monitoring parameters may be included based upon the type of vector, the particular transgene, or specific features of the disease or clinical population being targeted for treatment. These methods of assessing vector exposure are combined with more routine measurements of toxicity including clinical signs, body weight, food consumption, clinical pathology analytes, organ weights, macroscopic and microscopic pathology data (which may include localization of transgene expression via in situ hybridization [ISH] or immunohistochemistry [IHC]), and/or immune function (eg, humoral and cell-mediated immunity) to the vector components and transgene product. If warranted, safety pharmacology endpoints may also be included. Necropsy generally is suggested at an early time point, corresponding to the time of peak transgene expression in tissues, and one (or more) later time points to examine persistence and any long-term effects of transgene expression following the single GTx dose. 33 Biodistribution typically is evaluated in multiple tissues and usually should include at a minimum (per US Food and Drug Administration [FDA] guidance 34 ) nine tissues: the administration site, blood, brain, gonads, heart, kidneys, liver, lungs, and spleen. Localization to other tissues may be assessed on a case-by-case basis, depending on the vector, dose, transgenes, and ROA. The appropriate/necessary list of tissues/cells for biodistribution assessment should be customized to the product under development. Consultation with reviewing health authorities on such study design parameters is warranted.
Addressing the potential for vector shedding (through urine, feces, saliva, or other body secretions) into the environment, vector latency and reactivation (resuming productive viral replication) within the patient for non-AAV vectors, and the effects of long-term transgene expression are required by health authorities for clinical trials and application submissions, and these parameters may also need to be evaluated in nonclinical toxicity studies based upon the class of vector and activity of the transgene. Vector shedding can be assessed during nonclinical toxicity studies, 32,35 and the choice of samples and frequency of collection are influenced by several factors including the natural history of the parent viral vector, the ROA, and animal species. 18 The identification of significant levels of vector in the gonads may require additional techniques (eg, ISH or IHC) in order to identify the specific cell type transduced by the vector. Although not typically required, the presence of the vector in germline cells may dictate the conduct of developmental/reproductive toxicity studies including germline transmission to the next generation. The evaluation of vector DNA integration into the host cell genome is typically required for vectors that require integration to express their transgene (eg, lentivirus) in both clinical and nonclinical studies. The need to evaluate vector DNA integration for vectors that are classified as nonintegrating vectors (ie, AAV) is currently an evolving area of regulatory expectations and may be required in some cases. 36 Based on the collective knowledge of the authors, carcinogenicity studies have not been required for safety assessment of rAAV-GTx test articles but may be needed with integrating viral vectors like lentiviruses and retroviruses, where there may be an increased risk of tumorigenicity due to insertional mutagenesis or long-term expression of certain cell growth factor-regulating transgenes.
Pathologist Roles in the Discovery and Development of In Vivo GTx Products
Pathologists, both anatomic and clinical, play diverse roles in contributing to the discovery and development of in vivo GTx products. The emergence of GTx as a new modality in the biopharmaceutical industry and the pathologist’s skill set enables the pathologist to make significant contributions to GTx drug development and advancing candidate therapeutics to the clinic. Key pathologist roles span both discovery and development functions. Pathologists support the characterization of animal models of human disease and efficacy studies in these models, biodistribution studies of GTx test articles, evaluation of tissues to define the presence and extent of any GTx-related effects (desired or toxic), and they identify and qualify biomarkers that bridge the nonclinical program with clinical monitoring and data interpretation.
Anatomic pathologists provide expertise for tissue-based analyses. Development of GTx products often utilizes animal models of disease as the test system for evaluating the efficacy and occasionally toxicity of GTx products. Histopathologic characterization and validation of such animal models of disease using conventional H&E-stained tissue sections provide one pillar for assessing the efficacy of the GTx product as well as the necessary knowledge to interpret safety endpoints in combined efficacy/toxicity studies. Tissue-based analysis can support efficacy endpoints by demonstrating the reduction or reversal of histopathologic changes associated with the disease model following the administration of a GTx product. Tools like IHC or immunofluorescence (IF; to detect capsid or transgene protein) and ISH (to detect vector genome DNA or mRNA, especially when transgene protein products are untagged and indistinguishable from endogenous proteins) support morphology-based assessment for the biodistribution of the GTx product and provide spatial context for understanding the transgene expression and possible sites for test article-related effects (Figure 1). Furthermore, IHC and ISH may aid the pathologist in identifying the precise cellular tropism and subregional differentiation of transgene expression within the tissue. The pharmacodynamic evidence of GTx effectiveness is further complemented by the demonstration of concurrent and colocalized transgene expression in areas of tissue damage and recovery. Anatomic pathologists may employ a combination of histopathologic endpoints to differentiate findings that could be specifically associated with the administration procedure (eg, ROA and implanted device in vehicle-administered animals) or to discern the relevance of unexpected findings (eg, chronic and neoplastic findings that cannot be temporally associated with the administration of the GTx product).
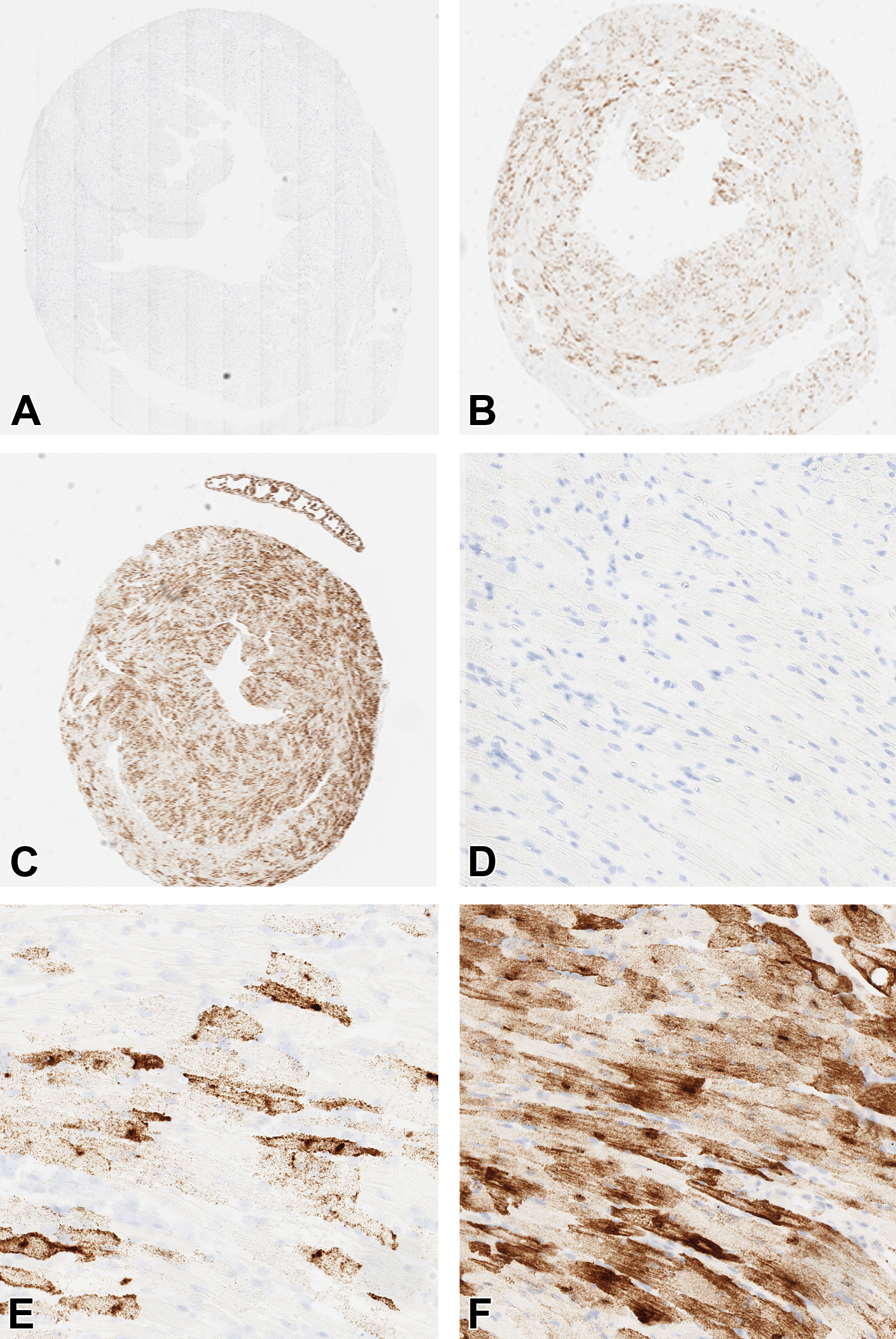
Morphology-based evaluation of the dose–response relationship for a recombinant adeno-associated virus (rAAV) vector. In situ hybridization (ISH) demonstrating vector dose-dependent increase in transgene messenger RNA (mRNA) expression per cardiomyocyte and increase in percentage area transduction in the heart of mouse. A, Saline control (1.5× objective). B, Low dose vector (1.5× objective). C, High dose vector (1.5× objective). D, Saline control (20× objective). E, Low dose vector (20× objective). F, High dose vector (20× objective).
Clinical pathologists provide expertise for fluid-based analyses. Biological fluids provide a unique portal for longitudinal acquisition (repeated access over time in individual animals) of analytes relevant to disease model characterization, disease progression, and assessment of efficacy leading to disease reversal and recovery. Fluids also are essential samples for in-life and terminal monitoring of toxicities associated with in vivo GTx. Clinical pathology data may be necessary to evaluate the pharmacology of secreted transgene products and to measure the dose–response relationship throughout the study. For example, in hemophilia A and B GTx programs, an essential endpoint of efficacy is the improvement of coagulation parameters and the decrease in bleeding rate, especially when correlated with Factor VIII (for hemophilia A) or Factor IX (for hemophilia B) transgene expression.
The combination of anatomic pathology and clinical pathology assessment for GTx product development often works synchronously to support risk identification, mechanistic toxicity studies, and the identification of dose–response relationships. For example, loss of transgene expression in cells or tissues coupled with reduced blood levels of a secreted transgene product may warrant investigating the potential of a humoral immune response to the transgene or cell-mediated immune response to the transgene and/or vector capsid. Useful anatomic pathology endpoints for this coordinated evaluation might seek to show leukocyte lineage-specific tissue infiltration by mononuclear inflammatory cells (eg, IHC for CD3/CD4/CD8 for different subsets of T-cells, B220 or CD20 for B-cells, and CD68 or Iba1 for macrophages); this approach is particularly powerful when using multiplex IHC or IF to demonstrate colocalization of inflammatory cells with transgene expression in cells of the affected tissue. Common clinical pathology endpoints that further support such investigations often include longitudinal acquisition of clinical chemistry, hematology, leukocyte subset analysis, and cytokine panels to demonstrate the potential immune-mediated tissue damage in response to vector uptake or transgene expression. Investigations of immune response may also include the evaluation of antidrug antibody response to the transgene protein or an evaluation of cell-mediated immune response to the transgene and vector capsid protein using an enzyme-linked immune absorbent spot assay (ELISpot) in isolated peripheral blood mononuclear cells (PBMCs) and/or spleen cells.
Taken together, the combined anatomic and clinical pathology analyses contribute substantially to the effectiveness of the study design and evaluation for efficacy studies as well as non-GLP and investigational new drug (IND)-enabling GLP toxicity studies. Moreover, these toxicologic pathology tasks for GTx products are comparable to those employed in performing the regulatory clinical and anatomic pathology analyses for animal toxicity studies for other categories of test articles.
Discovery and Early Development Considerations for rAAV-GTx
Key Elements of rAAV Vector Design
During the discovery and early development of an rAAV-GTx product, initial design and selection of the optimal rAAV vector are the first steps to success, and basic knowledge of rAAV vector design (capsid serotype and DNA construct) is helpful in assessing the efficacy and safety of novel rAAV-GTx products in animal studies. Research pathologists should assist in the initial design and selection of the optimal rAAV vector by advising on serotype selection, promoter design and selection, interspecies sequence similarity of the transgene/gene product, comparative biology, and early morphology-based biodistribution and transgene expression characterization. Accordingly, research pathologists should have some familiarity with principles of vector design.
Cell tropism of naturally occurring AAVs is often broad, but each serotype has a biased tropism profile depending on multiple factors such as localization of different cell surface AAV receptors on host cells, AAV serotype-specific interactions with the AAV receptor, and AAV capsid interactions with the transgene promoter. 37 –39 To date, 12 naturally occurring AAV serotypes from both human and nonhuman primates (NHPs), and over 100 AAV variants, have been identified. 23,40 Wild-type AAVs have a single rep gene encoding proteins necessary for viral replication, a single cap gene generating viral capsid proteins (VP1, VP2, VP3), and assembly-activating proteins necessary for viral capsid formation. The capsid proteins define the AAV serotype and help determine cell and tissue tropism, biodistribution, and intracellular trafficking following in vivo injection (Figure 2). The DNA of rAAV-GTx vectors retains the wtAAV inverted terminal repeats (ITR) yet is devoid of the rep and cap genes, which are replaced by the engineered transgene expression cassette placed between the ITRs. The choice of a serotype for a specific disease indication is guided by the tropism of the vector capsid for the cell types or tissue(s) involved in the disease process, the prevalence of preexisting antibodies against capsid proteins in the patient population (ie, seroprevalence) that might prevent tissue transduction by the vector, 41,42 and the ease of manufacture and scale-up for that particular serotype. Consequently, considerable efforts have been geared toward the identification of naturally occurring capsid serotypes or designer-engineered novel capsid variants with optimal cell and tissue tropism and improved immune evasion profiles. 43 –45
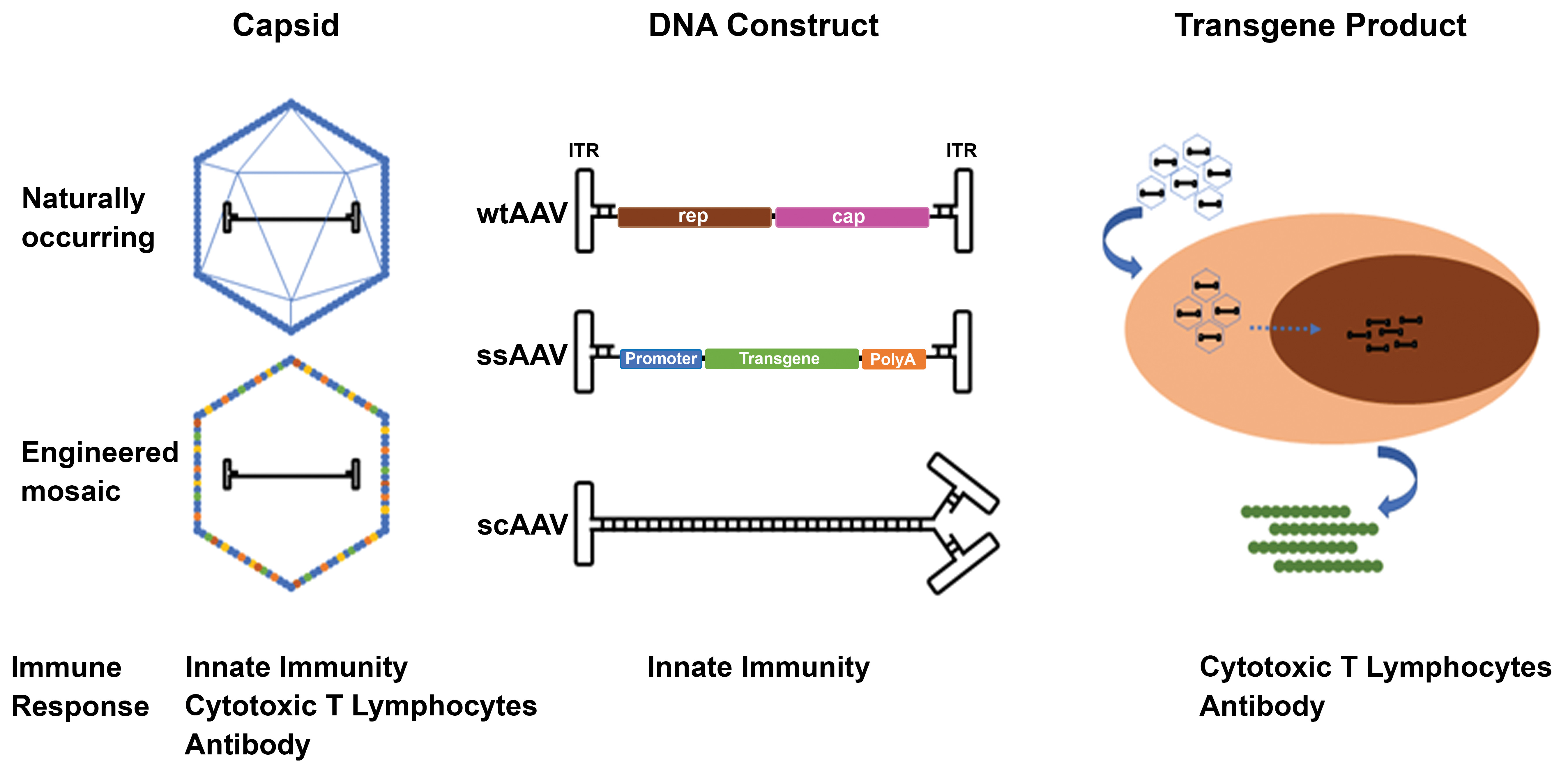
Adeno-associated viruses (AAV) are composed of an icosahedral capsid (composed of VP1, VP2, and VP3 subunits) enclosing a DNA construct capable of producing a transgene product (either protein or RNA, such as a microRNA [miRNA] that inhibits or modifies endogenous mRNA). The AAV capsid can be generated from preexisting wild-type AAVs (wtAAV) or engineered into mosaic variants to improve cellular tropism, transduction efficiency, or to evade preexisting immunity. For recombinant AAV (rAAV), DNA constructs are single-stranded DNA molecules (ssAAV); however, double-stranded, self-complementary DNA constructs (scAAV) are occasionally used to avoid the requirement of second-strand synthesis and to permit quicker transgene expression in vivo. All components of rAAV can induce different reactions by the host immune system such as humoral antibody response against capsid proteins, innate immune response against foreign DNA, or cytotoxic T-cell response against cells expressing transgene protein product.
Once a capsid serotype with a favorable profile is selected, effort is directed toward the design of a suitable transgene-bearing DNA construct to be packaged within the capsid of choice. Naturally occurring AAV contains single-stranded DNA with packaging capacity limited to 4.7 kilobases (Kb). Consequently, rAAV transgene-bearing DNA construct designs generally should be within this size range (∼4.5-4.8 Kb), inclusive of upstream promoter/enhancer elements, introns, and downstream polyadenylation (poly (A)) sequences required to drive optimal transgene expression and persistence (Figure 2). 31 For transgenes that are larger than 4.7 kb, carefully designed truncated transgenes that conserve crucial functional domains of the targeted gene can be used instead. For example, the ∼14 Kb dystrophin gene may be spliced down to ∼5 Kb to design microdystrophin constructs currently used in several rAAV-GTx clinical trials. 46 Alternatively, oversized vectors or regular-sized dual rAAV vectors have been used. 47,48 Single-stranded AAV (ssAAV) constructs that rely on cellular replication factors to synthesize the complementary strand of DNA, or self-complementary AAV (scAAV) constructs in which the complementary strand of DNA is provided in the construct, may be employed in the design of rAAV vectors (Figure 2). The advantages and disadvantages of ssAAV and scAAV are summarized in Table 1. It should be noted that multiple expression cassettes can be included in the same construct if more than one transgene product is needed and their combined sizes allows them to be accommodated within the rAAV packaging capacity.
Advantages and Disadvantages of Single-Stranded and Self-Complementary AAV Vectors.

Abbreviations: AAV, adeno-associated virus; scAAV, self-complementary AAV; ssAAV, single-strand AAV.
Although AAV capsid structure and target cell receptors determine cell and tissue tropism, the choice of promoter plays a critical role in controlling the specificity of transgene expression within a target cell type, the robustness of the transgene mRNA transcription and translation, and the persistence of transgene expression. Transgene expression in the desired target cells can be made more specific or enhanced in magnitude by modifying promoters. 53 For example, the 1.8 Kb neuron-specific enolase (NSE) promoter, the 470 base pair (bp) human synapsin-1 (SYN1) promoter, the 229 bp methyl CpG-binding protein 2 (MeCP2) promoter, and the 2 Kb herpes simplex virus 1 latency-associated transcript (HSV1-LAT) promoter can all drive neuron-specific transgene expression, in contrast with stronger promoters such as chicken β-actin (CBA) or cytomegalovirus (CMV), which drive more ubiquitous expression. Properties of selected ubiquitous and cell type-specific promoters are detailed elsewhere. 54
The design of the transgene can differ depending on the underlying disease mechanism. Transgenes may code for secreted proteins; nonsecreted membrane proteins; or intracellular proteins (eg, structural proteins), peptides, oligonucleotides, or inhibitory RNAs. For GTx aimed to correct monogenic disorders (eg, SMA or Friedreich’s ataxia), the transgene is composed of an open reading frame (ORF) expressing the gene product of interest in its native form or modified by codon optimization (to improve translation and reduce immunogenicity) or by deletions to allow the larger coding regions of the gene to fit within rAAV packaging capacity (such as microdystrophin for Duchenne muscular dystrophy). 55 For diseases with toxic gain-of-function mutations where gene suppression is needed to correct the disease phenotype (eg, Huntington disease), the transgene delivered may be a dominant negative transgene, an artificial primary microRNA (miRNA), or a short hairpin RNA (shRNA) that leads to the suppression of gene expression (ie, “knockdown”). 43 Post-transcriptional control elements can be engineered into the rAAV DNA construct to further fine-tune transgene expression, including regulatory sequences in untranslated regions or by codon optimization. 56 The poly(A) tail of a transcript is also an important element to consider as it is critical for nuclear export, translation, and mRNA stability. 54
Early Considerations Independent of rAAV Vector Design
Factors related to the host (patient or nonclinical animal species) and treatment regimen may influence the efficacy and safety profiles of rAAV-GTx products. Toxicologic pathologists are well versed in the importance of many parameters, including individual demographics (such as breed/strain, age, and sex), dose formulation, and selection of feasible doses and the optimal ROA. For example, sex has been shown to impact expression of transgenes following systemic delivery of rAAV vectors (eg, with greater expression in the liver of male mice). 57 –59 Similarly, host age can play a role. For instance, AAV serotypes 9, rh8, rh10, and rh43 (where “rh” stands for “rhesus” monkeys from which these serotypes were isolated) have both neuronal and glial tropism when administered systemically; however, differences in transgene expression occur, with predominantly neuronal expression in neonatal animals and glial cell expression in adults. 60 –64 Other factors such as basic concepts of methods of vector production are generally less familiar to the pathologist but should still be understood in principle. 53
Dose selection for GTx encompasses several additional nuances relative to other test article types. The dose of rAAV for in vivo administration is typically expressed as vector genomes (vg), given on a “per animal” (vg/animal) basis, particularly for intracranial or intrathecal (IT) ROAs; “per tissue/compartment” (vg/tissue) basis, as for intraarticular or intravitreal ROAs; or per unit weight (vg/kg) basis, as is commonly the case for intramuscular (IM) and intravenous (IV) ROAs. Depending on the animal size, ROA, and maximal feasible concentration of vector that can be achieved without causing vector aggregation, the dose can be formulated in small-volume sizes (μL) for systemic delivery in small animals or localized administration in large animals, or in large-volume sizes (mL) for systemic delivery in large animals. Dose extrapolation across species for weight-based doses (ie, vg/kg) is converted based on per kg body weight; however, tissue/compartment-based doses (ie, vg/eye or vg/brain volume) need to consider other factors such as differences in organ anatomy, tissue composition (eg, fluid or solid), and compartment volume. The tropism of rAAV may be influenced by the injected dose as well as the injection rate and volume. For local injections (brain [intraparenchymal], eye, etc), increasing the concentration of vector genomes in an injected dose increases the number of rAAV particles that can be endocytosed by all cells local to the injection site, thereby driving stronger and more focal gene expression within both intended and unintended cell populations. In contrast, lower vector genome concentrations may instead increase transduction specificity by reducing the number of vector particles endocytosed per cell. 53
The choice of ROA may offer some overall benefits for in vivo GTx. For example, when targeting the brain, direct brain intraparenchymal injection offers several advantages relative to either systemic delivery or central delivery directed into the CSF. Key benefits to intraparenchymal delivery include reduced exposure of peripheral organs, thus diminishing the likelihood of peripheral toxicity, and decreased amounts of vector needed to achieve high-level expression at the desired location. 65 Intraparenchymal injection or exposure of nerve termini (eg, brain, cornea, footpad) may have associated anterograde or retrograde axonal transport resulting in transgene expression in neurons distal to the site of exposure. 66 –68 Bolus administration of the same rAAV dose to mice by IT versus IV injections provides higher transduction efficiency in sensory neurons (of dorsal root and trigeminal ganglia) and central nervous system (CNS) tissues when given IT compared to IV, while comparable transduction efficiency is observed in peripheral organs for both ROAs. 69 In contrast, GTx treatments that require systemic target organ delivery typically will be administered by the IV route to ensure wider tissue transduction.
Initial in vivo pilot studies may be considered to assess rAAV transduction efficiency, vector DNA biodistribution, and cell tropism as well as differences in response based on animal species, age, ROA, disease-associated pathology, and approximate dose level, and so on. Pathologists will be essential players in evaluating such endpoints. These studies can be performed using a reporter transgene to provide data agnostic of the therapeutic transgene to speed data collection and interpretation. Localization of the transduced cells can be assessed quickly by standard molecular biology detection protocols performed on homogenized tissues or tissue sections to detect the products that are either reporter-tagged or that are specific for transduced cells. Common reporter genes for this purpose include green fluorescent protein (GFP) and β-galactosidase (lacZ). Expression of these reporter genes is typically driven by a ubiquitous promoter (eg, CBA, CMV) to assess the broadest potential of vector biodistribution. Although the distribution of vector DNA may not be impacted by the transgene selected for these pilot studies, the abundance of transgene expression may be influenced by the efficiency of transgene mRNA and protein expression; consequently, direct extrapolation of transgene expression levels between a reporter gene and an intended therapeutic transgene should be done with caution. 70 Additionally, some reporter transgenes express nonendogenous proteins that may also result in toxicity, immunogenicity, or both. 71 Some pilot studies may need to be conducted in large animal species such as dogs, mini-pigs, or NHPs to test novel delivery devices or anatomically constrained ROAs that are challenging or not feasible to assess in rodents due to body size limitations. The use of large animal species, more importantly, may help in assessing the potential for interspecies differences in transduction efficiency, promoter biology, and transgene expression.
Methods of vector preparation and purification may play a role in impacting the in vivo attributes of a particular rAAV (eg, tropism, biodistribution, and immunogenicity). For example, one report documented that an intraparenchymal brain injection of CsCl-purified AAV8 resulted in strong astroglial tropism of a CMV-driven GFP reporter gene, while intraparenchymal brain injection of iodixanol-purified AAV8 carrying the same CMV-GFP construct exhibited only neuronal transduction. 72 To our knowledge, no details have been discovered to explain the mechanism for such process-related differences in cell tropism.
Considerations in Designing Nonclinical Programs for rAAV-GTx
The design of development programs for evaluating the efficacy and safety of rAAV-GTx products varies on a case-by-case basis that is determined by disease indication and product attributes. Ultimately, nonclinical programs for rAAV-GTx products are similar in principle to those of other biologics. Key features will aid in identifying a biologically active dose range that can be translated into an initial clinical dose, understanding the safety profile using the proposed ROA and any novel delivery device, defining potential dose-related toxicities and the therapeutic index, and crafting a clinical monitoring plan. 19 –21
As noted earlier, rAAV-GTx transduction efficiency and product efficacy typically are assessed initially in proof-of-concept studies through a combination of in vitro cell-based assays (animal and human) and in vivo studies in rodent (typically mice) and/or large animal (often NHP) species. Early in vivo nonclinical efficacy studies in animal disease models may be modified to include safety endpoints to support clinical trials. 20,21 Indeed, the use of animal models of disease for toxicity or combined efficacy/toxicity studies for evaluating GTx products is encouraged in guidance documents from major regulatory bodies, including the European Medicines Agency (EMA) and the FDA. 20,21 However, when performing combined efficacy/toxicity studies, the animal model needs to be characterized sufficiently and the endpoints should be robust enough to distinguish between the disease model phenotype and the rAAV-GTx product-induced effects. 73 –75 This will require statistical power analysis or knowledge of the appropriate number of animals per group to permit both the robust analysis and to accommodate variability in disease phenotype, a requirement that might be challenging to obtain for some disease models. Endpoints such as transduction efficiency and expression in target cells (eg, percentage motor neuron transduction in the spinal cord for SMA or circulating Factor IX concentrations for hemophilia B) contribute to dose selection in the clinical trials.
Once proof-of-concept is achieved and initial efficacy is identified in the disease model, subsequent studies during the development phase are geared toward parameters related to the proposed human clinical trial design. Dose range-finding studies in one (or more) relevant animal models of disease, and in some cases wild-type animals, can aid in understanding the dose–response relationship of rAAV-GTx. Although these studies are ideally performed with the rAAV-GTx clinical candidate, surrogate capsids or DNA constructs may occasionally be used, if needed, as long as they express the same functional transgene product. Prior to clinical trials, however, the dose–response curve must be re-evaluated using the selected clinical rAAV-GTx candidate.
Dose Selection
Pathologists may be involved in dose selection for rAAV-GTx nonclinical studies by evaluating the semiquantitative endpoints of IHC or ISH (eg, percentage positive area or cells within the tissue, possibly by image analysis programs) and evaluating the efficacy in animal models, so the pathologist’s understanding of GTx dose selection principles is important to using these data to inform dose range estimations for toxicity studies. Nonclinical dose range-finding and efficacy studies for GTx products need to define the therapeutic (ie, pharmacologically effective) dose range. 64 The lower end of this range is referred to as the minimally effective dose (MED), which is the smallest dose with a discernable useful (efficacious) effect. The upper limit of the range is the maximum dose beyond which no additional therapeutic benefit is observed. The MED from nonclinical studies is often used to estimate the starting dose in clinical trials. Unlike other modalities where dose escalation can be done in the same individual, GTx can only be given once per patient because of the immune response that will neutralize a subsequent vector administration and sustained effect of the therapy. 23 Consequently, for ethical reasons, human clinical trials are initiated at a dose that is believed to have the potential for therapeutic benefit. The upper end of the therapeutic dose range in nonclinical studies provides a guide for clinical development regarding where the clinical dose will need to be raised to reach the optimal biological dose (ie, the dose that offers maximal therapeutic benefit with manageable risk). The nonclinical therapeutic dose range is only a guide for clinical development as the efficacious dose range may not directly extrapolate to humans. Nonclinical toxicity studies should be designed around the therapeutic dose range and a multiple thereof and should support the safety of the starting dose and the range within which the dose can be safely raised in determining the optimal biological dose in patients.
Like other therapeutic modalities, nonclinical toxicity studies for GTx should identify the potential toxicities that will need to be monitored in clinical studies and provide an assessment of the safety margin for the anticipated dose range that will be explored in clinical studies. A typical nonclinical toxicity study for GTx will have a low dose generally equivalent to the MED in efficacy studies. The middle dose is chosen based on the maximum effective dose determined in proof-of-concept studies; however, if there are known toxicity concerns, it may be set lower to define the optimal biological dose. The high dose is chosen to produce a toxic effect or provide an adequate safety margin (eg, 5- to 10-fold) relative to the estimated maximum dose in humans. The high dose may be limited to a maximum feasible dose, which is influenced both by what volume can actually be administered to an animal and the maximum concentration at which the vector can be formulated. Depending on risk–benefit considerations for a given disease condition and toxicity findings, a narrow therapeutic index (eg, 2-fold) may be acceptable for in vivo GTx products. In some instances, pivotal toxicity studies may be performed using only 2 doses. In these cases, the low dose is selected to mirror the maximum anticipated clinical dose that will be used. Studies using only 2 doses may be performed when there is a high degree of confidence that the low dose (maximum clinical dose) has an acceptable nonclinical safety profile or the resources for a 3-dose toxicity study are lacking. Prior experience with highly similar GTx products and modes of administration can help build confidence in predicting the likely outcomes of nonclinical studies once the GTx platform is well characterized.
Initial dose selection for human trials is defined using both empirical measurements for the novel GTx test article and interspecies scaling approaches. Following initial studies in rodents (usually mice but sometimes rats), the MED can generally be translated to larger animals (typically NHPs) and then to humans using a variety of scaling approaches such as dose/body weight, dose/surface area, vector concentration and/or volume/tissue area, dose/compartment volume, and so on. 76,77 Other factors that should be considered include differences in dose-related tropism of the vector in different species (rodents and nonrodents), the impact of the disease state on transduction, the comparative biology of the promoter, and so on. Biologically relevant principles and a well-considered scientific rationale should be applied in dose extrapolation across species and for first-in-human dose proposals. For example, administration of AAV8 expressing human coagulation Factor IX (AAV8-hFIX) in C57BL/6 mice at 4 × 1012 vg/kg resulted in approximately 2-log higher transgene expression compared with that achieved in rhesus macaques administered the same vector at a slightly higher dose of 5 × 1012 vg/kg. 78 Similarly, in a separate study, administration of AAV8 expressing hFIX in C57BL/6 mice and Wistar rats at 5 × 1012 vg/kg resulted in greater than 6-fold higher transgene expression in mice compared with rats. 79 Consequently, if vector tropism is similar in a rodent disease model and wild-type rodent but the transduction efficiency of the vector in an NHP is 10-fold less, then this difference needs to be taken into account in designing pivotal nonclinical safety studies and estimating the clinical dose and dose escalation plan.
The maximum concentration at which a vector can be formulated without precipitation or aggregation during production and shipment may be a limiting factor when choosing high doses for a nonclinical study and subsequent clinical use. The small size of rodents may necessitate dividing the dose volume to permit simultaneous administration at multiple sites (eg, IM injection). Vector concentration ultimately impacts the strength, specificity, and distribution of transgene expression, particularly when administered locally.
When determining doses for GTx products, the method used for calculating genome copies to be delivered is critical. The PCR-based assay (qPCR or droplet digital PCR [ddPCR]) is typically utilized to measure the total viral genomes. 80 However, PCR results can vary dramatically among laboratories, and efforts must be made to standardize and validate the nonclinical PCR methods with those that will be used to quantify viral genomes in the product destined for the clinic. Other appropriate analytical methods should be in place to ensure comparability of the vector employed in pivotal nonclinical efficacy and safety studies with that used in clinical trials. The FDA has provided guidance for this purpose that should be considered when validating a PCR assay. 34
Species Selection and Animal Models of Disease
As with other therapeutic modalities, the choice of nonclinical animal species plays a significant role in the development of rAAV-GTx. 81 The species of choice is selected on a case-by-case basis as determined by animals in which the test article is biologically active and are expected to be most sensitive to expected toxicological effects. 20,21 Animal models of disease commonly are employed for proof-of-concept studies and may serve, if the test article is pharmacologically active, as a stand-alone system for nonclinical safety testing of rAAV-GTx products. 20,21 There are no regulatory requirements that mandate the use of 2 nonclinical species (eg, rodents and nonrodents) in developing GTx products, 20,21 which is a substantial difference relative to the traditional 2-species approach generally required for nonclinical testing of small molecule drugs as well as nucleic acid-based products and (where relevant) large molecules (eg, protein and antibody-based therapies). The scientific rationale for the use of a particular nonclinical species, including the availability of wild-type (“normal”) versus disease models for evaluating efficacy and safety, and knowledge of rAAV biodistribution across species is often discussed in advance with the regulatory agency when a GTx product is initially being considered for development.
The animal species selected for testing an rAAV-GTx product should be pharmacologically relevant. The target cells and tissues in animals should be transducible by the rAAV-GTx vector, and the typically human-derived transgene product should be active in the animal species. Where feasible, rodents (mice or rats) are used most often due to the number of available genetic backgrounds, including engineered and spontaneous animal disease models, as well as the large catalog of reagents suitable for IHC evaluation of cell type-specific antigens (including those needed to characterize any immune response). However, this expansive set of tools for rodents does not guarantee success in predicting human responses to the test article. For example, initiating anti-rAAV T-cell immune responses in several mouse models does not reproduce the elimination of transduced hepatocytes as observed in clinical trials. 82,83 The same holds true for other nonclinical species, including NHPs. Another key consideration is that the animal species should be immunologically tolerant of the human transgene. Lack of immune tolerance typically necessitates the use of a transgene for an animal species-specific protein (ie, surrogate molecule) or immunosuppression. The prevalence of preexisting neutralizing antibodies to the AAV serotype used in the rAAV-GTx product is an important consideration in the selection of the nonrodent species and even individual animals as it may impact transduction efficiency. 84 That said, the loss of sustained transgene product expression or activity is more likely predicted by antibodies directed against the transgene product itself rather than by the existence of anti-AAV antibodies. 85 Importantly, the existences of anti-AAV and antitransgene antibodies have not been correlated with the extent of the cytotoxic lymphocyte response. 85 Toxicity of AdV GTx vectors is reportedly enhanced in human patients if a prior infection with wild-type AdV induces an immune response prior to administration of AdV-based GTx, 86 but a similar phenomenon has not yet been demonstrated for rAAV-GTx or other viral GTx vectors.
The animal species should be anatomically suitable for vector administration by the ROA intended for use in humans. Key elements to assess in comparing procedural details include factors such as the delivery rate (eg, bolus [“rapid”] injection vs infusion), injection volume, and number of injections (“dose splitting”). In some cases, a larger animal species (eg, dog or NHP) may be necessary if a delivery device (eg, chronically implanted catheter, large-bore hypodermic needle) will be employed clinically. In such cases, a comparison of the delivery device and administration procedures used in nonclinical studies should be made to the actual clinical delivery device and delivery procedure. This comparison should include any design modifications that are needed to accommodate anatomic differences between animals and humans (eg, for CNS indications, relatively smaller IT spaces and CSF volumes in rodents vs larger IT cisterns and CSF volumes in primates and especially in people 87 ). Such size constraints may dictate that nonclinical studies be performed in juvenile or adult animals, even for GTx products destined for pediatric use. Very young rodents at postnatal days 7 to 10 are approximately equivalent in physiologic terms to human neonates and, depending on the administration route, may be administered test article but are too small for many experimental manipulations. 88,89 Immature NHPs (<1 year of age) are larger but the supply of such immature animals is limited, particularly when maternal transfer of antibodies needs to be considered and infants need to be derived from mothers that are seronegative to the vector that will be administered.
It is recognized that biological responses of animal disease models may diverge from those of conventional (ie, wild-type “normal”) animals of the same species. The use of an animal model of disease for safety assessment may show evidence of toxicity that does not manifest in conventional animal species or vice versa. For example, the potential for toxicity induced by the rapid release of long-stored cell breakdown products after treatment of individuals with inborn errors of metabolism recently has been demonstrated in the acid sphingomyelinase deficiency (ASMD) knockout (KO) mouse model after treatment with recombinant human acid sphingomyelinase. 90 Treated ASMD KO mice display toxicity that is characterized by cardiovascular shock and death with liver inflammation, adrenal gland hemorrhage, and increases in serum ceramide and cytokine concentrations at rAAV vector doses 3-fold lower than those that do not elicit toxicity in wild-type mice, rats, and dogs. In contrast, overexpression of a transgene in conventional animals may result in apparent toxicity that would not manifest in species- and strain-matched animals with the disease. This possibility was recently illustrated by the development of severe neurologic signs and neuropathologic changes in cynomolgus monkeys after intracranial infusion of AAVrh8 vectors encoding α- and β-hexosaminidase transgenes. 91 The neurotoxicity was attributed to overexpression of β-N-acetyl hexosaminidase protein in healthy animals that already were expressing normal levels of hexosaminidases. In contrast, evidence of neurotoxicity was not observed in hexosaminidase-deficient mouse, cat, and sheep models of GM2 gangliosidosis administered AAVrh8 vectors encoding species-specific hexosaminidase after direct intracranial injection. These reports indicate that care will be necessary in designing nonclinical programs for rAAV-GTx products to permit evaluation and discrimination of both routine toxicities (ie, effects related to delivery of the rAAV vector and/or transgene) and the potential consequences of exaggerated pharmacology (ie, the release of toxic byproducts following successful transgene activity).
Possible disadvantages of using animal models of disease should also be considered when designing nonclinical studies. 30,92,93 One important consideration is the potential confounding effects of variable transgene transduction/expression and the inherent individual animal variability and divergent progression of the disease phenotype in the model. A second consideration may be the lack of full concordance of the disease phenotype in the animal models and human patients with the disease. A third drawback is the paucity of background pathology data for genetically engineered animals and species not commonly used in conventional toxicity studies, a deficit that may only be addressed through extensive phenotypic characterization with biostatistical support. A final consideration is the technical feasibility, increased costs, and extended timelines associated with generating sufficient diseased animals to perform a given study. These factors may require that safety assessment for an rAAV-GTx product be conducted in wild-type animals in parallel or instead of toxicity testing in an animal disease model and, as discussed previously, the decision on the number and types of studies should be made on a case-by-case basis that is determined by disease indication and product attributes.
Routes of Administration
The ROAs used in nonclinical studies should, whenever possible, reflect the route planned for use in the clinic. Evaluation of the preferred clinical route may not be feasible in all animal species due to size or other anatomical constraints. In such circumstances, utilizing more than one species or animal model will likely be necessary to demonstrate effective transduction of target cells using the chosen ROA. When evaluating the safety of GTx products administered locally, the tissues at the site of administration along with those of regional distribution (eg, lymph nodes via lymphatic drainage from the administration site, axonal transport) should be collected for histopathologic evaluation and biodistribution analysis to look for local expression and responses to the test article.
The target tissues and cell types of interest should be considered when choosing the ROA. For example, studies evaluating the ability of 4 different ROAs (intranasal, intratracheal, intubation, and modified intranasal) to effectively deliver pneumotropic rAAV6 vectors to respiratory tissues show that transgene expression depends on the choice of delivery method. 94 Transgene expression is consistently visible in the nasal cavity, trachea, and all branches of lung airways for all 4 methods, whereas transgene expression is consistently observed in the most distal aspect of lung lobes (alveolar epithelial cells) only after rAAV6 vectors were introduced via the intubation and intratracheal injection techniques. 94 In addition, rAAV vector genome copy numbers in lung tissues are approximately 4-fold lower in mice that received rAAV6 vectors via intranasal administration relative to the other 3 methods of vector delivery. 94 Investigatory studies using reporter constructs with appropriate detection such as IF or IHC techniques to localize vector biodistribution are often used to determine targeted delivery to a particular organ or cell type during lead candidate selection. In addition to anatomical localization, the volume of injection, effect of diluent, and the position of the nonclinical species during dose administration can all play a role in determining whether a GTx product can be delivered successfully using a given ROA to a nonclinical species for safety testing.
The ROA is a major determinant of potential biodistribution and target organ toxicity. For example, systemic rAAV-GTx administration (typically by IV injection or infusion) results in the liver as a major site of transduction by several rAAV serotypes. 95,96 Alternatively, local ROAs (eg, by intraocular, IM, and intraparenchymal brain injections) are intended to yield a high vector uptake at specific sites of disease with more constrained distribution to distant tissues. The primary benefits of localized dosing include a requirement for less vector as well as potentially decreased risk of systemic toxicity and/or immune response to the vector proteins. 97 For example, local administration by either IM injection or isolated limb infusion via a regional blood vessel results in high uptake in skeletal muscle. 98 Direct delivery into the CNS by either an intraparenchymal (eg, directly into a brain nucleus), IT (lumbar puncture into the subarachnoid space or into the cisterna magna), or intracerebroventricular (ICV) route primarily leads to higher transduction of brain and/or spinal cord neurons and the sensory neurons in the dorsal root ganglia (DRG). 99 –101 This increased CNS transduction capability reflects the direct delivery of the rAAV-GTx product via bypassing the existing blood–neural barriers that often limit or prevent neural transduction of IV-delivered agents. In addition, local delivery to more immunologically privileged sites (eg, CNS, eye) may generate a less robust systemic neutralizing antibody response, potentially allowing for repeat rAAV-GTx product dosing with a reduced need for immunosuppressive therapy. 102 –104 However, it is important to note that leakage from local ROA is common and often results in systemic unintended biodistribution of rAAV to remote organs. 97 For example, vector delivered to the CSF may result in significant systemic exposure. Importantly, vector genome numbers in remote organs may be similar to or even exceed those achieved with systemic administration or in the intended local target organs. 105 The ROA may also influence the location of rAAV delivery within an organ as well as the subsequent immune response. For example, when comparing ICV to intracisterna magna (ICM) inoculations of AAV9 vectors expressing GFP in dogs, both ROAs resulted in efficient transduction throughout the brain and spinal cord, but animals dosed by the ICV route developed encephalitis associated with a T-cell response to the transgene product possibly related to parenchymal exposure along the ICV injection tract. 106
Biodistribution
Conventional pharmacokinetic (PK) studies may not be relevant for rAAV-GTx assessment as many transgenes encode for membrane-bound or intracellular proteins, peptides, oligonucleotides, or interfering RNAs rather than circulating products. Instead, biodistribution studies replace many of the traditional PK studies conducted for small and large circulating molecules when evaluating “exposure” to GTx products. 20,107 The biodistribution study can be conducted as a stand-alone study, but biodistribution is often assessed in conjunction with combined efficacy and toxicity studies, where the time points for evaluation of biodistribution may be aligned with time points chosen for efficacy and toxicity evaluation in order to permit correlation of genome copy number and transgene expression levels with any pathology findings. The correlation of histopathologic findings to vector biodistribution is a common role for toxicologic pathologists who participate in nonclinical studies for GTx products.
Measuring the biodistribution of rAAV-GTx products is not as straightforward as measuring the PK profiles of small molecules and biologics. Assessments measure both viral copy numbers using a validated PCR-based assay (qPCR or ddPCR) as well as the extent of transgene expression utilizing RT-PCR for transgene mRNA, and enzyme-linked immunosorbent assay (ELISA) or liquid chromatography–mass spectrometry (LC-MS) for transgene protein. Quantification of vector amounts in circulation varies depending on whether the analyte is free vector in plasma or cell-associated vector in whole blood. In addition, different serotypes of rAAV can vary greatly in time to clearance. 41,108 –110 Once vector is taken up by tissues, it must undergo the processes of capsid uncoating and gene transcription within transduced cells, which, depending on the vector design (eg, ssAAV vs scAAV), can result in an additional delay in transgene expression. 49 For example, IM administration of scAAV versus ssAAV serotype 2 vectors expressing GFP in the cranial tibial (or tibialis anterior) muscle of mice results in strong transgene expression with scAAV at 1 week postdose versus no detectable expression using ssAAV. More than 50-fold higher expression was found in the scAAV group compared with the ssAAV group at 2 weeks and maximum transgene expression at 6 weeks with scAAV but at 6 months with ssAAV. 111 In the same study, delayed transgene expression in the liver was observed with ssAAV compared with scAAV. Species differences in receptor abundance and localization can affect the kinetics of transgene production and can influence species differences in response to the gene promoter. For example, an inverse zonation of hepatocyte transduction can be seen in mice versus NHPs transduced with AAV8-based GTx products, likely due to differences in receptor localization on target cells. 112 Total protein expression and protein localization resulting from vector administration also should be measured, in addition to viral genome copies, when calculating comparable dosages for humans. If the vector-derived protein cannot be distinguished from endogenous protein, then assessing mRNA expression may be an alternative. Such determinations typically are made using quantitative procedures (eg, spectrophotometric measurements) using fresh or frozen homogenized tissue rather than by digital image analysis of tissue sections assessing the intensity of IHC or ISH signals. In some instances, knowing the proportion of the target cell population and uniformity of distribution of the vector in the target cell population can be important for assessing the potential efficacy. In these cases, image analysis to assess the distribution of an IHC or ISH signal in tissue sections can be used (Figure 3). For example, an essential efficacy endpoint in SMA disease models is to demonstrate the percentage transduction of motor neurons in the spinal cord gray matter (ie, the target cell population for Zolgensma). Such cell type-specific assessment typically is conducted either on frozen or formalin-fixed, paraffin-embedded tissue sections by conventional IHC or ISH or alternatively on cells isolated by laser capture microdissection and subsequently homogenized for PCR-based assessment to demonstrate the presence and degree of transgene expression. These assays may also serve to identify and characterize the potential toxic findings in the target cells. Pathologists are essential participants in such biodistribution assessments.
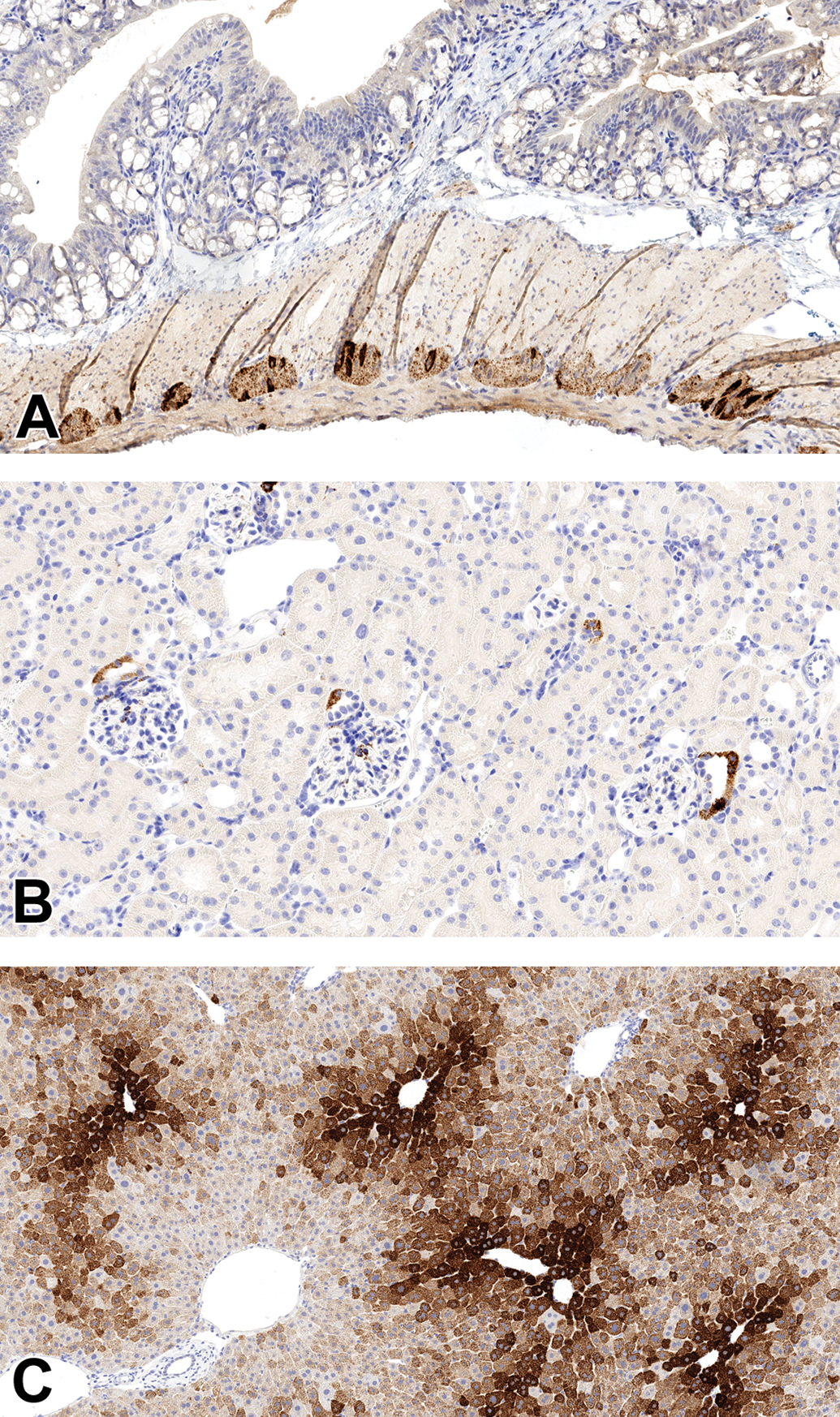
Morphology-based assessment of biodistribution in a mouse following systemic delivery of a recombinant adeno-associated virus (rAAV) vector. Immunohistochemistry (IHC) demonstrating transgene expression exclusively present within (A) neurons of the myenteric plexus in the colon (20× objective); (B) distal convoluted tubular epithelium of kidney, with sparing of cells in the macula densa of the juxtaglomerular apparatus (20× objective); and (C) hepatocytes, predominantly in the centrilobular regions with sparing of cells within portal triads (20× objective).
The FDA suggests a minimum of nine tissues for evaluating biodistribution for nonclinical studies of GTx products. 34 The recommended tissues include injection site(s), blood, brain, gonads, heart, kidney, liver, lung, and spleen. This minimum number may need to be supplemented if it does not include the specific target organs of interest (ie, where the transgene product is expected to exert its therapeutic effect). It may also need to be adapted to survey tissues related to the ROA by including organs such as draining lymph nodes (eg, cervical lymph nodes for intra-CNS injections) and tissues near the delivery site (eg, meninges and spinal nerve roots for IT injections, subcutis for subcutaneous injections, and vessel walls and perivascular tissues for IV injections). Although biodistribution does not necessarily predict safety, understanding the biodistribution of an rAAV-GTx product can guide the tissue selection in toxicity studies. For example, DRGs have not been collected historically in routine GLP toxicity studies, but recent experience indicates that DRG may be an important organ to evaluate for toxicity after CSF (ICM, ICV, or IT 99 ) or IV 100,113 administration of rAAV vectors.
At necropsy, special consideration needs to be given to tissue collection procedures for biodistribution studies in order to prevent cross-contamination among samples. Pathologists and necropsy personnel will share responsibility in ensuring this procedure is performed correctly. Animals should be exsanguinated to remove the blood and any residual circulating viral vector to allow for more accurate counts of successfully transduced vectors in organs. Procedures that minimize and ideally eliminate cross-contamination might include the use of a separate set of disposable sterile instruments for collection of each organ, or the use of reusable instruments that have been thoroughly cleaned, followed by inactivation of DNases and RNases and autoclaving prior to their use. Gloves should be changed between each animal or in the event of contamination. The organ collection order during the necropsy should be specified such that organs with low expected levels of vector are collected prior to organs with high-expected levels of vector.
Nonclinical Safety Endpoints for rAAV-GTx Studies
Ideally, animal toxicity studies should be devised to assess relevant endpoints of the planned clinical trial. The nonclinical studies are designed to identify, characterize, and quantify potential local and systemic toxicities associated with exposure to vector nucleic acid (vector DNA and transgene mRNA) and transgene protein. Toxicologic pathologists will be instrumental in this endeavor and often will generate and interpret critical data needed to define key target tissues and set the threshold values (eg, no observed adverse effect level [NOAEL]) used in risk assessment.
As with other therapeutic modalities, histopathologic evaluation coupled with clinical pathology analysis often is the “gold standard” for safety assessment in nonclinical studies for toxicity of rAAV-GTx products. 99,100,114 Key features to evaluate include the onset of dose-related toxic events (ie, acute vs delayed), the effect of dose level on the incidence and severity of these findings, the feasibility of the proposed GTx delivery system and procedure, and the nature of the immune responses (both humoral- and cell-mediated immune reactions to capsid proteins and the transgene product). Terminal time points generally are designed to capture the time of peak transgene expression (eg, 2-6 weeks postinjection) and a later time point (eg, 13 weeks postinjection or later) to measure vector persistence and monitor any long-term effects of transgene expression or immune response to the vector or transgene proteins. Potential risk mitigation strategies, such as the use of one or more immunosuppressive agents to control anti-rAAV immune responses that may limit rAAV-GTx efficacy or promote immunotoxicity, or mechanistic studies to distinguish various aspects of anti-rAAV immunogenicity, may be prospectively included in the nonclinical safety study design or set aside for evaluation in additional studies. 115
The endpoints and timing of the safety evaluations will be determined on a case-by-case basis that is determined by the factors such as the disease indication and product attributes and will depend upon prior characterization of the vector presence, persistence, and clearance from target cells and tissues. Safety evaluation of rAAV-GTx products generally occurs in conjunction with biodistribution studies to permit correlations between vector presence in a tissue and evidence of toxicity. As with other biopharmaceutical modalities, in-life endpoints typically include clinical signs, physical examinations, intermittent body weight measurements, and assessment of food consumption. Safety pharmacology endpoints may also be considered (depending on the target gene function, ROA, and known biodistribution) and may require a stand-alone study or additional animal species. Endpoints examined in specimens at or after necropsy typically include routine clinical pathology analytes (Table 2) and other biomarkers, organ weights, and macroscopic (gross) and microscopic pathology observations. Collection of a comprehensive battery of tissues is recommended (Table 3). Microscopic evaluation should include the standard list of tissues plus any other tissues that are known targets of the rAAV test article; since systemic function is integrated, transgene expression in a target organ should lead to evaluation of any downstream sites under its control (eg, pituitary gland and endocrine organs, respectively). Additionally, expanded tissue collections and specialized trimming techniques may be required depending on a priori knowledge of potential toxicities in organs infrequently collected or preferential vector tropism/biodistribution in such tissues. For example, recent reports have highlighted the potential of toxicity to occur in DRG, a tissue not typically examined in routine general toxicity studies. 99 –101 In the authors’ experience, 4- and 13-week-long, GLP-compliant, IND-enabling nonclinical toxicity studies may be sufficient to initiate clinical trials, although other time points may be added as needed at the sponsoring institution’s discretion. Retention of some animals for 26 weeks or longer after dosing may be considered, or requested by regulatory authorities, in some cases to assess transgene persistence and potential progression or attenuation of efficacy and toxicity over time. As with other therapeutic modalities, clinical pathology analysis and safety pharmacology assessment can be used at multiple time points in nonrodent studies to detect and monitor potential biomarkers of toxicity earlier during the in-life phase. Regional health authority variations are encountered with advanced therapeutic modalities where harmonized global regulatory guidance is not yet established. Sponsors should discuss the design and duration of the nonclinical program with regulatory health authorities as the nature of the rAAV-GTx product, disease indication, patient age, unmet medical need, and overall risk:benefit considerations will influence nonclinical study design, including study duration and dose levels.
Recommended Clinical Pathology Sampling in Developing rAAV Test Articles for In Vivo Gene Therapy.
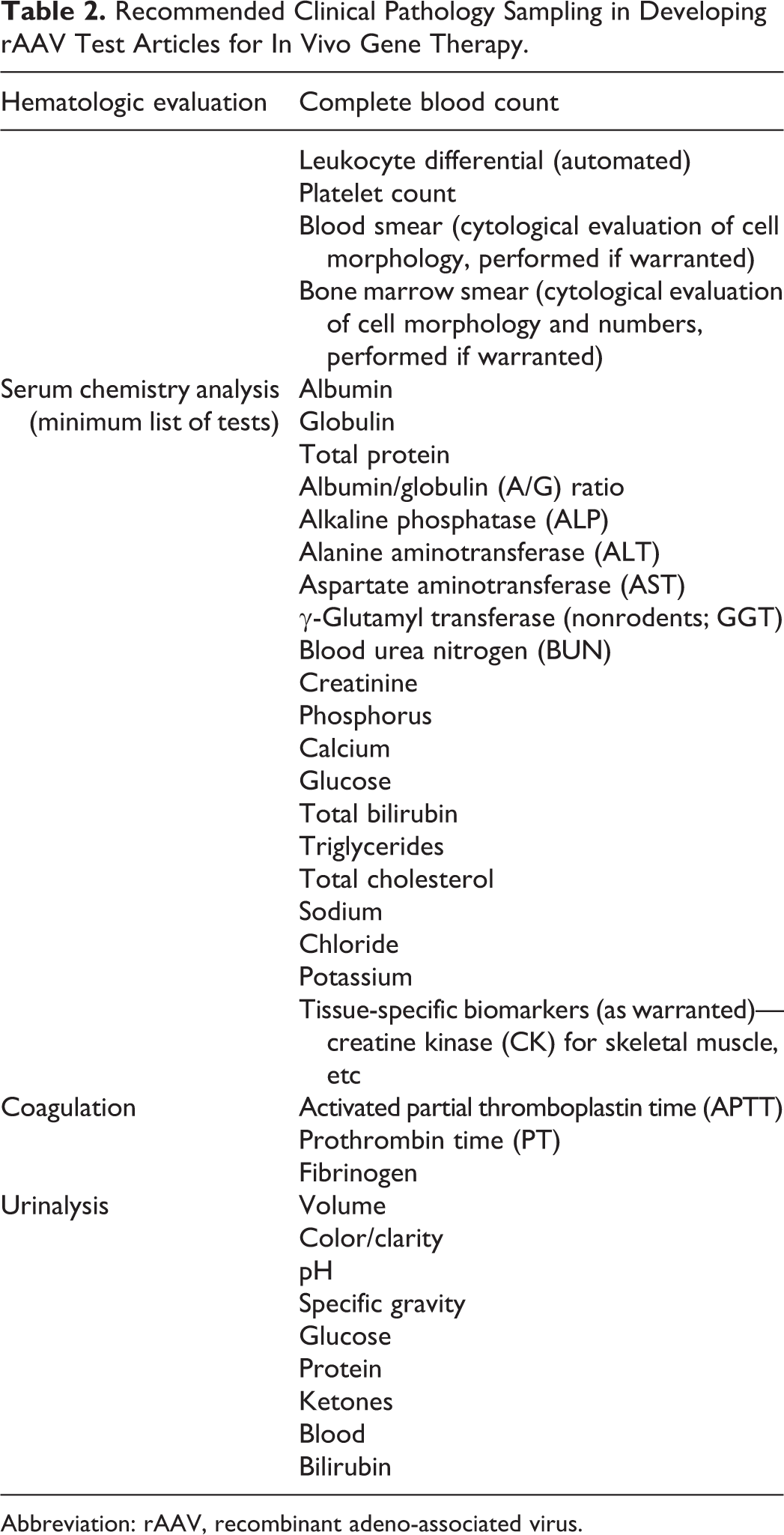
Abbreviation: rAAV, recombinant adeno-associated virus.
Recommended Tissue Sampling in Developing Test Articles for rAAV-Based In Vivo Gene Therapy.

Abbreviations: FDA, U.S. Food and Drug Administration; GLP, Good Laboratory Practice; rAAV, recombinant adeno-associated virus; STP, Society of Toxicologic Pathology.
a Minimal list of tissues (italicized) that is recommended by the FDA for evaluating biodistribution (along with blood) of gene therapy test articles. 34
b Minimal list of organs to be weighed.
c Dorsal root ganglia (DRG) should be collected per STP recommended best practice for peripheral nervous system sampling 116 in all nonclinical studies for rAAV test articles—although histopathologic evaluation may be performed at the sponsor discretion—since this organ is a sensitive target for the rAAV platform. Sacral DRG may be considered for collection as well, especially if the test article is administered by intrathecal (IT) injection into the lumbar cisterna.
Nonimmune Manifestations of rAAV-GTx Target Organ Toxicity
Target organ toxicity can be observed in any organ system, but certain target organs have become more commonly associated with AAV gene therapies. It is important, however, to first consider whether any particular finding is likely or plausibly related to the transgene biology and therefore may be considered pharmacologically mediated. Such toxicities may be interpreted as exaggerated pharmacology in nonclinical studies, and their safety implications should be considered in the context of the intended clinical use, as one would approach any other therapeutic modality. For example, exaggerated pharmacology in genetically replete animals with a gene replacement therapy intended to treat a genetic deficiency disorder should be interpreted in this specific context. On the other hand, a pattern of AAV-associated toxicities is emerging independent of the transgene products, potentially warranting a common rAAV platform/class effect such as those observed in liver and DRGs as well as immune-mediated findings (discussed in detail below). Findings may be observed at these sites following administration of the optimal efficacious dose. As noted above, the ROA may be a key determinant of target organ toxicity. Administration of rAAV-GTx by both the systemic (IV) route and direct local delivery to a specific CNS compartment (IT) can lead to transduction of hepatocytes and DRG neurons, with subsequent degeneration of hepatocytes, neurodegeneration, and axonal degeneration in the peripheral nerves, spinal nerve roots, and dorsal white matter tracts of the spinal cord. 99,100 Consequently, local rAAV delivery does not by default preclude occurrence of toxicities in distant tissues.
The liver is frequently highly transduced by rAAV-GTx products. High-dose exposures to rAAV-GTx test articles or high transgene expression driven by a strong promoter at moderate doses may induce hepatocellular degeneration and variable degrees of single-cell or multi-cell and focal or multifocal necrosis (Figure 4). These effects are mediated by vector DNA overload per hepatocyte, toxic transgene overexpression, or immune responses against rAAV components. 26,100,117 The extent of liver injury typically is transient and asymptomatic and is usually evident within days or weeks after rAAV-GTx administration as elevated serum levels of hepatocyte leakage enzymes (eg, alanine aminotransferase [ALT] and aspartate aminotransferase [AST]). 100,118 –120 That said, liver toxicity induced by high-dose rAAV-GTx administration occasionally has been linked to clinical illness in animals and human patients and can be sufficiently severe to cause death. 121,122
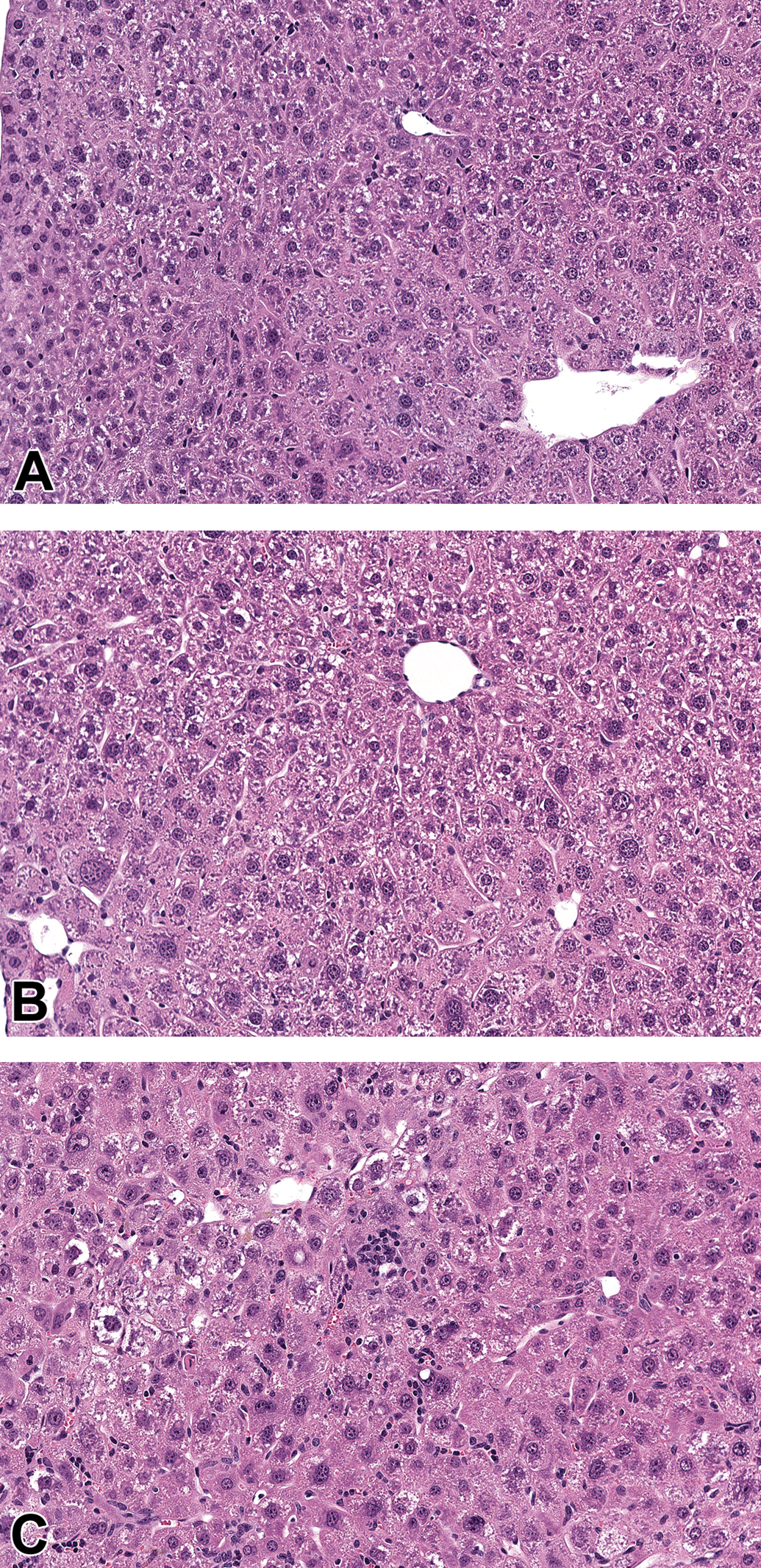
Recombinant adeno-associated virus (rAAV) hepatotoxicity in a mouse as shown by dose-dependent increase in cytokaryomegaly, with the additional findings of single-cell hepatocellular necrosis, mixed cell infiltrate, and increased mitoses in the high dose group. A, Saline control (20× objective). B, Low dose vector (20× objective). C, High dose vector (20× objective).
The DRGs may be highly transduced by rAAV-GTx products. Dorsal root ganglia lie outside the blood–brain barrier but appear to have direct communication with the CSF compartment and are preferentially exposed following CSF versus systemic delivery, but rAAV transduction is extensive following either central delivery or systemic administration. 104,113 Findings in DRGs (Figure 5) may include mononuclear cell infiltration (ie, accumulation of leukocytes without damage to DRG sensory neurons) or mononuclear cell inflammation (ie, aggregation of leukocytes with damage to the DRG parenchyma); neuronal degeneration and necrosis; and/or satellite glial cell proliferation. 113,123 Neuron numbers may or may not be decreased once the leukocyte influx recedes. In one review, the peak extent of the DRG pathology findings in NHP was reported to occur between 21 and 169 days postdose followed by reduced severity or progression by ≥180 days. 113 The occurrence of DRG toxicity at a later time point has been reported once in mice. Late-onset DRG neurodegeneration in SMA KO and wild-type mice occurred at 90 and 300 days postinjection of an rAAV GTx vector; this finding has been postulated to result from chronic transgene overexpression and progressive accumulation of the survival motor neuron protein at supraphysiological levels, related to transgene expression. 124 In a recent report, incorporation of target sequences for miRNA 183 in AAV DNA constructs resulted in reduced transgene expression in DRGs as well as reduced DRG toxicity in NHP. 125 In contrast, concurrent administration of steroids with vectors lacking the microRNA target sequences did not mitigate DRG toxicity. These reports suggest that DRG toxicity is primarily due to transgene overexpression, but an immune response against the vector and/or transgene product may also play a role. In-life neurological signs linked to rAAV-induced DRG pathology have generally not been observed in nonclinical or clinical studies (although in rare cases, ataxia and/or tremor has been reported following high-dose exposure in animals 99,100,113 and in one small clinical study in amyotrophic lateral sclerosis 126 ).
Inflammatory response in lumbar dorsal root ganglia (DRG) from a cynomolgus monkey is a class-specific finding following delivery of recombinant adeno-associated virus (rAAV)-based test articles. The lesion is characterized by multifocal neuronal degeneration/necrosis (shrunken, hypereosinophilic, and fragmented cells); infiltration by macrophages, lymphocytes, and fewer plasma cells that are sometimes removing necrotic neurons (ie, neuronophagia); and proliferation of satellite glial cells (multifocal nodules surrounding or replacing damaged neurons). Treatment: single injection of rAAV-mCherry at a dose of 1.5 × 1014 genome copies via the intracisternal (intracisterna magna [ICM]) route followed by a 14-day observation period. A, Dorsal root ganglion (10× objective). B, Dorsal root ganglion (20× objective).
Transgene-related toxicities with GTx may be associated with overexpression of the transgene or result from either reduction in the expression of an endogenous gene or rapid release of a metabolic product in response to correction of an inborn error in metabolism. Such toxicities are not unique to the use of GTx as a means of correcting the results of a genetic defect and might also be observed after repeated administration of a therapeutic protein.
Immune Responses to rAAV-Based GTx Products
Despite the relatively low immunogenicity of AAV and recent advancements with rAAV-GTx products, 25,127 the immune response to treatment remains a major limitation for both persistence of transgene expression following a single dose and the capacity for vector readministration. 44,120 In vivo rAAV-GTx can result in immune responses against capsid proteins, the vector DNA molecule, and/or transgene product (mRNA or protein), any of which may represent a source of potential immunotoxicity. Since all these components will be recognized as “non-self” by animal species, a variable but sometimes robust innate or adaptive immune response to the vector or human-derived transgene product is an expected occurrence in nonclinical studies.
Many reported toxicities after administration of rAAV-GTx products include stimulation of the innate and adaptive immune responses. 99,128 Affected target organs may exhibit acute inflammation or infiltration by mononuclear leukocytes, including numerous CD3+ T-cells with fewer macrophages and sometimes plasma cells. Inflammation may lead to degeneration or necrosis of transduced or bystander cells. Sentinel immune cells in target organs also may respond, leading to increased cell size (ie, hypertrophy, indicative of cell activation) and/or number (ie, reactive hyperplasia). Other common manifestations of tissue-specific immune reactions include more prominent sinusoidal macrophages (Kupffer cells) in liver, microgliosis and to a lesser extent astrogliosis in the brain and spinal cord, and increased satellite glial cellularity in DRG. 99,101 Pathologists are instrumental in assessing tissue responses indicative of these innate and acquired immune reactions to rAAV-GTx products.
The importance of potential anti-GTx immune responses cannot be overemphasized. For instance, liver toxicity (as shown by elevated serum activities of ALT and AST) has been reported in several clinical trials with concomitant increases in interferon-γ-positive, T-cell-mediated responses and decreased transgene expression. 26 This hepatotoxicity was reversible by glucocorticoid (prednisolone) administration, and the reversal coupled with rescued transgene expression implicates immune responses as potential, although inconsistently expressed, factors in the pathogenesis of rAAV-mediated liver toxicity in humans. 26
Innate Immune Responses to rAAV-GTx Products
The nonspecific capability of the innate immune system to attack microbial pathogens through the recognition of pathogen-associated molecular patterns (PAMPs), including rAAV elements, constitutes the first line of host defence. 129 Components of rAAV are recognized by 2 of the toll-like receptor (TLR) family members that are instrumental in mounting an antiviral innate immune response. The cell surface receptor TLR2 recognizes vector capsid proteins, while upon internalization into endosomes the vector DNA genome is recognized by TLR9. 130 In mice, activation of TLR9 in plasmacytoid dendritic cells allows the adapter molecule MyD88 to initiate an activation cascade that launches an interferon type I immune response that in turn drives CD8+ cytotoxic T-cell responses as well as the B-cell-mediated generation of neutralizing antibodies to both the transgene product and the AAV capsid proteins. 131 Elements of vector design, such as high CpG (cytosine and guanine linked by a phosphate group) content 132 and scAAV genomes, 133 may further enhance the TLR9-mediated innate immune response. Capsid recognition by TLR2 on the membranes of Kupffer cells and sinusoidal endothelial cells upregulates the production of inflammatory cytokines through activation of nuclear factor κB, as has been shown in primary human cell cultures. 134 These molecular mechanisms help drive the initial innate immune response that is mounted against a newly introduced GTx product.
In addition to TLR2 and TLR9, emerging data indicate that rAAV transduction may trigger the innate immune response via production of additional PAMPs. 135 For example, transgene expression at later time points following rAAV transduction activates the cytosolic double-stranded RNA sensor MDA5 in human hepatocytes grown in vitro and also in vivo in human hepatocytes engrafted into a chimeric mouse model. 136 In vivo, AAV capsid proteins can interact with components of the complement system and enhance vector uptake by macrophages without measurably impacting transgene expression in the target cell populations. 137 Recent data demonstrate the presence of AAV capsid-specific natural killer cells in AAV-seronegative individuals, and these have been posited to play a role in anti-AAV immune responses. 138 Although innate immune responses to vector nucleic acid and capsid proteins may be asymptomatic, signals generated by the innate immune system likely play a major role in shaping and potentiating the subsequent adaptive immune responses to vector capsid and transgene proteins. To that end, mice deficient in complement (C) receptor 1/2 and component C3 exhibit delayed humoral immunity against AAV2 vectors, thereby resulting in significantly lower neutralizing antibody titers compared with those of wild-type mice. 137 Similarly, 2 recent clinical trials by different sponsors for the treatment of Duchenne muscular dystrophy using a rAAV-based IV GTx product reported complement activation associated with reduced platelet and red blood cell counts as well as transient renal impairment. 139,140 Despite the successful removal of the clinical hold following inclusion of a modified steroid regimen and a limited course of eculizumab, a humanized monoclonal antibody inhibitor of complement activation, the trial was placed on a second clinical hold after another patient was reported with a similar serious adverse event secondary to complement activation. 141
Adaptive Immune Responses to rAAV-GTx Products
The onset of the adaptive (acquired) immune response against viruses and GTx viral vectors is delayed compared with the innate immune response. This later-onset reaction is mounted specifically against antigenic epitopes on the viral capsid proteins and/or transgene-derived protein and is undertaken in parallel by the humoral (antibody-based, B-cell-driven) and cell-mediated (T-cell-driven) branches of the adaptive immune system. Importantly, the initiation of the acquired reaction promotes the production of long-lived memory B-cells and memory T-cells that may limit the long-term efficacy of the transgene product and potentially pose a safety risk if subsequent therapeutic attempts are made using the same GTx product (eg, one constructed with an identical rAAV serotype).
Humoral immune response against rAAV
Wild-type AAVs are widespread, naturally occurring, nonpathogenic viruses that exist across all mammalian species. Prior exposure results in the presence of measurable preexisting antibodies against many AAV serotypes. For example, the prevalence of total anti-AAV2 antibodies is close to 70% in certain human populations 142 and nearly 100% in rhesus macaques 143 ; the prevalence of anti-AAV antibodies in humans for other serotypes ranges from approximately 40% to 45% for AAV6, AAV8, and AAV9, and up to nearly 70% for AAV1. 142 The incidence of anti-AAV neutralizing antibodies in humans progressively increases with age. 144 Neutralizing antibodies may prevent gene transfer using a particular AAV serotype 145 and occasionally may be able to cross-react and effectively neutralize other AAV serotypes, thus necessitating screening of NHP test subjects prior to their use in nonclinical safety or efficacy assessments and potentially limiting the therapeutic utility of that serotype to treat patients previously exposed to wtAAV infections. 84,146 Administration of rAAV triggers an anti-AAV humoral immune response in nonclinical test species and humans. 147,148 Neutralizing antibody titers as low as 1:5 have been shown to block AAV transduction completely in mice 149,150 and NHPs. 78 Expressed human-derived transgene protein may similarly induce a humoral immune response that also can neutralize the pharmacology of the transgene. 25 Accordingly, understanding the anti-capsid and antitransgene antibody responses is an essential element in conducting nonclinical efficacy and toxicity studies for rAAV-GTx.
Cell-mediated immune response against rAAV
Although humoral immunity against rAAV components captured the initial interest in rAAV-GTx, cell-mediated immunity has gained attention as an essential area of investigation. Cell-mediated immunity has gained importance following the observation that healthy individuals carry AAV capsid-reactive CD8+ cytotoxic T-cells that may expand upon administration of rAAV-GTx products. 151 Cytotoxic responses mediated by CD8+ T-cells and directed against rAAV2 capsid in patients with hemophilia B have been shown to destroy transduced hepatocytes and result in a gradual decline in expression of the transgene (Factor IX), thereby eliminating the benefits of treatment. 152 Capsid-specific memory T-cells are likely generated during childhood secondary to infections with wtAAVs, and these memory cells then persist in secondary lymphoid organs, such as the spleen, throughout the life of the individual. 151,153 Cell-mediated immune responses can similarly target the transgene-derived protein. Dystrophin-specific T-cells have been detected in patients who received an rAAV carrying a functional dystrophin transgene, which ultimately resulted in failure to establish sustained transgene expression. 154 Similarly, transgene-specific T-cells have been shown to reduce transgene expression in an α-1-antitrypsin (ATT)-deficient subject receiving rAAV1-AAT treatment. 155 Immune tolerance associated with induction of regulatory T-cells following IM gene transfer of AAT 156 or hepatic gene transfer of Factor IX 157 has been demonstrated to reduce the extent of inflammatory cell infiltrates directed against the rAAV capsid or transgene-derived proteins, which thus allows transgene expression to be prolonged (reviewed in the study of Biswas et al 158 ).
Nonclinical Assessment of Immune Responses Against rAAV-GTx Products
Assessment of humoral and cell-mediated responses to capsid and/or transgene may be useful for interpretation of findings in nonclinical studies. 26 However, it is difficult to predict whether immune responses mounted against rAAV capsid and/or transgene proteins interfere with the assessment of adverse pharmacology or result in adverse immune-mediated findings after vector administration. Therefore, a risk assessment strategy for evaluating nonclinical immune responses to rAAV-GTx products must be established on a case-by-case basis that is determined by disease indication and product attributes. Appropriate samples should be collected and retained to evaluate these immune responses.
This strategy typically employs the regulatory guidance principle that the study design should “obtain appropriate samples during the course of the study, which can subsequently be analyzed when warranted to aid in interpretation of the study results.” 159 In the event that immune response-related findings are observed in toxicity studies, previously collected and retained samples will aid in the interrogation of a potential immune response impacting pharmacology parameters (eg, peak expression level and persistence of transgene expression) or toxicity findings. In the authors’ experience, appropriate sampling and analysis for nonclinical efficacy and safety studies for GTx products should include retention and archiving of frozen and fixed tissues (including blood and serum) from a comprehensive list (Table 3). Pathologists are instrumental in understanding potential anti-rAAV-GTx immune responses via their roles in the analysis and interpretation of routine pathology endpoints (eg, clinical chemistry, hematology, microscopic tissue examination) and, in some cases, special immunopathology endpoints (eg, flow cytometric data, IHC, and ISH preparations).
To determine whether a nonclinical toxicity study has adequately evaluated the potential for adverse transgene pharmacology, the antibody response to the transgene-derived protein as well as the impact of such antibodies on pharmacologic activity of the transgene should be evaluated. Therefore, serum samples typically should be retained in nonclinical efficacy and toxicity studies so that the presence of antitransgene antibodies may be measured if nonclinical findings suggest the presence of an antigen–antibody complex mechanism or progressive loss of pharmacologic activity.
Cell-mediated immune responses to the rAAV capsid and transgene-derived proteins can occur in nonclinical studies (and clinical trials). Therefore, consideration should be given to retaining frozen lymphocytes, especially in nonrodents, so that the development of capsid- or transgene-specific, cell-mediated immune responses may be evaluated later if warranted. For example, PBMCs should be collected at a minimum of 2 time points: prior to vector administration (ie, baseline) and at necropsy. Paired PBMC samples allow each animal to serve as its own control. Additionally, tissue lymphocytes harvested at necropsy from freshly collected, unfixed specimens of bone marrow, lymph nodes (typically mesenteric and/or near the rAAV administration site), and spleen may aid in the identification of a cell-mediated immune response as antigen-specific responses may not always be identified in PBMCs. 26
Additional testing may be utilized, where feasible and warranted, to assess the potential for adverse innate immune responses that may occur shortly after rAAV-GTx administration. For this purpose, serum may be collected to measure such biomarkers of acute-phase immune responses as complement, proinflammatory cytokines, and routine clinical chemistry endpoints of acute hepatocyte damage (eg, ALT and AST activities), while whole blood may be obtained to evaluate hematologic parameters (especially counts of neutrophils, monocytes, and other cells of the innate immune response). The endpoints should be assessed prior to (baseline) and within hours to a few days (typically 2-7) following vector administration. The assessment of the innate immune responses to rAAV may have greater importance for indications where high doses of rAAV are required for efficacy. 100,139 In general, such additional testing is employed only to explain unexpected outcomes of prior studies rather than as a prospective part of all nonclinical studies for rAAV-GTx products. A reasonable compromise is to routinely collect and retain additional serum samples in case such mechanistic studies become desirable after the initial microscopic screening of tissues has been completed. These approaches are more commonly used in nonrodent species due to blood volume and tissue sample size limitations although inclusion of additional animals dedicated to such ancillary molecular testing may be useful in rodent studies.
Although immune responses against rAAV may occur in nonclinical species, the predictive value and translatability of immune-mediated findings in nonclinical studies should be critically evaluated as part of the risk assessment strategy. For example, the first T-cell-mediated immune response to rAAV capsid with parenchymal damage and loss of transgene expression observed in patients during a hemophilia B clinical trial was not observed in nonclinical safety studies in rodents or NHPs. 160 Similarly, a dog model of hemophilia B demonstrated sustained expression of the transgene for more than 8 years without evidence of an immune response. 161 Moreover, several mouse models induced to mount an anti-AAV T-cell immune response failed to eliminate transduced hepatocytes. 82,83 The interactions among rAAV components (capsid proteins and DNA molecules), transgene (mRNA and protein), and host genetics (eg, human leukocyte antigen [HLA] haplotype) are complex. Therefore, the implications of immune-mediated findings in nonclinical studies on safety assessment and translatability of animal-derived data to predicting human responses to rAAV remain a major area of investigation in developing gene therapies. 162
Minimizing Nonclinical Immune Responses Against rAAV-GTx Products
In the risk assessment strategy for developing rAAV-GTx products, several factors should be considered as a means of mitigating potential innate and/or adaptive anti-rAAV immune responses. Key parameters include basic biological attributes (both similarities and differences) of animals and humans, procedural elements of vector design and administration, the nature of the anti-rAAV immune response, and options for modifying the extent of this immune response. These factors may be viewed as individual components when designing the development program, but in fact they act in combination to influence the type and degree of anti-rAAV immunity as well as extent and persistence of transgene expression.
Biological characteristics represent the first key element to consider in devising a nonclinical strategy for mitigating immune response to rAAV-GTx. In principle, these considerations are identical to those encountered in developing any biologic-based therapeutic. For example, the degree of sequence and conformational similarity between the human transgene-derived protein and the corresponding protein for the nonclinical animal species can impact the incidence and magnitude of the immune response. Assessment of transgene product similarity between the human and nonclinical animal species is necessary in instances where immune responses against the transgene product are specific to the nonclinical animal species and thus prevent the evaluation of potential adverse pharmacology. In general, the amino acid sequence and conformation of proteins in NHPs are most closely conserved relative to human proteins; therefore, nonclinical efficacy and safety testing of GTx test articles often proceed in NHPs only, especially when the program cannot use a different nonrodent species (eg, a mutant dog model of disease does not exist). If warranted, a surrogate transgene that will express the animal species-specific orthologous protein can be used to preclude animal-specific immune responses to the human protein, thereby permitting protein function to be assessed in a setting where long-term transgene expression permits evaluation of the potential risk posed by exaggerated pharmacology. Alternatively, immunosuppression with one or several pharmacologic agents has been used in nonclinical studies. 163 The goal of immunosuppression is to transiently suppress the immune system in the particular host to allow the assessment of a pharmacological effect produced by an rAAV-GTx in the absence of a robust immune response. The nonclinical immunosuppression regimen is generally not being qualified for use in the clinical setting, so immunosuppressive regimens may be tailored to the nonclinical animal species as the specific experimental design but that do not have to directly transfer to the clinic. In some instances, immunomodulatory regimens are modeled in nonclinical studies to support the clinical trial design.
Second, prequalification of experimental animals may be used to mitigate anti-rAAV immune responses in nonclinical studies. The prevalence and titer of any preexisting anti-AAV immune response will impact enrollment of animals for nonclinical studies. Characterizing the antibody status (particularly the anti-rAAV neutralizing antibody titer) of nonrodent animal species is generally expected for selecting subjects that are seronegative for neutralizing antibodies. This step is crucial since seropositive individuals will potentially be resistant to full transduction and sustained transgene product levels necessary for nonclinical assessments of efficacy and safety. Prevalence of antibodies differs among animal species (for rodents and nonrodents) and among colonies from different animal suppliers (study by Wang et al 143 and authors’ unpublished data). Consequently, understanding the prevalence of antibodies in a particular animal species and for a specific animal supplier will suggest the number of subjects that should be prescreened for the study; neutralizing antibodies to the vector capsid greater than 1:5 have been shown to abrogate transduction when a vector is administered intravenously. 78,149,150 Finally, other aspects of the anti-AAV humoral immune response may have additional impact on the risk assessment strategy. For instance, animals and people may also have binding antibodies that do not neutralize cellular transduction by the vector. Indeed, binding antibodies recently have been shown to enhance vector uptake and transgene expression in hepatocytes. 164 The role of such binding (but non-neutralizing) antibodies in altering the biology or toxicity of the vector is poorly understood. Therefore, evaluation of binding antibodies and neutralizing antibodies as separate entities may be beneficial when developing rAAV-GTx products.
Finally, procedural factors represent another major element to consider in devising a nonclinical strategy for mitigating immune responses to rAAV-GTx. The design of the vector genome can impact the immunogenicity of rAAV-GTx. For instance, high CpG content 132 and self-complementary rAAV genomes 133 may enhance the TLR9-mediated innate immune response. The ROA can also influence the nature of the immune response. Preexisting neutralizing antibodies in blood can reduce rAAV transduction efficiency following IV and intra-articular injections where antibodies are readily available in the fluid phase. 145 On the other hand, subretinal, IM, IT, and intraparenchymal brain injections have barriers that limit vector exposure to circulating neutralizing antibodies and thus typically improve transduction following GTx (reviewed in detail in the study of Masat et al 145 ). However, studies cannot assume that rAAV administration to such barrier-protected sites will mitigate the impact of neutralizing antibodies on transduction efficiency. Indeed, neutralizing antibody titers in blood are strongly correlated to weak or absent transgene expression in primate retina, 84 and neutralizing antibody titers in CSF as low as 1:1 to 1:3 may impact rAAV transduction efficiency in the CNS of dogs. 165
Considerations Related to rAAV Chemistry, Manufacturing, and Control
Chemistry, manufacturing, and control (CMC) criteria of active pharmaceutical ingredients for any therapeutic modality constitute a continuum that seeks to achieve consistent product characteristics across the entire product life cycle (nonclinical studies as well as clinical trials through product registration and commercialization). There are several manufacturing platforms for producing rAAV-GTx products, most commonly DNA plasmid transfection of mammalian cells or infection of insect cells with recombinant baculoviruses. 166 The overall yield and quality of the rAAV vectors including product-related impurities (such as numbers of defective particles and full-to-empty capsid ratio) as well as process-related impurities (such as residual host cell DNA and proteins) vary with the manufacturing platform that is used.
Although pathologists typically are not intimately involved in CMC decisions, understanding fundamental variations in rAAV characteristics in GTx product formulations across all stages of the program is important in interpreting the results of nonclinical studies. Gene therapy vectors are highly complex products with evolving manufacturing and analytical standards. It is critical that the comparability of the test article used across the nonclinical and clinical programs be understood so that consistency of the doses and product characteristics can be assured. It is recommended that samples of each manufactured batch be retained to reconfirm product characteristics as analytical methods evolve during the program. Furthermore, as discussed above, except for the case of secreted transgene-derived products or the accessibility of tissues by biopsy for the assessment of transgene-derived product, most GTx programs target the expression of intracellular proteins that lack a robust biomarker for measuring efficacy and thus are challenging to assess in clinical trials. These are issues of a particular concern for rAAV-GTx since such products generally are administered once with the promise of delivering long-term biological benefit to the patient. Therefore, measures to assess the quantity of active vector per dose (ie, intact vector capsid containing the DNA construct and expressing the active transgene of interest) and a clear understanding of the dose–response relationship in nonclinical studies are necessary for accurately calculating the desired dose in human patients.
Current methodology to assess characteristics, vector composition, and titer of an rAAV-GTx product frequently relies on the application of qPCR to detect the vector genome. However, the qPCR assay is inherently variable, relative to the concentration methods of traditional biologic therapeutics. Furthermore, a single assay such as qPCR is incapable of distinguishing product-related impurities such as defective vector particles (ie, either empty particles or particles containing defective DNA constructs). The reliance on qPCR to compare administered dose among studies and associated outcomes is further complicated by the lack of reference standard material to calibrate assays among investigators. 167 To address several of these qPCR limitations, the utility of ddPCR has gained traction as a tool for assessing the absolute titer and quality of vector genomes without the need for a standard curve. 168 –170 Furthermore, additional orthogonal assays should be used to better characterize the proportion of defective vector particles and quantities of process-related protein and nucleic acid impurities (reviewed in the studies of Penaud-Budloo et al 166 and Wright 171 ). The numbers of defective viral particles present in the vector preparations may impact transduction efficiency, anticapsid immune response, and vector clearance. 172,173
Process-related impurities can differ among production methods. Residual DNA and proteins derived from mammalian or nonmammalian methods to produce vector (eg, HEK293 cells or baculovirus, respectively) or fetal bovine serum necessary for cell culture media are possible process impurities and potentially could induce an immune response to the foreign material independent of the rAAV serotype and transgene of interest. 174 The importance of this issue is demonstrated by the concern expressed by EMA in the review process of Glybera (alipogene tiparvovec, the first approved rAAV-based GTx in Europe), in which copackaged baculovirus DNA was not measured in the initial lots used during the clinical trials while variable but often large quantities of contaminating baculovirus DNA were present in subsequent lots generated by the commercial process. 175 Other examples of process-related impurities include surfactants, transfection reagents, and residual plasmid DNA. 166,171
Although impurities are expected to be present even in the most purified rAAV-GTx product, acceptable levels of impurities and best practices are not fully established. 171 Therefore, impurities need to be characterized, quantified, and assessed in nonclinical studies at levels equivalent to or higher than those proposed for testing in clinical trials (reviewed in the studies of Penaud-Budloo et al 166 and Wright 171 ). Safety testing in this regard typically involves conventional toxicity study designs including routine macroscopic, microscopic, and clinical pathology endpoints.
Regulatory Considerations
The discovery, development, and marketing approval of rAAV-GTx products for in vivo administration is governed by the same regulatory framework that applies to other biologics, but with additional considerations specific to GTx products owing to the unique challenges and risks inherent with long-term in vivo gene delivery. The nonclinical package required for rAAV-GTx programs is determined on a case-by-case basis, and early interactions with regulatory agencies are encouraged. The FDA’s INTERACT (INitial Targeted Engagement for Regulatory Advice on CBER producTs) program is intended to facilitate such interactions, typically early in the program when initial proof-of-concept data are available. On the other hand, the pre-IND meeting is generally later in the process when the design of pivotal toxicity studies is considered (Figure 6). Comparable procedures for seeking scientific advice exist in the European Union and other regulatory regions, and global health authorities are anxious to interact with GTx developers. A partial list of the FDA and EMA regulatory guidance documents on development of GTx products for in vivo administration is provided in Table 4.
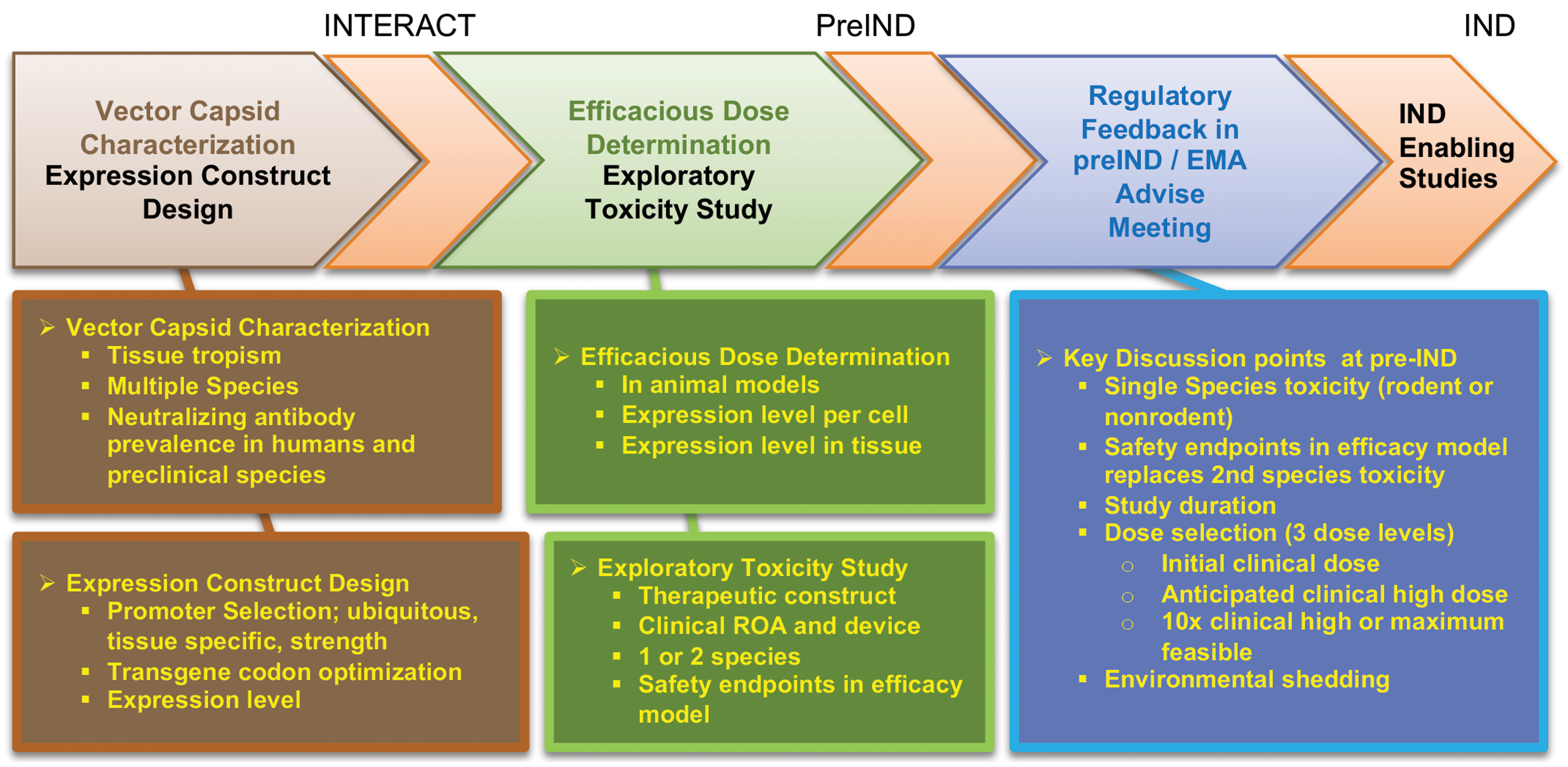
General summary and outline of major points to consider for in vivo gene therapy products during the design of a nonclinical safety package and the interaction with regulatory agencies. (Figure reproduced from Assaf and Whiteley, 114 by permission of Sage.)
Regulatory Guidance Documents on Development of In Vivo Gene Therapy Products.

Abbreviations: EMA, European Medicines Agency; FDA, U.S. Food and Drug Administration.
The unique concerns pertaining to nonclinical and clinical development of rAAV-GTx products are focused on issues related to the animal studies, product quality and consistency as discussed above, and clinical trial design principles in rare genetic diseases. For rAAV-GTx products, studies in animals ideally should be performed using a test system and ROA that mimics the intended clinical use in humans and should provide knowledge of the distribution and persistence of the rAAV vector DNA and transgene expression in target and nontarget tissues. The important topic of nonclinical biodistribution studies for GTx products was recently endorsed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), and an ICH expert working group has been formed to work toward preparing a harmonized guideline. 159 A comprehensive justification of the test species should address the test species permissiveness to viral transduction and responsiveness to the transgene protein product. If an animal model of disease is used in the nonclinical program, the discussion should include information regarding the onset, progression, and severity of disease in the model; the similarities and differences between the animal model and the human disease; and the timing of vector administration in the animal model relative to disease onset, progression in the patient population, and endpoints to be assessed. Additionally, as discussed above, suitable samples (eg, serum, PBMCs, or lymphoid tissues) should be retained so that potential immune responses to the rAAV capsid and transgene proteins in animal studies potentially driving an observed immunotoxicity may be characterized and their relevance (if any) to the target human population assessed. Endpoints for GTx safety assessment in which pathologists will be involved are typical of any other therapeutic modality and generally include clinical observation, necropsy, histopathology, clinical chemistry, hematology, coagulation and urinalysis as needed, and safety pharmacology, with emphasis placed on the endpoints that are most relevant to the GTx product under investigation.
To ensure consistent production of rAAV-GTx products of a standardized potency, a list of the vector lots used in each nonclinical study and a tabulated summary of the similarities and differences among the various lots of vector used in nonclinical studies and clinical trials should be provided. These lists should include detailed information on the assays and standards that were used to determine the concentration of each vector lot. If applicable, a comparison of each species-specific vector lot and the rAAV-GTx product intended for clinical use should be provided. This comparison should verify the ability of the delivery system to consistently supply the prespecified dose level of the product. If vector loss is observed during delivery, the actual vector dose level that was administered should be provided. If the assay differs between nonclinical and clinical lots, adequate material from each nonclinical lot should be retained so that it can be retested using the assay for the planned clinical lots in a side-by-side comparison. If warranted, the vector dose levels administered in the animal studies should be recalculated based on this post hoc reanalysis. The vector concentration and potency assays for the nonclinical lots should be comparable to the assays used to test the clinical lots. The dose extrapolation methodology, including a detailed description with mathematical formulas, sample calculations, and supporting data, should also be provided. Information on the potential for insertional mutagenesis, germline transmission, viral shedding, and an environmental assessment may also be required.
Summary
Advances in molecular biology and our understanding of the genetic basis of many diseases have led to a surge in pharmaceutical research efforts focused on discovering and developing safe and efficacious GTx platforms for treating previously intractable, progressive, and lethal diseases. Viral vectors have emerged as the leading in vivo gene transfer tool in GTx clinical trials due to their high transduction efficiency, and of these rAAV-derived constructs are preferred, for several reasons. Desirable attributes of rAAVs as GTx products include the availability of multiple rAAV serotypes with varying selective organ/cellular tropisms and transgene expression efficiencies, the lack of association of wtAAVs with any known illnesses in humans, the low risk of insertion of the rAAV transgene payload into the recipient genome, the long duration of transgene expression in postmitotic cells, and the general evidence of rAAV safety and tolerability in the clinical setting. Immune responses to the rAAV capsids can be a safety concern and detrimental to the transduction of desired tissues and can interfere with the sustained production of rAAV-GTx transgene products during therapeutic treatment.
Regulatory considerations specific to GTx products, including rAAV-GTx products and other in vivo GTx platforms, have been promulgated as a result of the unique challenges and risks associated with genetic manipulation of the recipient. This document has attempted to highlight key points to consider in the discovery and development of rAAV-GTx products; similar considerations as well as additional guidance documents (eg, safety assessment for tumorigenic potential for integrating lentiviral and retroviral test articles) will apply to other viral (integrating and nonintegrating) and nonviral GTx agents. These points emphasize that the nonclinical packages required for GTx programs are determined on a case-by-case basis determined by the disease indication and product attributes, and as such early interactions with regulatory agencies are encouraged. To be fruitful, such exchanges will require providing the scientific rationale and supporting data to justify the specific approach designed to successfully advance these exciting new modalities into the clinic. Toxicologic pathologists, with their abilities in evaluating and interpreting tissue changes and biomarkers of toxicity as well as identifying transgene expression in target cell populations using molecular pathology methods, will be integral in assessing the efficacy and safety of in vivo GTx products. The escalating popularity of GTx as a therapeutic modality therefore means that toxicologic pathologists must become familiar, even comfortable with parameters that must be considered in designing and performing effective nonclinical studies for developing novel gene therapies.
Supplemental Material
sj-docx-1-tpx-10.1177_01926233211041962 – Supplemental material for Scientific and Regulatory Policy Committee Points to Consider: Nonclinical Research and Development of In Vivo Gene Therapy Products, Emphasizing Adeno-Associated Virus Vectors
Supplemental material, sj-docx-1-tpx-10.1177_01926233211041962 for Scientific and Regulatory Policy Committee Points to Consider: Nonclinical Research and Development of In Vivo Gene Therapy Products, Emphasizing Adeno-Associated Virus Vectors by Julie A. Hutt, Basel T. Assaf, Brad Bolon, Joy Cavagnaro, Elizabeth Galbreath, Branka Grubor, Lisa M. Kattenhorn, Annette Romeike and Laurence O. Whiteley in Toxicologic Pathology
Supplemental Material
sj-docx-2-tpx-10.1177_01926233211041962 – Supplemental material for Scientific and Regulatory Policy Committee Points to Consider: Nonclinical Research and Development of In Vivo Gene Therapy Products, Emphasizing Adeno-Associated Virus Vectors
Supplemental material, sj-docx-2-tpx-10.1177_01926233211041962 for Scientific and Regulatory Policy Committee Points to Consider: Nonclinical Research and Development of In Vivo Gene Therapy Products, Emphasizing Adeno-Associated Virus Vectors by Julie A. Hutt, Basel T. Assaf, Brad Bolon, Joy Cavagnaro, Elizabeth Galbreath, Branka Grubor, Lisa M. Kattenhorn, Annette Romeike and Laurence O. Whiteley in Toxicologic Pathology
Footnotes
Authors’ Note
This “Points to Consider” article is a product of a Society of Toxicologic Pathology (STP) Working Group commissioned by the Scientific and Regulatory Policy Committee (SRPC) of the STP. It has been reviewed and approved by the SRPC and Executive Committee of the STP and endorsed by the Executive Committees of the British Society of Toxicological Pathology (BSTP) and European Society of Toxicologic Pathology (ESTP), but it does not represent a formal best practice recommendation of the Societies; rather, it is intended to provide key “points to consider” in designing studies or interpreting data from toxicity and safety studies intended to support regulatory submissions. The opinions expressed in this document are those of the authors and do not reflect views or policies of the employing institutions. Readers of Toxicologic Pathology are encouraged to send their thoughts on these articles or ideas for new topics to the editor. Drs. Hutt and Assaf contributed equally to this work.
Acknowledgments
The authors thank Ms. Beth Mahler for assistance with optimizing the figures and Dr. Page Bouchard for critical review of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.