
Abstract
Harmonization of diagnostic terminology used during the histopathologic analysis of rodent tissue sections from nonclinical toxicity studies will improve the consistency of data sets produced by laboratories located around the world. The INHAND Project (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice) is a cooperative enterprise of 4 major societies of toxicologic pathology to develop a globally accepted standard vocabulary for proliferative and nonproliferative lesions in rodents. A prior manuscript (Toxicol Pathol 2012;40[4 Suppl]:87S-157S) defined multiple diagnostic terms for toxicant-induced lesions, common spontaneous and age-related changes, and principal confounding artifacts in the rat and mouse central nervous system (CNS) and peripheral nervous system (PNS). The current article defines 9 new diagnostic terms and updates 2 previous terms for findings in the rodent CNS and PNS, the need for which has become evident in the years since the publication of the initial INHAND nomenclature for findings in rodent neural tissues. The nomenclature presented in this document is also available electronically on the Internet at the goRENI website (http://www.goreni.org/).
Keywords
Introduction
The INHAND Project (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice) is a combined venture of the British Society of Toxicological Pathology, European Society of Toxicologic Pathology, Japanese Society of Toxicologic Pathology, and Society of Toxicologic Pathology (based in North America) organized to devise standard nomenclature for proliferative and nonproliferative lesions in rodents that is recognized throughout the world. To date, the INHAND initiative has produced publications providing a set of internationally recognized diagnostic terms for categorizing lesions that occur in many organ systems of rodents (http://www.goreni.org/). The INHAND nomenclature generally is grounded on morphologic features that can be observed in hematoxylin and eosin (H&E)-stained sections, but ancillary diagnostic methods also are mentioned if considered relevant for confirmation. The growing importance of INHAND terminology is highlighted by the decision of the US Food and Drug Administration to utilize the INHAND lexicon as the foundation for Standard for Exchange of Nonclinical Data (SEND) nomenclature that is now required for nonclinical toxicity studies intended to be submitted in new product registration packages.
The original INHAND nomenclature for the rat and mouse central nervous system (CNS) and peripheral nervous system (PNS) defined 55 terms and included 136 figures. 1 The present article is an update to define 9 new terms for findings that were not included in the original manuscript; it also revises 2 previously defined INHAND terms to bring diagnoses for the nervous system findings into harmony with INHAND diagnostic terminology employed for other systems. These definitions acknowledge our improved understanding of cellular origins and neural lesion mechanisms that have been gained since the original INHAND publication.
Nomenclature
As in the initial INHAND paper on nomenclature for rodent neural lesions, this terminology is categorized based on lesion class (nonproliferative or proliferative), cell population (eg, neuron and glia), and—if warranted—by cellular target (eg, cell body, axon, myelin). All terms should be related specifically to their location in the CNS and/or PNS. An essential feature in providing diagnoses for neural findings in rodent studies will be to designate the CNS region(s) or PNS structure(s) that are affected, including any needed descriptors for particular sites (the fifth neuronal layer of the cerebral cortex, the Purkinje cell layer of the cerebellum, etc). The new terms are organized under the part of the nervous system that was damaged (neuron cell body, axon, myelin) or under a general category where the targeted element is another cell type or is not yet known. Terms under each of these categories are alphabetized for convenience.
Most images depict changes in tissues fixed by immersion in neutral buffered 10% formalin, processed routinely into paraffin, sectioned at approximately 5 μm, and stained with H&E. Processing conditions that vary from this procedure are noted where necessary in the figure legends.
Nonproliferative Lesions
These definitions describe basic morphologic features and diagnostic procedures that can be used to confirm their presence. Additional details have been listed in the previous INHAND publication on neural lesions in rodents. 1
Neuron—Cell Body
Autophagy, neuron, ganglion
Figure 1
Biological behavior: degenerative lesion.
Other term(s): none.
Nonpreferred (archaic) terms: Lysis, neuron; necrosis, neuronal.
Histogenesis: neuronal cell bodies of sensory ganglia, most commonly the dorsal root ganglia (DRG) and/or trigeminal (cranial nerve V) ganglia but on occasion in autonomic ganglia.
Pathogenesis: programmed cell death (though the nature of the fatal damage is not known).
Diagnostic features:
Cell pallor, typically appearing as stippled or granular, pale eosinophilic cytoplasm.
Loss of the nuclear membrane, with distribution of the residual chromatin as irregular amphophilic to deeply eosinophilic, central, often coalescing and/or winding clumps or islands that usually are located away from the cell center or near the cell membrane.
Narrow, circumferential (partial to complete), clear, colorless halo around chromatin, sometimes bounded by an outer, eosinophilic membrane.
No evidence of glial or inflammatory cell reactions to disintegrating neurons.
Special diagnostic techniques: The cell appearance is distinctive on H&E-stained sections. If special procedures are performed, the following features are evident:
Bielschowsky silver stain: The clumped chromatin residue appears as black debris.
Fluoro Jade: Disintegrating sensory neurons in ganglia do NOT stain.
Immunohistochemistry (IHC): In monkeys, affected neurons stain positively for peripherin and ATB16L1, indicating autophagy as an underlying process. Peripherin is a type III intermediate filament protein expressed mainly in ganglionic neurons, and ATB16L1 is an autophagy-related protein. 2
Ultrastructure: By transmission electron microscopy (TEM), autophagic neurons are filled with phagolysosomes, which gives the cells their granular cytoplasmic appearance by light microscopy; have disrupted nuclei comprised of angular to granular chromatin spicules often arranged in a vaguely circular array; and possess intact cell membranes. 2
Differential diagnoses:
Degeneration, neuron (a potentially reversible indication of substantial cell injury, which typically presents as cell and/or nuclear swelling).
Necrosis, neuron (an end-stage change indicating cell death, which is apparent as slightly shrunken, hypereosinophilic cells with small, condensed [“pyknotic”] or fragmented [“karyorrhectic”] nuclei).
Apoptosis (an end-stage finding indicating cell death, which presents as slightly shrunken cells possessing either nuclei with marginated crescents of chromatin or irregularly sized, globular pieces of the disintegrating nucleus).
Dystrophy, axonal (“spheroids”), a potentially reversible indication of disrupted axonal transport characterized by swollen axons with homogeneous, eosinophilic axoplasm.
Comment: Neuronal autophagy (Figure 1) is an incidental background finding in DRG and to a lesser extent trigeminal (cranial nerve V) ganglia and autonomic ganglia. In untreated and vehicle-treated control animals, the change is rare in rats and has not been reported in mice but is common in nonhuman primates (NHPs). 3 In rats, affected neurons usually are confined to a single ganglion, and only 1 or rarely 2 cells exhibit the finding. In contrast, in macaques (Figure 1) multiple ganglia may be affected, with the number of involved neurons per DRG ranging from 1 to several (typically 2-5, but sometimes up to 20-25 per affected ganglion). Ultrastructural features of this change suggest that it represents autophagy, but the initiating influence is not known. To date, this change has not been connected with in-life neurological signs or exposure to potentially neurotoxic test articles, though it is possible that a test article could increase the incidence of this background change. This incidental background finding historically has been diagnosed as “necrosis, neuronal” but going forward should be recorded as “autophagy, neuronal, ganglion,” so that it is not confounded with genuine test article-related neuronal necrosis.
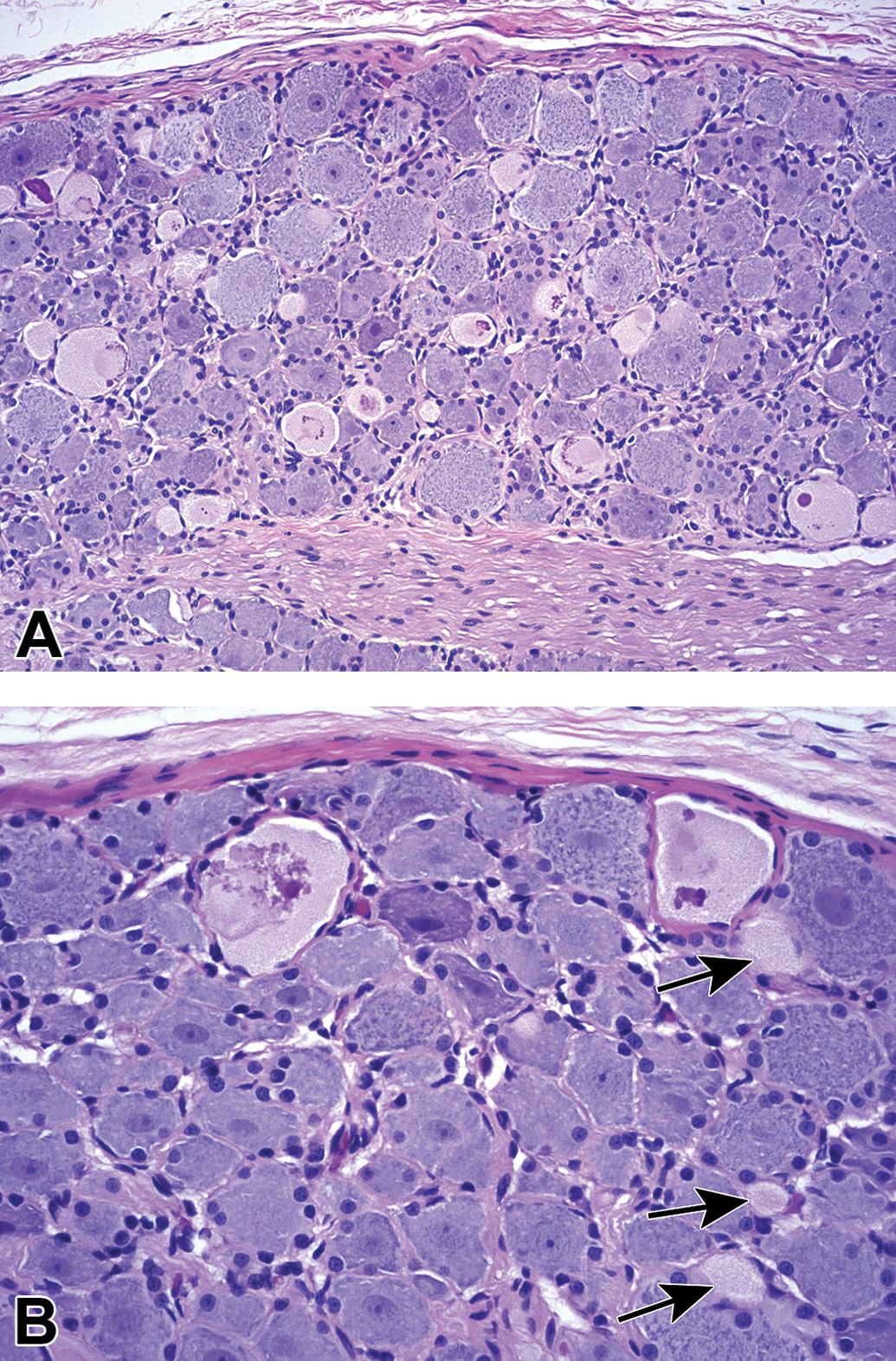
Autophagy, neuron, ganglion presents as few to many (A) neurons with pale eosinophilic, granular cytoplasm and variable amounts of dark eosinophilic, irregular chromatin clumps, globules, or spicules. Adjacent viable neurons have numerous basophilic cytoplasmic bodies (ie, Nissl substance) and sometimes large, centrally located nuclei with prominent basophilic nucleoli. Several smaller profiles of autophagic neurons are evident only as pale eosinophilic, granular cytoplasm with no nuclear debris (B, arrows). Dorsal root ganglion (DRG, lumbar), immature cynomolgus monkey, Hematoxylin and eosin.
Occasionally, neuronal autophagy may be difficult to distinguish from “dystrophy, axonal” (or spheroids), a potentially reversible indication of disrupted axonal transport, 4 or normal axon hillocks (ie, the enlarged proximal portion of the axon where it arises from the neuron cell body). The morphologic similarity between this neuronal change and these 2 axonal morphologies reflects a plane-of-section artifact where a neuron undergoing autophagy is cut tangentially through a peripheral portion of the cell body. Care is required in discriminating between neuronal autophagy (an incidental background lesion) and axonal dystrophy since these 2 findings arise by different mechanisms.
Neuron—Axon
Degeneration, nerve fiber
Figure 2
Biological behavior: breakdown of entire nerve fibers (both axons and their myelin sheaths).
Other term(s) (INHAND): radiculoneuropathy (nerve roots).
Preferred INHAND terms (when properly confirmed): degeneration, axonal; demyelination.
Histogenesis: mature lesion reflecting disintegration of an entire nerve fiber following initial primary injury to either axonal processes of neurons (INHAND term: “degeneration, axonal”) or myelin sheaths that surround the axons (INHAND term: “demyelination”), followed by secondary destruction of the initially spared element.
Pathogenesis: primary axonal injury with secondary myelin degradation or primary myelin damage with eventual secondary axonal disintegration, due to loss of trophic influences from the initially targeted cells.
Diagnostic features:
Axonal degeneration as a primary lesion appears first as eosinophilic swelling (spheroids), typically affecting multiple axons and occurring in the absence of myelin damage. 5
Late-stage axonal degeneration as a primary lesion results in axon fragmentation with generation of digestion chambers containing phagocytic macrophages (termed “gitter cells” in the CNS). Myelin degeneration may develop as a secondary change.
Myelin degeneration as a primary lesion usually presents as a clear space with ovoid-shaped myelin debris surrounding an intact centrally located axon. 5
Special diagnostic techniques:
Conventional histopathological examination of hard plastic (ie, resin)-embedded, toluidine blue-stained sections (mainly used for the PNS). 6,7
Demonstration of axonal fragmentation by histological stains (eg, silver impregnation [Bielschowsky’s, Bodian’s]) or IHC for cytoskeletal elements (eg, antineurofilament protein [NFP]).
Demonstration of myelin disruption by histological stains (eg, Luxol fast blue [LFB], Marchi technique, osmium impregnation) or IHC for myelin constituents (eg, antimyelin basic protein [MBP]).
Teased fiber preparations, applicable to PNS axons and myelin. 8,9 Demyelination can be demonstrated in teased fiber preparations in the form of paranodal or segmental demyelination.
Ultrastructural (TEM) evaluation to distinguish the main lesion: axonal degeneration versus demyelination. 10,11
Differential diagnoses
More specific diagnostic nomenclature is preferred where the precise mechanism of nerve fiber degeneration can be identified. The 2 INHAND terms that may be used where the lesion pathogenesis is known are:
Degeneration, axonal (ie, nerve fiber disintegration in which the primary lesion can be diagnosed definitively as affecting the axon), and
Demyelination (ie, nerve fiber degeneration where the primary lesion can be defined reliably as impacting either myelin or the myelin-generating cell [oligodendrocytes in the CNS, Schwann cells in the PNS]).
Comment: The catch-all diagnosis “nerve fiber degeneration” (Figure 2) is the generic term denoting degradation of entire nerve fibers, which are composed of axons and their enveloping myelin sheaths. This term may be employed as a “first-tier” finding, when microscopic examination of H&E-stained sections in the absence of special neurohistological methods cannot differentiate definitively between primary axonal loss with myelin retention (for which the INHAND term is “degeneration, axonal”) or primary myelin damage with axon preservation (encompassed by the INHAND term “demyelination”). When using “degeneration, nerve fiber,” the pathology report narrative should state that differentiating axonal from myelin degeneration was not possible on conventional H&E-stained, paraffin-embedded sections. When the incidence and severity of nerve fiber findings as well as their distribution among the groups indicate no relationship to treatment with the test item, the change may be considered as spontaneous and reported as “degeneration, nerve fiber.” Treatment-related, dose-dependent increases and/or unusual findings in nerve fibers should be further analyzed and characterized using special neurohistological techniques in the current or additional special studies so that the appropriate, more specific “second-tier” diagnostic term of “degeneration, axonal” or “demyelination” may be used. Where possible, the use of the appropriate second-tier term “degeneration, axonal” and “demyelination” is recommended, rather than “degeneration, nerve fiber,” for lesions in which the pathogenesis has been confirmed using neurohistological stains (eg, silver impregnation [Bielschowsky’s, Bodian’s] or IHC to detect cell type-specific cytoskeletal elements such as anti-NFP or anti-MBP) or other diagnostic methods (eg, electron microscopy, teased nerve fiber preparations) to unambiguously identify the structure (ie, axon or myelin sheath) that is the primary target of injury. 7,12
Nerve fiber degeneration as a spontaneous aging change in nerve roots of rodents sometimes has been termed “radiculoneuropathy” historically for chronic toxicity (≥26 weeks) and carcinogenicity studies (see below for the new INHAND definition of this historical term). However, in general for pathology reports the more precise descriptive term (“degeneration, nerve fiber”) accompanied by a comment that spinal nerve roots are the affected site should be used as the diagnosis in the data tables while radiculoneuropathy should be employed only as a summary interpretive term (like chronic progressive nephropathy [CPN]) in the narrative.
Degeneration, nerve fiber is a lesion in which a disintegrating nerve fiber is replaced by a “digestion chamber” containing variable quantities of axonal and/or myelin debris. A, Tibial nerve, Beagle dog. B, Dorsal spinal nerve root (lumbar region), adult Sprague-Dawley rat. Hematoxylin and eosin.
Radiculoneuropathy
Figure 3
Biological behavior: breakdown of entire nerve fibers (both axons and their myelin sheaths) in the dorsal and ventral spinal nerve roots.
Other term(s) (INHAND): degeneration, nerve fiber; degeneration, axonal; demyelination.
Histogenesis: spontaneous lesion reflecting nerve fiber disintegration following primary injury to either axonal processes of neurons (INHAND term: degeneration, axonal) or myelin sheaths that surround the axons (INHAND term: demyelination).
Pathogenesis: Primary segmental demyelination with secondary axonal degeneration in the large myelinated fibers of both dorsal and ventral roots of aging rats. 13 Primary axonal injury with secondary myelin degeneration is a possible lesion as well but has not been reported as a common pathogenesis of radiculoneuropathy.
Diagnostic features:
Blebbing (primary segmental demyelination) characterized by wide distention of the myelin sheath in myelinated fibers of various sizes, but more prominent in those of large diameter, due to interlamellar splitting.
13
∘ Blebbing involves nearly the entire internode (ie, the myelin sheath found between 2 nodes of Ranvier) when viewed in longitudinal sections and is frequently accompanied by macrophage invasion into the intramyelinic spaces and rarely inflammatory cells in the interstitium.
13
∘ Blebbing is accompanied by reduction of the axon diameter in affected fibers.
13
Distended sheaths invaded by macrophages sometimes show vesicular dissolution of myelin and phagocytosis of degraded myelin, resulting in reduced thickness of the myelin sheath. Thinly myelinated fibers are common in these lesions, indicating that active remyelination is coexistent with demyelination. 13
Focal accumulation of lipid debris, lipid-filled macrophages (following their arrival via local blood vessels), rhomboid clefts resembling cholesterol clefts, hemosiderin, and occasional hemorrhage also may be observed within the affected spinal roots. 14,15
Special diagnostic techniques:
Conventional histological examination of hard plastic (ie, resin)-embedded, toluidine blue-stained sections (mainly used for the PNS).
6
Cytoskeletal demonstration in axons by histological stains (eg, silver impregnation [Bielschowsky’s, Bodian’s]) or IHC (eg, anti-NFP). Myelin demonstration by histological stains (eg, LFB, Marchi technique, osmium impregnation) or IHC (eg, anti-MBP). Teased fiber preparations, applicable to PNS axons and myelin.
8,9
Differential diagnoses: degeneration, axonal; demyelination.
Comment: Posterior paralysis occurs spontaneously in senescent rats more than 2 years of age. 16 The causal degenerative changes within the nerve roots historically have been reported as radiculoneuropathy (Figure 3) in Sprague-Dawley, Wistar strain, Charles River CD strain, Brown Norway (BN/Bi), Glaxo Wistar (WAG/Rij), and (WAG/BN) FI rats. Similar changes though of milder severity may be seen rarely in aged mice. 17 The lesions are most prominent in the nerve fibers in the lumbar spinal nerve roots that contribute to the cauda equina, but lesions are not found in the nerve cells of the DRG and ventral horn of the spinal cord that serve as the origins of these nerve affected nerve fibers. 13
Radiculoneuropathy is an advanced stage of nerve fiber degeneration affecting the dorsal (A) and ventral (B) spinal nerve roots of aged rodents. This lesion is a spontaneous, usually bilateral background lesion commonly observed in many rat strains and fewer mouse strains. The change typically reflects primary demyelination of nerve fibers, indicated here by the presence of intact but shrunken axons but no myelin in several small to large vacuoles in the spinal nerve roots (A and B). Other medium-sized to large vacuoles contain granular debris and/or one to several plump, vacuolated macrophages but no axons. Numbers of dark Schwann cell nuclei scattered among nerve fibers are increased in several regions of the cross section. The incidence and severity of radiculoneuropathy occasionally may be increased by small molecule test articles that damage nerve fibers in the peripheral nervous system. This term generally is employed only for chronic studies (eg, rodent lifetime [2 year] carcinogenicity bioassays). Spinal nerve roots, 110-week-old, vehicle-treated, Sprague-Dawley rat. Hematoxylin and eosin.
The cause of this condition is not known, but it may be exacerbated by such factors as pressure on the nerves due to lesions in the adjacent spinal cord and hypoactivity. 15,17
Nomenclature recommendation: Although the well-known entity of radiculoneuropathy is associated with well-accepted morphologic features commonly seen in aged laboratory rats and occasionally mice on long-term studies (eg, 2-year carcinogenicity studies), this change by definition represents a mechanistically complex disease and not a morphologic descriptor. Therefore, best practice is to diagnose such lesions in young rodents for short-term studies (<26 weeks duration) using an appropriate INHAND morphologic descriptor such as “degeneration, nerve fiber” (see above for definition) with a comment that the affected site is the spinal nerve root. The more inclusive interpretation “radiculoneuropathy” is best reserved for much older animals in longer duration studies (eg, ≥26 weeks duration) where this term has been used historically.
Glia—Myelin
Accumulation, laminar, Schwann cell
Figure 4
Biological behavior: reparative Schwann cell hyperplasia in response to repeated episodes of demyelination.
Other term(s): onion bulbs; hypertrophic neuropathy; degeneration/regeneration, myelin; degeneration/regeneration, nerve fiber; demyelinating neuropathy.
Histogenesis: Schwann cells, by formation of concentric periaxonal layers comprised of intermingled Schwann cell processes plus collagen (from perineurial fibroblasts).
Pathogenesis: Repeated (cyclic) episodes of segmental demyelination and remyelination lead to the accumulation of supernumerary Schwann cells and collagen in concentric layers that surround axons of long nerves. The sequence of events in “accumulation, laminar, Schwann cell” is (1) partial or complete segmental demyelination of an internode; (2) mitosis of the Schwann cell associated with the demyelinated internode; (3) capture of the demyelinated internode by one of the 2 new Schwann cells, with centrifugal (outward) displacement of the other new Schwann cell; (4) circumferential orientation (influenced by the second-order basement membrane) and further mitosis of the displaced Schwann cell; and (5) successive centrifugal displacement of additional layers of basement membranes and Schwann cells due to repeated segmental demyelination and remyelination. Additional Schwann cells may be attracted to the demyelinating internode from other regions. 19
Diagnostic features:
Pathognomonic hallmark of hypertrophic neuropathy. Elongate accumulations of loosely whorled, concentric, mucin-rich, cell-poor tissue surrounding central axons of myelinated nerve fibers. Lamellae (onion bulbs) consist of circumferentially oriented, proliferating Schwann cells separated by longitudinally oriented collagen fibrils (laid down by fibroblasts in the nearby perineurium). The outermost lamellae may consist of fibroblasts or other cells (eg, macrophages). Structures tend to occur in a peripheral location within the nerve trunk. Onion-bulb formations surround only internodes that are undergoing segmental demyelination, that already have been demyelinated, and which may have been remyelinated, and not internodes undergoing Wallerian degeneration (ie, secondary demyelination following primary traumatic axonal injury). The centrally located, intact axon is often demyelinated or has a thin layer of myelin relative to the expected myelin sheath thickness for an axon of its caliber.
Special diagnostic techniques: Detection by IHC of key myelin elements, including Masson’s trichrome for collagen deposition. Peripheral myelin protein 22 (PMP22) for myelin integrity.
20
S100 for Schwann cell processes.
Differential diagnoses: demyelination (generalized).
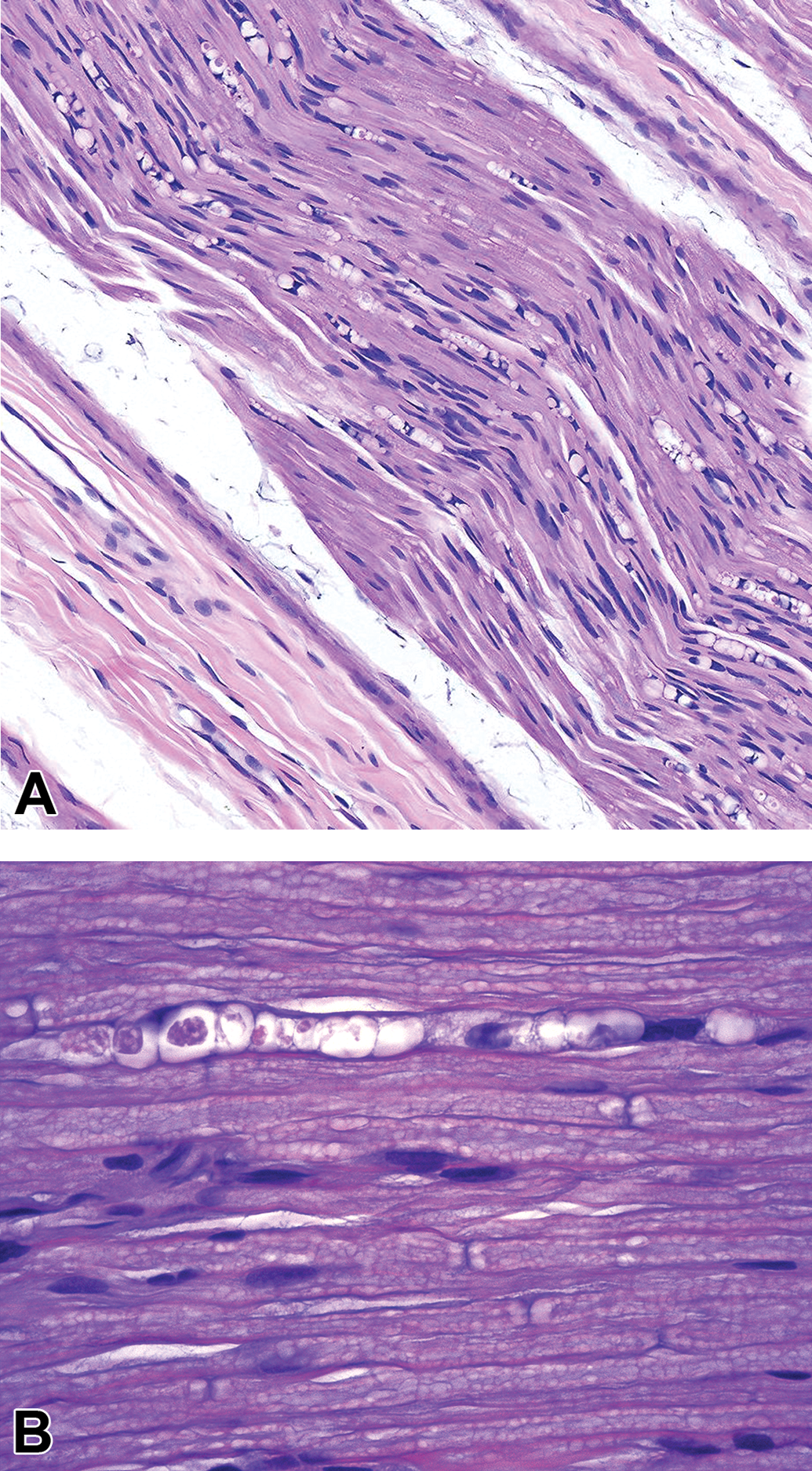
Comment: “Accumulation, laminar, Schwann cell” (onion bulb proliferation) is seen in several different clinical, genetic, and biochemical disorders in man. Onion bulbs are found most commonly in nerves showing the greatest evidence of segmental demyelination. The Schwann cell proliferation is a response to cyclic demyelination and regeneration of myelin and can cause thickening of nerves (hypertrophic neuropathy). 4
The Trembler (Pmp22Tr) mouse is an experimental model of hypertrophic interstitial neuropathy. This animal carries a spontaneous autosomal dominant mutation (Tr) of PMP22 on chromosome 11 that manifests as a Schwann cell defect characterized by severe hypomyelination and continuing Schwann cell proliferation throughout life. 21 The nerves of adult Trembler mice show a delay in initial myelination as well as ongoing cyclic segmental demyelination leading to development of an onion bulb neuropathy (Figure 4). Clinical symptoms manifest by 10 to 14 days of age as an action tremor affecting the head, neck, and limbs; convulsions, which decrease with increasing age; and weakness and rigidity of the limbs. 22
Onion bulb formations have been produced experimentally in rats by (1) injection of benzanthracene into nerves, 23 (2) feeding of lead carbonate, 24 and (3) repeated application of a tourniquet around a sciatic nerve. 25
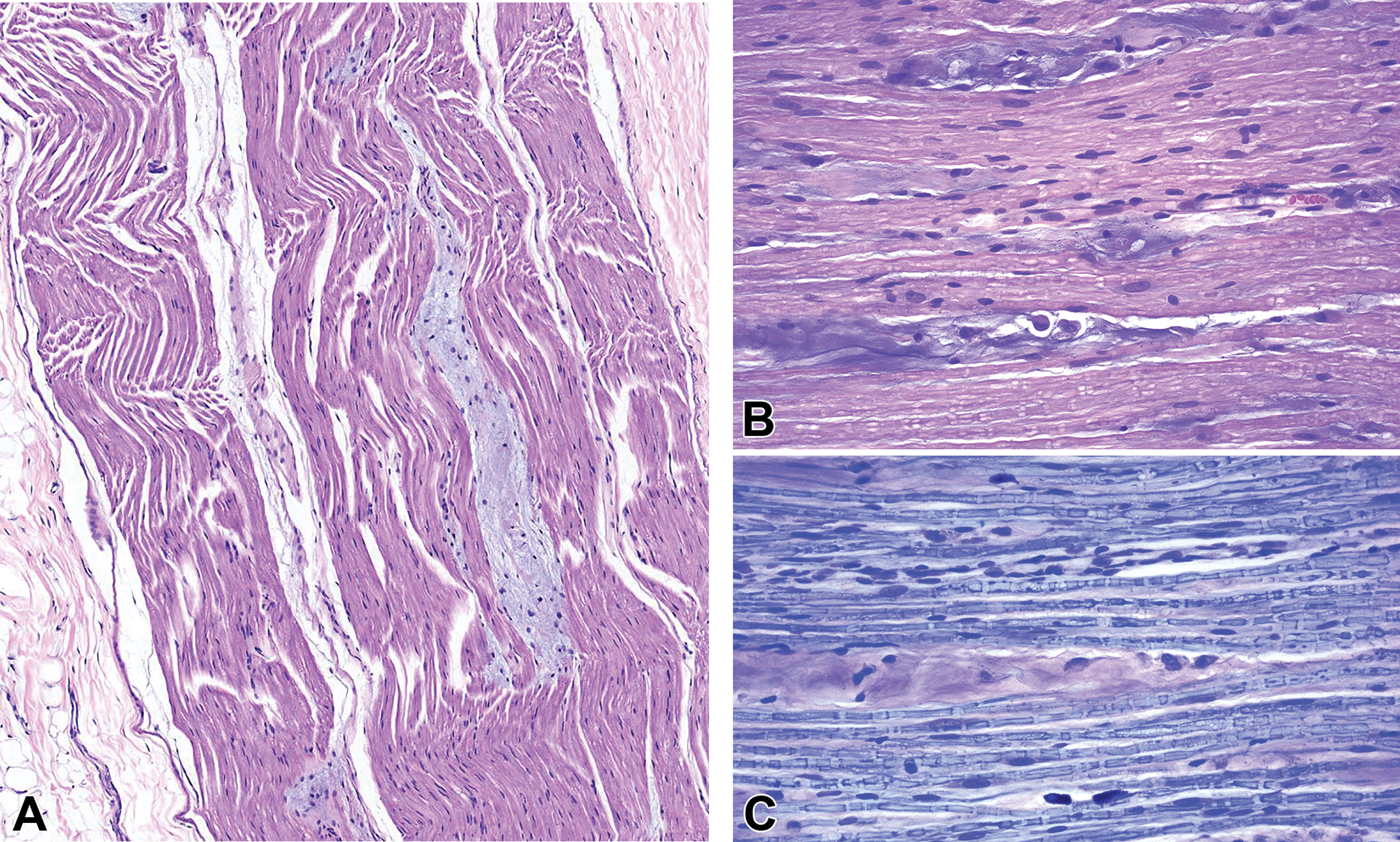
Accumulation, laminar, Schwann cell (“hypertrophic neuropathy”) appears as multiple concentric layers of Schwann cells intermingled with collagen (termed an “onion bulb” formation) surrounding intact pale, central axons. The variable but usually marked thinning of the dark myelin sheaths stems from repeated cycles of myelin degeneration and regeneration. Phrenic nerve, adult Pmp22 Tr-J (“Trembler”) mouse. Processing: immersion fixation in 2.5% glutaraldehyde, hard plastic resin embedding, semi-thin (1-µm-thick) section, Toluidine blue staining. [This image was provided courtesy of Dr. Lucia Notterpek, University of Nevada, Reno and is reproduced from Zhou et al. 18 with the permission of Elsevier.]
In humans, onion bulbs are the histological hallmark of Charcot-Marie-Tooth disease but are also seen in other hereditary neuropathies (Dejerine-Sottas disease, Refsum disease), in chronic diabetic neuropathy, and in chronic inflammatory demyelinating neuropathy. Cell constituents in and near the onion bulbs may need to be identified using IHC since the proliferating cells that form onion bulbs are morphologically similar to adjacent connective tissue cells and thus difficult to discriminate in routinely stained sections. 26 Onion bulbs also may be seen with the less common form of fulminant Guillain-Barré syndrome that occurs in association with Campylobacter jejuni infection. 27
General
Accumulation, matrix
Figure 5
Biological behavior: unknown.
Other term(s): none.
Histogenesis: unknown; presumably fibroblasts within the epineurium, perineurium, and/or endoneurium.
Pathogenesis: unknown.
Diagnostic features:
Accumulation of amorphous, paucicellular, pale basophilic (by H&E) or pink (by LFB) material as elongated elliptical deposits separating nerve fibers in peripheral nerves, including near DRG.
4
The few cells within these deposits have fusiform to oval nuclei (mainly large) with indistinct cell membranes and cytoplasm. Some deposits also contain small numbers of mast cells.
Special diagnostic techniques: none. Where desired, a Congo red stain may be applied to a serial section to confirm that the material is not amyloid.
Differential diagnoses:
Renaut body (which appear on H&E-stained sections as round, amorphous structures containing randomly oriented collagen fibrils and occasional spindle cells embedded in hyaline, pale eosinophilic matrix). Amyloid (which presents as homogeneous, generally acellular foci of pale eosinophilic and congophilic material in the endoneurial and epineurial connective tissues as well as in the walls of associated capillaries in somatic and autonomic ganglia and nerves
28,29
).
Comment: The finding may be identified readily (Figure 5), but its biological significance is not known. In the authors’ collective experience, the change is seen with some frequency in rodents and dogs and does occur in NHPs. Typical locations for this finding include spinal nerve roots and proximal somatic nerve trunks near DRG; occasionally, nerve trunks associated with trigeminal and autonomic ganglia also may be affected. The proximal location and lack of whorling within these deposits suggest that this change is not an early stage of the Renaut body.
Accumulation, matrix appears with a focal to multifocal accretion of cell-poor, pale basophilic (by H&E) or pink (by LFB), amorphous to fibrillar material as elongated islands separating adjacent myelinated nerve fibers (where normal myelin is eosinophilic by H&E and pale blue by LFB). A, Tibial nerve, Beagle dog, H&E. B and C, plantar nerve, adult Sprague-Dawley rat; H&E (B) and LFB (C). H&E indicates hematoxylin and eosin. LFB indicates Luxol fast blue.
Ectopic tissue
Figure 6
Biological behavior: self-limiting developmental aberration.
Other terms: Ectopia, heterotopia.
Histogenesis: normal non-neural tissue that is located in an unexpected site (more commonly in the CNS than the PNS).
Pathogenesis: abnormally positioned aggregate of normal non-neural tissue within the nervous system (eg, adipose tissue in the choroid plexus (Figure 6) or meninges) due to aberrations in early migration of precursor cells during development. (Note: Normal neurons present at abnormal locations in the CNS or PNS are referred to using the INHAND term “Heterotopia, neuronal.” 1 )
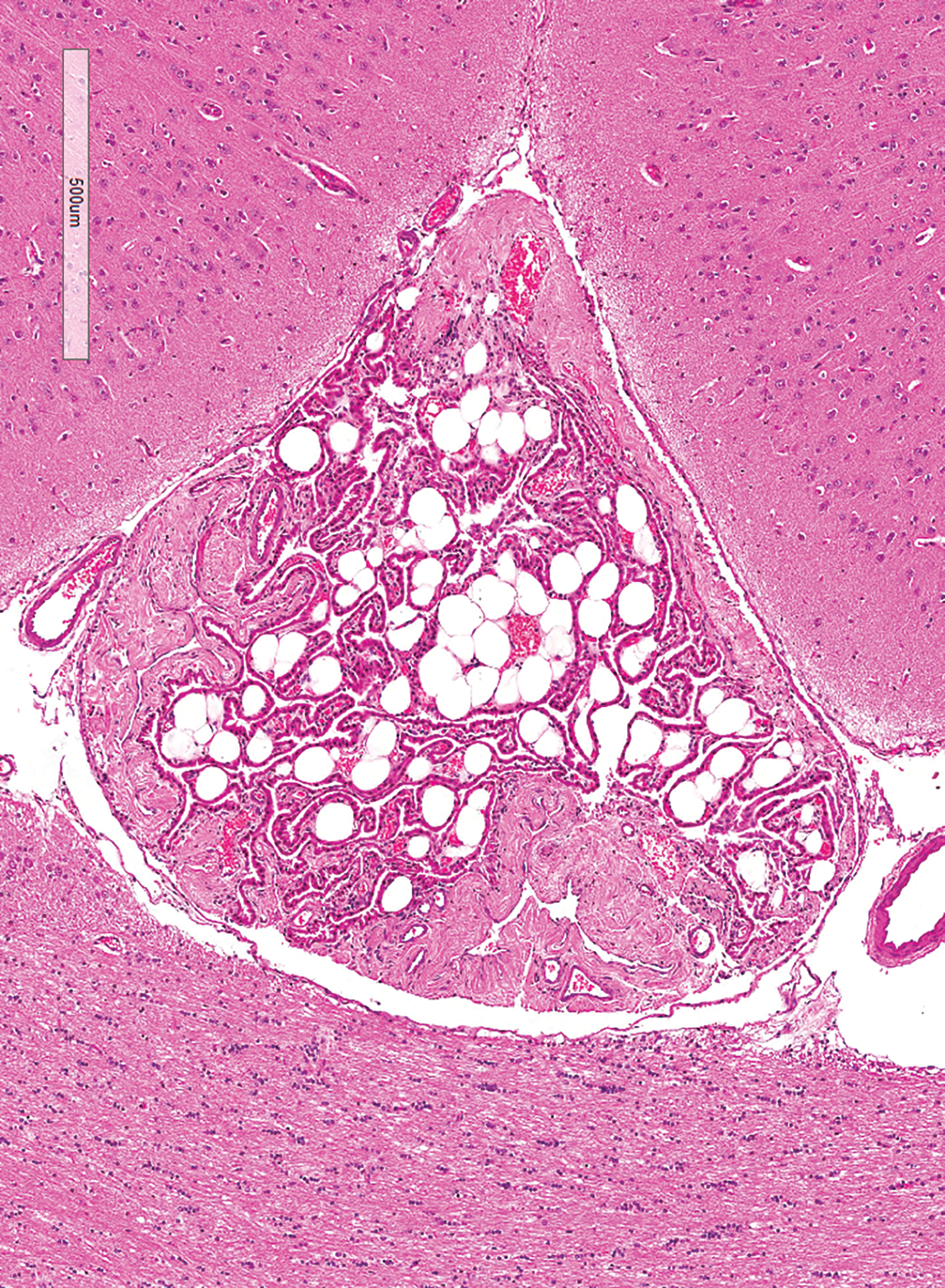
Ectopic tissue is recognized by the unexpected presence of normal cells derived from a non-neural lineage within the nervous system. Choroid plexus with ectopic adipose tissue. Brain (callosal sulcus), Beagle dog. Hematoxylin and eosin. Scale bar = 500 µm.
Diagnostic features:
Focal to multifocal distribution. Fully differentiated non-neural tissue in the brain (especially meninges), spinal cord, ganglia, or nerves. Usually circumscribed and not wholly integrated into the “host” tissue. No evidence of neural dysplasia or history of traumatic tissue injury.
Special diagnostic techniques: none.
Differential diagnoses:
Metaplasia: The altered tissue typically is well differentiated, is integrated intimately into the histological pattern of the host tissue, and is usually a result of tissue reaction and adaptation by non-neuronal components of the affected neural region. Neoplasia: Foci of minimally to mildly pleomorphic, non-neural tumor cells appear as variably invasive metastases that may involve neural parenchyma and/or nearby non-neural tissues (eg, choroid plexus, meninges). Vacuolation: Choroid plexus epithelium accumulates polyethylene glycol (PEG) in clear, colorless cytoplasmic vacuoles after administration of some PEG-conjugated (“pegylated”) biomolecules. If PEG retention is extensive, the vacuoles may be large enough to resemble white adipocytes (see figures 59-60 in the original INHAND paper on rodent nervous system findings by Kaufmann et al
1
).
Comment: The presence of any ectopic tissue is indicative of disturbances during early CNS development. A common example in the CNS is the “squamous cyst,” an entity common in mice but infrequent in rats (see figures 79-81 in the original INHAND paper on rodent nervous system findings by Kaufmann et al 1 ). Squamous cysts arise when misplacement of surface ectoderm at the time of neural tube closure leads to formation of a hollow, generally spherical nodule lined by stratified squamous epithelium and filled with keratin. Importantly, this non-neural tissue should be diagnosed using the more specific INHAND term “squamous cyst” rather than “ectopic tissue.” 1
Ectopic tissues in the CNS and PNS are usually a rare finding in toxicity studies performed in juvenile and young adult rodents, and care should be taken to properly interpret them as incidental findings and not metaplasia or neoplasia. Ectopic tissues may be a test item-related consequence if exposure occurs during early specification of neural regions (eg, embryonic and fetal periods in all species, and early postnatal life [up to approximately postnatal day 7] in rodents).
The term “xenobiotic tissue” typically should be used to describe ectopic tissue arising from deliberate engraftment of a cell-based test item into the CNS rather than as an induced or inherited developmental error. The reason for using “xenobiotic tissue” instead of ”ectopic tissue” for such injected exogenous biomaterials is to provide distinct diagnostic categories for abnormally positioned structures arising by disparate mechanisms.
Note: “Neuronal heterotopia” is an INHAND term that is distinct from ectopic tissue. This term should be used to describe the presence of normal-appearing neurons in an unexpected position (due to abnormal migration of precursor cells during development; see figures 9 and 10 in the original INHAND paper on rodent nervous system findings by Kaufmann et al 1 ).
Renaut body
Figure 7
Biological behavior: incidental finding.
Other term(s): Renaut corpuscle.
Histogenesis: fibroblasts of perineurial origin.
Pathogenesis: “normal” background structures in longer somatic nerves of all species and occasionally in autonomic nerves near various viscera (typically in the abdomen).
Diagnostic features:
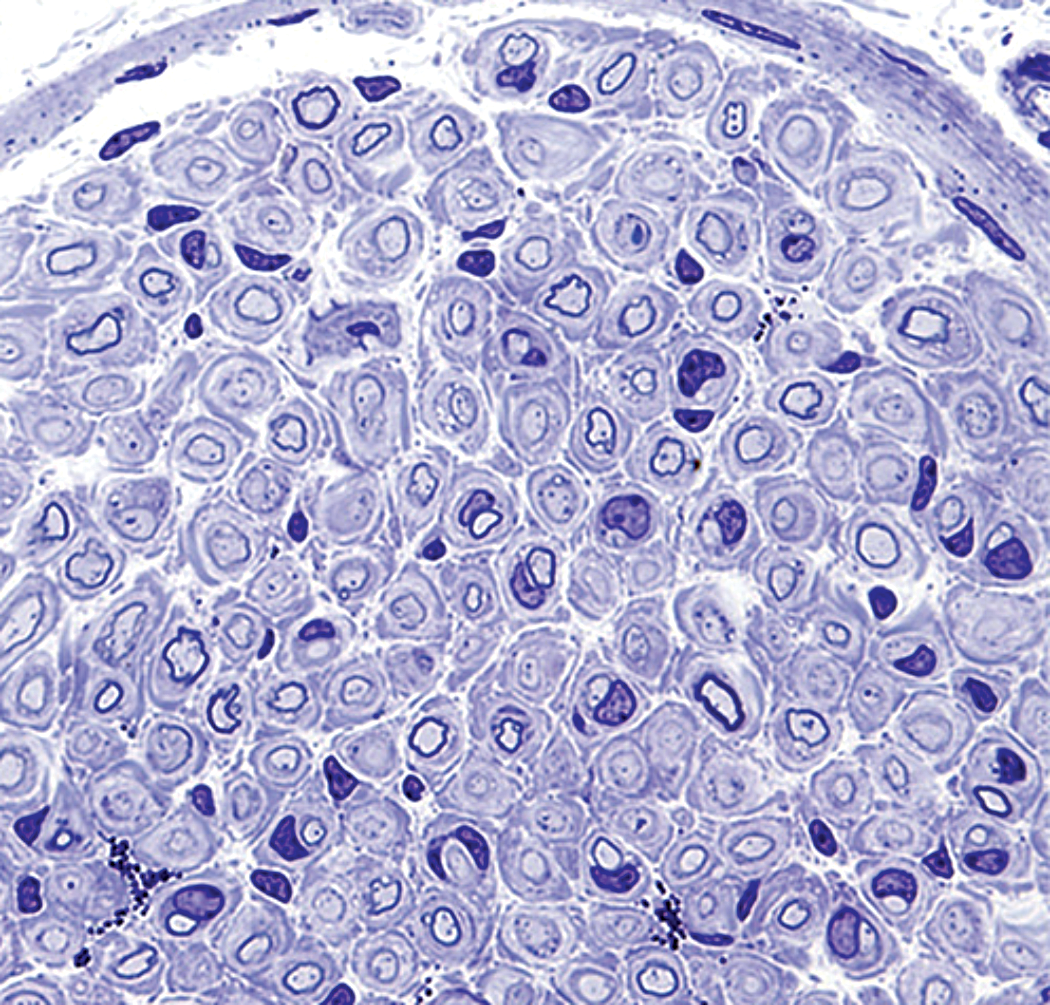
Eosinophilic, amorphous, hyaline structures attached to the inner layer of the perineurium but not penetrated by nerve fibers. Composed of fibroblasts and perineurial cells immersed in an extracellular matrix that contains randomly oriented collagen fibers and elastin precursors but not axons or Schwann cells. In nerve cross sections, Renaut bodies appear as round or ellipsoid, variably layered, pale structures located among nerve fascicles (Figure 7). In nerve longitudinal sections, they occur as elongated, pale bodies running parallel to the long axis of the nerve. Structures tend to occur in a peripheral location within the nerve trunk, mainly located at sites of normal anatomical nerve compression. Associated nerve fibers are morphologically normal.
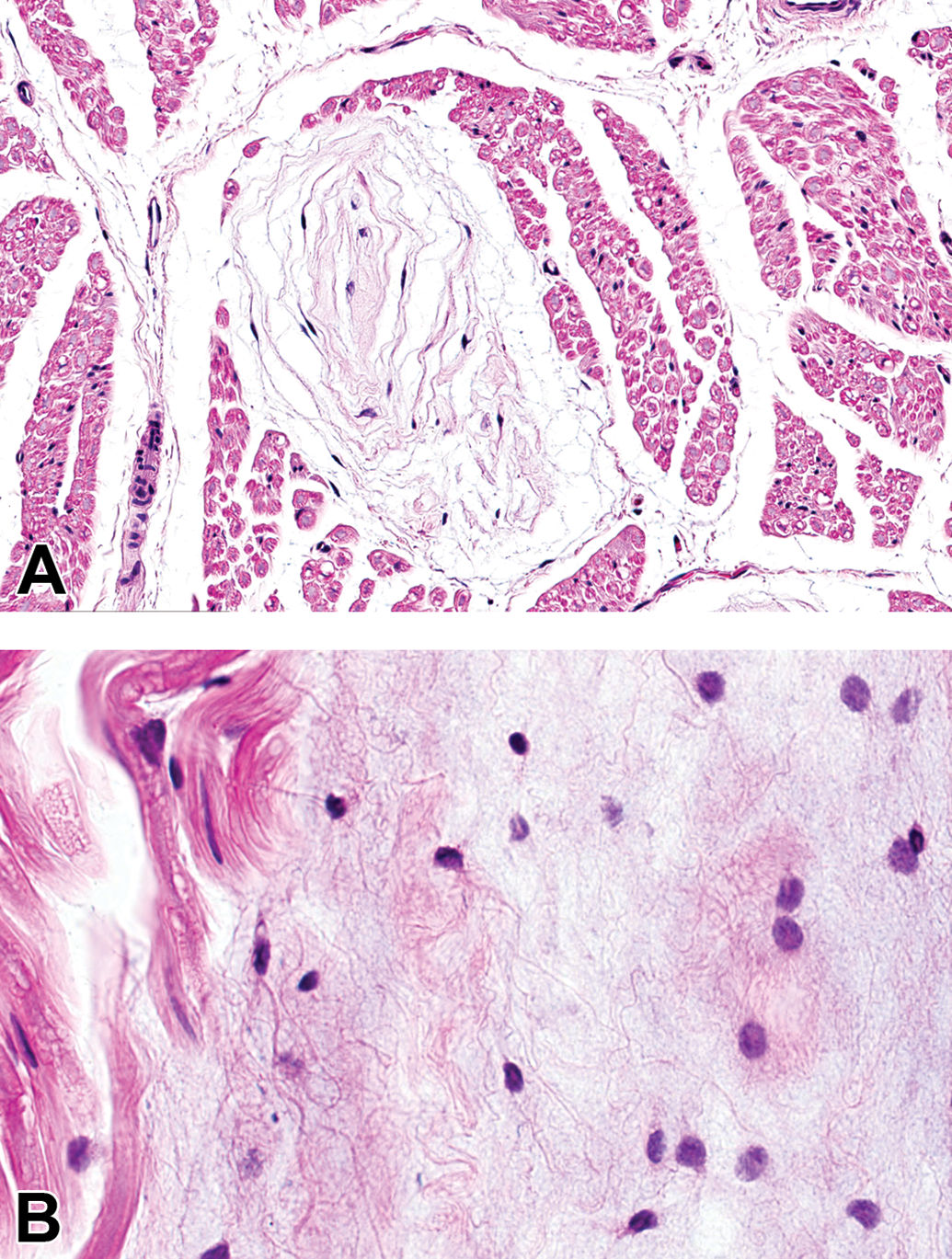
Renaut bodies are hyaline structures composed of concentric layers of fibroblasts and perineurial cells separated by a pale eosinophilic matrix. A, In cross section, Renaut bodies are round or elliptical, and the spindle fibers and cells are arranged in concentric laminae. B, In longitudinal section, Renaut bodies and their spindle cells and connective tissues are elongated along the longitudinal axis, and the pale matrix often is more prominent. Sciatic nerve, Beagle dog. Hematoxylin and eosin.
Special diagnostic techniques:
Alcian blue at pH 2.5 yields strong blue staining, suggesting that they contain acid mucopolysaccharides. Trichrome yields pale green staining. Fibroblast biomarkers like collagen, laminin, and vimentin may be demonstrated by IHC.
30,31
Perineurial cell markers such as epithelial membrane antigen, glucose transporter 1, and claudin-1 may be detected by IHC.
30,32
Ultrastructurally, the fibrous component is predominantly oxytalan,
30
which is a thin extracellular matrix fibril that contributes to connective tissue repair associated with mechanical loading.
33
Differential diagnoses (INHAND terms):
Accumulation, matrix (which appears as an accumulation of amorphous, paucicellular, pale basophilic material between nerve fibers on H&E-stained sections). Amyloid (which presents as homogeneous, generally acellular foci of pale eosinophilic and congophilic material in the endoneurial and epineurial connective tissues as well as in the walls of associated capillaries in somatic and autonomic ganglia and nerves
28,29
).
Comment: Renaut bodies have been reported in many mammals, including such common test species as rats but especially Beagle dogs (Figure 7). In rodents, the incidence of Renaut bodies as a spontaneous change is low, but their frequency may be augmented by experimental surgery 34 or manipulating housing conditions 35 to produce chronic nerve compression. In nonrodents, Renaut bodies have been observed in 2% of human superficial peroneal nerve biopsies 36 and in 36% or more of dogs in general toxicity studies, 31 suggesting that they may be a normal feature of nerve anatomy under certain conditions. Their numbers rise with age, 30,31 though to our knowledge this increase has not been confirmed in rodents. The peripheral location of these structures within nerve trunks has been hypothesized to imply that they act to cushion nearby nerve fibers from excessive mechanical trauma during the normal nerve compression of movement. 37 Renaut bodies are thought to arise from perineurial fibroblasts based on their expression of multiple collagen subtypes, laminin, and vimentin. 32
Renaut bodies are not known to be induced directly by exposure to neuroactive test articles, so pathologists often may choose not to record them in routine toxicity studies. However, the incidence of Renaut bodies in theory may be increased in a dose-dependent fashion during toxicity studies where the test article leads to prolonged recumbency. In such instances, the Renaut bodies are interpreted as sequelae of the extended inactivity rather than evidence of direct neurotoxicity (inferred by the absence of damage in nearby nerve fibers).
Proliferative Lesions
Routine rodent carcinogenicity studies do not allow for evaluation of lesion evolution over time, so the true biological behavior of neural neoplasms often is not known. While several morphologic forms of apparent glial cell neoplasia occur in rats and mice, their definitive identification using IHC and molecular characterization is seldom undertaken during routine animal toxicity testing. Some rodent glial cell neoplasms are not immunoreactive with IHC for glial fibrillary acidic protein (GFAP) and are immunoreactive with IHC for various macrophage biomarkers, suggesting that these CNS neoplasms may be derived from microglial cells. 38 Further morphological and molecular characterizations of rat gliomas using either genetically modified or chemical-induced rat glioma models are needed to address cellular composition, mutation landscapes, gene expression profiles, and, potentially, histogenesis of these neoplasms.
Hyperplasia, glial cell, not otherwise specified (NOS)
Figure 8
Other/previous terms: none.
Histogenesis: glial cell of undetermined origin.
Biological behavior: localized, noninvasive accumulation of preneoplastic glial cells.
Diagnostic features: Features are distinct from those defined for “glioma, not otherwise specified (NOS)”
39
: Circumscribed, small aggregate of oval to round glial cells, generally in deep regions of the brain. Lesion borders are indistinct. Low cell density, indicated by substantial neuropil between the glial cells. Occasional arrangement of glial cells near intralesional neurons (ie, satellitosis), but little or no perivascular cuffing. Glial cells may be larger (ie, hypertrophy) but do not exhibit unusual features such as increased cytoplasm (ie, gemistocytes) or debris consistent with phagocytic function (ie, “gitter” cells). No or rare mitotic figures. No reactive, degenerative, or necrotic elements with the associated parenchyma. Preneoplastic cells do not infiltrate the meninges. The following features indicative of a genuine neoplastic lesion (“glioma, NOS” as defined below) are absent: ∘ Intralesional foci of hemorrhage and necrosis. ∘ Reactivity of non-neoplastic glia in and around the preneoplastic cells (ie, no associated diagnosis of astrocytosis, microgliosis, and/or gliosis NOS). ∘ Cell pleomorphism (chiefly cellular and nuclear atypia).
Special diagnostic techniques: The following markers may be utilized in rodents to explore the lineage of the glial cells: Astrocytes ∘ Conventional staining: phosphotungstic acid hematoxylin (PTAH) ∘ IHC: GFAP Microglia ∘ IHC for macrophage lineage markers like: ▪ Mice: CD11b, F4/80 ▪ Rats: ED-1 (anti-CD68), OX42 ▪ All species: • Ionized calcium binding adaptor molecule 1 (Iba1), a conserved marker for macrophages and microglial cells • Mac-1 (CD11b/CD18) ∘ Lectin histochemistry (all species): Griffonia simplicifolia (GS-IB4), a monocyte/microglia marker.
Oligodendrocytes
Differential diagnoses: Differential diagnoses for small, non-neoplastic glial lesions may be used if the following characteristic features are observed:
Astrocytosis (“reactive gliosis”): discrimination may be difficult, but these foci of well-differentiated, non-neoplastic cells express GFAP widely and intensely. Typically associated with an area of parenchymal damage (necrosis, degeneration, edema, hemorrhage, etc) or compression from an adjacent neoplasm. Generally, no mitotic figures in the accumulated glial cells.
Microgliosis: a non-neoplastic response featuring localized, small collections of Iba1-positive cells with elongated irregular or rod-shaped nuclei that are associated with damage to the affected neural parenchyma (usually to a neuronal population 41 ).
Gliosis, NOS: a non-neoplastic tissue repair response that is characterized by reactive glial cells of uncertain lineage in areas of neuronal and/or neuropil damage.
Comment: The term “hyperplasia, glial cell NOS” is used for presumed preneoplastic glial cells representing a single CNS glial cell population (Figure 8). In general, morphologic features of the accumulating cells are suggestive of astrocytes (plump oval to round nuclei) or microglia (elongated or crescent-shaped spindle nuclei) rather than oligodendroglia (round nuclei in myelin-dense areas). “Hyperplasia, glial cell NOS” is an acceptable designation in instances when it is not possible to identify which population/populations of CNS glia is/are involved. However, where possible, more specific terms (eg, “hyperplasia, astrocytic” or “hyperplasia, microglial cell”) are preferred if the preneoplastic cell lineage can be confirmed using cell type-specific markers.
Hyperplasia, glial cell, not otherwise specified (NOS) is a preneoplastic lesion characterized by small, circumscribed, poorly demarcated aggregates of oval or spindle to sometimes round glial cells in deep brain regions (A). The cell density of these lesions is low, and mitotic figures are absent or very rare. The hyperplastic cells do not invade or induce reactive or degenerative changes in the adjacent parenchyma (B). Hypothalamus, adult Sprague-Dawley rat (These images are reproduced with the permission of the US National Toxicology Program).
Diagnostic criteria developed by the US NTP in devising the Nonneoplastic Lesion Atlas (https://ntp.niehs.nih.gov/nnl/nervous/index.htm) have been incorporated into the description above. Using these criteria, no clear-cut delineation based on cell features can be made for borderline glial lesions between the INHAND terms for non-neoplastic (ie, reactive) glial proliferation (“gliosis NOS”) and preneoplastic lesions (“hyperplasia, glial cell NOS”). In the authors’ collective experience, hyperplasia, glial cell NOS occurs mainly in chronic studies (where rodents are 12 or more months of age), while gliosis NOS can occur at any age as a reactive response to primary damage affecting nearby neurons and/or neuropil.
Glioma, not otherwise specified (NOS)
Figure 9
Other term(s)/previous and no longer preferred diagnosed entities: astrocytoma, atypical, benign; rat astrocytoma; rat astrocytoma, benign, low grade; malignant reticulosis.
Histogenesis: unknown (due to the absence of definitive cytoarchitectural features and IHC molecular signatures in tumor cells indicative of an origin from a specific glial cell lineage).
Diagnostic features:
Circumscribed but poorly demarcated lesion, usually of modest size, confined to one major area of the CNS (typically the cerebral cortex (Figure 9) or diencephalon). Moderate to dense cellularity, usually appearing as multiple small aggregates interspersed in the neural parenchyma (often in association with neurons) with no apparent intervening neuropil. Cells have small to moderate amounts of eosinophilic cytoplasm with indistinct cell borders. Cells have round to elongated, small, hyperchromatic nuclei. Variable numbers of mitotic figures in the accumulating glial cell population. Neoplastic cells can typically be found clustered around blood vessels (perivascular cuffing). ± Satellitosis (tumor cells aggregating around neurons). Neoplastic cells occasionally may infiltrate the nearby meninges. In rats, cellular differentiation of glioma NOS defines several morphological types.
42
-50
∘ Protoplasmic: stellate cells with delicate cytoplasmic processes that form a mesh-like matrix. ∘ Fibrillary: round cells with elongate nuclei. ∘ Gemistocytic: large, very plump cells with abundant cytoplasm encompassing an eccentric, round to oval nucleus. ∘ Pilocytic (pilloid): a rare variant composed of elongated, unipolar or bipolar cells arranged in bundles and bands. ∘ Oligodendroglial: dense round nuclei, clear pericellular halo of variable width, and sparse eosinophilic cytoplasm; may palisade around necrotic tumor tissue. ∘ Microglial: straight or curved, rod-like nuclei with no visible cytoplasmic features. ∘ Mixed: consisting of more than one glial component. In rats, glioma NOS may assume one or more unique patterns, including perineuronal satellitosis and perivascular cuffing at the neoplasm periphery (which may be indicative of a more aggressive biological behavior). In mice, glioma NOS is characterized by a monomorphic population of neoplastic cells (most similar to the protoplasmic variant in rats) that intermingles freely with the local neuronal population and, in some cases, with intralesional reactive astrocytes exhibiting a gemistocytic appearance.
51
-62
Features that may be present in rat glioma NOS neoplasms but that are usually absent in the mouse counterparts include: ∘ Foci of hemorrhage and necrosis. ∘ Palisading of neoplastic astrocytes around necrotic foci. ∘ Focal to multifocal pleomorphism (chiefly cellular and nuclear atypia).
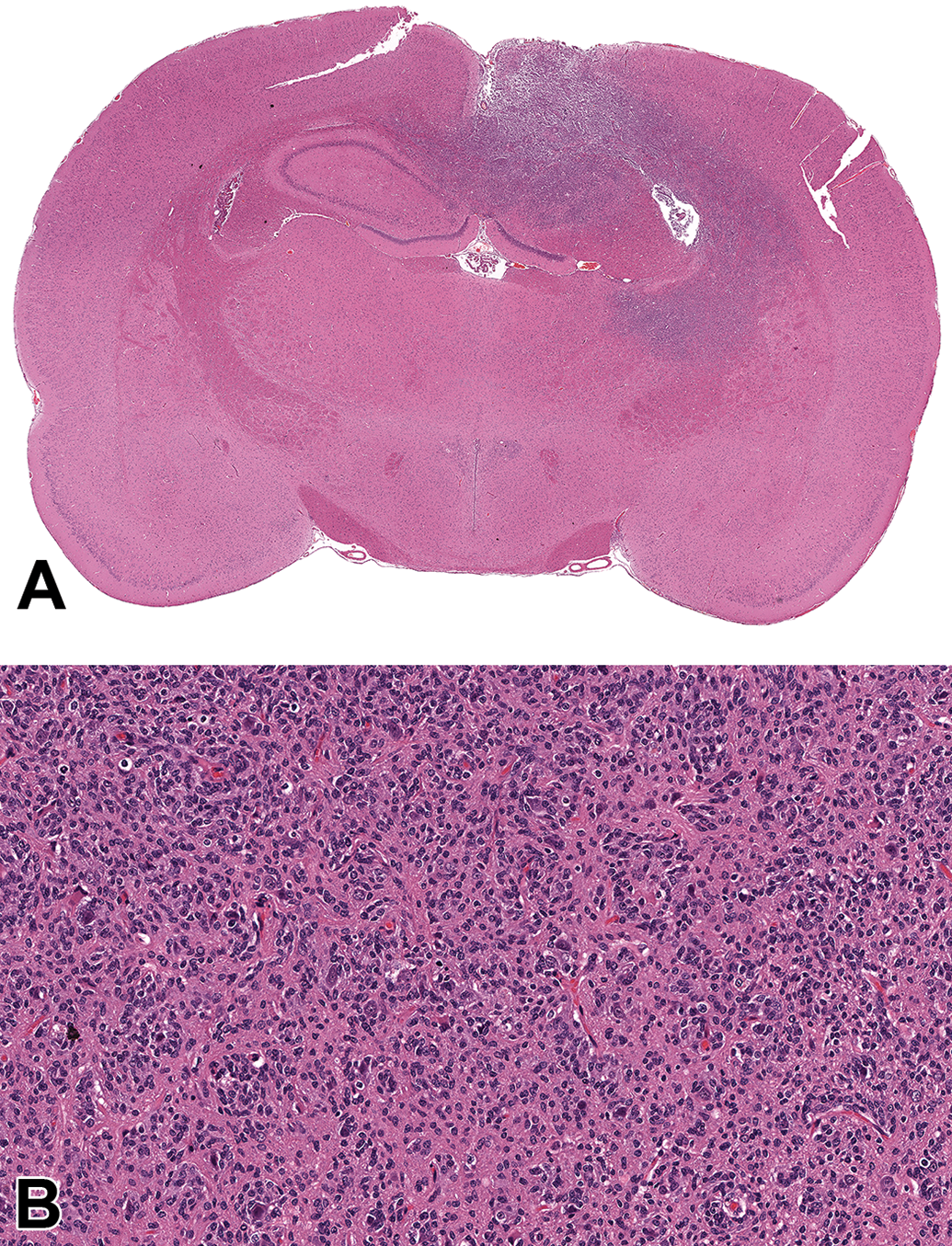
Glioma, not otherwise specified (NOS) occurs as a poorly demarcated glial cell neoplasm (A) of moderate to dense cellularity (B) that may invade the adjacent tissue and sometimes the meninges. Neoplastic cells may exhibit pleomorphism (mainly anisocytosis and anisokaryosis). Mitotic figures are visible though their number is variable. Tumor cells may form palisades around blood vessels (cuffing) or neurons (satellitosis). Additional features in some tumors include foci of hemorrhage and necrosis. Retrosplenial cortex and hippocampus, adult Sprague-Dawley rat (These images are reproduced with the permission of the US National Toxicology Program).
Special diagnostic techniques:
Conventional staining for the astrocyte marker PTAH.
Immunohistochemistry for the astrocyte marker GFAP may help to identify neoplasms of astrocytic lineage. However, glioma NOS in rats and mice generally lacks GFAP immunoreactivity. 42,60,63
IHC for cell lineage markers like:
∘ Mice: CD11b, F4/80
∘ Rats: ED-1 (anti-CD68), OX42
∘ All species:
Iba1, a conserved marker for macrophages and microglia
Mac-1 (CD11b/CD18)
Lectin histochemistry for the monocytic/microglial markers Griffonia simplicifolia (GS-IB4) or Ricinus communis 64 agglutinin 1 (RCA 1)
IHC evaluation of lymphocyte lineage markers like:
∘ CD3 (T cells)
∘ CD79a (B cells)
∘ Pax5 (B cells)
Differential diagnoses: Large glial masses typically can be diagnosed as a neoplasm based on such features as cellular pleomorphism, mitotic figures, and (in rats) foci of hemorrhage and/or necrosis within the tumors. Non-neoplastic differential diagnoses for smaller glial lesions, particularly if they do not form a discrete mass, may be considered if the following characteristic features are observed: Astrocytosis (reactive gliosis): discrimination may be difficult, but these foci of well-differentiated, non-neoplastic cells express GFAP widely and intensely in mice and rats. Microgliosis: a non-neoplastic response featuring localized, small collections of Iba1-positive cells with elongated irregular or rod-shaped nuclei that are associated with damage to the neural parenchyma (usually to a neuronal population
41
). Hyperplasia, glial cell, NOS: a presumed preneoplastic lesion characterized by localized, noninvasive accumulation of loosely packed, modestly enlarged, slowly proliferating glial cells with morphologic features consistent with a single cell lineage.
Comment: Low-grade astrocytomas from humans and domestic animals commonly exhibit strong GFAP labeling and PTAH staining. In contrast, rodent brain neoplasms historically designated as astrocytomas 42,60,62,65,66 consist of 2 glial cell populations—large numbers of GFAP-negative neoplastic cells and scattered GFAP-positive astrocytes interpreted to be reactive (ie, non-neoplastic) cells. Chemically induced gliomas in rats may contain GFAP-positive astrocytes representing both neoplastic and reactive populations. Recent work with chemically induced rat brain tumors has suggested that a diagnosis of “microglial tumor, malignant, high grade” could be considered for neoplasms previously categorized as “astrocytomas, malignant, high grade.” 38 This reclassification was made possible by the expanding battery of neural cell-specific biomarkers, which indicate that GFAP-negative malignant astrocytomas in rats actually may be Iba1-positive glial tumors. Work is currently underway at the US NTP to further characterize rat brain tumors including the histogenesis of these putative microglial tumors. Diagnostic criteria developed by the US NTP 39 are incorporated into the description above.
The “Histological Classification of Tumors of the Nervous System of Domestic Animals” released by the World Health Organization (WHO) in 1999 catalogs astrocytomas into low-grade (well-differentiated), medium-grade (anaplastic), and high-grade (glioblastoma or glioblastoma multiforme) neoplasms. 66 The high-grade astrocytoma is presumed to be the most malignant variant in animals, as is the case for astrocytomas in humans. In addition to having the pronounced pleomorphic features found in medium-grade (anaplastic) astrocytomas, high-grade glioblastomas have variable but prominent degrees of vascular proliferation and/or necrosis. A recent WHO classification for CNS tumors of humans now incorporates molecular profiles to more accurately categorize neoplasms, 67 clearly indicating the utility of IHC methods in identifying the histogenesis and potential sensitivity to therapy. To date, a comprehensive assessment of cell lineage markers in rodent tumors of the CNS (or PNS) has not been completed.
Reticulosis is a term that was introduced in human neuropathology by 1950 and then was discarded by 1980 with the reclassification of reticulosis as primary B-cell lymphoma. 68 In other species, “reticulosis” has been ascribed to cells arising from either a lymphocytic or alternative lineage using IHC markers. 69 This term was used to describe proliferation of “reticulohistiocytic cells” derived from vascular adventitia, leptomeninges, or the microglia or bone marrow-derived monocytic lineage cells. 70 In 1985, Krinke et al described diffuse, perivascular, multifocal tumors in the rat brain as “malignant reticulosis.” 71 Neoplastic reticulosis and related tumors, such as those formerly designated as “reticulum cell sarcoma” and “microgliomatosis,” are currently believed in humans to be B-cell lymphomas. Most cases of “gliomatosis cerebri” in humans are believed to be astroglial in origin, but oligodendroglial and microglial forms are also recognized. In dogs, some cases of “diffuse gliomatosis” were thought after IHC evaluation to be examples of microgliosis. That these canine tumors are B-cell lymphomas was not supported by the studies of Vandevelde et al, who examined 6 cases and found that they did not contain immunoglobulins. 69
In the absence of clear IHC findings and until further work is done to elucidate the cell lineage of these tumors, in a routine rodent carcinogenicity study these tumors should be diagnosed as glioma, NOS.
Revised Terms
These 2 updated terms have been renamed to conform to the INHAND format for defining lesions by morphologic descriptors. The revision brings diagnostic nomenclature for these findings into harmony with INHAND terminology for comparable changes when observed in other systems. Diagnostic features, special diagnostic techniques, and differential diagnoses as outlined for the original INHAND definitions 1 have not changed but are repeated here for the convenience of readers.
Neuron—Cell Body
Decreased cellularity, neuron
Figure 10
Biological behavior: Residual evidence of prior cell death or inadequate migration.
Original INHAND
1
term (now
Nonpreferred (archaic) terms: decreased neuronal numbers, decreased neuronal cellularity.
Pathogenesis: prior necrosis/apoptosis of neurons, or rarely a developmental defect in which a certain neuronal population partially or completely fails to form.
Figures in original INHAND publication 1 : nos. 1 to 6.
Diagnostic features:
Generally presents as a region-specific decrease in neuron numbers or expected cell density. Depending on when the process occurs, there may be no other finding than a decrease in cell density at a particular site. In relatively acute lesions, occasional residual fragments of apoptotic or necrotic neurons and/or reactive macrophages engaged in phagocytosis of debris may be present. In chronic lesions, activated astrocytes or multilineage gliosis (mainly astrocytes and microglia) may be evident near the area of previous neuronal damage or fill the sites where neuronal perikarya are missing, but neuronal debris is absent (has been removed).
Special diagnostic techniques: Morphometric/stereological analysis of perfusion-fixed tissue is a powerful tool for precise quantification of decreased numbers of neurons.
Differential diagnoses:
Region-specific variation in neuron numbers or neuronal density (ie, “within normal limits”). Hypoplasia, neuron (decreased formation leading to fewer numbers of neurons in the absence of a secondary reactive response). Necrosis/apoptosis, neuron (prominent residual fragments of dying neurons and/or reactive macrophages engaged in phagocytosis of debris, with visible disruption of the adjacent neuropil). Neuronal abiotrophy (see Comment below).
Comment: The term “decreased cellularity, neuron” is reserved for situations where the reduction in neuron numbers can be clearly documented (by any method). If applied in the absence of a method that controls bias, such as stereology, this diagnosis should be made only when there is a clear signal.
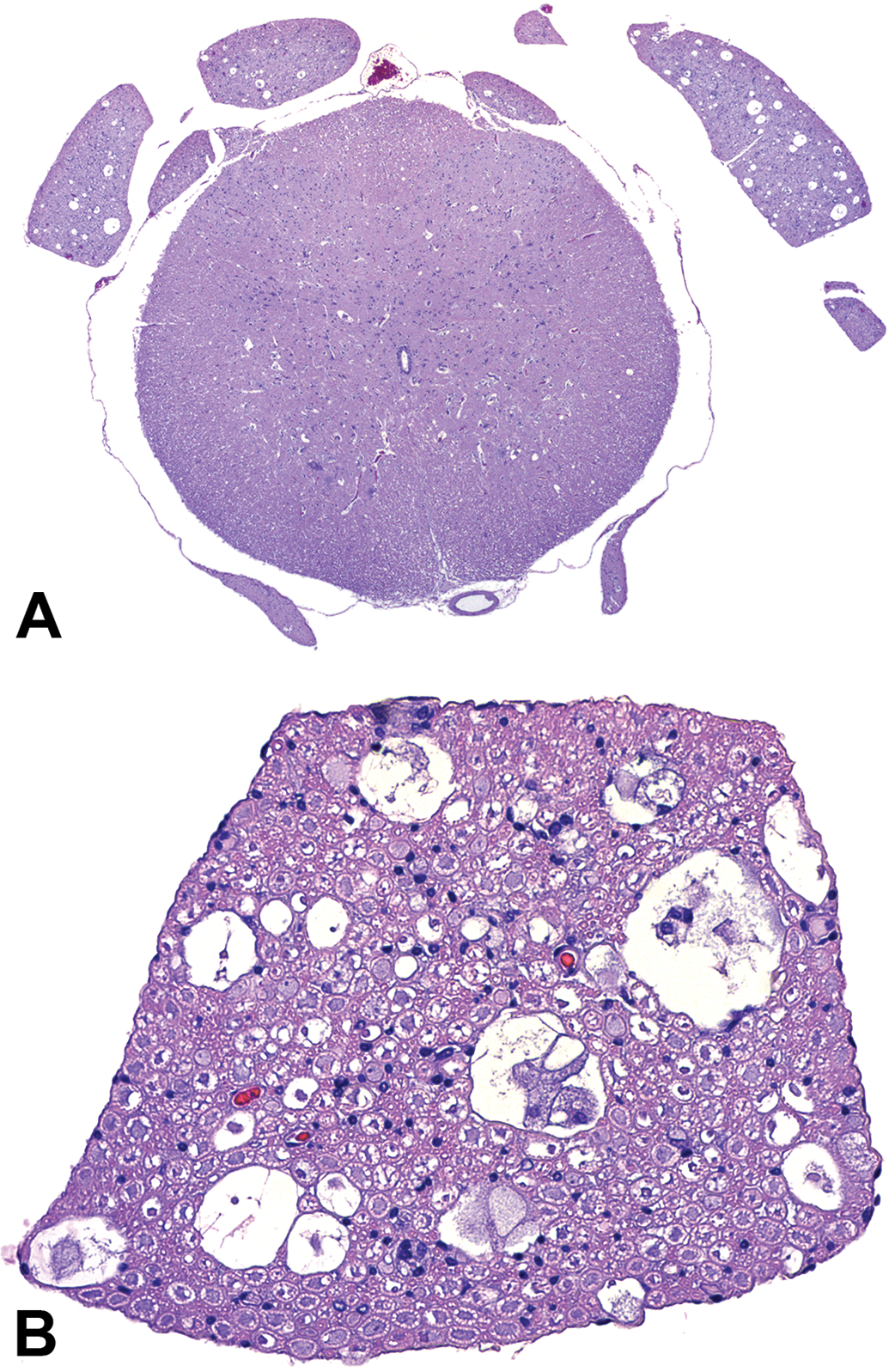
This histopathologic term refers either to a disappearance of neurons as the end-stage lesion of cell death (apoptosis or necrosis) within a focal area 72,73 (Figure 10A) or to a failure of adequate neuronal generation and/or migration during development 1,74 (Figure 10B). Degenerative changes within remaining neurons and/or reactive glial changes, such as an influx of macrophages or activation of astrocytes and/or microglia in surrounding tissue, may be present in situations where decreased neuronal cellularity follows neuron degeneration and therefore may aid in the diagnosis. Distinct evidence of dying neurons should always be described in specific terms (eg, necrosis). Very slow rates of neuronal cell loss that characteristically occur in various animal and human genetic disorders may be referred to as “neuronal abiotrophy,” indicating that cell loss arises from gradual atrophy/involution rather than abrupt degeneration.
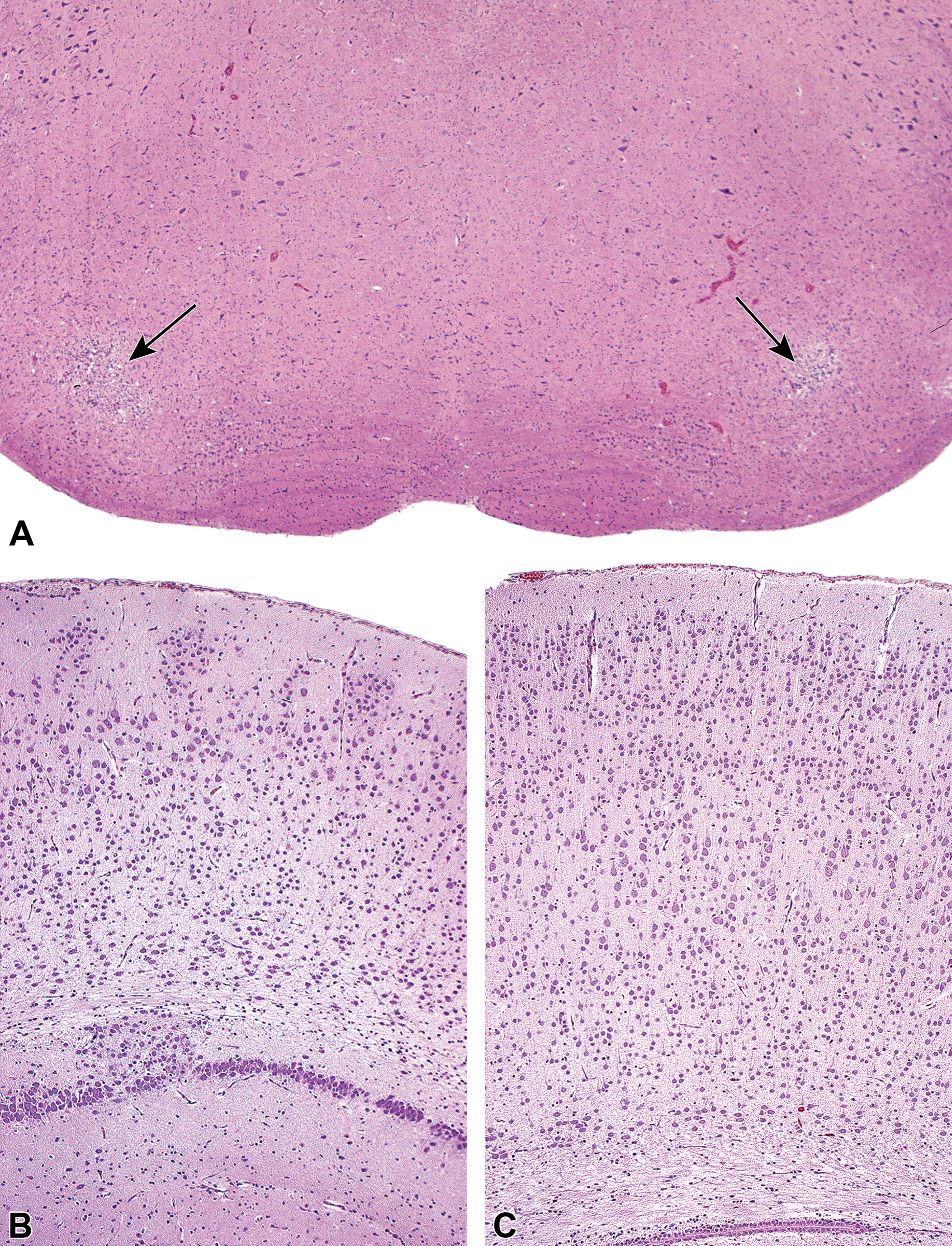
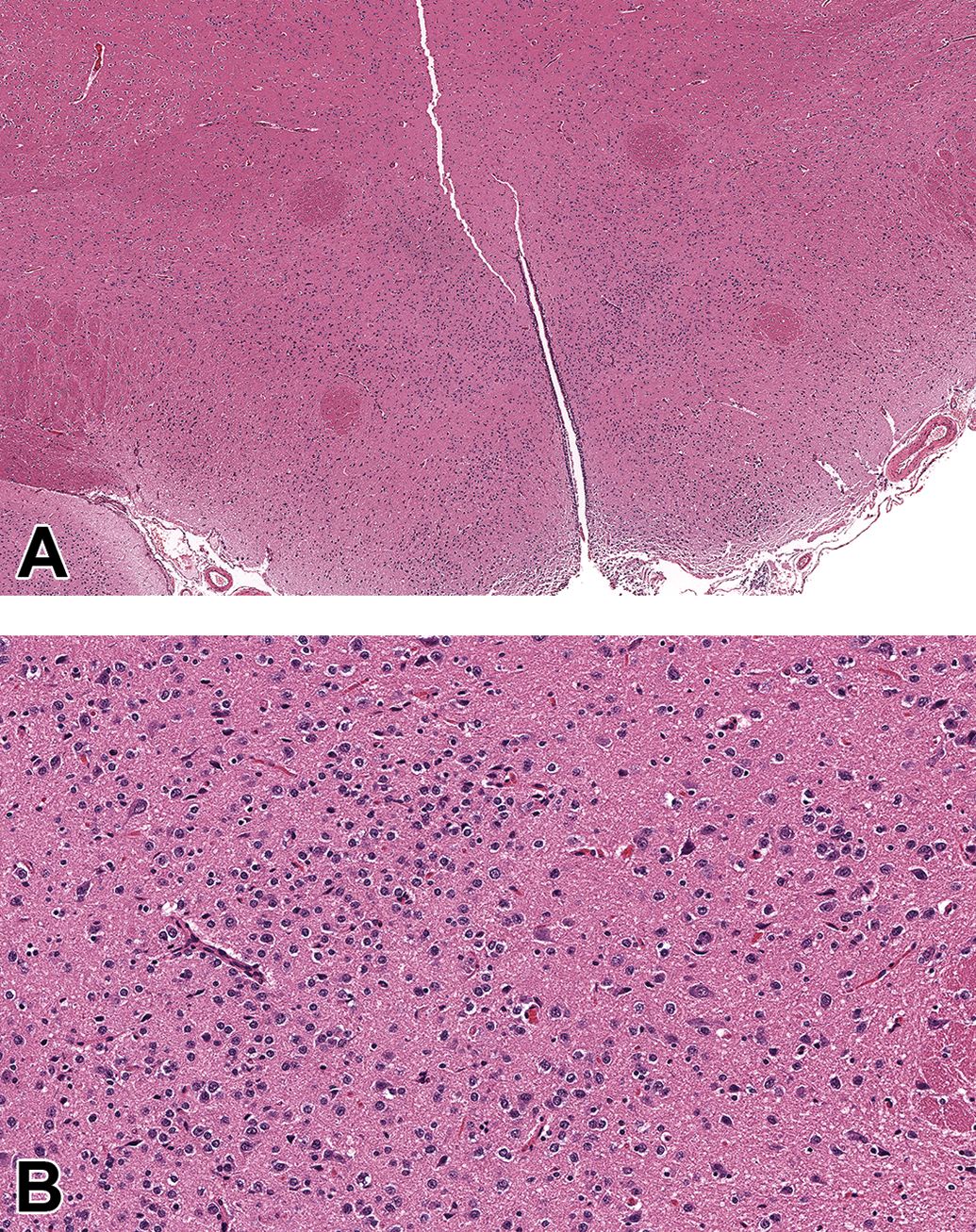
Decreased cellularity, neuron features a reduced number of neurons relative to the expected number for a given domain. This finding arises either secondary to the loss of previously formed neurons, as in these unique, bilaterally symmetrical foci of parenchymal necrosis with gliosis (arrows) affecting the rostral (superior) olivary nuclei of the ventral medulla oblongata (A), or as a consequence of abnormal neuronal degeneration during development, as in the widespread paucity of cerebrocortical neurons (B). A, Adult male F344/N rat following treatment with nitrobenzene (150 mg/kg) for 13 weeks (This image is reproduced with the permission of the U.S. National Toxicology Program). B, Young adult Wistar rat following maternal exposure to methylazoxymethanol (MAM, 30 mg/kg) on embryonic day 15 compared to (C) an age-matched control Wistar rat (These 2 images are reproduced from Kaufmann 74 with permission of John Wiley & Sons). Hematoxylin and eosin.
Although many industrial and environmental chemicals have been shown to directly destroy CNS neurons,
6
animal toxicity studies describing neuronal cell loss in rats and mice are relatively rare. Decreased neuronal cellularity has been induced in rats by carotid artery ligation
75
; treatment with alcohols (ethanol,
76
methanol,
77
and methylazoxymethanol
74
) and metals (trimethyltin
78
); neurotoxic analogs of excitatory amino acids that activate ionotropic glutamate receptors, such as kainic acid
79,80
and domoic acid
81
; or selective antagonists of N-methyl-
In rats, spontaneous, age-related decreases in neuronal cellularity affect Purkinje cells,
84
neurons in the suprachiasmatic nucleus,
85
and subcortical neurons.
86
Other reports describe spontaneous decreases in neuronal cellularity in the hippocampal CA3 region of rats in association with deficits in working memory.
87
The progressive, age-dependent loss of hippocampal neurons and subcortical nuclei can be delayed by treatment with acetyl-
Vascular
Necrosis/inflammation, media or wall, artery
Biological behavior: inflammation and fibrinoid necrosis of the arterial wall.
Original INHAND
1
term (now
Nonpreferred (archaic) terms: panarteritis nodosa, periarteritis, polyarteritis, polyarteritis nodosa.
Histogenesis: walls (especially the tunica media) of arteries (all sizes) and arterioles.
Pathogenesis: uncertain for spontaneous disease, presumably endothelial irritation or mural damage for agent-induced lesions.
Figures in original INHAND publication 1 : nos. 61 to 62.
Diagnostic features:
In the early stages, fibrinoid necrosis (incursion of eosinophilic, acellular material) of the tunica media occurs in conjunction with a mixed, but chiefly acute, inflammatory response.
Later stages may be characterized by several degenerative and inflammatory changes:
∘ Vessel proper: the inflammatory infiltrate contains many more mononuclear inflammatory cells and may be accompanied by intra-arterial fibrosis.
∘ Vessel proper: intimal proliferation and thrombosis may occur, resulting in narrowing and eventually obliteration of lumina. 90
∘ Perivascular connective tissue: expanded greatly by numerous mononuclear inflammatory cells and fibrosis.
Involvement of arteries in the CNS or adjacent to PNS structures may lead to compression atrophy and/or secondary inflammation in these tissues.
Special diagnostic techniques: Lesions on H&E-stained paraffin sections are characteristic, but may be confirmed by: Miller’s elastin stain, in which selective purple/black labeling of the arterial internal elastic lamina facilitates identification of disruption in this structure. Toluidine blue staining of perfusion-fixed tissue enhances visualization of vessel wall pathology.
Differential diagnoses: iatrogenic inflammation (associated with intrathecal placement of a cannula, often exacerbated by delivery of an antigenic or irritating pharmaceutical agent; recognized most readily by the presence of a narrow tract [following acute introduction of a needle] or circular cavity [associated with a chronically implanted cannula] within or adjacent to the inflamed tissue).
Comment: “Necrosis/inflammation, media or wall, artery” is the morphologic description of the neural manifestation of polyarteritis nodosa, a chronic, progressive, degenerative disease which most frequently occurs in aging male rats. 91 Inflammation and fibrinoid necrosis of the arterial wall may either begin in the endothelium and intima or else first affect the adventitia by extension from surrounding tissues and the vasa vasorum. This spontaneous disease usually affects the Sprague-Dawley and spontaneous hypertensive rat strains and is also rampant in rats with late-stage CPN. 91,92 In general, vasculature of the rat nervous system is spared. 93 A morphologically similar necrotizing polyarteritis presents as “Beagle pain syndrome” in young dogs due to involvement of the coronary arteries; other small- and medium-sized muscular arteries also are affected, but as with rats the blood vessels of the dog brain seldom are impacted. 94
The pattern of drug-induced vascular injury (DIVI) differs based on the nature of the test article and the test species. Small molecule-linked DIVI typically is confined to small- and medium-sized arterioles. In rodents, small molecule-induced inflammation and necrosis tends to impact medium-sized arteries in mesenteric and less commonly pancreatic and testicular vascular beds, while in dogs and NHPs small molecule DIVI affects cardiac vessels. 95 In contrast, biomolecule-related DIVI (which usually is evaluated in NHPs rather than rodents due to antigenicity considerations for human-based test articles) attacks a broader spectrum of vessels (small- and medium-sized arterioles but also venules and sometimes capillaries) spread among many organs—including choroid plexus in the brain. 95 These patterns may be helpful in distinguishing the likely mechanism of arterial wall inflammation and necrosis since blood vessels of neural organs are more likely to exhibit such lesions as a result of test article exposure rather than as a spontaneous background change.
Footnotes
Acknowledgments
The authors gratefully acknowledge the efforts of Dr. Georg Krinke, our now deceased colleague and friend, to the deliberations needed to produce this manuscript. We also appreciate the courtesy of Dr. Lucia Notterpek from the University of Nevada, Reno School of Medicine for kindly providing ![]() . Finally, the authors thank Ms. Beth Mahler for her consumate skill in optimizing the figures.
. Finally, the authors thank Ms. Beth Mahler for her consumate skill in optimizing the figures.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.