
Abstract
Harmonization of diagnostic nomenclature used in the pathology analysis of tissues from rodent toxicity studies will enhance the comparability and consistency of data sets from different laboratories worldwide. The INHAND Project (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice) is a joint initiative of four major societies of toxicologic pathology to develop a globally recognized nomenclature for proliferative and nonproliferative lesions in rodents. This article recommends standardized terms for classifying changes observed in tissues of the mouse and rat central (CNS) and peripheral (PNS) nervous systems. Sources of material include academic, government, and industrial histopathology databases from around the world. Covered lesions include frequent, spontaneous, and aging-related changes as well as principal toxicant-induced findings. Common artifacts that might be confused with genuine lesions are also illustrated. The neural nomenclature presented in this document is also available electronically on the Internet at the goRENI website (http://www.goreni.org/).
Keywords
Introduction
The INHAND Project (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice) is a joint initiative of the societies of toxicologic pathology from Europe (European Society of Toxicologic Pathology [ESTP]), Great Britain (British Society of Toxicological Pathologists [BSTPs]), Japan (Japanese Society of Toxicologic Pathology [JSTP]), and North America (Society of Toxicologic Pathology [STP]) to develop an internationally recognized nomenclature for proliferative and nonproliferative lesions in rodents. The present publication provides a set of standardized terms that are recommended for classifying lesions observed in the central (CNS) and peripheral (PNS) nervous systems of rodents. The diagnostic features are generally based on morphology in H&E-stained sections, but special diagnostic techniques are listed if applicable. The neural nomenclature presented in this document is also available electronically on the Internet at the goRENI website (http://www.goreni.org/). Standardized nomenclature for other organ systems (Renne et al. 2009; Thoolen et al. 2010) follows a similar anatomical approach and is or will be available at the same website in the future.
The global standardization and harmonization of nomenclature used in rodent studies for the diverse diagnostic features of proliferative and nonproliferative lesions will improve the comparability and consistency of study results from different laboratories located around the world, regardless of dissimilar cultural backgrounds and training practices and the length of time between studies. The global use of a harmonized nomenclature as a common scientific language in the pathology evaluation of rodent studies will assist all institutions and regulatory authorities that undertake hazard identification and risk assessment using rodent study data.
Morphology
The nervous system is an intricate cellular network with a complex three-dimensional organization whose anatomy and chemistry varies widely among regions (Bolon 2000) over very short distances (Switzer et al. 2011). As such, successful neuropathology analysis of the CNS and PNS typically requires advanced training and access to a variety of highly specialized reference materials (Bolon and Butt 2011; Bolon et al. 2011b). The key element for success in this endeavor is a detailed understanding of the intricate spatial and temporal diversity in the anatomic, functional, and molecular arrangement of major nervous system domains.
The two primary classes of functional cells in both the CNS and PNS are neurons and neuroglia (Summers et al. 1995). Other specialized cell lineages present in lesser amounts include choroid plexus, ependyma, meninges, and vascular endothelium. Diagnostic analysis in neuropathology is based on three consecutive, closely interrelated steps: (1) morphological assessment of the lesion, (2) topographical analysis of the lesions, and (3) integration of these findings. For a final etiological diagnosis, a subsequent examination using all available clinical, epidemiological, and molecular data informed by observations made at necropsy is necessary (Poirier et al., 1990).
Region-Specific Structure and Function
The CNS is commonly divided into two main divisions, the brain and spinal cord. The brain is then partitioned into three broad regions: forebrain, comprising the paired cerebral hemispheres and the diencephalon; midbrain, including the substantia nigra (SN) and the central components of the auditory and visual systems; and hindbrain, consisting of cerebellum, pons, and medulla oblongata. The “brain stem” consists of the midbrain and the ventral hindbrain (i.e., pons and medulla oblongata). The spinal cord is separated into four chief domains: cervical, thoracic, lumbar, and sacral. The PNS is generally divided into two systems, the somatic nerves (which carry motor and sensory signals) and the autonomic nervous system (which regulates internal homeostasis). Ganglia for cranial and spinal nerves are prominent at the intersection between the CNS and PNS.
The CNS has seven main parts (Amaral 2000; Kandel 2000). The most caudal part is the spinal cord. The gray matter contains the motor neurons responsible for both voluntary and reflex movements as well as interneurons responsible for reflex arc coordination, while the white matter includes longitudinal, ascending, and descending tracts of myelinated axons. The shape of the spinal cord varies along its length. Thickened zones at the cervical and lumbar intumescences contain the greater numbers of neurons required to supply the limbs, while the other regions that innervate the trunk are reduced in size as fewer neurons are required for this purpose. The second part is the medulla oblongata, which is the direct rostral extension of the cervical spinal cord and forms the caudal component of the brain stem. This region includes the centers responsible for vital autonomic functions (e.g., breathing, cardiac rhythmicity, digestion). The third part is the cerebellum, which modulates the range and force of movement as well as the ability to learn motor-related skills. It is connected to the rostral and caudal regions of the brain stem by several major fiber tracts termed peduncles. The fourth part is the pons, which is the middle portion of the brain stem. It is located ventral to the cerebellum. The pons contains the pontine nuclei, which relay information about movement and sensation from the cerebral cortex to the cerebellum, as well as centers involved in respiration, sleep, and taste. The fifth brain part, the midbrain, is the smallest and most rostral portion of the brain stem. It is located between the pons caudally and the diencephalon rostrally and contains many tiny but vital nuclei. One distinct midbrain nucleus of great clinical importance is the SN, which provides input to the basal ganglia (specifically the caudate nucleus and putamen) to regulate involuntary movement; attrition of the SN dopaminergic neurons is the characteristic neural lesion of Parkinson’s disease. The sixth main part is the diencephalon, the principal components of which are the thalamus dorsally and the hypothalamus ventrally. The thalamus plays a gating role to modulate sensory and motor information reaching the cerebral cortex from the rest of the CNS, while the many nuclei in the hypothalamus regulate autonomic, endocrine, and visceral functions. The last major brain part is made up of the cerebral hemispheres, which comprise the largest part of the mammalian CNS. This region consists of the cerebral cortex, the internal capsule (white matter tracts) immediately beneath it, and three deep neuron-dominated domains: the amygdala (which mediates social behavior and the expresison of emotions); the basal ganglia (which controls involuntary, fine movements); and the hippocampal formation (which supports memory). External surfaces of the brain and spinal cord are covered by a trilaminar set of meninges: the pia mater, the delicate inner layer; the arachnoid, the granular intermediate zone responsible for fluid regulation; and the dura mater, the tough outer jacket. The neuropil (i.e., the CNS parenchyma) is penetrated by numerous capillaries. The structural specialization of these vascular channels, such as an absence of fenestrations as well as numerous tight and adherens junctions, forms a major anatomic substrate for the blood–brain barrier (BBB; Willis 2011).
The CNS ventricular system is an interconnected series of fluid-filled reservoirs within the brain and the spinal cord. Two lateral ventricles located within the cerebral hemispheres are connected via foramina (of Monro) to the third ventricle within the diencephalon. This compartment is linked via the mesencephalic aqueduct (of Sylvius) to the fourth ventricle, which is located in the gap between the cerebellum above and the pons/medulla oblongata below. In rodents, this chamber drains either into the spinal cord as the central canal or else into the cisterna magna via the lateral apertures (of Luschka). The ventricles and central canal are lined by ependymal cells. The ventricles (especially the lateral and fourth chambers) contain prominent tufts of choroid plexus, which is specialized to generate cerebrospinal fluid (CSF; Johanson et al. 2011); choroid plexus epithelial cells are also predilection sites for a number of systemic disorders (Greaves 2000). The circumventricular organs are six special neuronal aggregates located in close proximity to the ependymal lining of the ventricular system (Garman 2011).
The PNS consists of ganglia and nerves. The characteristic neuronal and glial composition of ganglia varies by system (e.g., autonomic vs. somatic) and site. Nerves consist of axons (or “nerve fibers”) encased in thin layers of connective tissue. Myelinated axons are surrounded by numerous myelin layers, while unmyelinated axons (e.g., the majority of postganglionic axons in autonomic ganglia and axons of smaller neurons in sensory nociceptive ganglia) are insulated in the cytoplasm of glial cells.
Cytoarchitectural Features
The diagnostic nomenclature of the nervous system is based on the neural cell type that is primarily damaged, and on the specific cellular components that are altered in the principal target location/locations. Therefore, diagnostic pathologists first must know the normal features of the neurons, glia, and other cell lineages; their interrelationships; and their normal variations in shape and size in various regions of the nervous system (Garman 2011).
Neurons are the chief functional elements of the CNS and of ganglia in the PNS, and their axonal processes are the key functional components of the PNS. The typical neuron has four distinct parts: a cell body (soma or perikaryon), multiple dendrites (which receive incoming signals), a single axon (which carries outgoing signals), and presynaptic terminals (which participate in the formation of synapses). Neurons may be large, like motor neurons in the ventral horn of the spinal cord, or small, like granule cells in the cerebellar cortex; they may have a rich and extensive dendritic tree, such as cerebellar Purkinje cells, or very few dendrites, like neurons in dorsal root ganglia.
Glia support neural function in multiple ways. The macroglia consist of astrocytes, which perform multiple functions, and the myelinating cells: oligodendrocytes of the CNS and Schwann cells of the PNS. Astrocytes account for approximately 50% of all glia; the ratio of astrocytes to neurons is 60:40 in the rat (vs. 100:10 in humans; Montgomery 1994). The two main astrocyte phenotypes are protoplasmic (which predominate in normal conditions) and fibrous (which are increased in response to neural injury). Astrocytes repair neural damage by acting as phagocytes and filling cavities (i.e., forming scars), aid signal transmission by regulating neurotransmitter levels within synapses, promote neuronal survival and repair via production of growth factors, control the neural microenvironment via their role in the BBB and pia-derived glial membrane, sustain cerebral angiogenesis, and provide structure to the neuropil. In addition, astrocytes participate in detoxification (e.g., glutamate metabolism) and toxification (e.g., conversion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP] to the neurotoxic metabolite MPP+), and specialized radial glia act as conduits for neuronal migration and guides for axonal extension during development. Microglia are mesoderm-derived CNS glia that arise outside the nervous system from macrophages. Their primary function is protection of the CNS by immune surveillance and phagocytosis, but they also play a role in maintaining BBB function in injurious situations (Kofler and Wiley 2011). Following injury, resting microglia interact with neurons and astrocytes in becoming mobilized (i.e., activated), after which they migrate to sites of injury (especially necrosis), proliferate, and embark on their phagocytic careers. Microglia also act during neurodevelopment to remove nonviable and excessive neurons and glia that have undergone programmed cell death (Amaral 2000; Spencer 2000; Kierszenbaum 2002).
Morphologic Analysis of the Nervous System
Neuropathology screening during rodent preclinical toxicity studies is tailored to the nature of the experimental question. The number of regions evaluated during general toxicity studies (i.e., where a priori suspicion of neurotoxicity is not present) is abbreviated relative to the more expansive tissue list assessed in the course of specialized (i.e., neurotoxicity focused) studies. For general toxicity studies, neural tissues are often removed and fixed by immersion in neutral buffered 10% formalin or a similar fixative. This processing approach is relatively simple but commonly results in the generation of artifacts that are subject to misdiagnosis as neurotoxic lesions by inexperienced practitioners (Garman 1990; Jortner 2006). For specialized neurotoxicity studies, such artifacts are substantially reduced or eliminated by fixing neural tissues in situ using an intravascular perfusion approach (Fix and Garman 2000; Bolon et al. 2006; Jordan et al. 2011). Tissues from the PNS may require special processing techniques (e.g., plastic embedding) depending on the nature of the lesions to be evaluated and/or specific regulatory requirements (Bolon et al. 2011a; Jortner 2011).
The typical battery of neural tissues examined in both general toxicity studies and specialized neurotoxicity studies includes multiple sections taken through the forebrain, single sections across the midbrain and the hindbrain, sections through the cervical and lumbar levels of the spinal cord, and a single segment of a peripheral somatic nerve (e.g., OECD 1998; US EPA 1998a). Literature reports indicate that several areas of the rodent brain—cerebral cortex, hippocampal formation, and cerebellum—are particularly sensitive to toxic insults. In rodent studies, two coronal (cross) sections of forebrain are recommended (Solleveld and Boorman 1990; Radovsky and Mahler 1999) to include the cerebral hemispheres (with the frontal, cingulate, parietal, occipital, temporal and piriform cortices), the basal ganglia (caudate nucleus and putamen), corpus callosum (the prominent bridge formed of fibers connecting bilaterally symmetrical regions of the cerebral hemispheres), and the hippocampal formation. The caudal section of forebrain will include the hippocampus and diencephalon (thalamus and hypothalamus). The midbrain section should attempt to sample the SN. The hindbrain section usually incorporates the cerebellum and medulla oblongata; as a rule, pons is not examined in routine rodent studies (Solleveld and Boorman 1990, Radovsky and Mahler 1999). Spinal cord is sampled at the cervical and lumbar levels, ideally through the intumescences (which contain the neurons which supply motor axons to the somatic nerves that supply the fore and hind limbs, respectively), and optionally also at the midthoracic level (where the neurons in the lateral [intermediate] horn send axons to the sympathetic division of the autonomic PNS). The PNS is generally screened by examining a segment of the proximal sciatic nerve (the somatic nerve that carries axons from the motor neurons of the lumbar intumescence). Additional structures that are routinely sampled in the course of specialized neurotoxicity studies include pons, multiple dorsal root ganglia, and distal somatic nerves (OECD 1997; EPA 1998b; Bolon et al. 2006) and other structures as necessary (e.g., caudal colliculus; Morgan et al. 2004). This enhanced neural sampling strategy is employed, and generally mandatory, when a substance is suspected to have neurotoxic potential based on molecular similarities to known neurotoxicants, a putative mode of neurotoxic action, clinical data indicative of neural dysfunction, or a demonstrated capacity to produce structural lesions in the CNS and/or PNS.
Nomenclature
This lexicon has been organized in tiers based on lesion class (nonproliferative or proliferative), cell type (e.g., neuron, glia), and sometimes by cellular target (e.g., cell body, axon, myelin). The terms under each tier are alphabetized for easier retrieval. A final set of terms has been included to demonstrate common artifacts which have been misidentified as neurodegenerative or neurotoxic lesions by inexperienced diagnosticians.
All terms should be used with a clear relation to their topography within the CNS and/or PNS. Important considerations will be to define the major CNS area/areas or PNS structure/structures that are involved, including if necessary descriptors for specific subregions (the granule cell layer of the cerebellar cortex, the CA1 domain of the hippocampus, etc.). The subregion may be defined as a part of the diagnostic string for a neural lesion (e.g., “Brain: necrosis, diffuse, marked, Purkinje cells, cerebellum,” where “Brain” is the tissue), or the affected location may be accorded status as a tissue (e.g., “Cerebellum, Purkinje cell layer: necrosis, diffuse, marked,” where “Cerebellum, Purkinje cell layer” is the tissue). Either approach is acceptable as long as the designation is as specific as possible with respect to the damaged cell population/populations.
Nonproliferative Lesions
The most common nonproliferative anomalies in the CNS include damaged or dying cells (especially neurons, their axonal processes, and the myelin sheaths that insulate the axons) and the various reparative changes designed to minimize (in the CNS) or reverse (in the PNS) the damage. At the cellular level, the most important processes will involve neurons, various glial lineages, and blood vessels.
Neuron—Cell Body
Cell loss, neuronal (Figures 1–6)
Biological behavior: cell death
Synonym/synonyms: decreased neuronal numbers, decreased neuronal cellularity
Pathogenesis: prior necrosis/apoptosis of neurons, or rarely a developmental defect in which a certain neuronal population partially or completely fails to form
Diagnostic features:
generally presents as a region-specific decrease in neuron numbers; in relatively acute lesions, occasional residual fragments of dying neurons and/or reactive macrophages engaged in phagocytosis of debris may be present; in chronic lesions, activated astrocytes or multilineage gliosis may be evident near the damaged neurons or fill the sites where neuronal perikarya are absent, but neuronal debris has been removed.
Special diagnostic techniques: morphometric/stereological analysis of perfusion-fixed tissue is a powerful tool for precise quantification of neuronal cell loss.
Differential diagnoses: region-specific variation in neuron numbers (i.e., within normal limits); neuronal hypoplasia (decreased numbers with no secondary reactive response); neuronal necrosis/apoptosis (prominent residual fragments of dying neurons and/or reactive macrophages engaged in phagocytosis of debris, with visible disruption of the adjacent neuropil).
Comment: The term neuronal cell loss is reserved for situations where the reduction in neuron numbers can be clearly documented (by whatever method). This histopathologic term refers to disappearance of neurons as the end-stage lesion of cell death (apoptosis or necrosis) within a focal area. Degenerative changes within remaining neurons or reactive glial changes, such as an influx of macrophages or activation of astrocytes, in surrounding tissue aid in the diagnosis. Distinct evidence of dying neurons should always be described in specific terms (e.g., necrosis). Very slow rates of neuronal cell loss that characteristically occur in various animal and human genetic disorders may be referred to as neuronal abiotrophy, indicating that cell loss arises from gradual atrophy/involution rather than abrupt degeneration.
Although many industrial and environmental chemicals have been shown to directly destroy CNS neurons (Greaves 1990), preclinical toxicity studies describing neuronal cell loss in rats and mice are relatively rare. Neuronal cell loss has been induced in rats by carotid artery ligation (Bendel et al. 2005) and by treatment with trimethyltin (Little et al. 2002; Philbert, Billingsley, and Reuhl 2000) or neurotoxic analogs of excitatory amino acids that activate ionotropic glutamate receptors, such as kainic acid (Liang et al. 2007). Clinoquinol, a well-known cause of subacute myelooptic neuropathy in humans, induces moderate loss of neurons in the hippocampus of mice (Koga et al. 1988).
In rats, spontaneous, age-related, neuronal cell loss affects Purkinje cells (Rogers et al. 1984), neurons in the suprachiasmatic nucleus (Chee et al. 1988), and subcortical neurons (Sabel and Stein 1981). Other reports describe spontaneous neuronal cell loss in the hippocampal CA3 region in association with deficits in working memory (Kadar et al. 1990). The progressive, age-dependent loss of hippocampal neurons and subcortical nuclei can be delayed by treatment with acetyl-
In developmental (neurotoxicity) studies, the more specific term neuronal hypoplasia should be used to describe the morphological pattern in the offspring. This term indicates that the finding is a decrease in neuronal cell number only (without any other reactive cell response) of one or more major brain areas due to an impact on development.
Chromatolysis (Figures 7 and 8)
Biological behavior: reactive change to the perikaryon in response to axonal injury
Synonym/synonyms: axonal reaction, central chro-matolysis
Histogenesis: neurons
Pathogenesis: dispersion of clustered rough endoplasmic reticulum (RER) following cell injury to permit accelerated protein synthesis needed to complete repairs
Diagnostic features:
“central chromatolysis,” the most typical presentation in H&E-stained paraffin sections, is characterized by a substantial decrease or total loss of intracytoplasmic Nissl bodies (termed compact basophilia) in the cell center. Affected neurons have few to no Nissl bodies and eosinophilic cytoplasm, usually in conjunction with a round and swollen cell profile and peripheral translocation of the nucleus; enlargement of nucleoli may be observed; “peripheral chromatolysis” may be observed in partially repaired neurons, as Nissl bodies reform first near the cell center.
Special diagnostic techniques: stains for Nissl bodies (e.g., cresyl violet [or cresyl echt violet]) can facilitate detection.
Differential diagnoses: none
Comment: Chromatolysis is a reactive change in the perikarya of damaged medium- to large-sized neurons. Factors that can elicit this change include infections, ischemia, metabolic dysfunction, some toxicants, and trauma. The classic cause is transection of the axon (hence the synonym, axonal reaction). However, the change can also be engendered by direct harm to the cell soma or as a secondary response to primary demyelination (Duchen 1992).
In normal neurons, Nissl bodies composed of RER intermingled with myriad polyribosomes are widely dispersed in the cytoplasm. In injured neurons, the Nissl bodies undergo partial to complete dissolution, thus releasing ribosomes needed to manufacture new proteins required to repair the damaged cell infrastructure.
Central chromatolysis, the most typical presentation, is characterized by Nissl body loss in the center of the cell (perikaryon). This cell phenotype is most pronounced soon after the neuron has sustained injury. If the neuron survives the damage, the appearance of the perikaryon is restored to normal in reverse order once peripheral connectivity is reestablished. Ribosomes normally clustered in the cytoplasm as Nissl bodies are dispersed in the process of recovery and are reformed centrally to peripherally as the recovery process completes. Peripheral chromatolysis may also occur, particularly during later stages of neuron regeneration when the declining rate of protein production allows Nissl body ribosome depots to be reformed (McMartin et al. 1997). Ancillary changes that may accompany chronic chromatolysis include axonal atrophy (especially if the affected axon is confined to the CNS, where repair efforts are usually obstructed) and/or gliosis (via astrocytic or microglial proliferation; Summers, Cummings, and DeLahunta 1995a).
Heterotopia, neuronal (Figures 9 and 10)
Biological behavior: self-limiting developmental disturbance
Synonym/synonyms: ectopia
Histogenesis: neuronal precursors
Pathogenesis: abnormally positioned neuronal clusters due to aberrations in early neuronal migration and/or terminal differentiation
Diagnostic features:
neuronal clusters in atypical locations (e.g., within parenchyma, around ventricles, or beneath meninges), most commonly affecting the cerebral cortex, cerebellum, and hippocampus; loss of the normal laminar pattern of neuronal organization in the cerebral cortex; major heterotopiae are often associated with other brain malformations, such as hypoplasia of major brain areas or internal hydrocephalus (ventricular expansion); ectopic neurons generally display the morphologic features of the brain region in which they normally should have been found.
Special diagnostic techniques: none
Differential diagnoses: none
Comment: The presence of any heterotopic focus is indicative of disturbances during early CNS development. Severe heterotopiae has been linked to functional deficits, particularly hyperexcitability and related electrophysiological deficits, as a consequence of abnormal neural circuit organization (Gabel and LoTurco 2001).
Major heterotopiae are considered indicative of significant neurotoxic insults and are irreversible (Kaufmann 2000; Kaufmann 2011). Many patterns of cell displacement with profound neurobehavioral defects are characteristic of fetal alcohol syndrome (FAS) in humans; neuropathologic lesions and behavioral deficits in mouse and rat models of FAS exhibit a high degree of concordance with the human condition (Harper and Butterworth 1997). In developmental neurotoxicity (DNT) studies in rats, major heterotopiae are observed when dams are treated during neurogenesis with a single high dose of antimitotic agents, such as methylazoxymethanol (MAM, a common positive control agent in DNT studies). The induced pattern of lesions in the offspring varies with the gestational day on which the dam was treated. Heterotopiae with hyperexcitability is induced in rats by MAM (Kaufmann and Gröters 2006).
Minor heterotopiae may be seen spontaneously, may be reversible over time, and are of no reported clinical significance. In standard rodent studies, minor heterotopiae are not diagnosed, and thus either are not recognized or are not present. Incidences for minor heterotopiae are not reported in the current literature, but the experience of the working group members is that they are very low.
Necrosis, neuronal (Figures 11–18)
Biological behavior: degenerative lesion Synonym/synonyms: homogenizing cell change, ischemic cell change, metabolic arrest change, oncotic necrosis, “red dead” neurons Histogenesis: neuronal cell bodies Pathogenesis: recent cell death, typically affecting multiple adjacent cells of a given population or within a particular neural region Diagnostic features:
slightly shrunken, often angular neurons with hypereosinophilic cytoplasm in H&E-stained paraffin sections; nuclear consolidation, sometimes with shrinkage (early stages); karyorrhexis or karyolysis (later stages). Special diagnostic techniques: Necrotic neurons are specifically labeled by:
Fluorescent stains (e.g., Fluoro-Jade B or Fluoro-Jade C; Schmued et al. 2005) performed on 5- to 10-µm-thick (i.e., routine) perfusion-fixed, paraffin-embedded sections. Affected cells appear bright green against a dark field. (Note: erythrocytes that remain in blood vessels of immersion-fixed specimens will autofluoresce, which may make detection of necrotic neurons more difficult.) Cupric–silver stains (Switzer 2000) in 30- to 40-µm-thick frozen sections of unfixed tissue. Affected cells are black against a pale yellow background. dark neuron artifact (evident as spiky basophilic neurons due to shrinkage of both nucleus and cytoplasm with contracture of the neuronal cell body, often associated with a prominent, tortouous, basophilic apical dendrite)
Differential diagnoses:
Comment: Neuronal necrosis is a common end-stage cellular response to irreversible injury. This lesion may be induced by many causes, the most common of which are ischemia, metabolic dysfunction, or exposure to certain toxicants (chemicals, drugs, or metals). Many different mechanisms can initiate the intracellular biochemical changes that ultimately lead to neuron destruction, but as typically used in neuropathology the term neuronal necrosis refers to the pathway by which disruption of cellular energy systems results in fluid accumulation within organelles (microvacuolation) and eventually the entire soma (swelling, or oncosis) rather than to the apoptotic cascade (Levin et al. 1999). Gliosis (astrocytic or microglial or both) often serves as an ancillary, nonspecific indicator of prior neuronal necrosis in chronic lesions (McMartin et al. 1997).
Neuronal cell death includes a spectrum of changes in addition to the well-known shrunken eosinophilic (red dead) profile described here. Other lesions in the continuum include neuronal swelling and lysis as well as “ghost forms” (i.e., empty plasma membranes, representing the last remaining evidence of dead cells).
Genuine necrotic lesions comprising basophilic neurons have been demonstrated following peracute neuronal damage, such as that induced by electrical or mechanical damage (Csordás, Mázló, and Gallyas 2003; Zsombok, Tóth, and Gallyas 2005). This finding is sometimes referred to in human neuropathology as chronic nerve cell change. Soon after injury, lethally affected cells will exhibit shrunken, pale basophilic cytoplasm, sometimes in conjunction with a slightly darker nucleus. This change progresses in hours to the more typical hypereosinophilic appearance that is characteristic of cells that have been dead for several days.
The major differential diagnosis for neuronal necrosis in H&E-stained paraffin sections is dark neuron artifact, a consequence of pressure applied to unfixed or inadequately fixed neural tissue (see definition of dark neuron artifact in this glossary). This change usually presents as clusters of moderately shrunken, angular neurons having dense basophilic cytoplasm, a dark condensed nucleus, and often a spiraling apical dendrite (Duchen 1992; Jortner 2006). Major predilection sites are cerebral cortex (especially the middle layers), hippocampus (CA1 and CA3), cerebellum (Purkinje cells), and spinal cord (large motoneurons of the ventral [anterior] gray horn). Dark neuron artifact usually, but not always, may be differentiated from the acute (basophilic) variant of neuronal necrosis by the the more condensed and dark appearance of the cytoplasm, nucleus, and apical dendrite as well as the time course of the neural damage.
Neuronophagia (Figures 19–22)
Biological behavior: a microglial response to neuronal damage, culminating in phagocytosis of the degenerating neurons
Synonym/synonyms: microglial cell nodule
Histogenesis: resident microglial cells and their circulating precursors (monocyte lineage)
Pathogenesis: microglia respond to proinflammatory signals (chemokines and cytokines) or altered cell surface markers present on degenerating or virus-infected neurons.
Diagnostic features: Infiltrates of microglia located adjacent to neurons are readily recognized in H&E-stained sections by their characteristic cytoarchitectural features:
microglia nuclei in early stages of activation are pale, elongated, and sometimes irregularly contoured, whereas nuclei of fully activated cells are larger and have more rounded profiles; the ramified cytoplasm of microglia is indistinguishable without special stains early in activation, but in older lesions the cells come to resemble macrophages, complete with debris-filled cytoplasm; associated neurons typically have morphologic features consistent with degeneration;
Special diagnostic techniques: Although neuronophagia is readily recognized in H&E-stained sections, detection of markers specific for activated microglia may be used for confirmation.
Immunohistochemistry options: CD11b/c (mice); CD45; ionized calcium-binding adapter molecule 1 (Iba1, a marker for activated macrophages/microglia); OX-42, the rat counterpart of CD11b; as well as more general markers for macrophages and monocytes such as CD68, ED-1 (a rat marker for activated macrophages/microglia), ED-2 (specific for rat peripheral macrophages), F4/80 (a mouse macrophage marker), Mac-1, and RM-4 (a marker for rat dendritic cells and macrophages; Fix et al. 1996; Gehrmann et al. 1995; Ito et al. 2001; Mander and Morris 1995; Nagatani et al. 2009). Lectin histochemistry option: Griffonia simplicifolia (GS-IB4).
Differential diagnoses: none
Comment: Neuronophagia represents the process by which microglia (the resident phagocytic cells of the CNS) are activated to remove the degenerating neurons (McMartin et al. 1997; Summers, Cummings, and DeLahunta 1995). Neuronophagia may be encountered in any lesion characterized by neuronal death—most typically eosinophilic neuron degeneration, which is the most common cytomorphologic pattern of neuronal necrosis (Kelley, Lifshitz, and Povlishock 2007). Depending on the causes and circumstances surrounding neuronal injury, activated microglia may contribute to the degenerative process, strip neurons of their synaptic processes, or even play a protective role (Neumann et al. 2006; Stoll and Jander, 1999).
Neuronophagia is most prominent in lesions in which dying neurons have altered or foreign surface antigens, or have surface antigen/antibody complexes. In these latter conditions, early stages of neuronophagia may be characterized by microglia that surround normal-appearing neurons. Nevertheless, evidence of frank neuronal degeneration will typically be found elsewhere in the sections. In addition, microglial nodules may also be found without any recognizable neurons in association with them.
In toxicant-associated CNS lesions, infiltrating microglial cells commonly exist in relatively close proximity to degenerating neurons but often do not show the prominent degree of neuronophagia that is seen in immune-mediated and viral-induced encephalitides. Therefore, the principal diagnosis in cases of neurotoxic neuronal degeneration is likely to be “acute neuronal necrosis/neuronal cell loss,” with neuronophagia merely being accepted as an expected secondary component of the degenerative process.
Vacuolation, neuronal (Figures 23–26)
Biological behavior: expansion of intraneuronal cytoplasm or membrane-bound organelles
Synonym/synonyms: none
Histogenesis: neurons (cell bodies or processes)
Pathogenesis: retention of fluid or metabolic by-products inside a subcellular compartment
Diagnostic features: cytoplasmic vacuolation (usually clear or pale eosinophilic) of neurons in CNS gray matter or PNS ganglia
Special diagnostic techniques: Neuronal vacuoles in some storage diseases or in induced phospholipidosis may be confirmed by electron microscopy or using special stains (e.g., Luxol fast blue [LFB], periodic acid-Schiff [PAS], or Sudan black) to detect a specific biochemical component within vacuoles
Differential diagnoses:
demyelination (see definition in this glossary); vacuolation of white matter (spongy change or edema; see definition in this glossary); vacuolation artifact due to postmortem autolysis; improper collection, handling, or fixation of neural tissue at the time of necropsy; or prolonged immersion (e.g., over the weekend) in ethanol baths during tissue dehydration. An identified artifactual vacuolation should not be recorded in the pathology findings data set.
Comment: Vacuolation is among the most frequent changes detected in CNS tissue. However, it is not always easy to distinguish pathologic (real) from artifactual vacuolation. The distribution and colocalization of other structural abnormalities—such as reactive gliosis, axonal spheroids, or cellular debris—aid in differentiating genuine vacuolar lesions from artifactual changes.
Vacuolation of the neuron cell body and neuropil is a characteristic lesion of spongiform encephalopathies such as Creutzfeldt-Jacob disease in humans (Dearmond and Prusiner, 1997) and comparable naturally occurring conditions reported in many domestic animal species but not in rodents (Summers, Cummings, and DeLahunta 1995). In the neuropil, vacuoles are seen in neuronal perikarya, dendrites, and axons. They may be shown to be bounded by single or double membranes via electron microscopy.
Neuron—Axon
Atrophy, axonal
Biological behavior: an adaptive, transient, or progressive morphological feature of regressing cell processes
Synonym/synonyms: somatofugal atrophy
Histogenesis: axonal processes of neurons
Pathogenesis: disturbed anterograde (central to peripheral) transport of essential structural molecules
Diagnostic features:
diminished average axonal diameter with distended interaxonal spaces; may exist in conjunction with axonal swelling.
Special diagnostic techniques: Axonal atrophy may be evaluated directly by examining axonal structure or indirectly by assessing the morphology of the myelin that ensheathes shrunken axons. Options include:
routine histological stains to demonstrate axons (e.g., silver impregnation stains [Bielschowsky’s, Bodian’s]) or myelin (e.g., LFB) in the CNS and PNS; common immunohistochemical methods to reveal axons (e.g., anti–neurofilament protein [NFP]) or myelin sheaths (e.g., antimyelin basic protein [MBP]) in the CNS or PNS; teased fiber preparations applicable to PNS axons and myelin (Krinke, Vidotto, and Weber 2000a).
Differential diagnoses: endoneurial edema (widened interaxonal spaces without significant lessening in the average axonal diameter (McMartin et al. 1997))
Comment: Axonal atrophy as a pathologic process is generally most prominent in the PNS (McMartin et al. 1997). The basis for this site predilection is that the entire length of the axon must be sustained by proteins transported from the cell body (soma). Thus, the most vulnerable portions of CNS neurons following disruptions in active transport are the distal elements of long axons. The main mechanisms for primary axonal atrophy are inadequate anterograde transport of neurofilaments, the main proteins in the axoplasm and thus a major determinant of axonal caliber (Summers, Cummings, and DeLahunta 1995), and inhibited retrograde transport of target-derived trophic signals to the neuron soma (Gold, Griffin, and Price 1992). Axons may shrivel in the absence of axonal loss or neuronal lesions (Elder et al. 1999), although axonal atrophy may also be a precursor lesion to eventual axonal fragmentation or loss (Gold, Griffin, and Price 1992). Axonal atrophy also occurs as a secondary consequence to primary demyelinating diseases (Hanemann and Gabreels-Festen 2002). Finally axonal atrophy evolves during normal neurodevelopment in axonal branches that fail to reach an appropriate target tissue (a withdrawal [retraction reaction] rather than degeneration; Bernstein and Lichtman 1999).
Atrophied axons are best observed following a widespread insult (e.g., chemical exposure, surgical manipulation) because numerous axons in multiple nerves are affected. However, axonal atrophy can occur focally if an adjacent axon is so engorged that it impinges on its neighbors (McMartin et al. 1997). In general, axonal atrophy is most evident in cross sections of neural tissue; in the PNS, the lesion is often best revealed in plastic sections. Ancillary indicators of axonal atrophy may be the presence of infolded myelin loops (loss of myelin circularity; Krinke et al. 1988), which represents an acute secondary accommodation by fully functioning myelin sheaths to the pathologic decline in axonal size or astrogliosis as a chronic response (Andersson et al. 2005).
Degeneration, axonal (Figures 27–31)
Biological behavior: breakdown of axonal structure
Synonym/synonyms: axonopathy, dying-back axonopathy, nerve fiber degeneration, Wallerian-type degeneration
Histogenesis: axonal processes of neurons (mainly affecting cells in the CNS)
Pathogenesis: primary axonal injury, or a secondary response to primary myelin damage.
Diagnostic features:
the characteristic early finding is multiple, swollen, eosinophilic axons (spheroids; McMartin et al. 1997). Myelin sheaths are typically unaffected in such acute lesions; the principal late-stage lesion is axonal fragmentation with the formation of digestion chambers containing phagocytic macrophages (gitter cells) and central axonal fragments. Ovoids of degenerated myelin may develop secondarily; these changes are often accompanied in the PNS but not the CNS by attempted nerve fiber repair: axonal regeneration, proceeding from central to peripheral, in association with Schwann cell proliferation.
Special diagnostic techniques:
routine histological evaluation of plastic- or resin-embedded, toluidine blue-stained sections (mainly used for the PNS; Greaves 2007); cytoskeletal labeling of CNS or PNS axons by histological stains (e.g., silver impregnation [Bielschowsky’s, Bodian’s]) or immunohistochemistry (e.g., anti-NFP); teased fiber preparations, applicable to PNS axons and myelin (Krinke, Vidotto, and Weber 2000a).
Differential diagnoses: axonal dystrophy (see definition in this glossary)
Comment: The term axonal degeneration is the preferred term for this change as it is the most general form. The alternate designations are specific to certain disease processes or pathogeneses.
“Axonopathy” implies a primary axonal injury that results in loss of the distal axon without a major impact on the cell body; it can be modified to denote the location of the lesion along the length of the axon (e.g., central or peripheral, proximal or distal). For example, distal axonopathy (e.g., induced by exposure to organophosphorus esters) typically involves the largest and longest axons, such as those of peripheral nerves, the proprioceptive and motor tracts of the spinal cord, the optic tract, and other long peripheral nerves.
Other terms imply knowledge of the lesion pathogenesis. For instance, the term Wallerian degeneration has attained such frequent use that it is now employed for nearly any type of axonal disintegration. However, in the strict sense this term refers to active dissolution of the distal extremity of a myelinated axon following surgical transection. Where axonal degeneration is thought to occur through a similar event (i.e., chemical transection by a neurotoxicant), the term Wallerian-like or Wallerian-type degeneration is preferable (Grant Maxie and Youssef 2007). Similarly, “dying-back” axonopathy/neuropathy implies that the focus of toxicity is the neuronal cell body and that degeneration begins at the synapse and then progresses back up the distal axon (Cavanagh 1964). Clearly this term must be used with caution as this mechanism will not apply to all cases of axonal degeneration.
Axonal degeneration is commonly observed in all age groups of rats as an occasional spontaneous finding, e.g. in histologic sections of the spinal cord from rats of 15 months of age or older (Mufson and Stein 1980) or in spinal roots and peripheral nerves in subchronic neurotoxicity studies (Eisenbrandt et al. 1990). An inherited spongy degeneration characterized by vacuoles in the periaxonal or intramyelinic spaces as well as the cytoplasm of some oligodendrocytes or astrocytes in the pons and thalamus is described in zitter rats (Kondo et al. 1995). All these changes in rats become more prominent with aging.
Dystrophy, axonal (Figures 32–35)
Biological behavior: disruption of axonal structure
Synonym/synonyms: axonal swelling, neuroaxonal dystrophy
Histogenesis: axons, often within the terminals and preterminals of longer fibers (Grant Maxie and Youssef 2007)
Pathogenesis: intracellular accumulation of cytoskeletal elements
Diagnostic features:
large, eosinophilic, fusiform, or torpedo-shaped swellings (spheroids) in axons, best visualized in longitudinal sections. In cross sections, the spheroid diameters are larger than the profiles of nearby unaffected axons; spheroid contours may be granular, smooth, or vacuolated; spheroids are most commonly observed in and around relay nuclei in the brain and in axonal endings in the PNS; occasional basophilia due to mineralization can be observed within spheroids; the g-ratio (the ratio of axonal diameter to nerve fiber [axon + myelin] diameter) is reduced (McMartin et al. 1997).
Special diagnostic techniques:
spheroids appear black using routine silver impregnation techniques (e.g., Bielschowsky’s or Bodian’s stains); by electron microscopy, spheroids contain accumulations of normal and degenerating organelles as well as abnormal membranous and tubular structures.
Differential diagnoses: axonal degneration (see definition in this glossary)
Comment: A single unifying pathogenesis of axonal dystrophy has not been determined, but the mechanism is likely to involve a disturbance of retrograde axonal transport leading to regional accumulation of neurofilaments and entrapped organelles in spheroids located at sites of axonal constriction (e.g., nodes of Ranvier). In contrast to neuronal degeneration (see definition in this glossary), the spheroids of axonal dystrophy (1) tend to persist for long periods and (2) are not usually associated with an inflammatory reaction because they rarely undergo fragmentation and dissolution.
Axonal dystrophy may occur as an age-related background finding or as a neuropathologic change in certain neural diseases, including some neurotoxicity conditions. Spontaneous lesions have been reported in the relay nuclei of the caudal brain stem (e.g., gracile and cuneate nuclei and rostral portions of the dorsal funiculus in rats older than 6 months of age (Farmer, Wisniewski, and Terry 1976; Fujisawa and Shiraki 1978) and also in the autonomic ganglia of aged rats (Schmidt, Plurad, and Modert 1983)). Axonal dystrophy is also characteristic of many neuronal storage diseases, some gene-targeted (knockout) mice (e.g., gad −/−, Saigoh et al. 1999; Sepp −/−, Valentine et al. 2005), vitamin E deficiency, and in diabetic rats (Schmidt et al. 2000; Sima and Yagihashi 1986). Classic neurotoxicants associated with widespread induction of axonal swellings include acrylamide; carbon disulfide; 3,3′-iminodipropionitrile (IDPN; Griffin et al. 1982); and γ-diketones (LoPachin and Lehning 1997). These agents produce structurally similar lesions that appear to arise by different molecular mechanisms (Graham 1999).
Glia—Cell Body
Type II astrocytes (Figures 36–39)
Biological behavior: cytotoxic response
Synonym/synonyms: astrocytic swelling, Alzheimer type II astrocytes
Histogenesis: resident astrocytes in the brain
Pathogenesis: expansion of astrocytes and/or their organelles (epecially nuclei) following exposure to increased quantities of circulating metabolic by-products
Diagnostic features:
swollen nuclei characterized by central clearing and marginated heterochromatin; nucleoli (typically one or two in number) may be increased in size; the cytoplasm of affected astrocytes is not apparent (which contrasts with the appearance of other reactive astrocytes in H&E-stained sections); astrocytes may cluster in pairs or triplets; most frequently encountered in the globus pallidus but may be found in diverse brain regions (including the neocortex, basal ganglia, and hippocampus)
Special diagnostic techniques: The distinctive cytologic appearance is pathognomonic, but additional confirmation is afforded by the weak pattern of labeling by anti–glial fibrillary acidic protein (GFAP) immunohistochemistry.
Differential diagnoses: none
Comment: Type II astrocytes represent the cytomorphologic correlate of cytotoxic astrocyte swelling and are the hallmark brain lesion of hepatic encephalopathy (Agamanolis 2005; Fuller and Goodman 2001; Harris et al. 2008; Jayakumar et al. 2006; Norenberg 1981 and 1996; Norenberg et al. 1974; Summers, Cummings, and DeLahunta 1995). Cell dysfunction leads to disruption of the cytoskeleton, thereby explaining the weak GFAP-labeling pattern. If the underlying causes are adequately treated, type II astrocytes will revert to a normal cytomorphologic pattern over time; if hepatic disease continues unabated, astrocyte swelling may become sufficiently severe and generalized to produce brain stem herniation and death (Agamanolis 2005; Norenberg, Rama Rao, and Jayakumar 2005).
The proposed toxicant is ammonia, a by-product of protein catabolism in the liver and urease-producing colonic bacteria. Severe hepatic insufficiency or portosystemic shunting of venous blood from the intestinal tract permits very high levels of ammonia to reach the brain, where they rapidly cross the BBB. Ammonia is efficiently converted into glutamine within the cytoplasm of astrocytes; this action protects adjacent neurons from toxicity at the expense of poisoning the astrocytes (Albrecht and Norenberg 2006; Jayakumar et al. 2006; Norenberg, Rama Rao, and Jayakumar 2005). Potential mechanisms of glutamine-induced astrocytic swelling include osmosis of parenchymal water down a steep solute gradient, resulting in intracellular swelling, and translocation of cytosolic glutamine into the mitochondria where it is converted into glutamate and ammonia (Albrecht and Norenberg 2006) and promotes free radical production (i.e., oxidative/nitrosative stress) as well as induction of the mitochondrial permeability transition (Albrecht and Norenberg 2006; Jayakumar et al. 2006; Norenberg, Rama Rao, and Jayakumar 2005 and Norenberg et al. 2007).
Type II astrocytes are usually not observed in perfusion-fixed tissues from experimental animals in which hyperammonemic states have been induced (M.D. Norenberg, personal communication). Therefore, this pattern of altered astrocytic morphology may represent an artifact (albeit a helpful and consistent one) of immersion fixation.
Astrocyte swelling/vacuolation (Figures 40 and 41)
Biological behavior: expansion of astrocytic cytotoplasm or membrane-bound organelles
Synonym/synonyms: acute gliopathy, astrocyte swelling, astrocyte vacuolation, glia syndrome, glio-vascular lesion
Histogenesis: resident astrocytes
Pathogenesis: retention of fluid and/or metabolic by-products
Diagnostic features:
astrocyte swelling leads to prominent neuropil vacuolation and sometimes compression of adjacent neurons; predominantly gray matter distribution; bilaterally symmetrical.
Differential diagnoses: none in rodents. (Thiamine deficiency in ruminants results in astrocytic swelling due to intra-astrocytic edema [associated with pericapillary and perineuronal vacuolation within a spongy neuropil] adjacent to ischemic neurons in necrotic zones of the cerebral cortex (Summers, Cummings, and DeLahunta 1995).)
Comment: This astrocytic lesion is generally thought to represent acute energy deprivation resulting from impaired glucose utilization via the glycolytic pathway (Cavanagh 1993; Forsyth 1996; Krinke and Classen 1998). The vascular endothelium appears to be involved secondarily, possibly because swollen astrocytic end feet compress the adjacent capillaries (Ito et al. 2011).
Several toxicants induce astrocyte swelling and vacuolation in the brain, including 6-aminonicotinamide (6-AN; Sasaki 1982; Krinke and Classen 1998), chlorosugars (Jacobs and Ford 1981), dinitrobenzene, and tribromoimidazole (Cavanagh 1993). The topography and cellular changes vary due to site-specific vulnerability to each toxicant. Cerebral astrocytes are not the sole target, because dogs exposed to 6-AN develop similar lesions in perineuronal satellite cells within the dorsal root ganglia and cranial (superior) cervical autonomic ganglia (Krinke and Classen 1998).
Spontaneous, multifocal, spongiform encephalopathy associated with astrocytic reaction has been reported in neuroglia of the cerebral cortex of aged rats (Krinke and Eisenbrandt 1994). This rat lesion becomes more prominent with age.
Astrocytosis (Figure 42)
Biological behavior: Tissue repair (scar)
Synonym/synonyms: astrogliosis, gemistocytic astrocytes, gemistocytosis, glial hyperplasia, glial hypertrophy, reactive astroglia
Histogenesis: Resident astroglia (some may come from stem precursor cells)
Pathogenesis: proliferation of astrocytes to fill or encircle defects
Diagnostic features:
increase in astrocyte numbers near or in damaged CNS regions; common cytoarchitectural features in H&E-stained sections include large, pale nuclei with readily apparent cytoplasm; increase in pale eosinophilic cytoplasm (in “gemistocytes”) and swollen cell processes (McMartin et al. 1997; Grant Maxie and Youssef 2007); eccentric nuclei in “gemistocytes.”
Special diagnostic techniques: Immunhistochemical procedures to define astrocyte markers:
cell lineage: cytoskeletal proteins such as GFAP and vimentin (Fix et al. 1996; Grant Maxie and Youssef 2007); lesion character: cell proliferation markers such as 5-bromo-2-deoxyuridine (BrdU), Ki67, or proliferating cell nuclear antigen (PCNA) to differentiate increased cell size (hypertrophy) from enhanced cell number (hyperplasia).
Differential diagnoses: Gliosis NOS (see definition in this glossary)
Comment: The terms astrocytosis and astrogliosis are often used interchangeably. Some pathologists apply the two terms in a strict sense, where astrogliosis implies an increase in the numbers and/or size of filament-rich cell processes (astrocytic hypertrophy), whereas astrocytosis implies cell proliferation (astrocytic hyperplasia) only (Montgomery 1994; Summers, Cummings, and DeLahunta 1995). Plump-reactive astrocytes with rounded profiles and prominent processes are sometimes termed gemistocytes.
Glial cells increase spontaneously in the aging rodent brain in common with other species (Mandybur, Ormsby, and Zemlan 1989).
Gliosis, Not Otherwise Specified (NOS; Figures 43–46)
Biological behavior: Tissue repair (scar)
Synonym/synonyms: astrocytosis, astrogliosis, gemistocytosis, glial hyperplasia, glial hypertrophy, microgliosis, oligodendrocyte satellitosis
Histogenesis: CNS glia, especially resident astrocytes and microglia
Pathogenesis: defect renovation by cell hypertrophy and/or hyperplasia of any or multiple glial cell lineages
Diagnostic features:
cells are identified as glia (rather than neurons) using cytoarchitectural characteristics and location; in general, cell lineage should be further defined in H&E-stained sections using cell type-specific features for each category of reactive cell: astrocytes—large cells with ample eosinophilic cytoplasm, large oval nuclei, and several engorged cell processes; microglia—small cells with spindle-shaped, sometimes wavy nuclei (hence the designation rod cells) and little if any cytoplasm; oligodendrocytes—small cells with round nuclei and thin rims of pale cytoplasm, typically arranged in rings around a damaged neuron (i.e., satellitosis).
Differential diagnoses: astrocytosis/astrogliosis, microgliosis, satellitosis (see appropriate definitions in this document)
Comment: The term gliosis NOS is a common, nonspecific reactive response of CNS glial cells, chiefly astrocytes and microglia (resident elements of the immune system) rather than oligodendroglia (myelinating cells). Gliosis NOS is an acceptable designation in those cases when it is not possible to identify which population/populations of CNS glia is/are involved. However, where possible, more specific terms (e.g., astrocytosis, microgliosis, or satellitosis) should be preferred. Comparable terms used to describe reactive glial lesions in the PNS include “Schwann cell proliferation” and “bands of Büngner.”
Gliosis may result from increased size (hypertrophy) and/or number (hyperplasia) of glial cells. Studies of glial cell responses to a range of stimuli suggest that hyperplasia is more characteristic of microglia than astrocytes. This reaction may develop in the CNS following many forms of injury including inflammation and neurotoxicity (e.g., methylmercury; Nagashima 1997).
Microgliosis (Figures 47–49)
Biological behavior: inflammation-promoting reaction
Synonym/synonyms: microglial cell proliferation, reactive microglia, gitter cells
Histogenesis: resident microglia
Pathogenesis: proliferation of cells engaged in immune survellience (e.g., antigen processing and presentation) and effector activities (e.g., phagocytosis)
Diagnostic features:
focal accumulation/accumulations of microglia; the key cytoarchitectural feature of microglia in H&E-stained sections is an elongate, spindle-shaped appearance (i.e., rod cells; Summers, Cummings, and DeLahunta 1995; Grant Maxie and Youssef 2007).
Special diagnostic techniques: detection of markers specific for microglia may be used for confirmation:
Immunohistochemistry options: CD11b/c (mice); CD45; complement type 3 receptors; Iba1; keratin sulphate proteoglycan, which is not present in tissue macrophages; OX-42, the rat counterpart of CD11b; as well as more general markers for macrophages and monocytes such as CD68, ED-1 (a rat marker), ED-2 (specific for rat peripheral macrophages), F4/80, and Mac-1 (Fix et al. 1996; Gehrmann et al. 1995; Ito et al. 2001; Mander and Morris 1995); Lectin histochemistry option: Griffonia simplicifolia (GS-IB4).
Differential diagnoses: none
Comment: Microgliosis typically develops in response to a localized CNS injury (usually to a neuronal population). The resident cells respond by becoming activated (to better serve their functions as antigen-presenting cells and phagocytes), which usually entails hypertrophy and some hyperplasia. Activated microglia may be generated in advance of overt lesions in neuronal constituents (LaVoie, Card, and Hastings 2004). In times of intense demand, circulating monocytes may be recruited into the neuropil to serve as stem cells for microglia (Stoll and Jander 1999). In cases of significant brain damage, microglia are the main phagocytizing cell population, typically developing the characteristic morphology of “gitter cells” (i.e., large, round cells packed with numerous small, clear vacuoles; Figure 40).
Microgliosis in rodents develops in response to many different insults, including such neurotoxicants as carbonyl sulfide (Morgan et al. 2004), methamphetamine (Escubedo et al. 1998; LaVoie, Card, and Hastings 2004), and trimethyltin (Kuhlmann and Guilarte 2000). Microgliosis appears to wax and wane in advance of the astrogliotic response (Kuhlmann and Guilarte 2000).
Satellitosis
Biological behavior: possibly an effort to more efficiently support adjacent neurons
Synonym/synonyms: reactive oligodendroglia
Histogenesis: resident oligodendroglia
Pathogenesis: response to primary neuronal degeneration (Franklin and Kotter 2008)
Diagnostic features: rings or clusters of oligodendroglia near a degenerating neuron cell body
Special diagnostic techniques: Immunohistochemistry for cell type-specific markers (i.e., myelin proteins) such as 2′3′-cyclic nucleotide 3′-phosphodieserase (CNP; Summers, Cummings, and DeLahunta 1995), myelin oligodendrocyte glycoprotein (MOG), and Nogo-A (which labels mature cells; Kuhlmann et al. 2007)
Differential diagnoses: none
Comment: Oligodendrocytes are the least reactive glial population to CNS injury (Summers, Cummings, and DeLahunta 1995). In general, the term satellitosis should only be used after a critical comparison to the degree of satellite cells located near neurons in site-matched structures from control animals. The lesion has been described following such insults as lead acetate (Ozsoy et al. 2010) and thiram (dimethylcarbamothioylsulfanyl N, N-dimethylcarbamodithioate, a fungicidee; Lee and Peters 1976).
Glia—Myelin
Demyelination (Figure 50)
Biological behavior: disintegration of intact myelin
Synonym/synonyms: myelinolysis, myelinopathy
Histogenesis: myelin, or myelinating cells (oligodendrocytes in the CNS, Schwann cells in the PNS)
Pathogenesis: destruction of a normally formed myelin sheath without a primary impact on the ensheathed axon
Diagnostic features
early on, primary demyelination may be differentiated from secondary demyelination by the presence of intact denuded axons in the former condition. In both cases, myelin ovoids may be present (McMartin et al. 1997); reduced myelin staining in demyelinated or hypomyelinated fibers; during remyelination (which occurs effectively only in the PNS), myelin segments of variable thickness appear along the affected nerve fiber, and ovoid nuclei of dividing Schwann cells form linear rows (Büngner’s bands) in close proximity to the axon.
Special diagnostic techniques: Procedures may directly probe myelin integrity or indirectly explore the mechanism of demyelination by assessing axonal structure.
Conventional stains for myelin (paraffin sections): LFB or solochrome cyanine for myelin (used independently or in combination with a cresyl violet counterstain for axons), or osmium tetroxide; in the later stages of demyelination, recently phagocytosed and partially digested myelin debris within macrophages may be identified using LFB/PAS staining (Grant Maxie and Youssef 2007). Marchi technique (Strich 1968). Immunohistochemical stains (paraffin sections) for specific myelin proteins in: Myelin sheaths—MBP, MOG, and proteolipid protein (PLP; Sato et al. 2003); Myelinating cells—CNP and Nogo-A for oligodendroglia (CNS) (Kuhlmann et al. 2007); S-100 for Schwann cells (PNS). Conventional stains for axons (paraffin sections), to indicate whether demyelination is primary (i.e., myelin is lost, but the axon remains intact) or secondary (i.e., the axon is lost first and myelin degenerates secondarily) silver impregnation techniques (Bielchowsky's, Bodian's) label the axonal cytoskeleton. Ultrastructural analysis (plastic- or resin-embedded sections), which enables precise identification of the numbers and thicknesses of myelin lamellae that surround axons, and is particularly useful in the identification of remyelinated axons versus normal axons (McKay, Blakemore, and Franklin 1998; Smith and Jeffery 2006); Teased fiber preparations (PNS) to compare internodal distance (i.e., myelin segment length) as well as axon and myelin integrity (Krinke et al. 2000a).
Conventional stains for myelin (frozen sections or tissue blocks):
Differential diagnoses:
Intramyelinic edema (see definition in this glossary)
Comment: “Primary demyelination” develops when an initiating insult (frequently a toxicant) is directed against myelin. Primary demyelinating lesions spare axons and should not result in Wallerian-type (secondary) axonal degeneration in the distal nerve fibers. In contrast, “secondary demyelination” results when a primary axonal degenerative lesion leads to subsequent myelin loss. An axon can survive if it is deprived of its myelin sheath, but the myelin sheath cannot survive if its central axon disintegrates.
Spontaneous primary demyelination occurs in the spinal nerve roots (especially ventral) in the lumbar spinal cord of aging rats (Krinke 1983). The most extensively studied animal model of induced primary demyelination is experimental autoimmune encephalomyelitis (EAE), which induces CNS lesions similar to multiple sclerosis (MS) in humans (Ryffel 1988). Myelin loss is produced by inoculation of animals with either homogenized complete myelin or purified myelin components that are carried in appropriate adjuvants or by passive transfer with T-cells sensitized to respond to myelin antigens. Some chemicals, such as ethidium bromide (Suzuki 1988) and lysolecithin (Hall 1988), elicit primary demyelination after direct injection into the CNS or PNS. These focal to multifocal lesions bear some resemblance to the demyelinated lesions of MS. An unusual mechanism of demyelination in the PNS following tellurium exposure seems to result from altered cholesterol metabolism in Schwann cells (Anthony et al. 2001; Jortner 2000).
Remyelination occurs effectively and completely in the PNS by Schwann cells but to only a limited extent in the CNS (Zawadzka and Franklin 2007; Patrikios et al. 2006; Franklin and Kotter 2008). However, ongoing investigations suggest that CNS remyelination is a natural sequela to demyelination and may be more extensive than is currently believed. In both the CNS and PNS, regenerated myelin sheaths are thinner and have shorter segments than the originals.
Intramyelinic edema (Figures 51–58)
Biological behavior: disruption of myelin lamellae
Synonym/synonyms: leukoencephalopathy, myelin edema, myelin vacuolation
Histogenesis: lamellae of the myelin sheath (CNS or PNS)
Pathogenesis: influx of fluid between myelin layers
Diagnostic features:
the myelin sheath surrounding axons is disrupted by small-to-large vacuoles, which may be empty or contain small amounts of membranous material; the appearance and topographic distribution depends on the mechanism/mechanisms of injury and the causative agent; in later stages of long lasting intramyelinic edema, secondary degeneration of myelin and the axon may develop.
Special diagnostic techniques: Possible techniques are described in detail in the definition for “demyelination” (above).
Ultrastructural evaluation (ideally of perfusion-fixed tissue) is the principal diagnostic procedure for demonstrating the separation of myelin lamellae and/or cytoplasmic swelling in myelin-producing cells. Electron microscopic evaluations are particularly valuable when vacuoles involve the neuropil rather than being restricted to white matter tracts. Immunohistochemistry for GFAP is often helpful, not only revealing the presence (or absence) of secondary astrocytic reactions but also demonstrating that the vacuoles are not localized to an intra-astrocytic compartment. Magnetic resonance imaging (MRI) has been used successfully to detect the presence and distribution of intramyelinic edema in human patients and animal models (Peyster et al. 1995).
Differential diagnoses:
cytoplasmic vacuolation (of oligodendroglia, of astrocytes, or—if localized to the gray matter—of neurons; typically presents as one or a few, variably sized [but often large], clear vacuoles within the cell body); artifactual vacuolation (due to inappropriate histological processing; see definition for “white matter vacuolation” in this glossary).
Comment: Intramyelinic edema most frequently develops as a result of separation of myelin lamellae along the major dense lines (intraperiod lines), which represent the fused outer layers of the myelinating cell membranes (Hirano and Llena 2006; McMartin et al. 1997; van Gemert and Killeen 1998). At the light microscopic level, a diagnosis of intramyelinic edema is indicated when vacuoles are found to separate individual lamellae within myelin sheaths. However, ultrastructural evaluations are typically required to demonstrate whether there is separation of the myelin lamellae along the intraperiod lines or if the edema involves the cytoplasm of the oligodendrocytes (or Schwann cells, in the case of the PNS; Hirano and Llena 2006).
Intramyelinic edema is a common neurotoxic consequence. This lesion may arise from a direct effect of a chemical on the myelin sheath or may result from injury to the myelin-producing cells (oligodendrocytes or Schwann cells; Bouldin 2000; McMartin et al. 1997; Summers, Cummings, and DeLahunta 1995; van Gemert and Killeen 1998). When oligodendroglia or Schwann cells are injured, there may be swelling of the cytoplasmic processes (in addition to separation of the myelin lamellae).
Intramyelinic edema is classically associated with exposure to lipophilic compounds (e.g., hexachlorophene, triethyltin) that rapidly penetrate the BBB and have an affinity for myelin (Krinke 2000; Steinschneider 2000). The distribution of intramyelinic edema induced by these lipophilic chemicals (viz. widespread vacuolation of prominently myelinated regions of the brain and spinal cord) is quite different from that seen in association with high-dose vigabatrin treatment, which produces vacuolation of the neuropil within selected neuroanatomic regions (Schaumburg 2000). Nevertheless, the appearances of the individual vacuoles in these two situations may be similar. Early stages of intramyelinic edema may not be associated with either myelin or axonal degeneration and therefore, may be completely reversible. However, prolonged edema may result in secondary degeneration of the myelin sheaths or axons. For example, chronic exposure to hexachlorophene has been associated with axonal degeneration, and phagocytosis of myelin has been demonstrated ultrastructurally in rabbits treated with triethyltin (Krinke 2000; Steinschneider 2000). Degenerative changes may be seen in oligodendrocytes (e.g., in cuprizone toxicity), and hydrocephalus has also been reported to develop secondarily to aqueductal stenosis resulting from edema within myelin sheaths in the midbrain (Bouldin 2000).
Choroid Plexus
Vacuolation (Figures 59–60)
Biological behavior: incidental
Synonym/synonyms: none
Histogenesis: choroid plexus epithelium
Pathogenesis: retention of fluid, metabolic by-products, or insoluble foreign materials
Diagnostic features: clear, round, variably sized, cytoplasmic vacuoles fill or distend choroid plexus epithelium; distribution is typically multifocal to diffuse.
Special diagnostic techniques: Postfixation with lipid-preserving agents (e.g., 1% osmium tetroxide) followed by transmission electron microscopy may help to define the affected organelles.
Differential diagnoses: physiologic vacuolation (usually occurs as a few to many, variably sized [but generally small], clear vacuoles within the cell body of choroid plexus epithelium)
Comment: Genuine vacuolar change in xenobiotic-treated rodents arises from intracellular accumulation of lipids (e.g., phospholipidosis), undigested material (e.g., membrane degradation products, polyethylene glycol [PEG, conjugated to biopharmaceuticals to extend their circulating half-life]), and/or water (i.e., hydropic change) within lysosomes. For example, F344 rats given bis(4-amino-3-methylcyclohexyl)methane, an amine-curing agent for epoxy resin, develop varying degrees of vacuolar change in the choroid plexus due to water uptake and, less frequently, lamellar inclusion bodies (Shibata et al. 1990). Treatment with diisobutamide, a piperidine ring compound with antiarrhythmic activity, induces marked vacuolation of the choroid plexus epithelium as well as many peripheral organs in rats (and to a lesser degree in dogs and monkeys) due to intralysosomal accumulation of lamellated phospholipid inclusions (myelin figures; Koizumi et al. 1986; Greaves 2000; Johanson et al. 2011).
Vascular
Arteritis (Figures 61 and 62)
Biological behavior: inflammation and fibrinoid necrosis of the arterial wall
Synonym/synonyms: panarteritis nodosa, periarteritis, polyarteritis, polyarteritis nodosa
Histogenesis: arteries (all sizes), arterioles
Pathogenesis: uncertain for spontaneous disease, presumably endothelial irritation or mural damage for agent-induced lesions
Diagnostic features:
in the early stages, fibrinoid necrosis (incursion of eosinophilic, acellular material) of the tunica media occurs in conjunction with a mixed, but chiefly acute, inflammatory response; later stages may be characterized by several degenerative and inflammaotry changes: vessel proper: the inflammatory infiltrate contains many more mononuclear inflammatory cells and may be accompanied by intraarterial fibrosis; vessel proper: intimal proliferation and thrombosis may occur, resulting in narrowing and eventually obliteration of lumina (Rubin et al. 2000); perivascular connective tissue: expanded greatly by numerous mononuclear inflammatory cells and fibrosis. involvement of arteries in the CNS or adjacent to PNS structures may lead to compression atrophy and/or secondary inflammation in these tissues.
Special diagnostic techniques: lesions on H&E-stained paraffin sections are characteristic but may be confirmed by:
Miller’s elastin stain in which selective purple/black labeling of the arterial internal elastic lamina facilitates identification of disruption in this structure; Toluidine blue staining of perfusion-fixed tissue enhances visualization of vessel wall pathology.
Differential diagnoses: iatrogenic inflammation (associated with intrathecal placement of a cannula, often exacerbated by delivery of an antigenic or irritating pharamceutical agent; recognized most readily by the presence of a narrow tract [following acute introduction of a needle] or circular cavity [associated with a chronically implanted canula] within or adjacent to the inflamed tissue).
Comment: Polyarteritis nodosa is a chronic, progressive, degenerative disease which most frequently occurs in aging male rats. Inflammation and fibrinoid necrosis of the arterial wall may either begin in the endothelium and intima or else first affect the adventitia by extension from surrounding tissues and the vasa vasorum. This spontaneous disease usually affects the Sprague-Dawley and spontaneous hypertensive rat (SHR) strains and is also rampant in rats with late-stage chronic progressive nephropathy (CPN; Percy and Barthold 2001; Suzuki, Oboshi, and Sato 1979). In general, however, the vasculature of the nervous system is generally spared (Cutts 1966). This may be a helpful distinguishing feature for the identification of compound-induced arteritis. The rodent, in contrast to the dog, appears to have a particular predisposition to develop agent-induced arteritis in medium-sized arteries in mesenteric and pancreatic vascular beds in response to the administration of drugs that act on the cardiovascular system.
Although the term arteritis has been widely used in the classical form of the rodent disease, it may be more suitable to use descriptive terminology of inflammation or degeneration in compound-induced changes (please refer to the INHAND Cardiovascular System manuscript when available).
Infarct (Figures 63 and 64)
Biological behavior: degenerative lesion of the CNS parenchyma
Synonym/synonyms: regional necrosis
Histogenesis: medium-to-large-sized artery or vein
Pathogenesis: disruption of blood flow to a discrete area, leading to ischemia in a single region
Diagnostic features:
neural damage is generally localized to a single region near one major vessel; early on, generalized involvement of cells (from many lineages, but especially neurons) having pyknotic nuclei and hypereosinophilic cytoplasm; in later stages, widespread karyorrhexis and karyolysis; the end-stage lesion is cystic degeneration of the neuropil with accumulation of many activated microglia and recruited macrophages (gitter cells) to scavenge necrotic debris, occuring in conjunction with myriad glial cells (mainly reactive astrocytes) in the adjacent viable neuropil.
Special diagnostic techniques: Characteristic features are readily recognized using routine methods. Immunohistochemical detection of reactive astrocytes (anti-GFAP), or possibly proliferating capillaries (anti–factor VIII-related antigen) can be used to help locate the boundaries of the glial and vascular scars in older lesions.
Differential diagnoses: hemorrhage (which is often multifocal and localized near capillaries located at a distance from larger vessels but is not associated with necrosis; see definition in this glossary)
Comment: When applied to the nervous system, the term infarct (i.e., stroke) implies that generalized cell death in a circumscribed region results from vascular disruption, with attendant reduction in oxygen delivery to metabolically active neural cells, especially neurons (McMartin et al. 1997). The main cause of neural infarcts is usually an occlusion, although other vascular incidents such as vessel rupture (spontaneous or traumatic) or systemic hypotension (i.e., from profound blood loss or prolonged shock) can also interrupt CNS circulation. The site and cause of the vascular accident are rarely visible in histologic sections; instead, the diagnosis is inferred by the presence of focally extensive, universal necrosis limited to the neuropil supplied by a specific vascular field. Early infarcts (less than 24 hr old) exhibit an abrupt transition from apparently normal neuropil to a region where all cells exhibit some or all of the structural features characteristic of necrotic cells: nuclear pyknosis, increased cytoplasmic eosinophilia, and possibly cytoplasmic vacuolation and modest cell shrinkage. Irregular hemorrhages may surround some or many vessels near the margin of the infarct. After several days, activated microglia and recruited macrophages (i.e., phagocytes [gitter cells]) begin entering the infarct in large numbers to remove necrotic debris. Eventually, removal of the dead tissue produces a large, fluid-filled, unlined cavity whose walls are densely infiltrated with mixed glial cells (mainly astrocytes) and new capillaries.
Thrombus (Figures 65 and 66)
Biological behavior: vascular narrowing or complete blockage in one or more arteries
Synonym/synonyms: thrombosis, vascular occlusion
Histogenesis: intravascular aggregation of coagulated blood containing platelets, fibrin, and other incidentally entrapped blood cells
Pathogenesis: Three principal factors are involved: damage to the endothelium, alterations in blood flow, and increased coagulability of the blood.
Diagnostic features:
focal to multifocal presence of an irregular, eosinophilic (but mottled), intralumenal mass containing entrapped blood cells (especially platelets and erythrocytes); often adheres to some portion of the vascular wall; may be associated with an area of infarction (locally or at a distance).
Special diagnostic techniques: Elastin stains may help delineate the internal elastic lamina of arteries, thereby clearly defining the boundary between the thrombus and the vessel wall.
Differential diagnoses: postmortem clot (see comment below)
Comment: In rats and mice, thrombosis is more likely due to endothelial cell damage following exposure to an intravascular toxicant (e.g., bacterial endotoxins, xenobiotic small molecules). Some pharmaceuticals may produce conditions conducive to a hypercoagulable state by several mechanisms that impact the normal structure and function of the vessel wall and/or different blood constituents, or by altering blood flow dynamics (Ramot and Nyska 2007). Vascular inflammation may be a contributing factor, but less so in rodents than in domestic animal species.
Thrombosis and subsequent brain infarction are rare in rats and mice. In the F344 rat, it can be associated with mononuclear leukemia. An infarct resulting from arterial obstruction is initially visible as acute cellular (especially neuronal) necrosis (see definition in this glossary), with or without edema and hemorrhage (see definition in this glossary). Over time, the neuropil may be lost and/or replaced by glial and vascular proliferative responses, both of which persist after necrotic neurons have disintegrated.
Thrombi must be differentiated from “postmortem clots.” The latter findings are homogenous in color (relative to the variegated color pattern of genuine thrombi), consist chiefly of fibrin and platelets with very few entrapped blood cells and do not adhere to the vessel wall.
General
Cholesterol clefts (Figures 67 and 68)
Biological behavior: depending on size and location, either an incidental finding or a space-occupying mass that can occlude circulation of the CSF or compress the adjacent parenchyma
Synonym/synonyms: cholesteatoma, cholesterol granuloma
Histogenesis: cell membranes
Pathogenesis: extracellular accumulation and crystallization of lipoprotein by-products released from sites of necrosis, inflammation, or hemorrhage
Diagnostic features:
flat, thin, rhomboid spaces intermingled in random but often quasi-parallel planes; the typical cleft is approximately 50 µm in length and bulges slightly at the middle; in paraffin sections, the clefts are empty spaces left by crystals that were dissolved by the solvents used for histological processing.
Special diagnostic techniques: cholesterol crystals in frozen sections are birefringent; Oil Red O will stain clefts if they contain esterified cholesterols (Kruth 1984).
Differential diagnoses: none
Comment: Most cells use cholesterol for the synthesis of cell membranes. In the CNS, normal myelin catabolism results in the formation of triglycerides and cholesterol esters. However, neither extracellular (cholesterol clefts) nor intracellular accumulation of cholesterol or cholesterol esters is common as its metabolism is highly regulated.
Accumulation of cholesterol or cholesterol esters occurs in several pathologic processes in the CNS including necrosis, inflammation, and hemorrhage. Foamy macrophages may be found in damaged white matter tracts where there is significant myelin breakdown, which results in phagocytosis of lipids and cholesterol from the disintegrating cell membranes. In horses, cholesterol crystals thought to arise secondary to localized hemorrhage induce a granulomatous response in the choroid plexus. The resulting granuloma (or cholesteatoma) can obstruct the outflow of CSF through the interventricular foramen, thus resulting in obstructive hydrocephalus. Comparable lesions in rodents are very rare.
Hemorrhage (Figure 69)
Biological behavior: depending on size and location, either no clinical signs or a space-occupying mass that compresses the adjacent CNS parenchyma
Synonym/synonyms: hematoma, hemorrhagic lesions
Histogenesis: parenchymal or meningeal blood vessels
Pathogenesis: vascular disruption
Diagnostic features:
hemorrhage in the CNS is usually restricted to the meninges (epidural or subdural) and/or the parenchyma; scattered petechial foci or large contusions of focal, multifocal, or diffuse distribution may be found in the neuropil at a distance from the vascular system; hemorrhage is often in close proximity to capillaries; inflammatory cell infiltrates may be present, especially as demarcating reactions near mature hematomas; hydrocephalus and hematomyelia (copious amounts of blood in the CSF) may occur secondary to large hematomas near the ventricular system; chronic lesions may be associated with “cholesterol clefts” (see definition in this glossary).
Special diagnostic techniques: Acute hemorrhage is readily apparent in neural tissue as the bright red color of erythrocytes is unique to this cell type. Special stains for intracellular iron accumulation (e.g., Prussian blue) are often useful for finding hemosiderin depostion at sites of old hemorrhage, especially in the case of microhemorrhages (Wilcock and Colton 2009).
Differential diagnoses: infarct (hemorrhagic variants, which usually appear as circumscribed zones of necrosis with hemorrhage in the watershed near a single medium-to-large-caliber artery or vein)
Comment: Hemorrhage within the nervous system manifests in several fashions. Erosion or rupture of vascular walls in the meninges or parenchyma commonly arises from pathologic processes (like thromboembolic showers during disseminated intravascular coagulation [DIC] or systemic infections, or from hemorrhagic diatheses; Jubb and Huxtable 1992; Summers, Cummings, and DeLahunta 1995) or trauma. Careful observation is necessary to define whether extravasations were genuine in life processes as they also can be produced as postmortem artifacts; terminal hemorrhages provoke little or no tissue reaction (Jubb and Huxtable 1992). The pinpoint spots of blood seen on the cut surface when fresh brain is sliced usually mark the cut ends of congested capillaries, not petechial hemorrhages (Jones, Hunt, and King 1996).
Diapedesis (movement through intact vascular walls) of red blood cells is a common event in the sudden death of many causes and is frequently mimicked by postmortem neural trauma. The white matter of the cerebrum and the cerebellum is particularly susceptible to this process. Pathologic petechiae produced by this mechanism can by produced by anoxia, DIC-related microembolisms, or infectious diseases (Jubb and Huxtable 1992).
Subarachnoid hemorrhages as a consequence of large vessel disruption may develop into space-occupying masses that compress the adjacent brain (Jones, Hunt, and King 1996). Secondary events to hematoma include widespread brain edema, areas of neural ischemia, herniation, and/or lethal brain stem compression. Small lesions develop astrocytic scars, while larger foci liquefy and form cysts lined by hemosiderin-laden macrophages (Summers, Cummings, and DeLahunta 1995). Isolated hematomas are quite rare in rodents (Jubb and Huxtable 1992). However, some intracerebral neoplasms (e.g., mononuclear cell leukaemia, pituitary adenomas, or primary brain tumors) may cause disruption of the vasculature in rats that results in substantial perilesional hemorrhage, edema, and necrosis (Solleveld and Boorman 1990). Perivascular microhemorrhages are common features in transgenic mouse models of Alzheimer’s disease as a secondary consequence of cerebral amyloid angiopathy (Wilcock and Colton 2009).
Induced models of CNS hemorrhage typically employ rodents with spontaneous hypertension (Lee and Berry 1978) as a precursor for vascular accidents (Nagatani et al. 2005) or explore the impact of vascular wall lesions (e.g., atherosclerosis; Shiraya et al. 2009) and compounds that target vessel elements (Skold, Risling, and Holmin 2006) on the genesis and evolution of CNS hemorrhage.
Hydrocephalus (Figures 70–72)
Biological behavior: depending on the extent and chronicity, either no clinical signs or a compression of the adjacent CNS parenchyma
Synonym/synonyms: ventricular dilatation
Histogenesis: not applicable
Pathogenesis: variable causes (see comment below), all leading to dilation of one or brain ventricles via long-standing fluid accumulation
Diagnostic features:
dilation (macroscopic) of the lateral ventricles and sometimes the third ventricle; atrophy (or hypoplasia, if present during development) of the adjacent brain (generally the cerebral cortex); attenuation (flattening and cilial loss) of the ependymal lining in enlarged ventricles.
Differential diagnoses:
artifactual ventricular dilation (due to supraphysiological vascular pressure during perfusion fixation).
Comment: Hydrocephalus is a chronic change that represents a common final presentation resulting from any of several etiologies. One mechanism is compensatory, where ventricles become dilated due to atrophy or hypoplasia of the brain parenchyma during development. Another mechanism is obstruction, due to a stricture (e.g., cholesterol clefts, neoplasm) that impedes CSF circulation (Miller and Ironside 1997); the most critical segment of the ventricular system in this respect is the mesencephalic aqueduct (of Sylvius), the narrow channel connecting the third and fourth ventricles (Summers, Cummings, and DeLahunta 1995). This variant is sometimes termed noncommunicating due to the inability of fluid in the rostral ventricles to move caudally. Additional albeit rare mechanisms include excessive production of CSF by the choroid plexus epithelium or insufficient resorption of CSF at the arachnoid granulations. These two forms are sometimes called communicating since CSF flow is not affected.
The compensatory form of hydrocephalus is typically congenital, reflecting developmental disruption of CNS evolution during gestation. Teratogens, including MAM (Kaufmann and Gröters 2006), are a known cause of this defect (Figure 66). An inherited aqueductal stenosis leading to congenital hydrocephalus has been described for some strains of rats and mice (D’Amato et al. 1986), but congenital hydrocephalus is generally very rare, usually with an incidence of less than 1% in most rat strains.
The obstructive form of hydrocephalus typically occurs in rodents during the course of long-term carcinogenicity studies in which aged animals develop intracerebral neoplasms (e.g., gliomas, pineal gland tumors, or pituitary gland tumors—see Figure 67) or inflammatory masses (Solleveld and Boorman 1990; Radovsky and Mahler 1999) that distort the brain parenchyma and occlude either the interventricular foramen (of Monro) connecting the lateral ventricle to the third ventricle (leading to hydrocephalus of one lateral ventricle) or the aqueduct (leading to dilation of both lateral ventricles and often the third ventricle).
Inflammation
Biological behavior: depending on the extent and distribution (localized or widespread), either no clinical signs or additional damage to neural tissues that may already be affected by another etiologic process
Synonym/synonyms: encephalitis, meningitis, myelitis, neuritis (i.e., site-specific terms used to more precisely describe the distribution of the lesion)
Histogenesis: resident glia (astrocytes and microglia), resident and/or circulating leukocytes (of all classes), vascular endothelium and pericytes
Pathogenesis: multiple (see comment below), all representing responses by local or systemic immune cells to a noxious stimulus
Diagnostic features:
the cardinal features are a recognizable leukocyte infiltrate in association with other indicators of tissue damage (e.g., edema, fibrosis, gliosis, hemorrhage, necrosis [neuronal or glial], vascular congestion; Summers, Cummings, and DeLahunta 1995); the leukocytes may be a homogeneous population consisting of a single-cell lineage or a conglomeration (termed mixed) with multiple inflammatory cell types, but in all cases cells are well differentiated; inflammation predominantly comprising granulocytes (mainly neutrophils) may have an additional modifying terms, e.g. suppurative. A mixed infiltrate comprising chiefly lymphocytes, macrophages, and plasma cells may have simply “mononuclear cell” as a descriptor. An infiltrate predominantly of macrophages, possibly including multinucleated giant cells, is designated as “granulomatous,” while “pyogranulomatous” implies mostly neutrophils and macrophages; axonal fragmentation and/or myelin degeneration may be features of inflammation in the nervous system; following direct CNS delivery of antigenic or irritating compounds, it is necessary to distinguish the inflammation associated with cannula insertion (i.e., a foreign body reaction) from that induced by the compound (Butt 2011).
Special diagnostic techniques: the presence of inflammation is readily recognized in H&E-stained sections, but immunohistochemistry for cell type-specific markers may be used to identify particular leukocyte lineages (Eltayeb et al. 2007; Randall and Pearse 2008).
Differential diagnoses: infiltrate, inflammatory cell (see definition in this glossary); lymphoma, malignant (see definition in this glossary); reticulosis, malignant (see definition in this glossary).
Comment: Inflammation can be a normal host defense mechanism, an excessive misdirected response (e.g., autoimmune disease), or the drug-induced sequal to immune dysfunction or neural cell injury. Certain animal models of neural inflammation are characterized by a distinct pattern of lesions (e.g., multifocal, often perivascular influx of mononuclear cells for EAE; Eltayeb et al. 2007). In general, inflammation is distinguished from other highly cellular leukocytic reactions in the CNS (e.g., neoplasms like lymphoma and malignant reticulosis) by having more well-differentiated cells from multiple lineages.
It is important to distinguish the biologically distinct processes “inflammation,” which implies that the active influx of leukocytes is central to the pathogenesis, from “inflammatory cell infiltration.” The latter term implies either an innocuous focal collection of leukocytes as part of the incidental background histopathology, or a modest infiltration of inflammatory cells secondary to another primary xenobiotic-induced tissue response. Inflammatory cell infiltration is not associated with ancillary tissue responses like edema, fibrosis, gliosis, hemorrhage, necrosis, or vascular congestion.
Infiltrate, inflammatory cell (Figure 73)
Biological behavior: incidental
Synonym/synonyms: none
Histogenesis: resident and/or circulating leukocytes (of all classes)
Pathogenesis: uncertain, but presumably a self-limiting immune response designed for minor surveillance or tissue repair activities
Diagnostic features:
small, focal to limited multifocal aggregation of leukocytes (usually mononuclear cells) in the CNS parenchyma (commonly in gray matter), choroid plexus, or the meninges (often near blood vessels); no association with other indicators of tissue damage typical of an active or resolving inflammatory process (e.g., edema, fibrosis, gliosis, hemorrhage, necrosis [neuronal or glial], vascular congestion); no association with axonal fragmentation and/or myelin degeneration.
Special diagnostic techniques: none
Differential diagnoses:
inflammation (see definition in this glossary);
Comment: Inflammatory cell infiltration implies a small, focal (or occasionally multifocal) collection of leukocytes as part of the incidental background histopathology or a limited secondary infiltration of inflammatory cells in response to another primary xenobiotic-induced tissue response. In contrast, inflammation implies a primary role for the leukocytes in damaging neural tissue, and it is accompanied by a plethora of related destructive changes (e.g., edema, fibrosis, gliosis, hemorrhage, necrosis, and/or vascular congestion).
Lipofuscin accumulation (Figure 74)
Topography: gray matter, CNS nuclei
Biological behavior: age-associated intracellular accumulation of cell degradation products
Synonym/synonyms: lipofuscinosis
Histogenesis: neurons, astrocytes, oligodendrocytes
Pathogenesis: Lipofuscin represents residual bodies derived from autophagosomal lysosomes and is composed of polymers of lipid and phospholipid complexed with protein.
Diagnostic features:
buildup of brown pigment granules in neuronal cytoplasm, generally of medium-to-large-sized cells; the color of pigment granules varies from faint yellow to dark brown; some are eosinophilic (ceroid).
Special diagnostic techniques:
lipofuscin granules may be detected using several special stains: pink using PAS, pale red to red by Oil Red O, dark blue to purple by LFB, and pale blue to blue by Schmorl’s technique for melanin, as well as being acid fast by Ziehl-Neelsen. Lapham’s method (Lapham, Johnstone, and Brundjar 1964) is highly specific for neural lipofuscin; autofluorescence using ultraviolet light (365 nm) is a valuable adjunct to lipofuscin identification.
Differential diagnoses:
neuromelanin granules (a by-product of catecholamine synthesis), which occur characteristically in the SN and other brain nuclei of humans and nonhuman primates but not in rats and mice.
Comment: Lipofuscin accumulation results from age-related reductions in the efficiency with which neural cells eliminate degradation by-products (Kreutzberg, Blackmore, and Graeber 1997). Lipofuscin arises in the cell bodies of neurons, astrocytes, and oligodendrocytes by lipid peroxidation in cell membranes. Lipofuscin has been reported in capillary endothelium and pericytes in old monkeys (El-Ghazawi and Malaty 1975), but not in these cells in rats and mice. Lipofuscin seems to accumulate without deleterious effects to the cell.
Spontaneous lipofuscin accumulation occurs with age in both rats and mice but is rare. In aged rats, pyramidal neurons in the hippocampus and cerebellar Purkinje cells exhibit the most prominent accretion of lipofuscin (Riga and Riga 1974, Amenta et al. 1988). Buildup of lipofuscin in the CNS of rodents during carcinogenicity studies (18 months for mice and 24 months for F344 rats) is milder than its accrual in nonneural tissues (e.g., adrenal cortex, renal tubular epithelium, thyroid follicular cells). Electron-dense residual bodies consistent with lipofuscin may be detected more frequently by electron microscopic examination than by light microscopic assessment (Sturrock 1996). Strain differences in lipofuscin buildup within the CNS have been reported for both rats, where Sprague-Dawley animals are more affected (Zeng et al. 1994), and mice, where pigment accumulates in multiple brain regions of 25+-month-old ASH/TO mice (Sturrock 1996).
Induced accumulation of lipofuscin in the rodent CNS is produced by certain neurotoxic agents. Exposure to alcohol (Paula-Barbosa et al. 1991) or lead (Selvin-Testa et al. 1995) enhances lipofuscin accretion in rats, as does chronic vitamin E deficiency (Towfighi 1981). Engineered deletions of nuclear factor κB p50 (Lu et al. 2006) or cathepsins D and F (Koike et al. 2000; Tang et al. 2006) have been shown to accelerate lipofuscin buildup in neurons. Transgenic mice that overexpress interleukin 6 in astrocytes exhibit a constitutive BBB defect, accumulate excessive iron within the CNS, and exhibit elevated lipofuscin formation (Casteinau et al. 1998). Unlike age-related (physiologic) lipofuscin, induced forms of the pigments seem to be cytotoxic, as indicated by neuronal cell loss in ceroid lipofuscinosis.
Mineralization (Figures 75–78)
Biological behavior: incidental
Synonym/synonyms: calcification, calcospherite
Histogenesis: variable
Diagnostic features:
variably sized, often irregular, purple/ lue foci in H&E-stained CNS sections; vascular walls may be involved (especially the tunica media); laminated appearance (alternating dark and light zones) is typical.
Special diagnostic techniques:
the classic von Kossa stain may be useful in paraffin sections, though the chelation chemistry (silver nitrate) is not specific for calcium salts; Alizarin red S may also be used; the best fixatives for tissues containing calcium deposits are nonacidic fixatives such as neutral buffered 10% formalin or alcohol (Bancroft and Gamble 2002).
Differential diagnoses:
basophilic bodies (see comment), which have to be distinguished from dystrophic calcification that has occured at a site of neural tissue damage (Solleveld and Boorman 1990).
Comment: Mineralization in the CNS is presumed to represent a repair response, occuring as either a primary vascular wall lesion (the incidence of which varies by age) or a secondary dystrophic event in necrotic areas.
Mineralization in the CNS is a rare phenomenon in animals. However, some mice strains may show a higher incidence. Although affected vessels may be encrusted so heavily that the lumen is narrowed, thrombosis or areas of ischemia are rare. Spontaneous vascular mineralization without evidence of prior vascular injury is occasionally seen. The most common calcium salts in CNS mineralization are carbonates and phosphates (Summers, Cummings, and DeLahunta 1995). Calcium salts in the neuropil are monorefringent. Compounds that interfere with calcium and/or phosphorus homeostasis and cause mineralization in multiple organs (Brown et al. 2005a, b; Spencer 1998) may potentially cause mineralization in the brain as well.
Amorphous basophilic bodies are occasionally found in the thalamus of aged mice (B6C3F1 and other strains; Morgan et al. 1982) and rats (Yanai et al. 1993). These irregular laminated bodies are particularly numerous in aging mice lacking vitamin D receptor (Kalueff et al. 2006). Similar foci may be seen less frequently in the rat cerebellum (Yanai et al. 1993). The concretions are bilaterally symmetrical, contain calcium and phosphorus, are located outside cells in association with vascular basement membranes, and are not associated with a cellular reaction in the adjacent neuropil. The deposits may interrupt the blood supply, thus inducing focal necrosis with eventual astroglial scarring, capillary proliferation, and (in large lesions) cavitation (Maronpot, Boorman, and Gaul 1999). The pathogenesis and significance of these deposits are unknown (Solleveld and Boorman 1990).
Squamous cyst (Figures 79–81)
Biological behavior: depending on size and location, either an incidental finding or a space-occupying mass that compresses the adjacent CNS parenchyma
Synonym/synonyms: epidermoid cyst, epidermal inclusion cyst, epithelial inclusion cyst
Histogenesis: surface ectoderm that is misplaced during neural tube closure (Grant Maxie and Youssef 2007)
Pathogenesis: developmental anomaly (Boorman, Montgomery, and MacKenzie 1990)
Diagnostic features:
cyst are generally lined by stratified squamous epithelium and filled with concentric layers of keratin (Boorman, Montgomery, and MacKenzie 1990; Goldschmidt et al. 1998); all four layers of the normal epidermis, including a granular cell layer, are present; granulomatous inflammation to the keratin may be present near and within ruptured cysts.
Special diagnostic techniques: none.
Differential diagnoses: dermoid cyst (a term used if the stratified squamous epithelium also includes adnexae such as sweat glands, hair follicles, and/or sebaceous glands)
Comment: Squamous cysts are rare in rats and are considered to be incidental findings (Boorman, Montgomery, and MacKenzie 1990). A common predilection site appears to be the spinal meninges (Levine 1966). In mice, squamous cysts are relatively common CNS lesions (Maronpot, Boorman, and Gaul 1999; Nobel et al. 1987). They are usually located on the midline of the brain (especially near the fourth ventricle) and increase in size with advancing age (Maronpot, Boorman, and Gaul 1999). Spinal squamous cysts are also a frequent finding in the lumbosacral leptomeninges of mice. Although compression of the adjacent CNS tissue can be present and even at times pronounced, the animals rarely exhibit neurological signs (Summers, Cummings, and DeLahunta 1995).
Syringomyelia/Hydromyelia
Biological behavior: anomalous development or a degenerative lesion
Synonym/synonyms: none
Histogenesis: not applicable
Pathogenesis: uncertain, but presumed to arise from system-wide pressure increases in CSF reservoirs with resultant increases in pressure within the central canal
Diagnostic features:
syringomyelia: cavitation of the spinal cord parenchyma (typically in the dorsal funiculi and/or medial portions of the dorsal horn); hydromyelia: dilation of the central canal of the spinal cord.
Special diagnostic techniques: none
Differential diagnoses: none
Comment: Syringomyelia (parenchymal cavitation) and hydromyelia (central canal dilation) often occur in tandem and generally affect more than one spinal cord segment. In most cases, the communication between the syrinx (cavity) and the dilated central canal is difficult to discern. Primary syrinx (cavity) formation is usually a consequence of aberrant axial development; it often occurs in conjunction with congenital hydrocephalus and bony malformations of the skull and cervical vertebral bodies. Syrinx establishment may also present as parenchymal edema (Summers, Cummings, and DeLahunta 1995) secondary to primary hydromyelia as well as infection (Virelizier, Dayan, and Allison 1975), inflammation, neoplasia, certain toxicants (e.g., quisqualic acid; Yang et al. 2001), trauma, or vascular compromise. Such lesions can expand with time if the increase in CSF pressure is sustained.
The syrinx is typically lined by frayed parenchyma, reactive glia, and possibly ependyma (near the opening of the conduit that connects the cavity to the canal). The absence of a glial reaction implies an acute lesion, while an ample glial response is considered indicative of a chronic lesion (Harding 1992). In general, inflammation and glial proliferation in the spinal cord parenchyma adjacent to the cavity is minimal (Summers, Cummings, and DeLahunta 1995).
Proliferative Lesions
Neural neoplasms are an important but typically infrequent finding in rodents examined at the end of 18- to 24-month carcinogenicity studies. Conventional rodent studies do not permit repeated assessment for lesion progression over time, so an appreciation for the true future biological behavior of rodent neural neoplasms is unclear. Thus, in this lexicon we predict the impact on host function as “benign” or “malignant” based only on morphologic characteristics such as cellular differentiation, invasiveness, and proliferative rate. Certain lesions that have well-differentiated features but are biologically aggressive over time (e.g., glial neoplasms) have been termed malignant and low grade rather than benign to better address the usual clinical outcome of this category of neural tumors. Innovations in noninvasive small animal imaging should help resolve questions regarding the biological behavior of these lesions in the future.
Many classification systems have been defined for neuroproliferative lesions in genetically engineered (Weiss et al. 2002) and toxicant-treated (Koestner et al. 1999; Krinke et al. 2000b; Solleveld, Gorgacz, and Koestner 1991; Weber et al. 2011) rodents. The current recommendations have been designed to define suitable terms for common proliferative terms in mice and rats.
Neuronal
Medulloblastoma (Figures 82–85)
Biological behavior: malignant neoplasm
Synonym/synonyms: cerebellar neuroblastoma, primitive neuroectodermal tumor (PNET) of cerebellum
Histogenesis: neuroepithelial tissue
Diagnostic features:
localized totally or chiefly within the cerebellum; highly cellular mass comprising neuroepithelial stem cells exhibiting mainly neuronal differentiation; generally uniform cell appearance resembling the granular cell layer of the cerebellar cortex (Becker and Hinton 1983; Cardesa et al. 1996; Gould et al. 1990; Gullotta 1990; Krinke et al. 2000b; Mennel 1988; Solleveld, Gorgacz, and Koestner 1991; Solleveld and Boorman 1990; Yamate et al. 1987), characterized by small, round, or elongated (carrot-shaped) profile; round to elongated, hypochromatic nucleus; prominent nucleoli; indistinct cytoplasm and cell borders. Formation of whorls and pseudorosettes (i.e., concentric arrangement of tumor cells encircling small blood vessels) is common; pseudorosettes may have central fibrillary material; bizarre mitotic figures are frequent; invasive growth pattern, often replacing cerebellar folia; can metastasize within the CNS via CSF-filled cavities.
Special diagnostic techniques: Specific immunohistochemical markers have not been identified in the rat and mouse. Human tumors demonstrate coexpression of more than one type of intermediate filament in the same neoplasm, indicating their undifferentiated primitive neuroectodermal status. Synaptophysin and neuron-specific enolase (NSE) may be useful in nonhuman primates and dogs. Expression of GFAP depends on the presence of astrocytic islands within the tumor. Reactivity is reduced in anaplastic tumors.
Differential diagnoses:
ependymoma, malignant (see definition in this glossary) typically located near the ventricular system (e.g., mesencephalic aqueduct of the midbrain, the fourth ventricle beneath [rather than in] the cerebellum, or the central canal of the spinal cord); polygonal cells arranged in rows and circles. Pinealoma, malignant: located on the midline of the dorsal midbrain surface.
Comment: medulloblastoma is a member of the PNET family. Derived from the cells of the cerebellum, this neoplasm is a variant of neuroblastoma.
The lesion is rarely observed in mice as a primary tumor but may be induced by direct implantation of pellets containing carcinogenic compounds into the vermis or lateral lobes of the cerebellar cortex, or via transplacental and neonatal induction with the alkylating agent ethylnitrosourea (ENU). Medulloblastoma may also be induced experimentally in mice, rats, and hamsters by intracranial inoculation with various primate or human viruses (Rapp et al. 1969; Padgett et al. 1977; Ogawa 1989) or in mice by genetic engineering (Huse and Holland 2009).
Neuromyoblastoma, malignant (Figures 86 and 87)
Biological behavior: malignant neoplasm
Synonym/synonyms: none
Histogenesis: oligopotent neuronal precursors
Diagnostic features:
topography: locally invasive mass found in the ventral cranial cavity in the region of the pituitary gland and adjacent spinal nerves; two cell populations, the proportion of which varies among different tumors and among distinct areas within the same tumor (Ernst et al. 1993; Maekawa et al. 1989; Miller, Westwood, and Jackson 1992; Shirota, Itoh, and Kagiyama 1986):
Neuroblastic cells occur in irregular groups of relatively uniform cells with round or slightly oval nuclei showing an even chromatin stippling and no nucleolus, poorly defined cytoplasm with an eosinophilic, fine fibrillary appearance, and few mitotic figures.
Myoblastic cells are pleomorphic with single or multiple vesicular nuclei containing prominent nucleoli, abundant eosinophilic cytoplasm, shapes ranging from extended (strap-shaped) to large spheres, with variable numbers of intracytoplasmic fibers showing cross striations. Mitotic figures are variable but are often common and occasionally bizarre.
Special diagnostic techniques: Both immunohistochemistry and electron microscopy may be useful for confirming the diagnosis, especially with respect to the myoblastic phenotype.
Immunohistochemistry neuroblastic cells are NSE positive, while myoblastic cells are NSE negative; striations in myoblastic cells are readily demonstrated using either phosphotungstic acid hematoxylin (PTAH) or Heidenhain’s iron hematoxylin stains; both cell types are negative for GFAP and show no increase in S-100 protein staining. Ultrastructural examination of myoblastic cell types show bundles of irregularly orientated myofilaments with occasional primitive Z-banding. Occasionally, more differentiated cells have a more normal myofilamentous orientation and band pattern for striated muscle.
Differential diagnoses:
medulloblastoma—a cerebellar PNET showing neuroectodermal and mesodermal elements (see definition in this glossary); peripheral nerve sheath tumor, malignant—a mixed tumor containing well-differentiated striated muscle fibers intermingled with neural elements (chiefly Schwann cells that have increased positive S-100 staining in association with axons; see definition in this glossary); teratoma, malignant—a mixed tumor containing tissue with ectodermal, endodermal, and mesodermal differentiation.
Comment: Brain tumors showing multilineage differentiation are rare in rodents. The neuromyoblastoma reported in the Alderley Park (Wistar-derived) rat arises in the brain stem or adjacent cranial nerves, and neuronal and myoid differentiation are consistent features.
Glial/Schwann Cell
Astrocytoma, malignant, low grade (Figures 88 and 89)
Biological behavior: minimally aggressive neoplasm
Synonym/synonyms: astrocytoma, benign, astrocytoma, low grade; glioma, astrocytic, benign
Histogenesis: resident astrocytes
Diagnostic features:
poorly demarcated lesion, usually of modest size, confined to one major area of the CNS; moderate-to-dense cellularity; neoplastic cells may infiltrate the meninges; cells exhibit uniform (i.e., well-differentiated) features including round or oval nuclei, a variable but typically modest amount of eosinophilic cytoplasm, and indistinct cell borders; in rats, cellular differentiation defines several types (Cardesa et al. 1994; Fitzgerald, Schardein, and Kurtz 1974; Gopinath 1986; Jaenisch 1990; Maekawa and Mitsumori 1990; Solleveld, Gorgacz, and Koestner 1991; Yamate et al. 1987; Zwicker et al. 1992): protoplasmic: stellate cells with delicate cytoplasmic processes that form a mesh-like matrix; fibrillary: round cells with elongate nuclei; gemistocytic: large, plump cells with abundant cytoplasm encompassing an eccentric, round to oval nucleus; Pilocytic (pilloid): a rare variant composed of elongated, unipolar, or bipolar cells arranged in bundles and bands. in rats, neoplastic astrocytes may assume one or more unique patterns, including perineuronal satellitosis and perivascular cuffing at the neoplasm periphery (which are indicative of a more aggressive nature); in mice, this lesion is characterized by a monomorphic population of neoplastic cells (most similar to the protoplasmic variant in rats) that intermingles freely with the local neuronal population and, in some cases, reactive gemistocytic astrocytes (Faccini, Abbott, and Paulus 1990; Fraser 1971, 1986; Frith and Ward 1988; Krinke and Kaufmann 1996; Krinke et al. 2000b; Morgan and Alison 1988; Morgan et al. 1984; Radovsky and Mahler 1999; Swenberg 1982; Walker et al. 1994; Zimmerman and Innes 1979); features that may be present in rat neoplasms but that are usually absent in the mouse counterparts include: foci of hemorrhage and necrosis; palisading of neoplastic astrocytes around necrotic foci; focal to multifocal pleomorphism (chiefly cellular and nuclear atypia). neoplastic astrocytes in the rat and mouse brain generally lack GFAP reactivity (Cardesa et al. 1994; Krinke and Kaufmann 1996); in ENU-induced gliomas in rats, most astrocytomas were negative for GFAP and Leu-7 but positive for S-100 and (usually) vimentin (Zook, Simmens, and Jones 2000; Raju et al. 1990); immunohistochemistry for macrophage/microglial markers like ED-1, Iba1, and RM-4 may help differentiate astrocyte-like neoplasms of histiocytic lineage from true astrocytomas (Nagatani et al. 2009). astrocytoma, malignant “high grade” (see definition in this glossary): extends over multiple regions of brain (or spinal cord) either as a single continuous lesion or as multicentric lesions; causes extensive damage to the CNS parenchyma; typically includes multiple foci of necrosis and hemorrhage, cellular atypia and pleomorphism, and invasive growth as prominent features. glioma, mixed, low grade: a mixed tumor with neoplastic astrocytes and neoplastic oligodendroglia (the latter representing 20% or more of the mass; see definition in this glossary); astrocytosis (reactive gliosis): differentiation may be difficult, but these well-differentiated nonneoplastic cells express GFAP widely and intensely (see definition in this glossary).
Special diagnostic techniques:
Differential diagnoses: Several glial lesions should be considered, the characteristic features of which are listed here:
Comment: Astrocytomas from humans and domestic animals commonly express GFAP and are labeled by PTAH. In contrast, neoplastic astrocytes of the rodent brain do not label with GFAP or PTAH (Kleihues and Cavenee 1997; Koestner et al. 1999; Krinke et al. 2000b), and GFAP-positive astrocytes within spontaneous gliomas of the rat appear to represent reactive rather than neoplastic cells. Chemically induced gliomas in rats contain GFAP-positive astrocytes representing both neoplastic and reactive populations. Multinucleated giant cells and vascular endothelial cell proliferation are not features of naturally occurring astrocytomas in the rat.
The World Health Organization (WHO) international classification of human CNS tumors uses four grades based on predicted biological behavior: grades 1 and 2 for low grade (more benign) tumors, and grades 3 and 4 for high grade malignant tumors (Kleihues and Cavenee 1997).
A further subclassification according to the cellular differentiation may be decided on a case-by-case basis at the discretion of the study pathologist.
Astrocytoma, malignant, high grade (Figures 90 and 91)
Biological behavior: malignant neoplasm
Synonym/synonyms: glioma, astrocytic, malignant
Histogenesis: resident astrocytes
Diagnostic features: Primary sources for these features are given above in the definition for astrocytoma, low grade:
focally extensive, multicentric, or diffuse lesion with no discernible boundaries that spreads over two or more major areas of the CNS; high cellularity; neoplastic cells often surround neuronal cell bodies that persist within the field (i.e., satellitosis), palisade along foci of necrosis, and spread in Virchow-Robin spaces along radiating blood vessels; extensive infiltration of the meninges and ependyma is common; cells have polymorphic (i.e., poorly differentiated to anaplastic) features including variably shaped (usually round to fusiform) nuclei, indistinct cell borders, and may exhibit protoplasmic or fibrillary differentiation; reactive astrocytes (gemistocytes positive for GFAP) may be present; hemorrhage and necrosis may occur.
Special diagnostic techniques:
Neoplastic astrocytes in the rat and mouse brain generally lack GFAP reactivity (Cardesa et al. 1994; Krinke and Kaufmann 1996; Krinke et al. 2000b). In ENU-induced gliomas in rats, most astrocytomas were negative for GFAP and Leu-7 but positive for S-100 and (usually) vimentin (Zook, Simmens, and Jones 2000; Raju et al. 1990).
Differential diagnoses: several glial lesions should be considered, the characteristic features of which are listed here.
astrocytoma, malignant, low grade (see definition in this glossary): confined to a single region of the CNS and usually of modest size; lesion has a monomorphic appearance due to a low degree of cellular atypia and pleomorphism; necrosis and hemorrhage are normally absent. glioma, mixed, malignant: a mixed tumor with neoplastic astrocytes and neoplastic oligodendroglia (the latter representing 20% or more of the mass; see definition in this glossary); reticulosis, malignant: a neoplasm (thought to arise from mesenchyme) characterized by spindle cells that infiltrate along and in blood vessels and meninges of the CNS. The histiocytic character of this lesion indicates that future efforts at tumor diagnosis will need to employ a broad panel of cell type-specific glial and macrophage markers to define the lineage of rodent CNS lineages (see definition for microgliosis in this glossary). Comment: The “WHO International Histological Classification of Tumors of Domestic Animals—Tumors of the Nervous System” (Koestner et al. 1999) classifies astrocytomas into low-grade (well differentiated), medium-grade (anaplastic), and high-grade (glioblastoma or glioblastoma multiforme [GBM]) neoplasms. The high-grade astrocytoma is the most malignant variant. In addition to having the pronounced pleomorphic features found in medium-grade (anaplastic) astrocytomas, high-grade glioblastomas have vascular proliferation and/or necrosis. The term astrocytoma, malignant, high grade described in this article includes both the medium-grade and high-grade categories of malignant astrocytoma in the WHO classification.
A malignant astrocytoma described in a Sprague-Dawley rat exhibited binucleated granular cell differentiation. The distinctive feature of this phenotype was the presence of scattered binucleated cells that contained numerous hypereosinophilic cytoplasmic granules with a diameter of 1 to 2 µm. The neoplastic astrocytes stained positively for lysozyme, PTAH, and vimentin. Within the binucleated cells, granules stained with PAS and PTAH both before and after diastase digestion. This neoplasm has features resembling the human granular cell astrocytoma (Pruimboom-Brees et al. 2004)
Astrocytomas are the predominant neoplasm of glial origin described in the VM and BRVR mouse strains. Neoplasms occur at a 1% incidence mainly in males and spread throughout the CNS, including the fore-, mid- and hindbrain as well as the spinal cord (Fraser 1986).
Glioma, mixed, malignant, low grade
Biological behavior: tumor does not show aggressive tumor growth pattern
Synonym/synonyms: oligoastroglioma, benign
Histogenesis: astrocytes and oligodendrocytes
Diagnostic features: lesion characteristics are drawn from multiple publications for both mice (Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Walker et al. 1994; Krinke and Kaufmann 1996; Radovsky and Mahler 1999) and rats (Fitzgerald, Schardein, and Kurtz 1974; Gopinath 1986; Yamate et al. 1987; Jaenisch 1990; Maekawa and Mitsumori 1990; Solleveld, Gorgacz, and Koestner 1991; Zwicker et al. 1992; Cardesa et al. 1994); the lesion is confined to one major area of the CNS; the mass consists of a sheet of neoplastic oligodendrocytes and astrocytes exhibiting one of two arrangements: cell types intermingled in various proportions, or separate large regions containing predominantly a single cell type located adjacent to one another; each glial cell provides at least 20% of the neoplasm; vascular endothelial cell hypertrophy and hyperplasia may be present; the border of the tumor with surrounding normal tissue is well delineated; necrosis and hemorrhage are usually absent. astrocytes neoplastic astrocytes of rat and mouse brain generally lack GFAP reactivity; in ENU-induced gliomas in rats, most astrocytomas were GFAP and Leu-7 negative, most were S-100 positive, and nearly all were vimentin positive (Zook, Simmens, and Jones 2000; Raju et al. 1990). oligodendrocytes positive immunostaining for MBP has been reported in human and rat oligodendrogliomas and may be of use to confirm a diagnosis in the mouse; CNP may be a useful aid in diagnosing less differentiated tumors; some human oligodendrogliomas express S-100 and Leu-7, but expression is not specific for oligodendrogliomas; in ENU-induced gliomas in rats, most oligodendrogliomas are Leu-7 positive but are negative for GFAP and usually S-100; neoplastic cells are generally vimentin negative but may be focally positive; neoplastic oligodendrocytes stain positively for galactose cerebroside and carbonic anhydrase C. glioma, mixed, malignant, high grade (see definition in this glossary): extends over multiple regions of the brain (or spinal cord) either as a single continuous lesion or as multicentric lesions; mixture of neoplastic astrocytes and neoplastic oligodendroglia, which may be differentiated from the malignant astrocytoma because the number of each neoplastic cell population exceeds 20% of the mass; may have foci of necrosis and hemorrhage, exhibit pronounced cellular atypia and pleomorphism, and demonstrate invasive growth. astrocytoma, malignant, low grade (see definition in this glossary): neoplastic astrocytes comprise more than 80% the lesion. oligodendroglioma, malignant, low grade (see definition in this glossary): neoplastic oligodendrocytes comprise more than 80% the lesion.
Special diagnostic techniques:
Differential diagnoses:
Comment: Some oligodendrogliomas may contain a considerable population of astrocytes that are reactive rather than neoplastic. The GFAP immunocytochemical stain can be used in rodents to distinguish reactive astrocytes (GFAP positive) from neoplastic astrocytes (GFAP negative). Ependymomas often include glial elements (Gopinath 1986) because the phenotype of embryonic ependymal cells may resemble that of glial precursor cells.
Glioma, mixed, malignant, high grade (Figures 92–95)
Biological behavior: malignant tumor
Synonym/synonyms: glioma, anaplastic
Histogenesis: astrocytes and oligodendrocytes and/or precursor cells
Diagnostic features:
lesion characteristics are drawn from multiple publications for both mice (Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Walker et al. 1994; Krinke and Kaufmann 1996; Radovsky and Mahler 1999, and Krinke et al. 2000b) and rats (Fitzgerald, Schardein, and Kurtz 1974; Gopinath 1986; Yamate et al. 1987; Jaenisch 1990; Maekawa and Mitsumori 1990; Solleveld, Gorgacz, and Koestner 1991; Zwicker et al. 1992; Cardesa et al. 1994); this diffusely infiltrating lesion has an indistinct border and is present in multiple areas of the brain (and/or spinal cord); the lesion consists of neoplastic astrocytes and neoplastic oligodendrocytes in variable proportions, but each cell type constitutes at least 20% of the neoplasm; cellular atypia and pleomorphism are widespread; astrocytic or oligodendrocytic differentiation may not be obvious in some areas and some neoplasms; both types of neoplastic glial cells may either be diffusely intermingled or form areas composed predominantly of a single cell type; occasional tumor giant cells may be present (generally of astrocytic lineage); Foci of necrosis, marked vascular proliferation, edema, and hemorrhage may occur.
Special diagnostic techniques:
astrocytes
neoplastic astrocytes of rat and mouse brain generally lack GFAP reactivity; in ENU-induced gliomas in rats, most astrocytomas were GFAP and Leu-7 negative, most were S-100 positive, and nearly all were vimentin positive (Zook, Simmens, and Jones 2000; Raju et al. 1990). oligodendrocytes positive immunostaining for MBP has been reported in human and rat oligodendrogliomas and may be of use to confirm a diagnosis in the mouse; CNP may be a useful aid in diagnosing less differentiated tumors; some human oligodendrogliomas express S-100 and Leu-7, but expression is not specific for oligodendrogliomas; in ENU-induced gliomas in rats, most oligodendrogliomas are Leu-7 positive but are negative for GFAP and usually S-100; neoplastic cells are generally vimentin-negative but may be focally positive; neoplastic oligodendrocytes stain positively for galactose cerebroside and carbonic anhydrase C.
Differential diagnoses:
glioma, mixed, malignant, low grade (see definition in this glossary):
the lesion is confined to one region of the brain (spinal cord); the tumor consists of well-differentiated astrocytes and oligodendroglia with little to no cellular atypia in either tumor cell population, with each cell type comprising at least 20% of the mass. astrocytoma, malignant, high grade (see definition in this glossary): neoplastic astrocytes comprise more than 80% of the lesion. oligodendroglioma, malignant, high grade (see definition in this glossary): neoplastic oligodendrocytes comprise more than 80% of the lesion.
Comment: Experimental studies have indicated that gliomas in adult rats are initially composed of either differentiated astrocytes or oligodendrocytes. The cellular composition becomes mixed and anaplastic as the neoplasms increase in size.
The malignant mixed (anaplastic) glioma in rodents shares some histological features with the human lesion designated GBM. Some pathologists consider a diagnosis of GBM specific to human neuro-oncology and do not use the term in diagnosing neoplasms in the rat.
Oligodendroglioma, malignant, low grade (Figures 96–98)
Biological behavior: minimally aggressive tumor
Synonym/synonyms: glioma, oligodendrocytic, benign; oligodendroglioma, benign
Histogenesis: oligodendrocytes
Diagnostic features:
lesion characteristics are drawn from multiple publications for both mice (Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Walker et al. 1994; Krinke and Kaufmann 1996; Radovsky and Mahler 1999, Krinke et al. 2000b) and rats (Fitzgerald, Schardein, and Kurtz 1974; Gopinath 1986; Yamate et al. 1987; Jaenisch 1990; Maekawa and Mitsumori 1990; Solleveld, Gorgacz, and Koestner 1991; Zwicker et al. 1992; Cardesa et al. 1994); circumscribed lesion with a distinct border, confined to one major area of the brain or spinal cord; composed of sheets, rows, or nests of small, uniform neoplastic cells with round, central, hyperchromatic nuclei; clear to lightly stained cytoplasm (perinucelar halo); and distinct cell borders; the pronounced clear perinuclear halo is a common artifact of delayed fixation in this tumor type and results in the classic “honeycomb” or “fried-egg” pattern; sheets of neoplastic cells are intersected by fibrovascular stroma; prominent microvascular proliferation with atypical capillary endothelial hyperplasia can be extensive, especially at the periphery of the neoplasm; necrosis with cystic changes and hemorrhage with hemosiderosis may be present; other glial cells, such as astrocytes and transitional forms between oligodendrocytes and astrocytes, may be present in varying numbers.
Special diagnostic techniques:
positive immunostaining for MBP has been reported in human and rat tumors and may be of use to confirm a diagnosis in the mouse; oligodendrocyte transcription factor 1 (Olig-1) is a potential oligodendrocyte marker in humans; CNP may be a useful aid in diagnosing less-differentiated tumors; some human oligodendroglioma express S-100 and Leu-7, but this pattern is not specific for oligodendrogliomas; in ENU-induced gliomas in rats, most oligodendrogliomas are Leu-7 positive but are negative for GFAP and usually S-100; neoplastic cells are generally vimentin negative but may be focally positive (Zook, Simmens, and Jones 2000); neoplastic oligodendrocytes stain positively for galactose cerebroside and carbonic anhydrase C.
Differential diagnoses:
oligodendroglioma, malignant, high grade (see definition in this glossary): may extend over multiple regions of the brain or spinal cord; causes extensive damage, has foci of necrosis and hemorrhage, exhibits pronounced cellular atypia and pleomorphism and demonstrates invasive growth. ependymoma, benign (see definition in this glossary): neoplastic cells are polygonal and arranged in rows and rosettes (i.e., a halo of cells surrounding an empty lumen); oligodendroglial cells within the neoplasm are infrequent; tumor is confined to ventricles and periventricular regions of the brain (and spinal cord). glioma, mixed, malignant, low grade (see definition in this glossary): lesions containing at least 20% neoplastic astrocytes and 20% neoplastic oligodendroglia are considered to be mixed gliomas; if more than 80% of neoplastic cells are of a single cell type (astrocytes or oligodendroglia), the tumor is designated as a low-grade tumor of the predominant cell type (astrocytoma or oligodendroglioma).
Comment: The “WHO International Histological Classification of Tumors of Domestic Animals—Tumors of the Nervous System” (Koestner et al. 1999) divides oligodendroglial tumors into (1) oligodendroglioma (benign) and (2) anaplastic (malignant). Macroscopically, both categories are red, pink-red, or gray-pink and appear as solid or sometimes soft gelatinous masses. Cavities or friable regions (foci of necrosis) and/or dark red areas (hemorrhage) may also be evident during gross inspection. These lesions are found primarily in cerebral hemispheres, basal ganglia, and corpus callosum. Well-differentiated oligodendrogliomas are readily recognized at the microscopic level by the typical “honeycomb” pattern created by well-defined cell membranes and clear perinuclear halos of unstained cytoplasm in the neoplastic cells.
In many mouse strains, low-grade oligodendrogliomas are the predominant chemically induced neoplasm of glial origin. They are rarely seen to occur naturally, although they have been described as a spontaneous lesion in the BALB/c strain.
Oligodendroglioma, malignant, high grade (Figures 99–103)
Biological behavior: malignant tumor
Synonym/synonyms: glioma, oligodendrocytic, malignant; oligodendroglioma, anaplastic
Histogenesis: oligodendrocytes
Diagnostic features:
lesion characteristics are drawn from multiple publications for both mice (Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Walker et al. 1994; Krinke and Kaufmann 1996; Radovsky and Mahler 1999; Krinke et al. 2000b) and rats (Fitzgerald, Schardein, and Kurtz 1974; Gopinath 1986; Yamate et al. 1987; Jaenisch 1990; Maekawa and Mitsumori 1990; Solleveld, Gorgacz, and Koestner 1991; Zwicker et al. 1992; Cardesa et al. 1994); well-circumscribed lesion with distinct borders, extending over multiple areas of the brain (or spinal cord); neoplastic cells exhibit focal or diffuse anaplasia as evidenced by high cellularity, pronounced cellular atypia and pleomorphism, nuclear polymorphism, prominent proliferation of glomeruloid vessels at the tumor margins, increased mitotic index, necrosis, and/or meningeal infiltration. Some neoplastic cells commonly exhibit more typical oligodendroglial features: round, central, hyperchromatic nuclei, and clear-to-lightly stained cytoplasm (perinucelar halo) with distinct cell borders; neoplastic cells are typically arranged in sheets, rows, or nests, although cells with larger round to oval nuclei may also form circles; atypical capillary endothelial hyperplasia (garlands), especially at the tumor periphery, is extensive and is a characteristic feature; foci of necrosis with cystic cores and/or hemorrhages are frequent.
Special diagnostic techniques:
positive immunostaining for MBP has been reported in human and rat tumors and may be of use to confirm a diagnosis in the mouse; Olig-1 is a potential oligodendrocyte marker in humans; CNP may be a useful aid in diagnosing less-differentiated tumors; some human oligodendroglioma express S-100 and Leu-7, but this pattern is not specific for oligodendrogliomas; in ENU-induced gliomas in rats, most oligodendrogliomas are Leu-7 positive but are negative for GFAP and usually S-100; neoplastic cells are generally vimentin negative but may be focally positive (Zook, Simmens, and Jones 2000); neoplastic oligodendrocytes stain positively for galactose cerebroside and carbonic anhydrase C.
Differential diagnoses:
oligodendroglioma, malignant, low grade (see definition in this glossary):
confined to one region of the CNS; neoplastic cells are a monomorphic population of well-differentiated oligodendroglia (e.g., central nuclei and perinucelar halo and distinct cell borders) with no cellular atypia. ependymoma, malignant (see definition in this glossary): neoplastic cells are polygonal and in generally arranged in rows and rosettes; oligodendroglial cells within the neoplasm are infrequent; tumor is confined to ventricles and periventricular regions of the brain (or spinal cord). glioma, mixed, malignant, high grade (see definition in this glossary): lesions containing at least 20% neoplastic astrocytes and 20% neoplastic oligodendroglia are considered to be mixed gliomas; if more than 80% of neoplastic cells are of a single cell type (astrocytes or oligodendroglia) and exhibit modest to marked atypia, the tumor is designated as a malignant (high grade) tumor of the predominant cell type (astrocytoma or oligodendroglioma).
Comment: Remarks listed under the definition “Oligodendroglioma, malignant, low grade” are applicable here.
Schwannoma, benign
Biological behavior: benign neoplasm in any organ containing peripheral nerves Synonym/synonyms: neurilemmoma, neurinoma Histogenesis: Schwann cells (myelinating cells of peripheral nerves) considered to be of neuroectodermal origin but capable of facultative differentiation to express mesenchymal properties. Diagnostic features:
Characteristics were compiled from the following references: Koestner, Swenberg, and Wechsler 1971; Stewart et al. 1974; Mandybur and Brunner 1982; Swenberg 1982; Gough et al. 1986; Alison et al. 1987; Laber-Laird, Jokinen, and Jerome 1988; Landes et al. 1988, 1990; Rice and Ward 1988a, 1988b; Cardesa et al. 1989, 1990; Russel and Rubinstein 1989; Maekawa and Mitsumori 1990; White et al. 1990; Yoshitomi and Brown 1990; Yoshitomi and Boorman 1991; Greaves, Faccini, and Courtney 1992, 2004; Jensen et al 1993; Walker et al. 1994; Krinke 1996; Kleihues and Cavenee 1997; Ernst et al. 2001; Ikeda, Sato, and Sueyoshi 2003; Stemmer-Rachamimov et al. 2004; Teredesai and Wöhrmann 2005; expansive, compressing, usually encapsulated lesions located near a peripheral nerve or nerve plexus, commonly growing without inducing clinical signs; two basic patterns are characteristically observed:
Antoni A pattern: anaxonal Schwann cells are elongated with indistinct cell borders and form nuclear palisades (i.e., cell nuclei arranged in parallel bands). Adjacent palisades and the intervening cytoplasms form “Verocay bodies” (a characteristic Antoni A arrangement in which the palisades form parallel rows separated by homogeneous, anuclear, eosinophilic intercellular material); Antoni B pattern: sparsely cellular regions, with a clear matrix, sometimes containing cystic cavities. Antoni A and B patterns are not always both apparent in a given neoplasm, so that a tumor can be composed of predominantly one pattern; several tumor variants are defined by their morphologic characteristics: cellular variant: composed mainly of cellular Antoni A tissue, with no Verocay bodies; granular cell variant: contains cells with cytoplasmic granules comparable to those in granular cell tumors of the meninges; melanotic variant: some tumor cells contain melanosomes; plexiform variant: grows in a multinodular (“plexiform”) pattern, presumably involving the various branches of a nerve plexus. Special diagnostic techniques: Schwann cell differentiation can be confirmed using several techniques:
positive immunohistochemistry for S-100, PLP or peripheral myelin protein 22 kDa (PMP22); demonstration by electron microscopy of convoluted cytoplasmic processes lined by a continuous basal lamina. Differential diagnoses:
fibroma: benign spindle cell neoplasm arising from adjacent connective tissue; tumor cells express vimentin but not Schwann cell markers. leiomyoma: eosinophilic spindle-shaped cells with elongated but blunt-ended nuclei and positive immunostaining for desmin; bundles of cells in leiomyomas are usually oriented perpendicular to each other. schwannoma, malignant (see definition in this glossary): exhibits cellular atypia, high mitotic (proliferative) activity, and locally invasive growth and/or distant metastases. neuroma: Nonneoplastic proliferative lesion of a peripheral nerve containing increased numbers of Schwann cells that invest regenerating axons. Comment: In the rat, characteristic lesions occur in the heart (endocardial schwannoma, schwannomatosis), near the ear pinna, inside the eye (intraocular) and orbit, and in the mandibular salivary gland. The incidence is quite low in all strains tested (Novilla et al. 1991).
Reporting of tumor variants or specific patterns may be decided on a case-by-case basis and is not mandatory in routine standard study.
Schwannoma, malignant
Biological behavior: malignant neoplasm in any organ containing peripheral nerves
Synonym/synonyms: neurilemmoma, malignant; neurinoma, malignant
Histogenesis: Schwann cells (myelinating cells of peripheral nerves), considered to be of neuroectodermal origin but capable of facultative differentiation to express mesenchymal properties.
Diagnostic features:
Characteristics were compiled from the following references: Koestner, Swenberg, and Wechsler 1971; Stewart et al. 1974; Mandybur and Brunner 1982; Swenberg 1982; Gough et al. 1986; Alison et al. 1987; Laber-Laird, Jokinen, and Jerome 1988; Landes et al. 1988, 1990; Rice and Ward 1988a, 1988b; Cardesa et al. 1989, 1990; Russel and Rubinstein 1989; Maekawa and Mitsumori 1990; White et al. 1990; Yoshitomi and Brown 1990; Yoshitomi and Boorman 1991; Greaves, Faccini, and Courtney 1992, 2004; Jensen et al. 1993; Walker et al. 1994; Krinke 1996; Kleihues and Cavenee 1997; Ernst et al. 2001; Ikeda, Sato, and Sueyoshi 2003; Stemmer-Rachamimov et al. 2004; Teredesai and Wöhrmann 2005; expansive, compressing, unencapsulated lesions located near a peripheral nerve or nerve plexus; commonly asymptomatic unless compression and invasion of the CNS or other tissues produces functional changes; high mitotic rate, cellular or mitotic atypia, and/or locally invasive growth or distant metastases are malignant features; two basic patterns are characteristically observed:
Antoni A pattern: anaxonal Schwann cells are elongated with indistinct cell borders and form nuclear palisades. Adjacent palisades and the intervening cytoplasms form Verocay bodies (a characteristic Antoni A arrangement in which the palisades form parallel rows separated by homogeneous, anuclear, eosinophilic intercellular material); Antoni B pattern: sparsely cellular regions, with a clear matrix, sometimes containing cystic cavities; both Antoni A and B patterns are not always apparent in a given neoplasm, so that a tumor can be composed of predominantly one pattern; tumor variants (defined above under Schwannoma, benign) may be used when appropriate.
Special diagnostic techniques: Schwann cell differentiation can be confirmed using several techniques:
positive immunohistochemistry for S-100, PLP, or PMP22; demonstration by electron microscopy of convoluted cytoplasmic processes lined by a continuous basal lamina. fibrosarcoma: malignant spindle cell neoplasm arising from adjacent connective tissue and often encompassing or effacing the affected nerve; tumor cells may express vimentin but not Schwann cell markers. leiomyoma: eosinophilic spindle-shaped cells with elongated but blunt-ended nuclei and positive immunostaining for desmin; bundles of cells in leiomyoma are usually oriented perpendicular to each other. neuroma: Nonneoplastic proliferative lesion containing increased numbers of Schwann cells that invest regenerating axons. schwannoma, benign (see definition in this glossary): Lacks cellular atypia, has low mitotic (proliferative) activity, and exhibits no invasive growth or distant metastases.
Differential diagnoses:
Comment: In the rat, characteristic lesions occur in the heart (endocardial schwannoma, schwannomatosis), near the ear pinna, inside the eye (intraocular) and orbit, and in the mandibular salivary gland.
Schwannomas may be induced in rats by direct-acting alkylating agents such as N-nitrosoethylurea or methylmethane sulfonate, which act as transplacental carcinogens. Schwannomas have also been induced in rats following postnatal exposure to 7,12-dimethylbenz[α]anthracene, or N-nitrosomethylurea. Malignant schwannomas have been described in double transgenic mice expressing simian virus 40 large tumor antigen and prokaryotic β-galactosidase (LacZ) under the control of the MBP promoter (Jensen et al. 1993). Genetically engineered mouse models of neurofibromatosis, with manipulation of genes NF1 or NF2, develop peripheral nerve sheaths tumors, including schwannomas (Stemmer-Rachamimov et al. 2004).
Reporting of tumor variants or specific patterns may be decided on a case-by-case basis and is not mandatory in routine standard study.
Meninges
Aggregates, granular cell (Figures 104 and 105)
Biological behavior: nonneoplastic proliferation
Synonym/synonyms: none
Histogenesis: uncertain, but presumably meningeal cells of neural crest origin
Diagnostic features: characteristic features are distinctive (Mitsumori et al. 1987; Mitsumori, Maronpot, and Boorman 1987; Mitsumori, Stefanski, and Maronpot 1988; Radovsky and Mahler 1999; Krinke et al. 2000b):
granular cells are polygonal with distinct borders and often but not always packed with variably sized, eosinophilic cytoplasmic granules; this finding presents as either scattered single cells or small clusters, particularly in the meninges; granular cell aggregates do not compress adjacent neural tissue.
Special diagnostic techniques: Cytoplasmic granules are highlighted using the PAS stain.
Differential diagnoses:
tumor, granular cell, benign (see definition in this glossary):
meningeal mass composed of densely packed clusters of granule cells;
unless particularly small, the mass results in noninvasive compression of the underlying brain.
Comment: This rare lesion of the rodent meninges is characterized by its discrete size and lack of compression of the adjacent brain parenchyma. As granular cells are no resident cells of the meninges, the term “hyperplasia, granular cell” should not be used.
Tumor, granular cell, benign
Biological behavior: benign neoplasm
Synonym/synonyms: benign granular cell tumor; granular cell tumor, benign
Histogenesis: uncertain, but presumably meningeal cells of neural crest origin
Diagnostic features: Characteristic features are distinctive (Mitsumori et al. 1987; Wright et al. 1990; Yoshida et al. 1997; Radovsky and Mahler 1999; Krinke et al. 2000b; Vang et al. 2000):
masses are solid, round to plaque-like, usually micronodular proliferations with a fine vascular stroma that are confined to the meninges;
these masses usually result in noninvasive compression of the underlying brain;
the tumor consists of a relatively homogeneous population of polygonal cells with central to eccentric, round to oval nuclei with little anisocytosis. Tumor cells usually are closely associated but may lie free in more dispersed tumor tissue. The nuclei are typically clear with a fine, dispersed chromatin pattern. The cytoplasm typically contains eosinophilic granules by H&E staining but not all cells display this feature;
other less-dominant cell types within the tumor include elongated irregular nuclei (reminiscent of microglial nuclei) and small round cells with sparsely granular cytoplasm and chromatin-dense nuclei;
mitotic figures are generally absent.
Special diagnostic techniques: granular cell differentiation can be confirmed using several techniques:
PAS staining highlights the granular cytoplasmic contents of many of the tumor cells (Krinke et al. 2000);
electron microscopy reveals two cell types, one with dense lysosomal bodies and the second with intermediate filaments (Yoshida et al. 1997).
Differential diagnoses:
aggregates, granular cell (see definition in this glossary): small cell foci where the elements have the histologic features of granular cells; hyperplastic foci do not compress the neuropil.
tumor, granular cell, malignant (see definition in this glossary): gross and histologic appearance resembles that of benign granular cell tumors; however, malignant masses invade the underlying brain, typically as one or more small micronodular clusters of infiltrating neoplastic cells.
Comment: The benign granular cell tumor (GCT) of the rat meninges is a relatively common neoplasm in chronic studies. Similar tumors are rare in mice (Radovsky and Mahler 1999). A review of 107 rat meningeal tumors identified a transition from meningothelial meningiomas to granular cell tumors in 21 of 26 cases, suggesting that all rat meningeal tumors might be related and derived from an arachnoidal cell precursor (Mitsumori et al. 1987). This conclusion was supported by a later electron microscopic study (Yoshida et al. 1997).
Benign GCT of the rat does not invade the brain, but in some cases the plane of section may present with tumor islands artifactually appearing to be within the brain without any apparent connection to the meninges. Further sectioning is necessary to demonstrate the noninvasive (i.e., superficial) character of these masses.
Human GCT with a similar morphology occurs in soft tissues such as tongue and subcutis and more rarely in brain (Vang et al. 2000). Their relationship to the rat meningeal granular cell tumor is not resolved, although current evidence favors neural crest origin for both human and rat tumors (Wright et al. 1990).
Tumor, granular cell, malignant (Figures 106–110)
Biological behavior: malignant neoplasm
Synonym/synonyms: malignant granular cell tumor; granular cell tumor, malignant
Histogenesis: uncertain, but presumably meningeal cells of neural crest origin
Diagnostic features: characteristic features are distinctive (Mitsumori et al. 1987; Wright et al. 1990; Yoshida et al. 1997; Radovsky and Mahler 1999; Krinke et al. 2000b; Vang et al. 2000):
masses are solid, round to plaque-like, usually micronodular proliferations with a fine vascular stroma that are confined to the meninges;
the mass both compresses and invades the adjacent brain, typically as small, often multiple, micronodular clusters of infiltrating neoplastic cells. Frequently there is no intervening evidence of normal meningeal tissue;
despite its malignant growth pattern, the mass consists of a relatively homogeneous population of polygonal cells with central to eccentric, round to oval nuclei with mild-to-moderate anisocytosis and, rarely, megakaryocytosis. Tumor cells usually are closely associated but may lie free in more dispersed tumor tissue. The nuclei are typically clear with a fine dispersed chromatin pattern. The cytoplasm usually contains eosinophilic granules, but not all cells display this feature;
other less dominant cell types within the tumor include elongated irregular nuclei (reminiscent of microglial nuclei) and small round cells with sparse granular cytoplasm and chromatin-dense nuclei;
mitotic figures are generally absent.
Special diagnostic techniques: granular cell differentiation can be confirmed using several techniques:
PAS staining highlights the granular cytoplasmic contents of many of the tumor cells (Krinke et al. 2000b);
electron microscopy reveals two cell types, one with dense lysosomal bodies and the second with intermediate filaments (Yoshida et al. 1997).
Differential diagnoses: tumor, granular cell, benign (see definition in this glossary): gross and histologic appearance resembles that of the malignant GCT benign masses may compress but do not invade the underlying brain.
Comment: GCTs of rat meninges are relatively common neoplasms in chronic studies. Similar tumors are rare in mice (Radovsky and Mahler 1999). Although all GCTs of rat meninges are commonly classed as benign, differing growth patterns indicate the existence of benign and malignant variants. Malignant GCTs differ from their benign counterparts chiefly in their micronodular invasive character. Some benign masses may be mistaken as invasive if the plane of section does not show a connection to the meninges.
Meningioangiomatosis
Biological behavior: benign proliferative condition, considered a vascular malformation or hamartoma (i.e., disorganized mixture of cells normally found in the affected tissue) rather than a neoplasm.
Synonym/synonyms: none
Histogenesis: perivascular, mesenchymal, pluripotent cell with fibroblastic or meningothelial differentiation
Diagnostic features:
plaque-like thickening of the cerebral meninges (which may be evident grossly), frequently affecting the dorsal surface of the forebrain, with bilateral distribution involving the midline;
perivascular penetration of Virchow-Robin’s spaces occurring in the cortex and/or the brain stem;
both the plaque-like thickening of the meninges and the perivascular cells penetrating the brain are composed of spindle-shaped cells with slender nuclei and occasional polygonal, probably meningothelial cells;
proliferating cells exhibit no features of atypia, pleomorphism or high mitotic activity;
the perivascular spread may progress from the meninges into the cranium, around the vascular foramina in the skull.
Special diagnostic techniques: This lesion is reported to consistently be immunoreactive for vimentin. Some cells may be positive for α-smooth muscle actin, indicating possible myofibroblastic differentiation.
Differential diagnoses:
meningioma, benign (see definition in this glossary): perivascular penetration is not a feature of benign meningioma.
meningioma, malignant (see definition in this glossary): characteristic features of malignant meningioma include cellular atypia, pleomorphism, and high mitotic activity; perivascular spread without such features does not warrant the diagnosis of malignant meningioma.
Comment: Meningioangiomatosis is a rare condition. Few cases have been reported in humans and sporadic cases only in animals. In humans, most instances are observed in children and young adults, where they may be asymptomatic but typically present with seizures. In dogs, the observed clinical signs are those characteristic of local brain compression, such as ataxia, circling, rotatary nystagmus, mild tetraparesis, muscle atrophy, or proprioreceptive deficits (Ribas, Carpenter, and Mena 1990; Pumarola et al 1996).
In the mouse, this lesion was correctly characterized only recently (Balme, Roth, and Perentes 2008). It has probably been mistaken for malignant meningioma in the past based on its perivascular distribution. Previous misidentified cases likely include reports of a mouse meningeal tumor that resembled benign fibrous meningioma but was classified as a malignant meningioma because it “exhibited extensive infiltration of the ventral brain along the adventita of small blood vessels” (Morgan et al. 1984) and a “meningeal sarcoma” (Krinke and Kaufmann 1996). No characteristic clinical signs were reported in mice.
The hamartomatous character of meningioangiomatosis is evident by its frequent occurrence in human patients with neurofibromatosis type 2 (Stemmer-Rachamimov et al. 1997). Meningiomas arising in the background of meningioangiomatosis in humans have a benign clinical course, although they pathologically and radiologically mimic invasive meningiomas (Kim et al. 2002).
meningioma, benign (Figures 111 and 112)
Biological behavior: benign neoplasm
Synonym/synonyms: none
Histogenesis: stromal cells of the meninges
Diagnostic features:
apparent grossly as clearly defined masses, plaques, or meningeal thickenings overlying the surface of the brain, cranial (usually optic) nerves, or spinal cord;
in the brain, usually dorsal or dorsolateral over the cerebral hemispheres but may be basilar in areas such as the sella turcica;
well-demarcated;
varying compression but no invasion of the underlying parenchyma;
several variants are possible (Rubinstein 1972; Gopinath 1986;Mitsumori et al. 1987; Mitsumori, Maronpot, and Boorman 1987; Mitsumori, Stefanski, and Maronpot 1988):
fibroblastic type: elongated (spindle or fusiform) cells with pale eosinophilic cytoplasm. Nuclei are small, elongated, and have a reticular or hyperchromatic chromatin pattern. The cells form interwoven bundles in loose or fascicular patterns, with varying amounts of collagen separating individual cells; may show irregular palisading or myxomatous areas;
meningothelial type: larger epithelioid cells with homogenous eosinophilic cytoplasm and a vesicular nucleus arranged in sheets or lobules separated by a fibrous stroma;
mixed type: contains both fibroblastic and meningothelial components.
psammoma bodies (intraneoplastic laminar calcified concretions) are an uncommon feature of rodent meningiomas;
mitotic figures are rare and of normal configuration;
granular cells occur in some syncytial meningiomas in rats.
Special diagnostic techniques: positive immunohistochemistry for vimentin, collagen, and reticulin.
Differential diagnoses:
granular cell tumor, benign (in rats; see definition in this glossary):
meningeal mass comprising homogenous polygonal cells with distinct borders and abundant PAS-positive cytoplasmic granules;
compresses but does not invade the underlying parenchyma;
meningioma, malignant (see definition in this glossary): invasive growth, cellular atypia, pleomorphism, frequent mitotic figures (some abnormal), and multinucleated cells.
meningioangiomatosis (see definition in this glossary): plaque-like thickening of the cerebral meninges with perivascular extension, usually over the dorsal cerebral cortex; comprising well-differentiated spindle (meningothelial) cells.
histiocytic sarcoma (in mice): well-differentiated histiocytic, multinucleated giant cells with little fibroblastic differentiation; pseudopalisading arrangement of tumor cells around necrotic areas.
reticulosis, malignant (see definition in this glossary): pleomorphic lymphoid to histiocytic-type cells and prominent stroma infiltrating the meninges as well as perivascular and periventricular spaces; mass contains abundant reticulin fibers; multinucleated giant cells and mitotic figures may be numerous.
Comment: Benign meningiomas are fairly common in mice (and dogs) but are also found in rats. Lesions may be induced in dogs and nonhuman primates with Rous Sarcoma virus and methylcholanthrene.
Reporting of tumor variants or specific patterns may be decided on a case-by-case basis and is not mandatory in routine standard studies.
Meningioma, malignant (Figures 113–118)
Biological behavior: malignant neoplasm
Synonym/synonyms: meningeal sarcoma
Histogenesis: stromal cells of the meninges
Diagnostic features: apparent grossly as poorly circumscribed solid masses, plaques, or meningeal thickenings applied to the surface of the brain, cranial (usually optic) nerve, or spinal cord. Clear attachment or proximity to the meninges; invasive growth into the underlying brain parenchyma, typically with extensive infiltration along the adventitia of small radiating blood vessels, with no clear demarcation between the neoplasm and normal tissue; highly cellular, with cells arranged in interlacing or irregular sheets; mitotic figures numerous and sometimes abnormal; distinct variants based on the primary differentiation pattern (Becker and Hinton 1983; Maekawa 1984; Krinke et al. 1985; Maekawa et al. 1987; Yamate et al. 1987; Mennel 1988; Mitsumori, Stefanski, and Maronpot 1988; Gould et al. 1990; Gullotta 1990; Maekawa and Mitsumori 1990; Solleveld and Boorman 1990; Solleveld, Gorgacz, and Koestner 1991; Janisch and Schreiber 1994; Cardesa et al. 1996; Rempel, Ge, and Gutierrez 1999, Krinke et al. 2001): fibrous type: spindle cells with eosinophilic and fairly abundant cytoplasm as well as elongate nuclei associated with abundant extracellular collagen matrix; spindloid type: spindle cells with scant quantities of basophilic cytoplasm and elongate nuclei, with little extracellular collagen; undifferentiated type: pleomorphic cells with sometimes bizarre nuclei and occasional multinucleated giant cells.
Special diagnostic techniques: positive immunohistochemistry for vimentin, collagen, and reticulin.
Differential diagnoses: meningioma, benign (see definition in this glossary): expansive but well-demarcated mass encompassing or adjacent to the meninges which may compress but does not invade the underlying brain parenchyma; spindle cell morphology is uniform; mitotic figures are infrequent and of normal configuration. histiocytic sarcoma: well-differentiated histiocytic, multinucleated giant cells with little fibroblastic differentiation; pseudopalisading arrangement of tumor cells around necrotic areas.
Comment: This lesion is rare as a spontaneous tumor in rats and mice but may occur spontaneously in hamsters more frequently. Malignant meningiomas have been induced by implantation of carcinogenic chemicals into the leptomeninges of mice (e.g., methylcholanthrene) or by intracerebral injection of Rous sarcoma virus (strain Bratislava) in newborn rhesus monkeys (Macaca mulatta).
Reporting of tumor variants or specific patterns may be decided on a case-by-case basis and is not mandatory in routine standard study.
Ependyma
Ependymoma, benign
Biological behavior: benign neoplasm
Synonym/synonyms: none
Histogenesis: ependymal cells lining the ventricular system of the brain and the central canal of the spinal cord
Diagnostic features: characteristic features have been detailed for both mice (Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Walker et al. 1994; Krinke and Kaufmann 1996; Radovsky and Mahler 1999, Krinke et al. 2000b) and rats (Dagle, Zwicker, and Renne 1979; Gopinath 1986; Yamate et al. 1987; Jaenisch 1990; Kaneko et al. 1990; Maekawa and Mitsumori 1990; Solleveld and Boorman 1990; Solleveld, Gorgacz, and Koestner 1991; Cardesa et al. 1994; Schmitt et al. 1997; Spassky et al. 2005):
the neoplasm is located near the ventricles and aqueduct of the brain and/or the central canal of the spinal cord;
the lesion consists of polygonal cells with round to oval, hyperchromatic nuclei having delicate chromatin and indistinct cellular borders that are arranged in rows and rosettes (a halo of cells surrounding an empty lumen);
luminal rosettes, cilia, and basal bodies (blepharoplasts) are found in well-differentiated ependymomas;
perivascular pseudorosettes (cells arranged around blood vessels rather than empty lumens) or tubular structures may be present;
the neoplasm may include other glial elements.
Special diagnostic techniques:
mouse ependymal cells express S100β and
blepharoblasts are especially prominent when labeled using the PTAH stain.
Differential diagnoses: ependymoma, malignant (see definition in this glossary): this neoplasm invades the neuropil adjacent to the ventricular system; other histological features of malignancy include pronounced cellular atypia and pleomorphism, necrotic foci, and multinucleated giant cells; subcellular organelles indicative of well-differentiated ependyma (e.g., cilia and associated basal bodies) are uncommon. papilloma, choroid plexus (see definition in this glossary): this tumor consists of papillary formations upon a fibrovascular stroma lined by a single layer of cuboidal to columnar epithelial cells. rosette and pseudorosette formation is absent. medulloblastoma (see definition in this glossary): this neoplasm occurs in the cerebellum but not in association with the ventricular system; rosettes with a central lumen are common and typically contain eosinophilic fibrillary material.
Comment: Ependymomas are rare neoplasms in the CNS of rats. Rat ependymomas often include other glial elements (Gopinath 1986), because the histologic appearance of embryonal ependymal cells resembles that of glial precursor cells. One spontaneous rat ependymoma has been reported in the tentorium cerebelli, which bore no anatomical relation to the ventricular system (Yamate et al. 1987).
In human adults, these neoplasms are generally negative in immunohistochemical studies for epithelial marker proteins (intermediate filaments and secretory proteins) but do express the astroglial marker GFAP. Some ependymomas have unique cytoarchitectural features such as vacuoles and papillary projections; the tanycytic ependymoma, originating from the subcommissural organ, consists of specialized ependymal cells (tanycytes), which have long basal processes projecting into the hypothalamus. These variants have been reported in humans but not in rats or mice.
Ependymoma, malignant (Figures 119–124)
Biological behavior: malignant neoplasm
Synonym/synonyms: anaplastic ependymoma
Histogenesis: ependymal cells lining the ventricular system of the brain and the central canal of the spinal cord
Diagnostic features: characteristic features have been detailed for both mice (Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Walker et al. 1994; Krinke and Kaufmann 1996; Radovsky and Mahler 1999, Krinke et al. 2000b) and rats (Dagle, Zwicker, and Renne 1979; Gopinath 1986; Yamate et al. 1987; Jaenisch 1990; Kaneko et al. 1990; Maekawa and Mitsumori 1990; Solleveld and Boorman 1990; Solleveld, Gorgacz, and Koestner 1991; Cardesa et al. 1994; Schmitt et al. 1997; Spassky et al. 2005):
the neoplasm is located near the ventricles and aqueduct of the brain and/or central canal of the spinal cord;
polygonal cells are arranged in rows, rosettes about empty central lumens, and/or pseudorosettes surrounding blood vessels. Rosettes and pseudorosettes may be uncommon;
subcellular organelles indicative of well-differentiated ependyma (e.g., cilia and associated basal bodies [blepharoplasts]) are uncommon;
features of malignancy include pronounced cellular atypia and pleomorphism, invasive growth, foci of necrosis, and multinucleated giant cells;
the neoplasm may include other glial elements (reflecting the glial origin of ependymal cells);
mitotic activity and cellularity are relatively high.
Special diagnostic techniques:
mouse ependymal cells express S100β and GLAST (Spassky et al. 2005). GLAST is also a marker for rat ependymal cells (Schmitt et al. 1997);
blepharoblasts, if present, are especially prominent when labeled using the PTAH stain.
Differential diagnoses: ependymoma, benign (see definition in this glossary): this neoplasm is confined to the ventricular lumen; neoplastic cells are well differentiated and tend to form rows, rosettes around empty lumens, and pseudorosettes surrounding blood vessels; cilia and basal bodies (blepharoplasts) are common. oligodendroglioma, malignant (see definition in this glossary): this neoplasm presents as sheets of neoplastic cells with condensed round nuclei, clear (nonstaining) cytoplasm (i.e., fried egg appearance), distinct cellular borders, and atypical capillary endothelial hyperplasia; this mass may be found in extraventricular locations.
Comment: Ependymomas, in common with oligodendrogliomas, are located near the ventricular system and may contain areas in which neoplastic cells are arranged in rows and rosettes. Because ependymomas in rats are rare, adequate diagnostic criteria have not been defined to differentiate the malignant variants of these two tumors from malignant gliomas.
Malignant glioma seems to be the diagnosis preferred by some pathologists rather than malignant ependymoma for certain CNS neoplasms induced by ENU. These tumors apparently originate from the cells of the subependymal matrix and differentiate into glial cells as well as ependymal cells; some pathologists classify these neoplasms as glioependymomas. Among the ENU-induced CNS tumors, malignant ependymomas are the most common type of spinal cord tumor. They are characterized by a direct connection to the ependymal lining of the central canal. In contrast, ENU-induced malignant ependymomas in the brain are rare and lack any direct connection to the ependymal lining of the ventricular system (Koestner, Swenberg, and Wechsler 1971).
Choroid Plexus
Papilloma, choroid plexus (Figures 125 and 126)
Biological behavior: benign neoplasm
Synonym/synonyms: none
Histogenesis: epithelial cells of the choroid plexus
Diagnostic features: characteristics were compiled from Thompson et al. 1961; Wechsler, Rice, and Vesselinovitch 1979; Morgan et al. 1984; Dickson et al. 1985; Marks et al. 1989; Chandra, Riley, and Johnson 1992; Koestner et al. 1999; Pace 1998; Radovsky and Mahler 1999:
the neoplasm is located close to anatomic sites of choroid plexus;
well-developed papillary formations occur in an arboriform (intricately branched) pattern;
a single layer of cuboidal to columnar epithelial cells with round to oval nuclei and abundant eosinophilic cytoplasm line narrow fibrovascular stromal cores;
epithelial cell pseudostratification (piling up) is absent;
mitotic figures are absent.
Special diagnostic techniques: choroid plexus epithelium in the rat expresses transthyretin at very high levels. Choroid plexus epithelium in rats shows positive immunohistochemistry for cytokeratin antibodies.
Differential diagnoses: carcinoma, choroid plexus (see definition in this glossary): mass arises from the choroid plexus and exhibits the following cytoarchitectural features of malignancy: invasive growth into the adjacent brain, cellular atypia and pleomorphism, and prominent pseudostratification (piling up); mitotic rate is variable, but mitoses are present. ependymoma, benign (see definition in this glossary): mass arises from the ependyma, usually at a distance from sites of choroid plexus attachment; instead of a papillary pattern, neoplastic cells are arranged in rows, rosettes (about small, empty central lumens), and pseudorosettes (about capillaries); neoplastic ependymal cells have cilia and a basal body (blepharoplast).
Comment: Papillomas of the choroid plexus are among the neuroectodermal tumors observed less commonly in rats. They are also extremely rare in mice with the exception of the cases of chemically induced tumors (intraventricular administration) or in transgenic mice transfected by simian virus 40.
In dogs, papillomas stain with cytokeratin antibodies. While some human choroid plexus tumors demonstrate multifocal immunoreactivity for GFAP, similar staining has not been reported in nonhuman tumors of the choroid plexus.
Carcinoma, choroid plexus
Biological behavior: malignant neoplasm
Synonym/synonyms: none
Histogenesis: epithelial cells of the choroid plexus
Diagnostic features: characteristics were compiled from Zimmerman and Innes 1979; Swenberg 1982; Morgan et al. 1984; Gopinath 1986; Solleveld et al. 1986; Solleveld, Gorgacz, and Koestner 1991; Yamate et al. 1987; Frith and Ward 1988; Morgan and Alison 1988; Faccini, Abbott, and Paulus 1990; Maekawa and Mitsumori 1990; Solleveld and Boorman 1990; Cardesa et al. 1994; Walker et al. 1994; Krinke and Kaufmann 1996, Krinke et al. 2000b:
the neoplasm is located close to anatomic sites of choroid plexus attachment;
the neoplasm is composed of well to poorly developed papillary formations in an arboriform pattern;
multiple rows of atypical and pleomorphic epithelial cells line a fibrovascular stromal core;
epithelial cell pseudostratification (piling up) is a frequent feature;
invasion into the adjacent brain is usual.
Special diagnostic techniques: choroid plexus epithelium in the rat expresses transthyretin at very high levels
Differential diagnoses: papilloma, choroid plexus (see definition in this glossary): papillae consist of a single layer of cuboidal to columnar epithelial cells with round to oval nuclei and abundant eosinophilic cytoplasm lining narrow fibrovascular stromal cores; neoplastic cells do not invade the underlying brain; cytoarchitectural features of malignancy (cellular atypia and pleomorphism, epithelial pseudostratification) are absent. ependymoma, malignant (see definition in this glossary): a papillary, fibrovascular stroma lined by nonciliated epithelial cells is not a feature of ependymomas. Comment: Neoplasms of the choroid plexus are rare to extremely rare in rats and mice.
Other Cell Lineages
Hamartoma, lipomatous (Figures 127 and 128)
Biological behavior: nonneoplastic, space-occupying mass derived from a resident cell type
Synonym/synonyms: none
Histogenesis: adipocytes (white fat cells)
Diagnostic features: features have been described in the following references: Budka 1974; Morgan et al. 1984; Morgan and Sheldon 1988; Adkison and Sundberg 1991; Brander and Perentes 1995; Krinke et al. 2000b:
predominantly located in the midline or ventricles of the brain;
well-demarcated mass;
composed of single or multiple clusters of mature white adipose cells containing one large fat droplet;
may be associated with dysgenesis of the corpus callosum and lateral displacement of adjacent blood vessels and brain tissue.
Special diagnostic techniques: none, as the cells have typical features of well-differentiated white fat cells
Differential diagnoses: teratoma, benign or malignant: complex tumors containing fat as well as other tissues (derived from all three germ cell layers)
Comment: This extremely rare tumor-like nodule represents an overgrowth of a cell population that normally occurs at the site (as apposed to a choristoma [heterotopic cell rest], which is a collection of normal mature cells located in an aberrant site). This lesion is not a neoplasm, but its biological behavior is that of a space-occupying benign tumor. Lipomatous hamartomas have been described in C57BL and C3H/HeJ mice (Adkison and Sundberg 1991). It has been reported in rats only once (Brander and Perentes 1995).
Reticulosis, malignant (Figures 129–133)
Biological behavior: malignant neoplasm
Synonym/synonyms: lymphoreticulosis; microgliomatosis; primary (malignant) lymphoma of the nervous system; primary histiocytic sarcoma of the brain
Histogenesis: uncertain (a mixed origin of mononuclear and glial cells has been proposed in other species, including humans)
Diagnostic features: characteristics were compiled from Rubinstein 1972; Koestner 1974; Vandevelde, Fankhauser, and Luginbühl 1985; Garman, Snellings, and Maronpot 1985; Bigner et al. 1986; Gopinath 1986; Garman 1988; Zwicker et al. 1992; Cardesa et al. 1994; Favara et al. 1997; Thio et al. 2006:
the lesion is composed of pleomorphic cells and prominent stroma containing abundant reticulin fibers;
cells diffusely infiltrate meninges as well as perivascular and periventricular areas;
the cellular population consists of variable numbers of lymphoid to histiocytic-type cells with pleomorphic nuclei;
multinucleated giant cells may be prominent in some lesions;
mitotic figures may be numerous.
Special diagnostic techniques: The classic method is to employ a reticulin stain to reveal the abundant reticulin fibers surrounding neoplastic cells. In rats, the tumor cells generally show strong positive immunohistochemistry for the macrophage marker CD68 (clone ED1).
Differential diagnoses: astrocytoma, malignant (see definition in this glossary): the neoplasm may infiltrate along blood vessels and into the meninges, resulting in a secondary desmoplastic reaction; this tumor is composed of round to fusiform cells with indistinct cellular borders; poorly differentiated neoplastic cells may be positive for GFAP but only on extremely rare occasions. Consequently, it may be difficult to differentiate malignant astrocytoma from malignant reticulosis. sarcoma, histiocytic: multicentric lesion inflammation, granulomatous: certain neural lesions have a granulomatous appearance and are considered inflammatory rather than neoplastic; these lesions predominantly exhibit a perivascular pattern of infiltration and are composed of various cell types including lymphocytes, plasma cells, and macrophages. Comparable lesions are recognized in other animal species, particularly dogs.
Comment: Malignant reticulosis was originally used as a diagnostic term for lesions of supposed “microglial” (mesenchymal) and lymphoproliferative cell origin. However, the term today has sometimes been used as a diagnosis for lesions best described as malignant lymphoma. The term has been discarded by some pathologists as the existence of microglial neoplasms has yet to be established.
This term is often applied to lesions of the CNS in rats and dogs that are neither lymphomas nor typical gliomas in terms of their histological characteristics. Lesions produced in F344 rats by exposure to acrylonitrile have been described as microgliomas and were diffusely infiltrative, resulting in marked perineuronal satellitosis and perivascular aggregates. Others have proposed that neoplasms occurring in the brain of Sprague-Dawley rats characterized by prominent meningeal invasion, submeningeal infiltration, satellitosis, and perivascular cuffing should be considered astrocytomas.
In human cases, the cell type in malignant reticulosis has been identified by immunochemical studies as an immunoblast, implying that the lesions are malignant extranodal lymphomas. Definitive identification of the cell lineage in rodents will require further characterization to differentiate cells of lymphoid (CD3-positive T-lymphocytes, CD45RA B-lymphocytes) and monocytic (positive for CD68 (clone ED1) [rat] or F4/80 [mouse]) origin.
Common Artifacts
Certain incidental changes are commonly misidentified as neuropathologic lesions by inexperienced researchers. The three changes described here are the most common such findings in rodents. Identified artifacts should be not reported in the pathology data set. However, systematic artifacts where one dose group only is involved may be recognized and noted as a comment to the organ/tissue at the discretion of the study pathologist.
Dark neuron artifact (Figures 134–136)
Synonym/synonyms: basophilic neurons, dark “spiky” nerve cells, neuronal hyperchromatosis
Diagnostic features: characteristics were compiled from Cammermeyer 1960, 1961, 1972, 1973, 1978; Garman 1990; Summers, Cummings, and DeLahunta 1995; Jortner 2006:
usually abundant in cerebral cortex, hippocampus, cerebellar cortex, and large-sized neurons in the brain stem;
shrinkage of both nucleus and cytoplasm with contracture of the neuronal cell body from the surrounding neuropil, sometimes resulting in clear, polygonal, perineuronal retraction spaces;
tortuous, “corkscrew-shaped” dendrites are occasionally present, especially within the cerebral cortex;
primarily dense basophilic staining of the cytoplasm (cell body and dendrite), usually obscuring a shrunken, dark-stained nucleus due to blending of the nucleus into the cytoplasm;
loss of clarity of Nissl substance;
can occasionally be associated with slight eosinophilia, giving a dark blue-red tint (i.e., amphophilic) on H&E-stained sections;
all affected neurons have comparable features.
Mechanism: the exact pathogenesis is unknown, but certain factors have been confirmed:
rough handling of unfixed tissue (e.g., traumatic dissection, pressure) promotes this change (Cammermeyer 1978; Garman 1990);
ischemia is an integral part of the process (Cammermeyer 1973);
a role for neuronal excitotoxicity is suggested by the demonstration that treatment of rats with glutamate antagonists prior to formaldehyde fixation abolishes dark neuron formation following cortical biopsy (Kherani and Auer 2008).
Differential diagnoses:
necrosis, neuronal (see definition in this glossary): necrotic neurons typically have bright eosinophilic cytoplasm (“red dead” neurons) with dark, condensed nuclei; in their acute stages, ischemic neurons are stated to be indistinguishable from basophilic neurons (Cammermeyer 1972), but such early lesions are usually characterized by the presence of adjacent neurons in different stages of degeneration (in contrast to the monomorphic traits of dark neuron clusters); necrotic lesions of some duration are often associated with glial changes such as reactive glia and/or activated microglia (Jortner 2006).
Comment: The “dark neuron” is a common histological artifact that has often been interpreted by inexperienced investigators as a degenerating or dead cell (Jortner 2006). This finding is most common in immersion-fixed tissues but can also be observed in adequately perfused tissues (Cammermeyer 1978; Garman 1990); the extent of this artifact is least when CNS tissue is fixed by perfusion and then left in situ (i.e., not handled) for several hours prior to removal (Garman 1990).
Myelin Bubbles
Synonym/synonyms: adaxonal vacuoles, myelin spaces, myelin vacuoles
Diagnostic features (Yao et al. 1994):
The finding typically presents as an enlarged but focal space surrounding an intact axon and thus is most readily recognized in longitudinal sections of nerve; typical bubbles are irregular, elliptical expansions with thin walls; several adjacent bubbles may be separated by thin septae (presumably representing the apposed cell membranes of adjacent Schwann cells); larger myelinated fibers in peripheral nerves are more likely to be affected; the change mainly affects paraffin-embedded tissue, not plastic-embedded specimens.
Mechanism: The pathogenesis is unknown
Differential diagnoses: (Collan et al. 1980) degeneration, axonal (see definition in this glossary): the myelin swellings extend for some distance (i.e., form chains); the axon inside swollen Schwann cell chains may be disintegrating; macrophages (gitter cells) may be present within the myelin swellings to remove debris (i.e., myelin digestion chambers); intramyelinic edema (see definition in this glossary): the myelin sheath surrounding axons is disrupted by small to large vacuoles, which may be empty or contain small amounts of membranous material; in later stages of long lasting intramyelinic edema, secondary degeneration of myelin and the axon may develop.
Comment: This finding is a common artifact in nerve biopsies in humans and likely reflects manipulation of the tissue prior to fixation (Collan et al. 1980).
White Matter Vacuolation
Synonym/synonyms: vacuolar artifact
Diagnostic features (Radovsky and Mahler 1999):
focal to widespread, irregular, fine to large vacuoles confined to densely myelinated regions;
predominantly noted in the corona radiata of the cerebrum, corpus callosum, internal capsule, cerebellar deep white matter, and brain stem;
vacuoles have smoothly rounded outlines and are generally empty.
Mechanism: since the nervous system has a high lipid content, histological processing for conventional paraffin embedding results in solvent-associated lipid extraction and often the formation of fine vacuoles in the white matter
Differential diagnoses: age-associated vacuolation: occurs in the cerebral white matter; affects very old female mice (Wells and Wells 1989). astrocytic swelling and vacuolation: the vacuoles are intracellular and, therefore, much smaller. intramyelinic edema (see definition in this glossary): the myelin sheath surrounding axons is disrupted by small-to-large vacuoles which may be empty or contain small amounts of membranous material; in later stages of long lasting intramyelinic edema, secondary degeneration of myelin and the axon may develop. spongiform encephalopathy (Summers, Cummings, and DeLahunta 1995; Wells and Wells 1989): vacuoles occur within neuronal perikarya or processes; in experimental scrapie (the transmissible spongiform encephalopathy affecting sheep) in mice, vacuolation is noted only in perfusion-fixed, paraffin-embedded brain and not in fresh-frozen cryostat sections (Betmouni, Clements, and Perry 1999).
Comment: Artifactual vacuolation occurs randomly in densely myelinated structures, while vacuolar lesions (i.e., neuropathologic changes) commonly exhibit bilateral symmetry (Hooper and Finnie 1987).
Artifactual vacuolation is exacerbated by holding samples of formalin-fixed tissue in 70% alcohol for prolonged periods (i.e., over the weekend; Wells and Wells 1989). Vacuolation is also enhanced in autolyzed tissues and may be accompanied by mild astrocytic swelling and condensation of nuclei accompanying mild neuropil vacuolation in the gray matter (Summers, Cummings, and DeLahunta 1995).
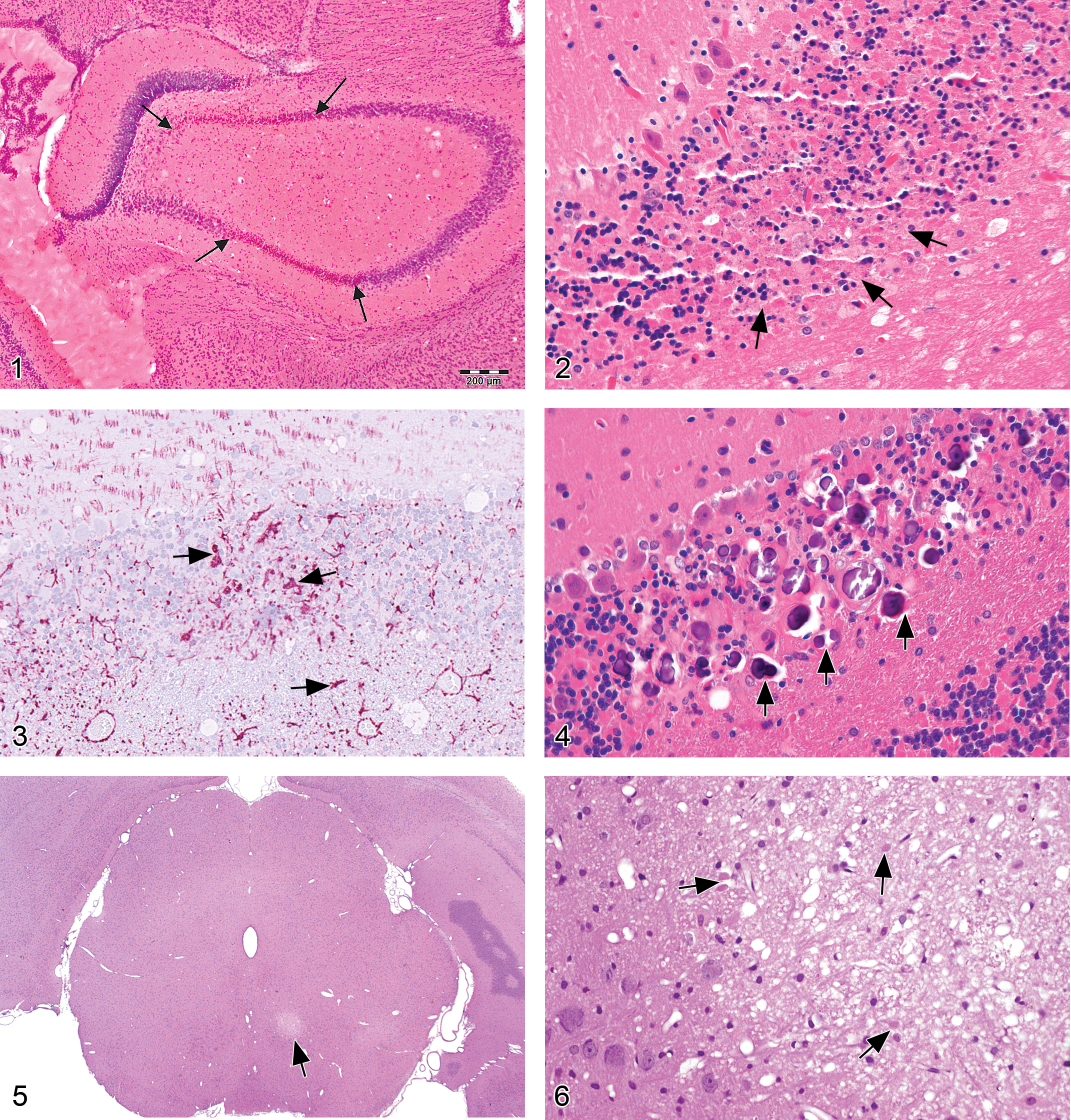
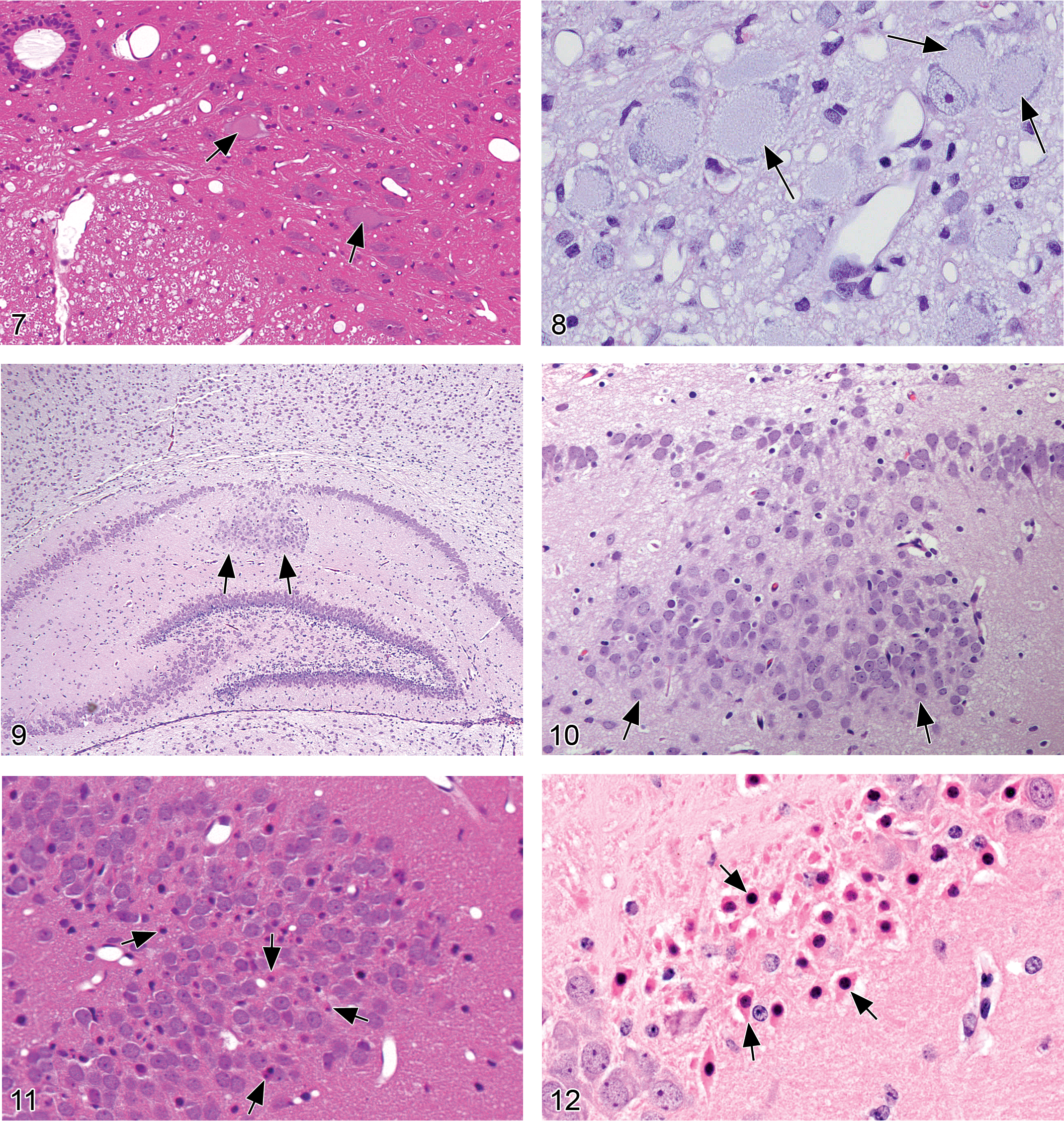
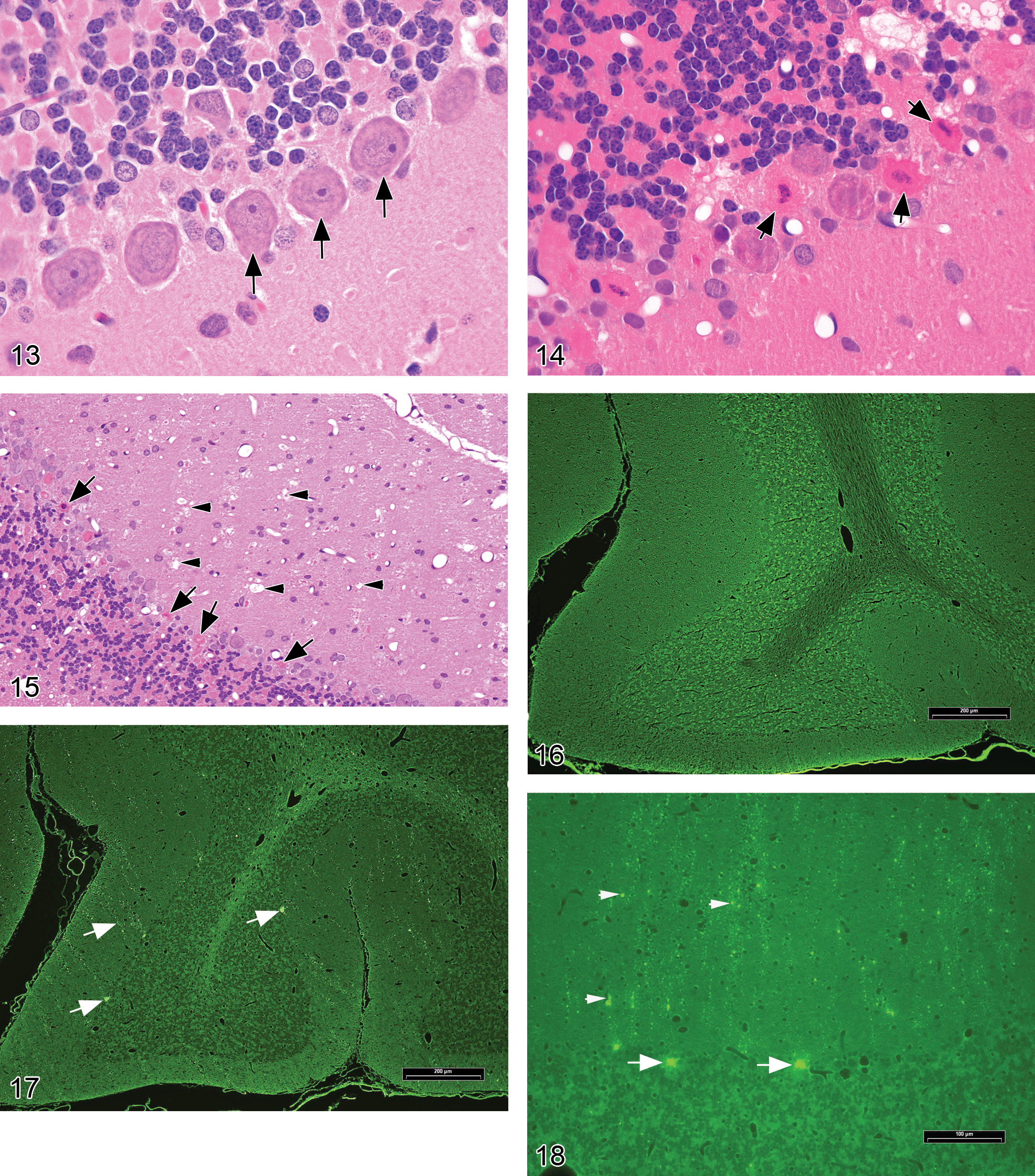
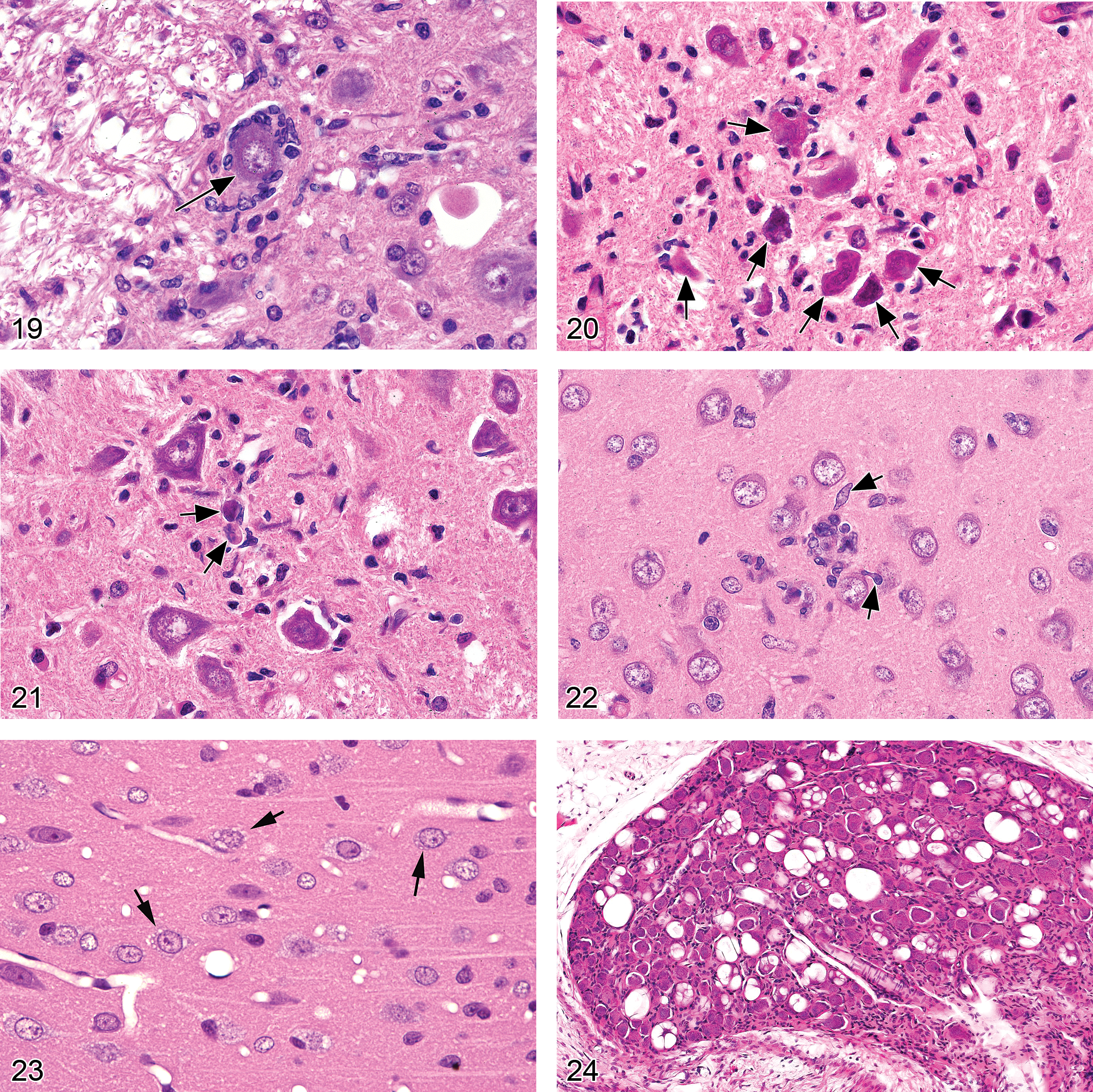
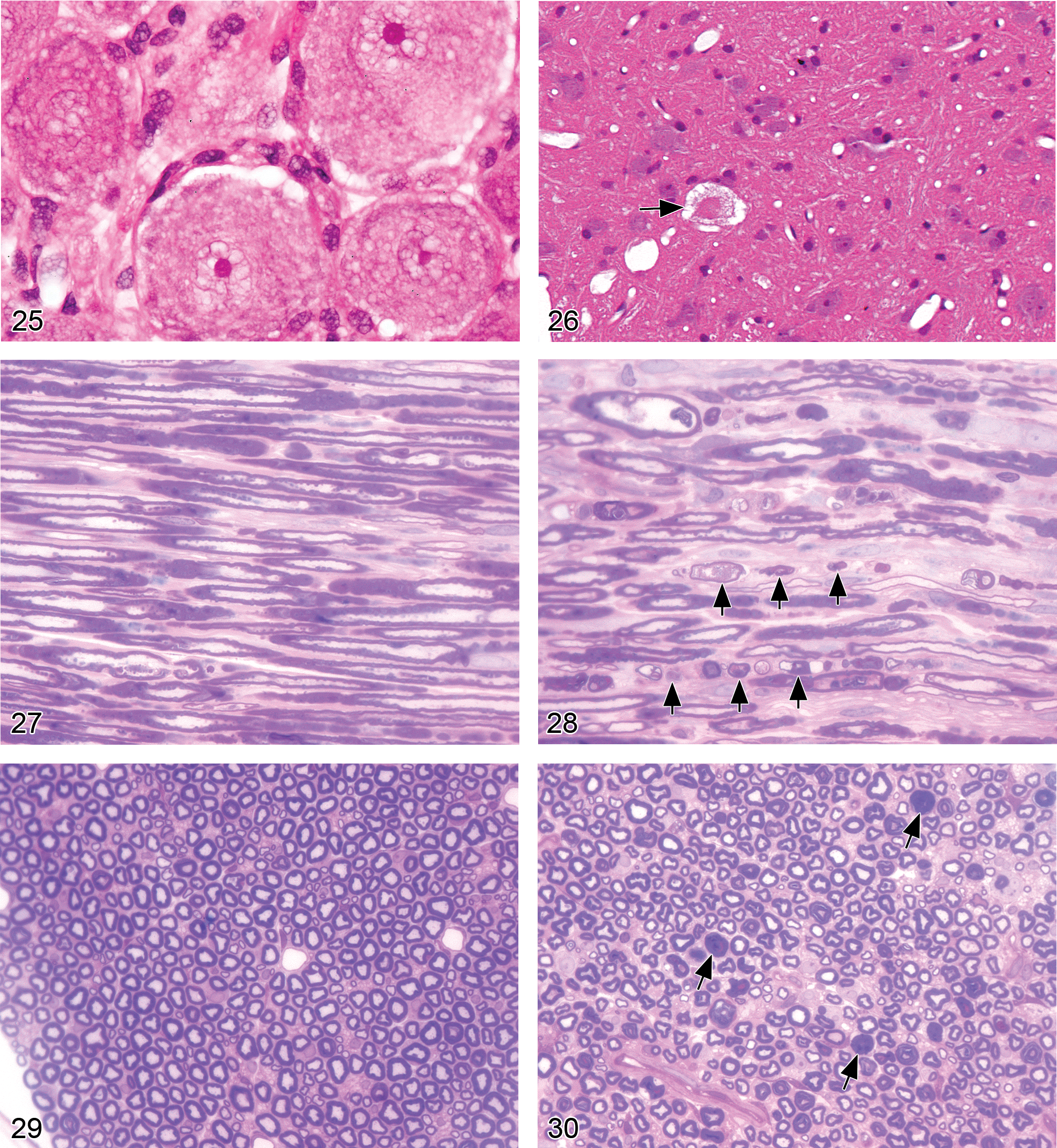
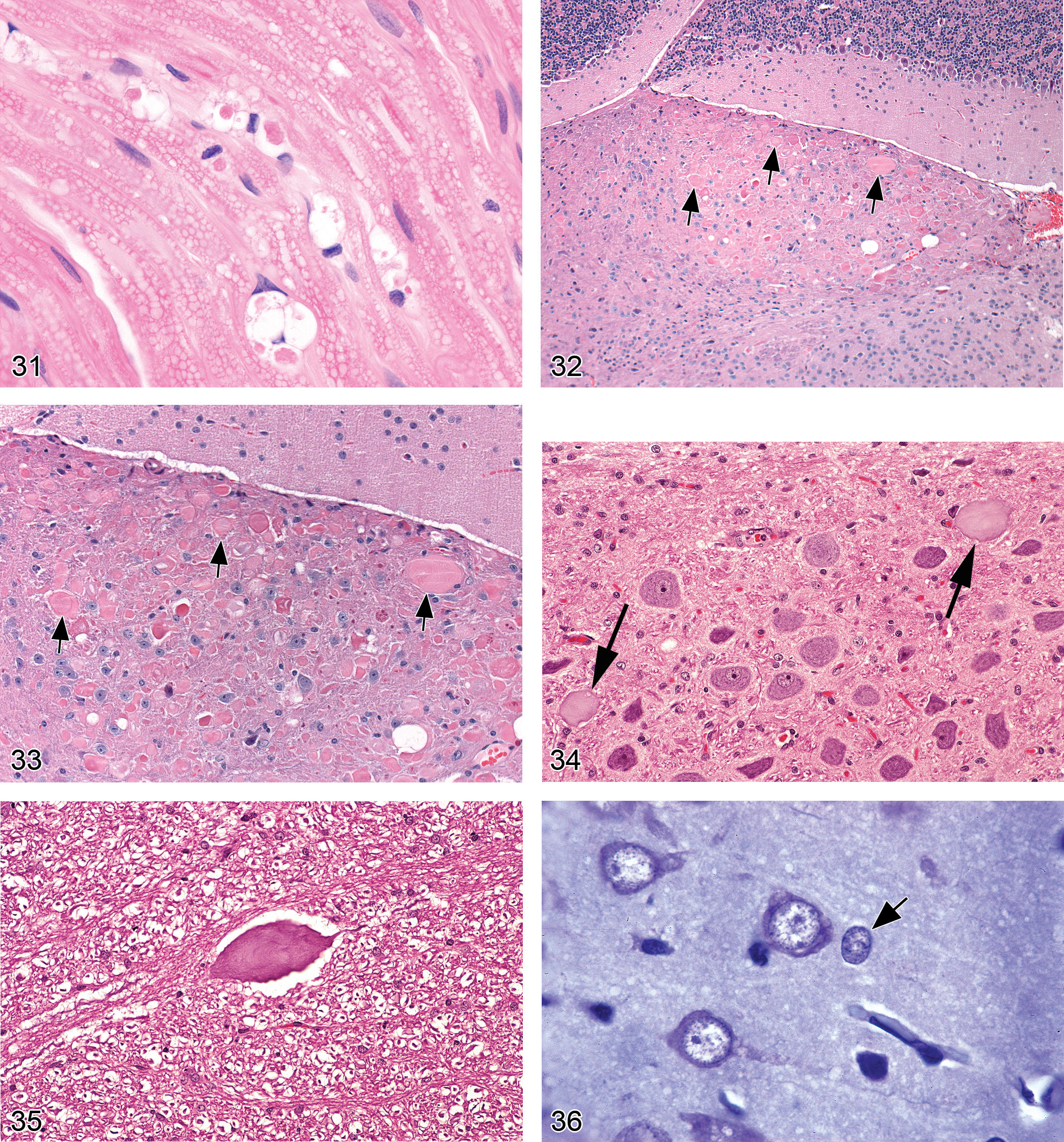
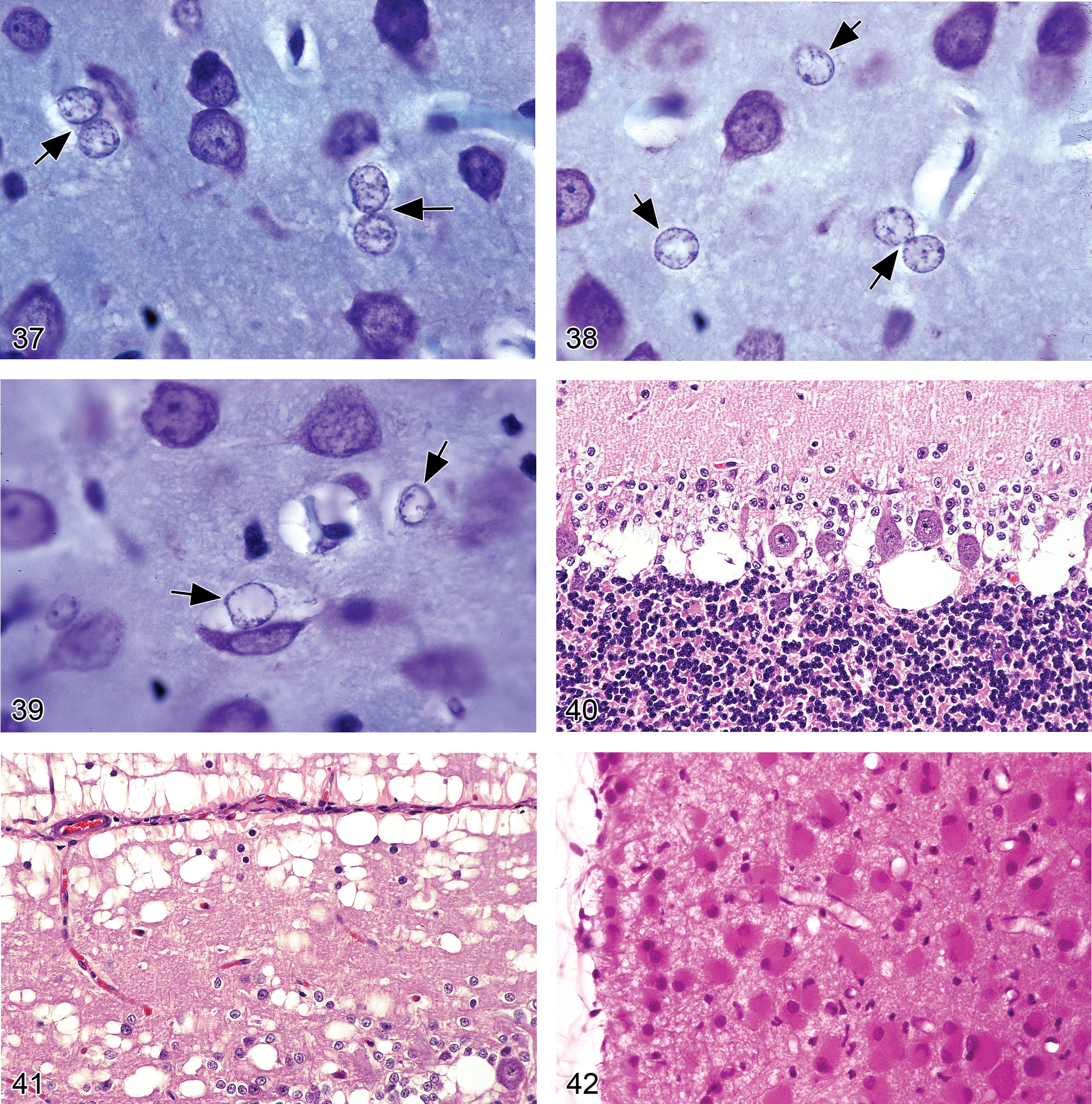
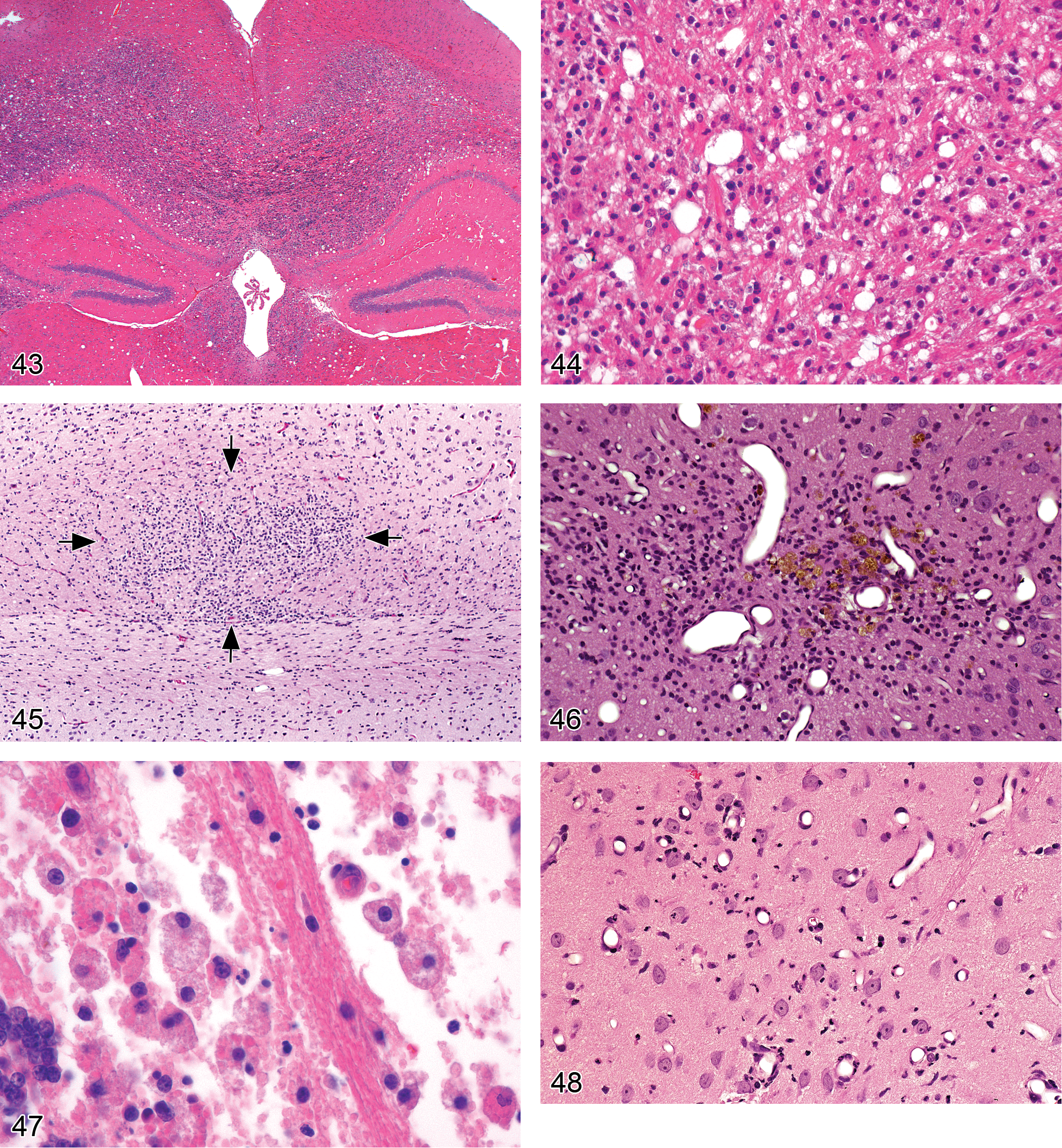
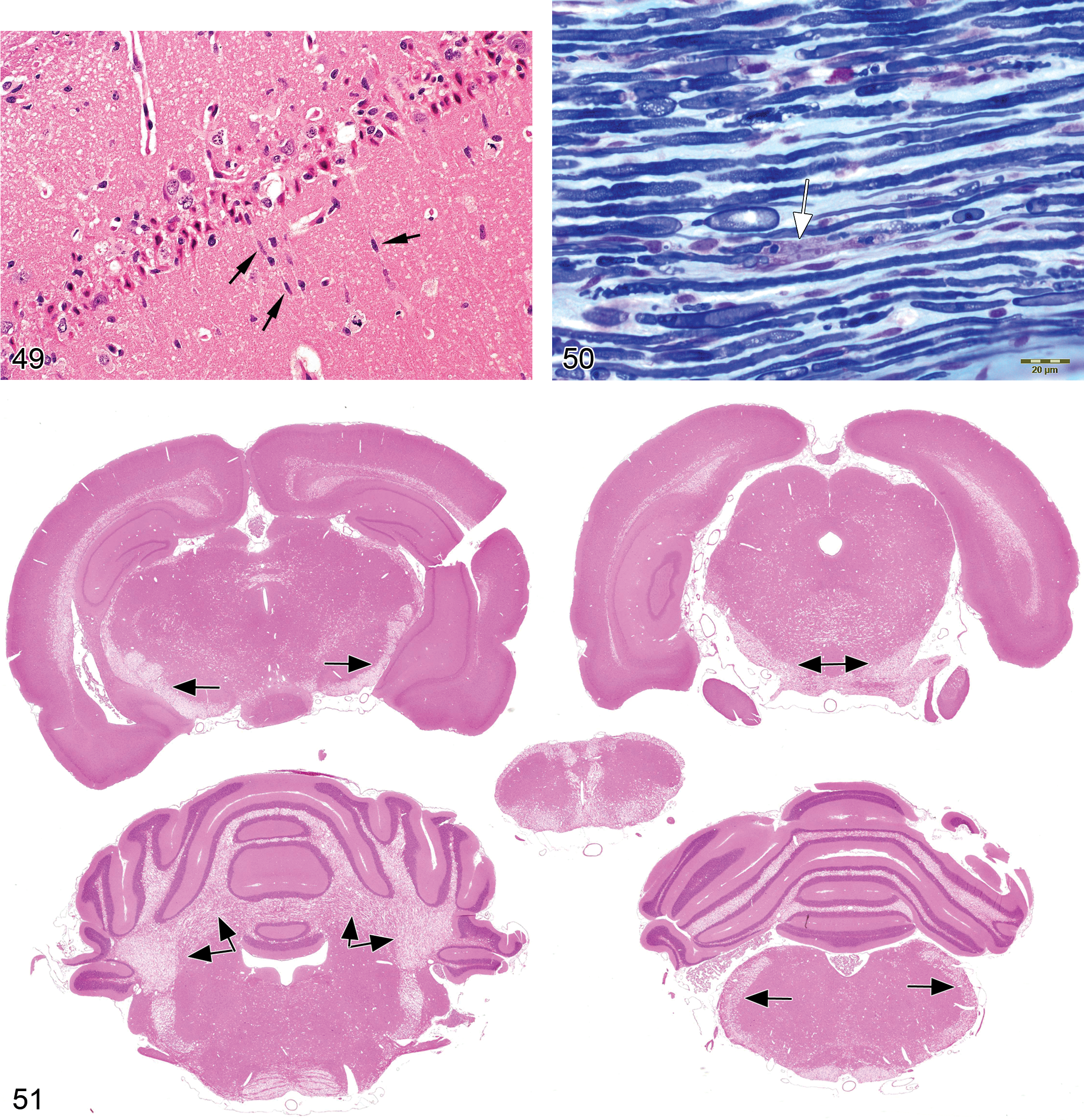
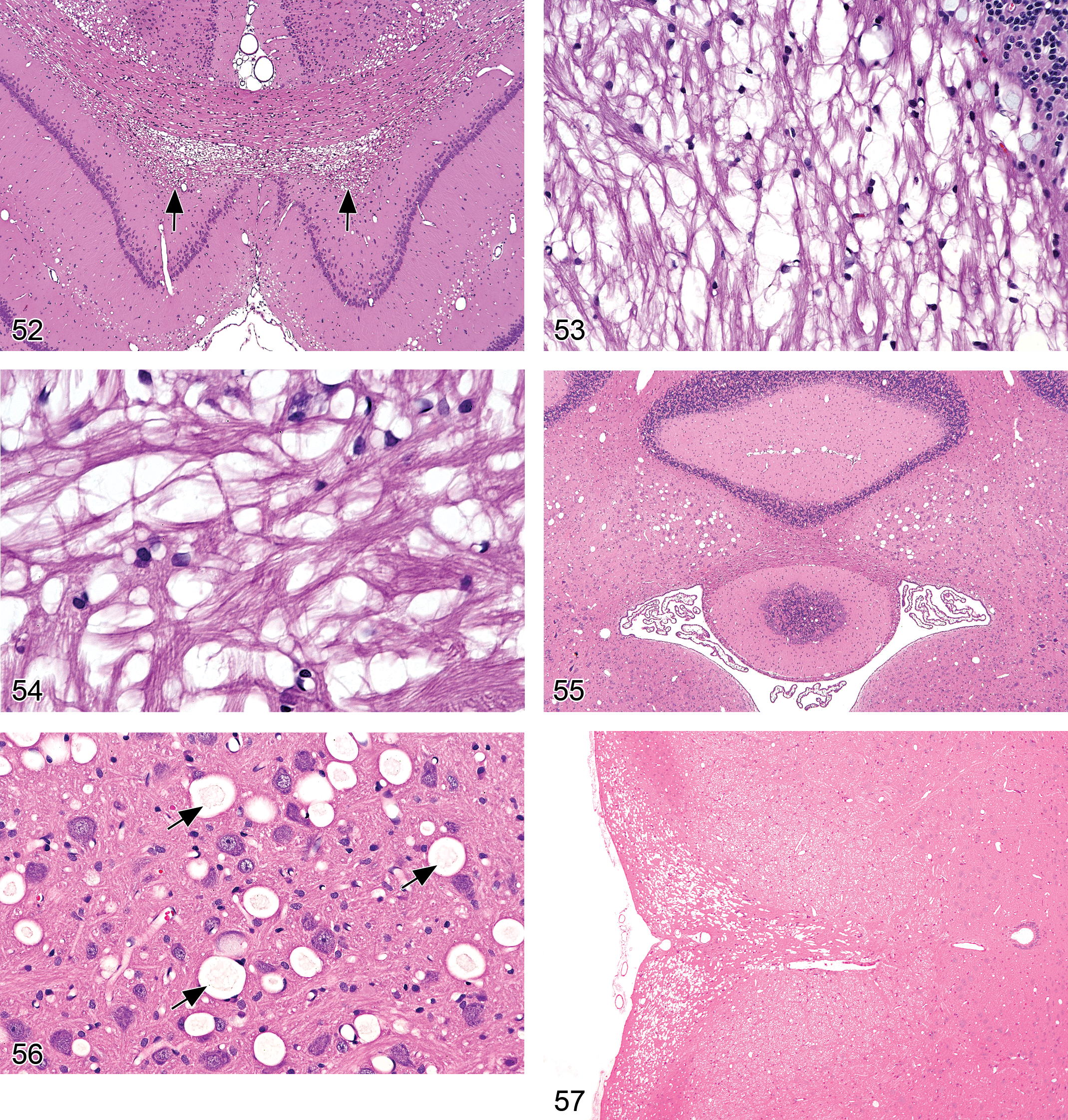
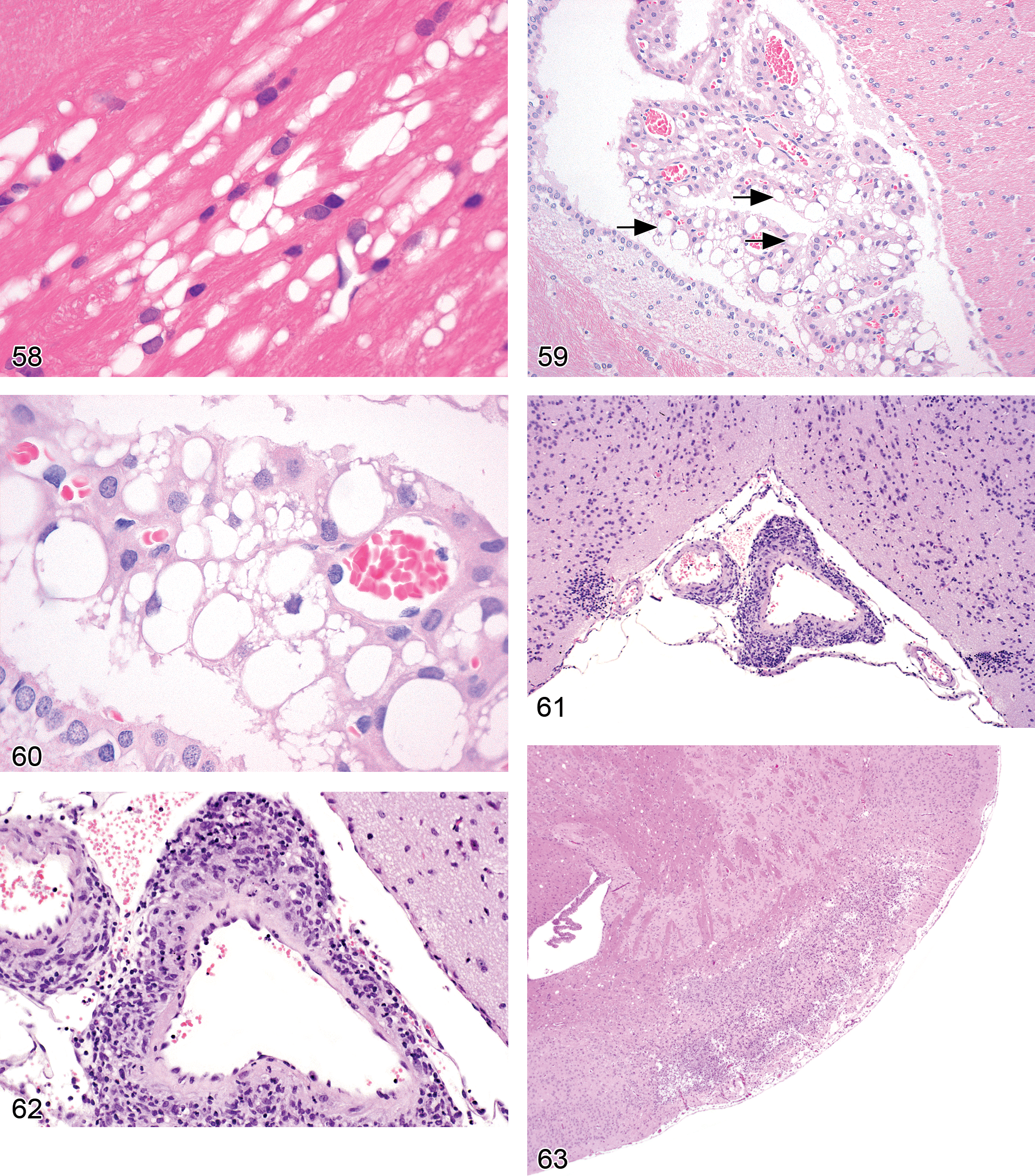
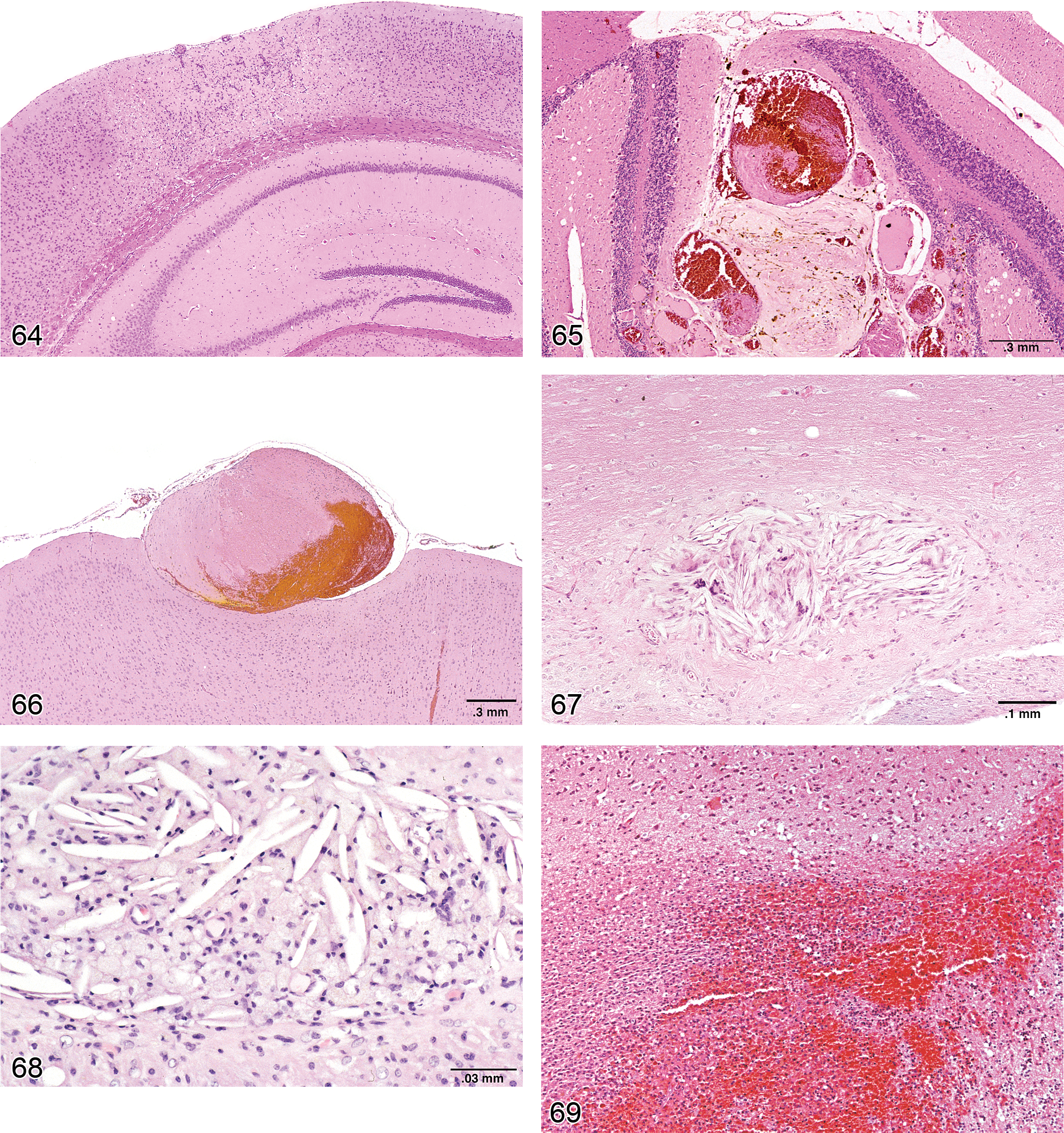
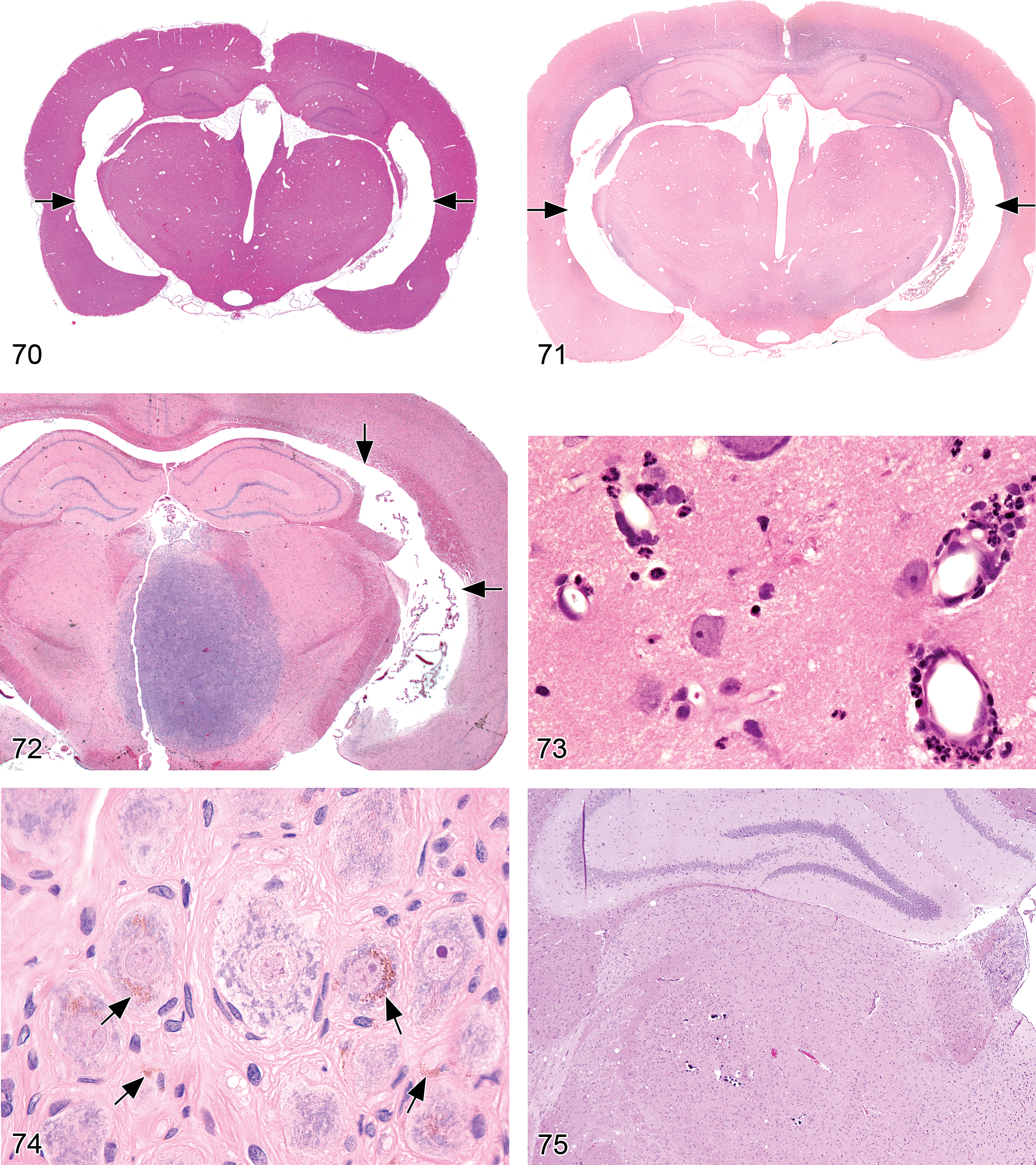
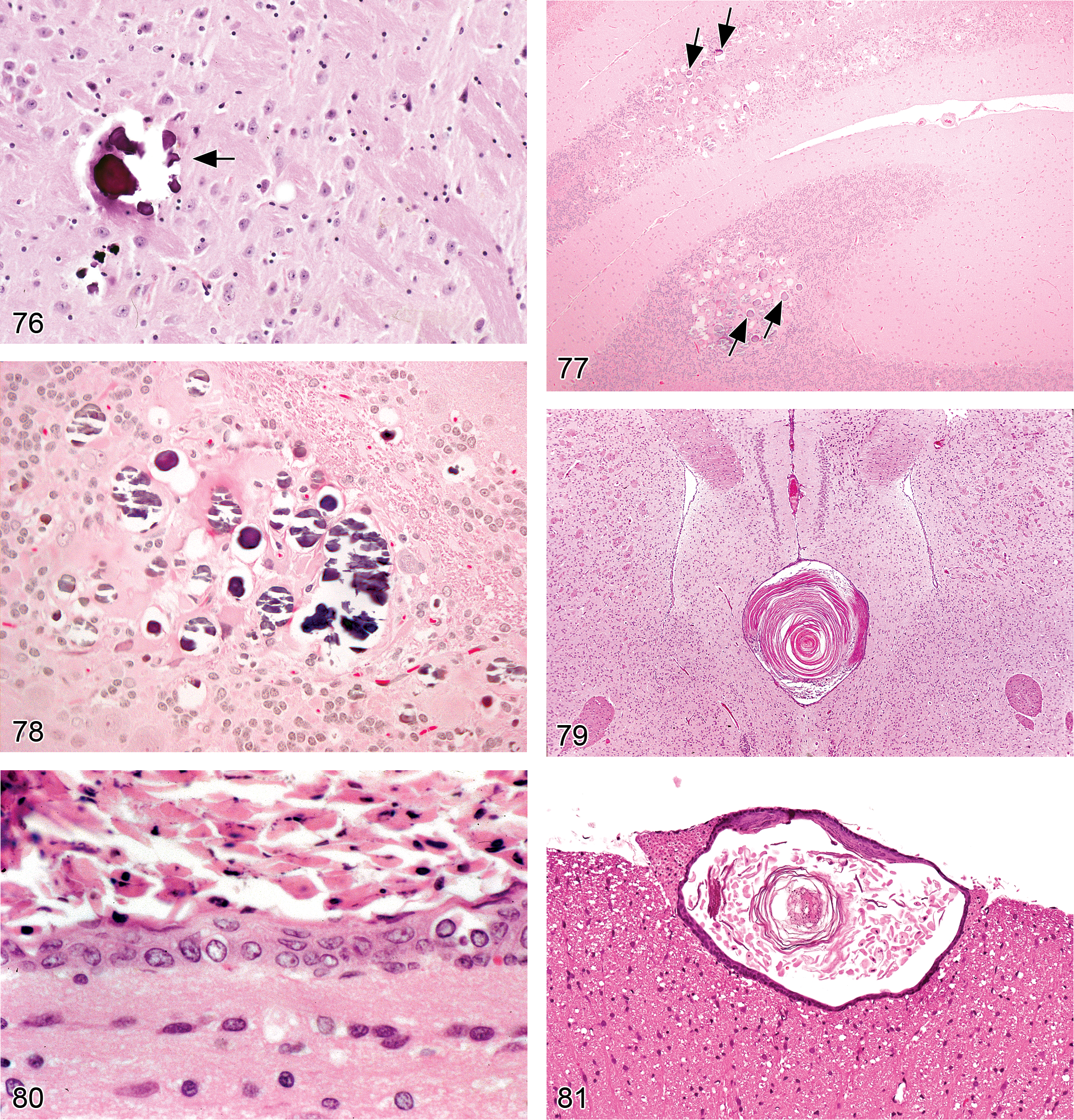
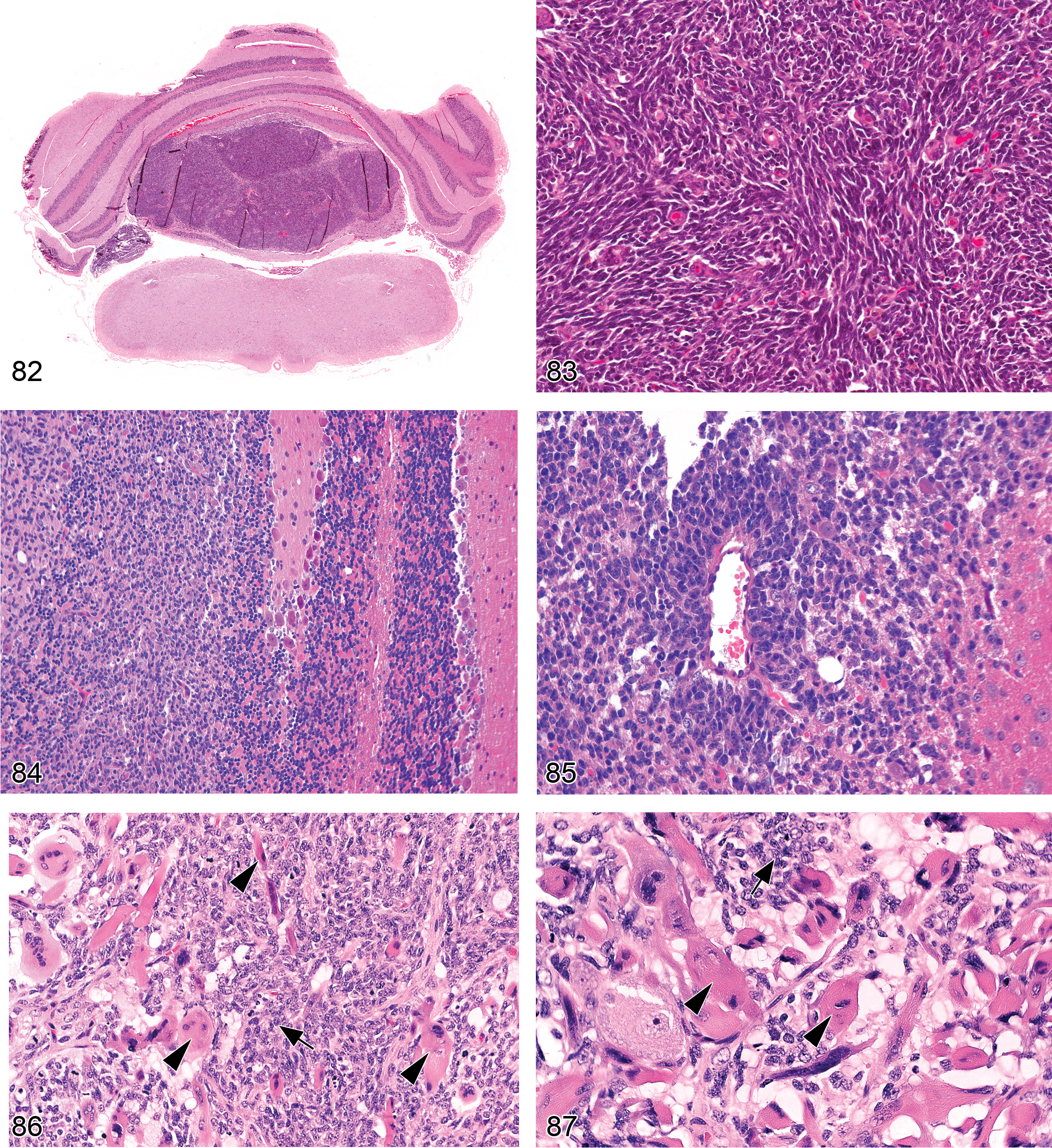
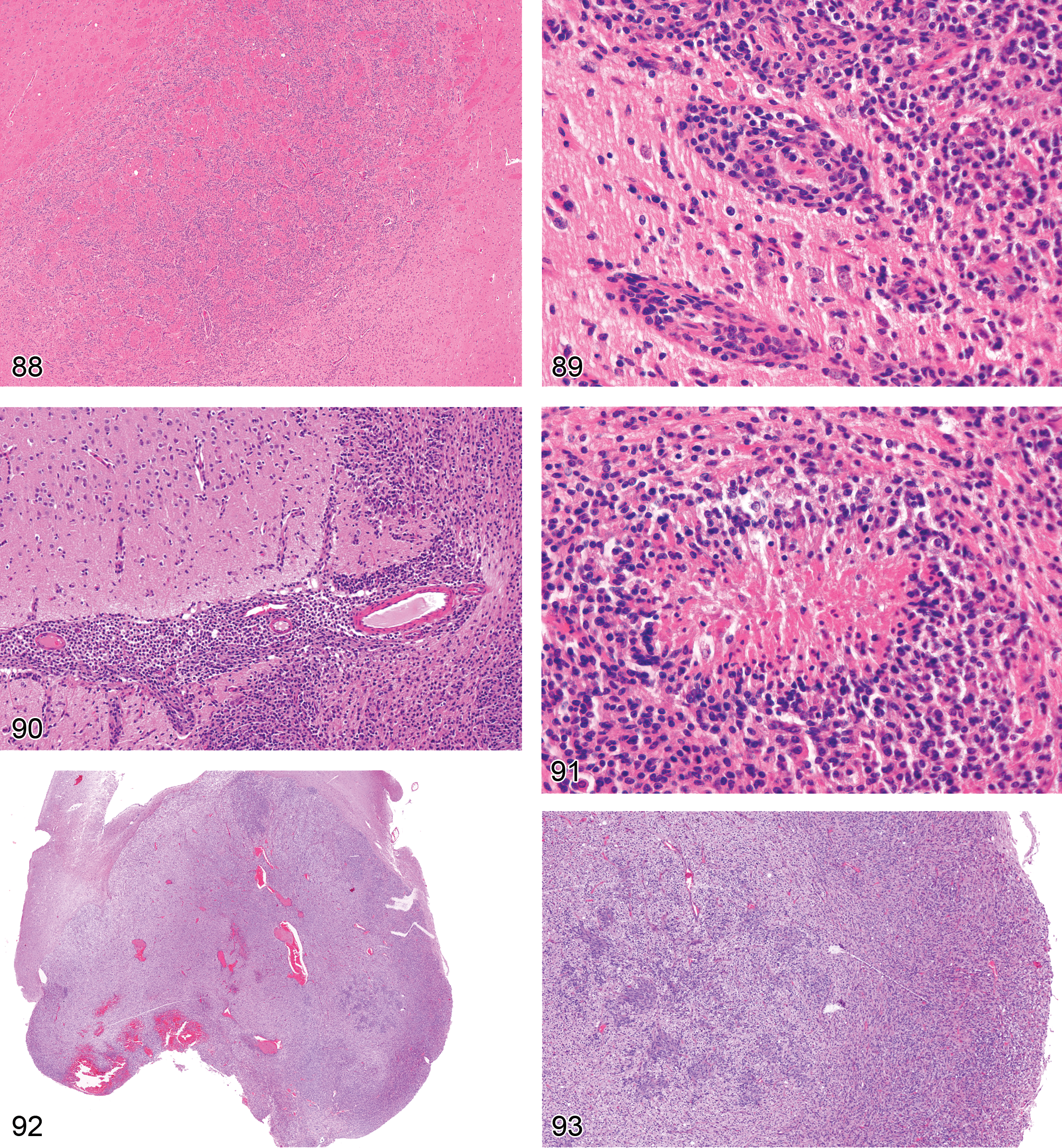
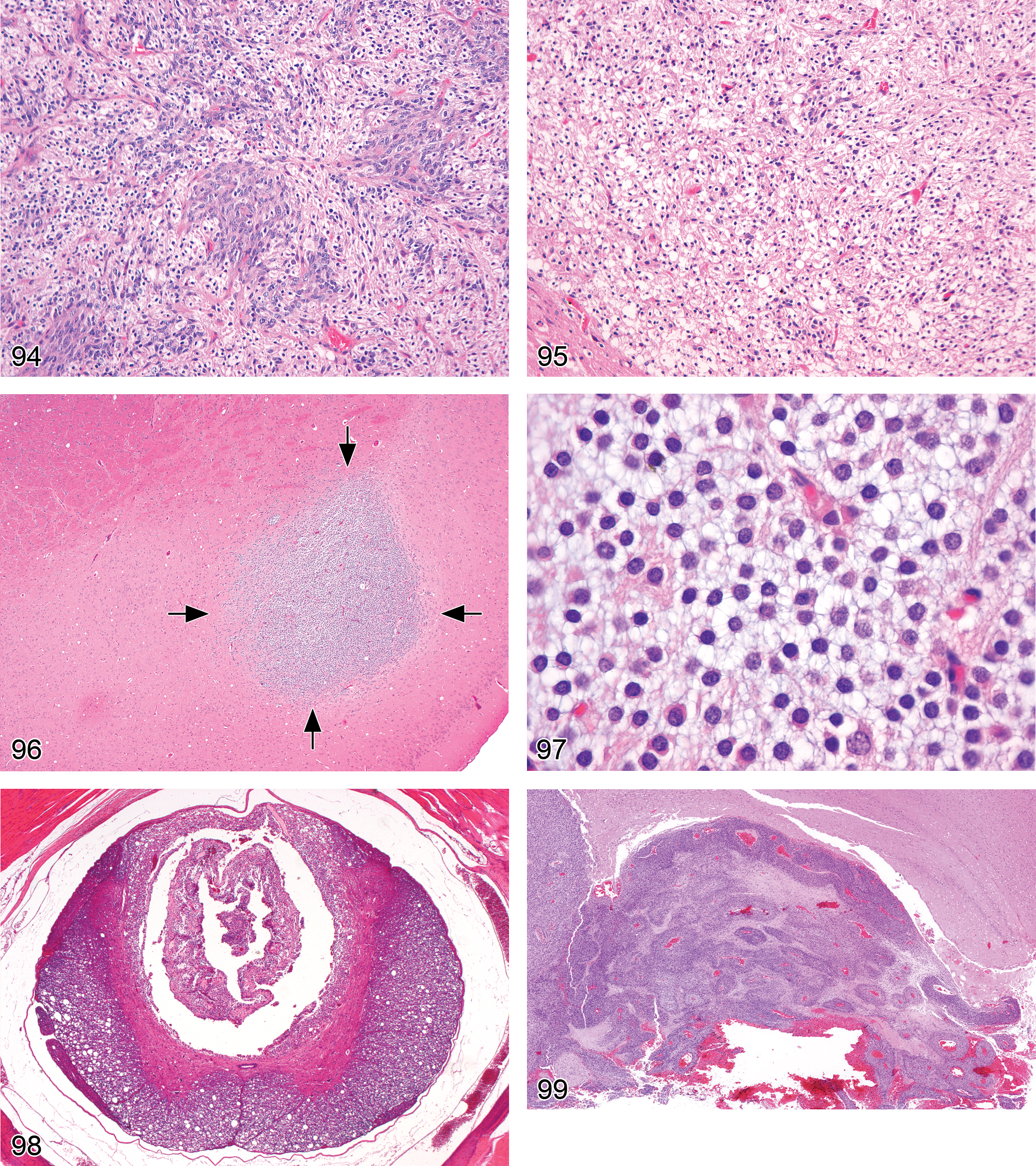
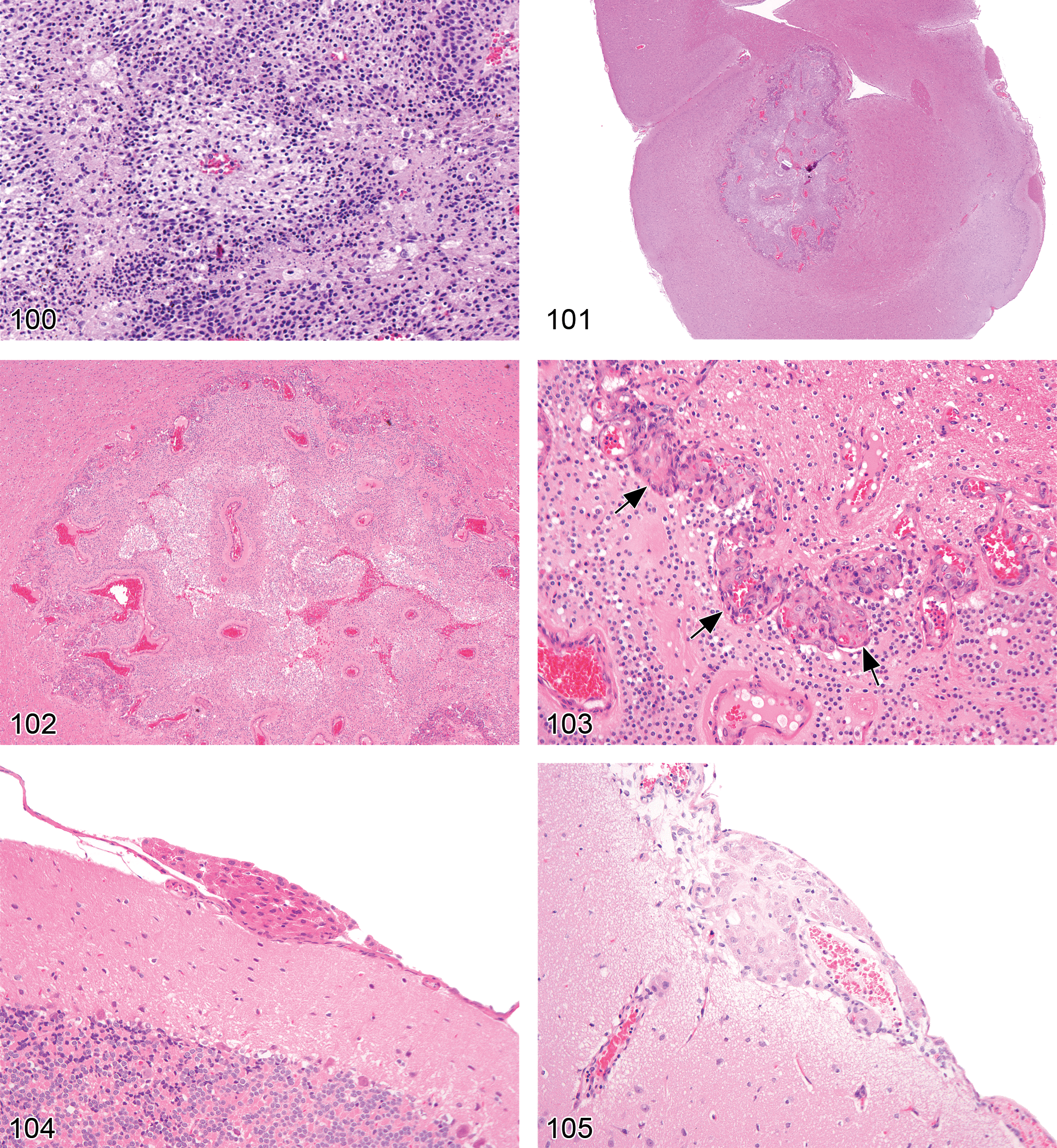
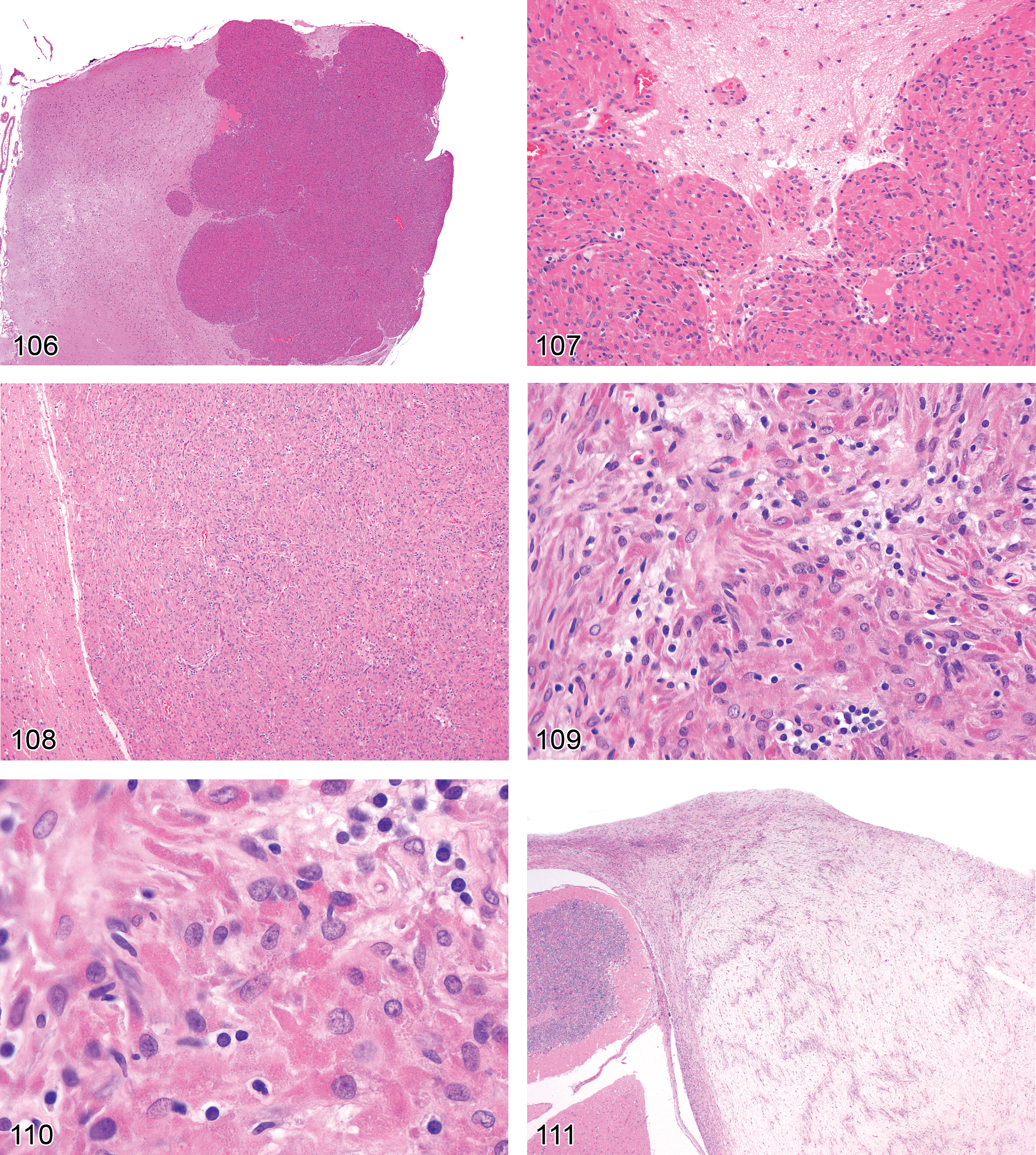
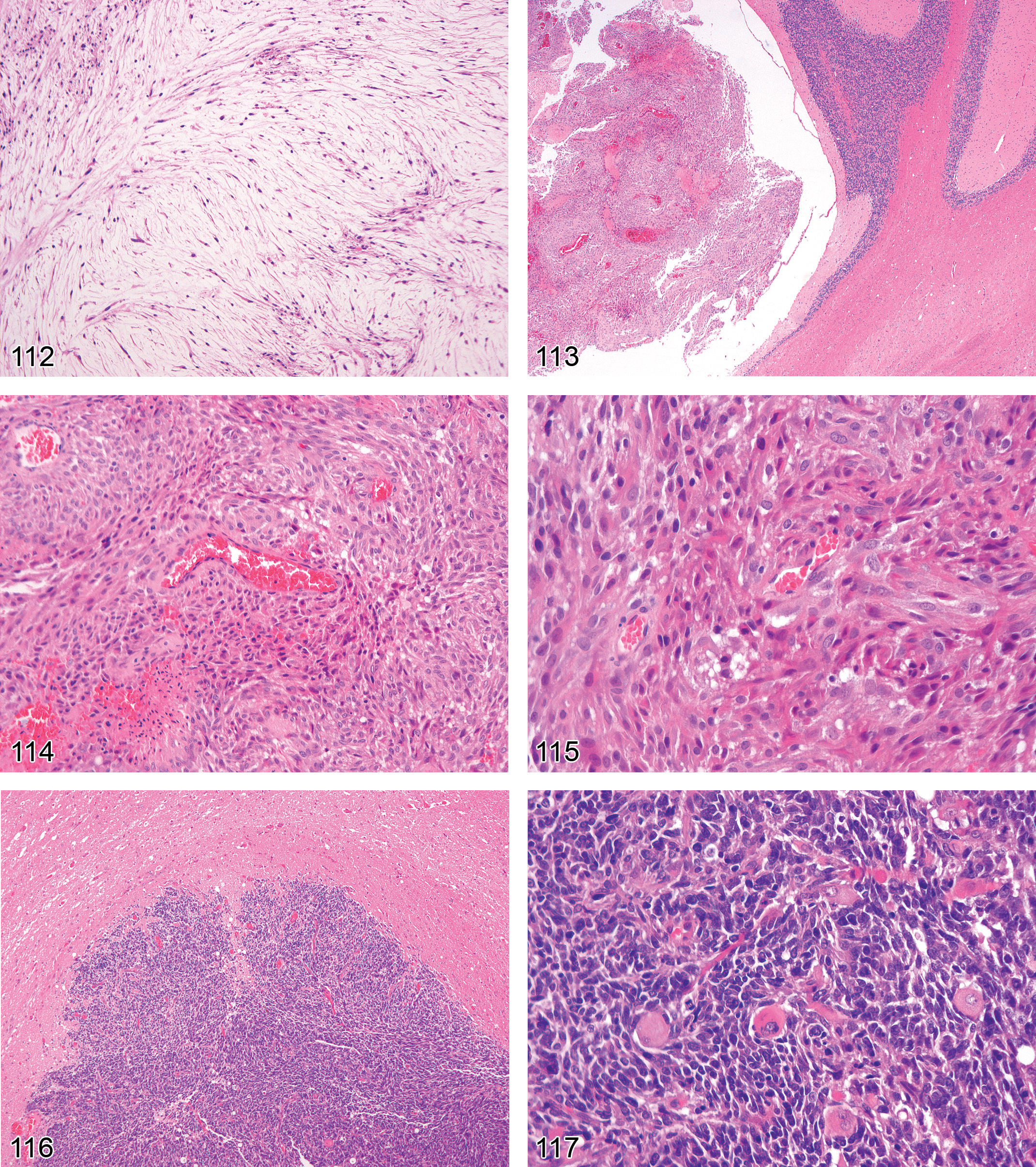
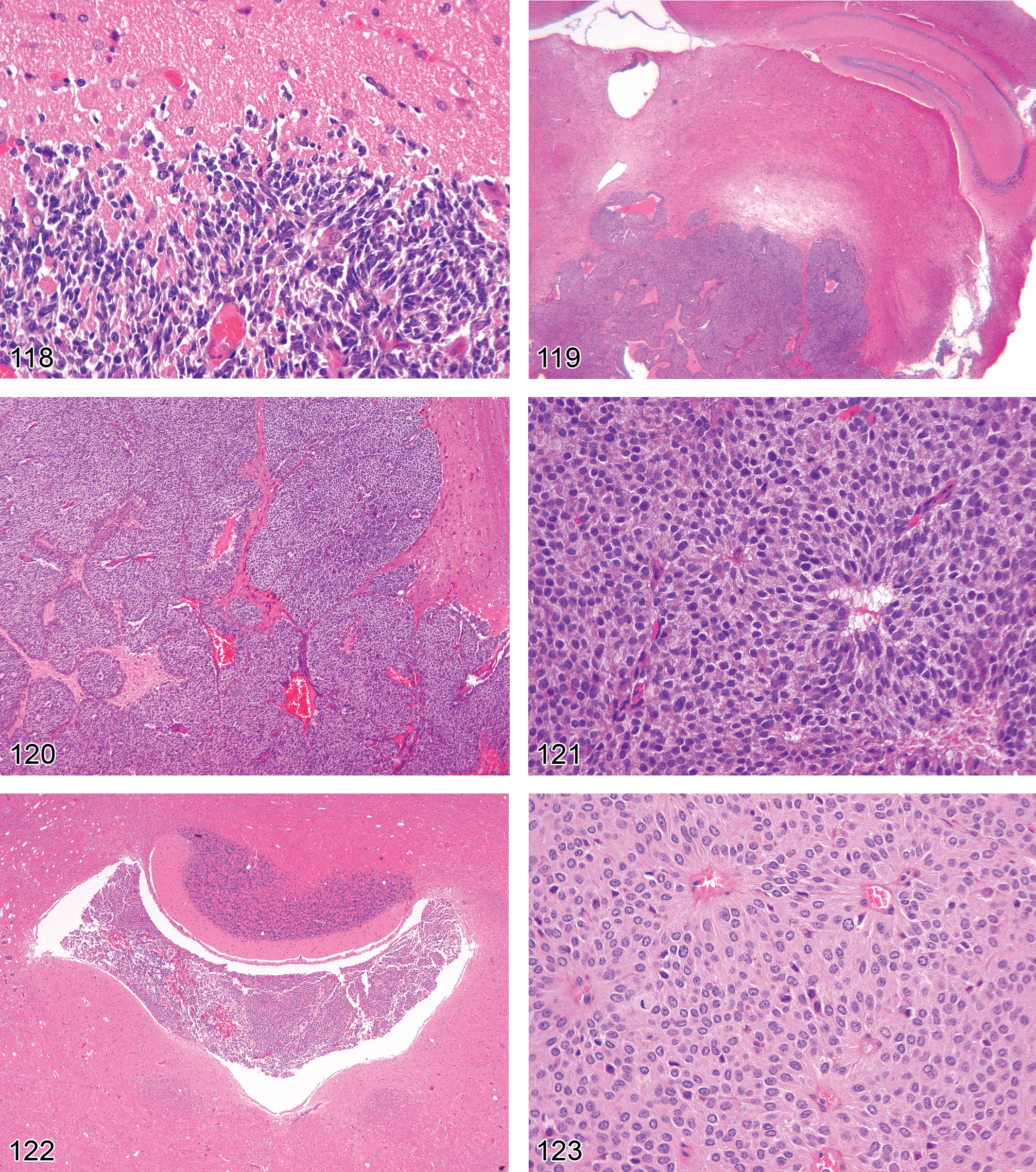
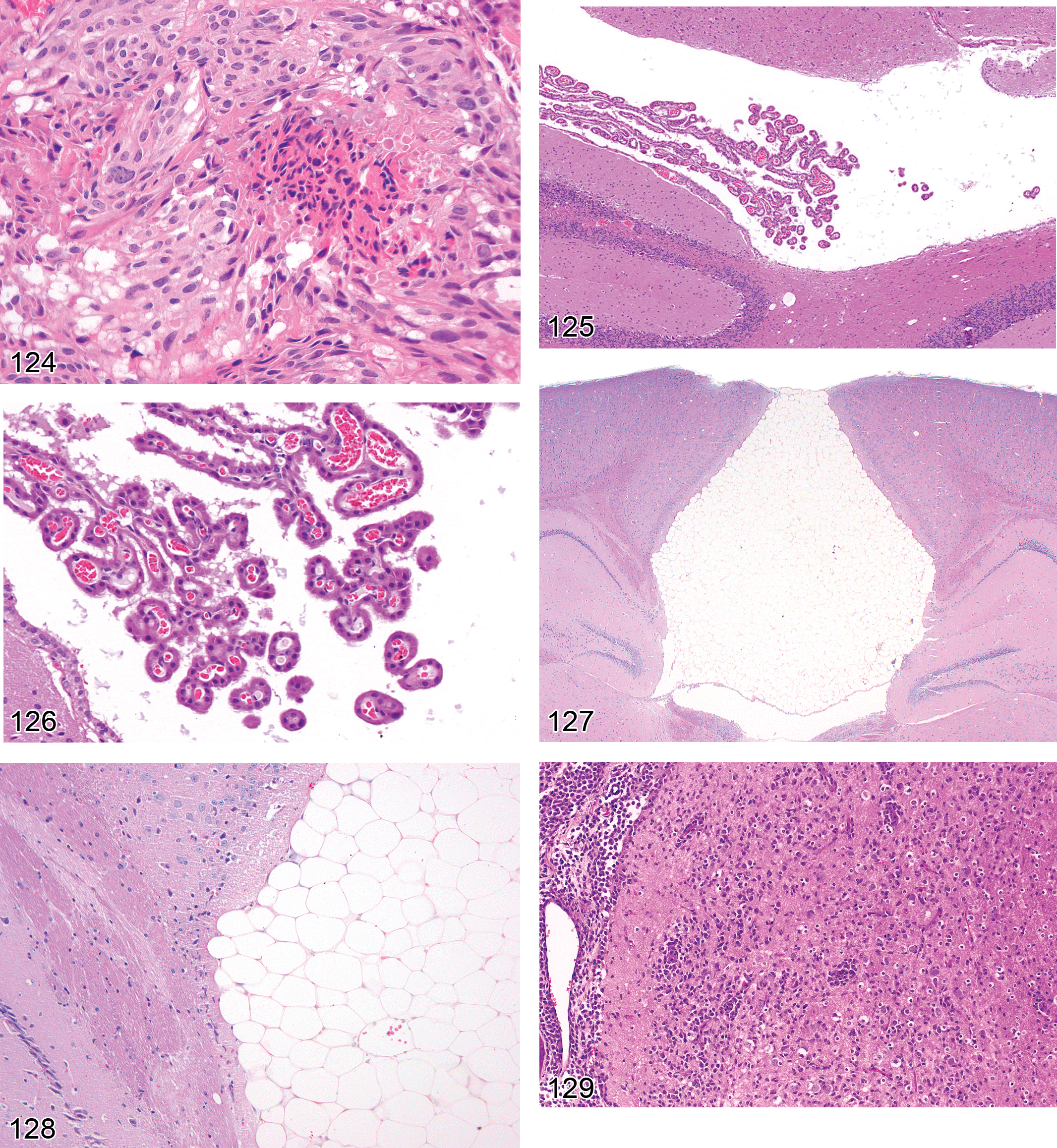
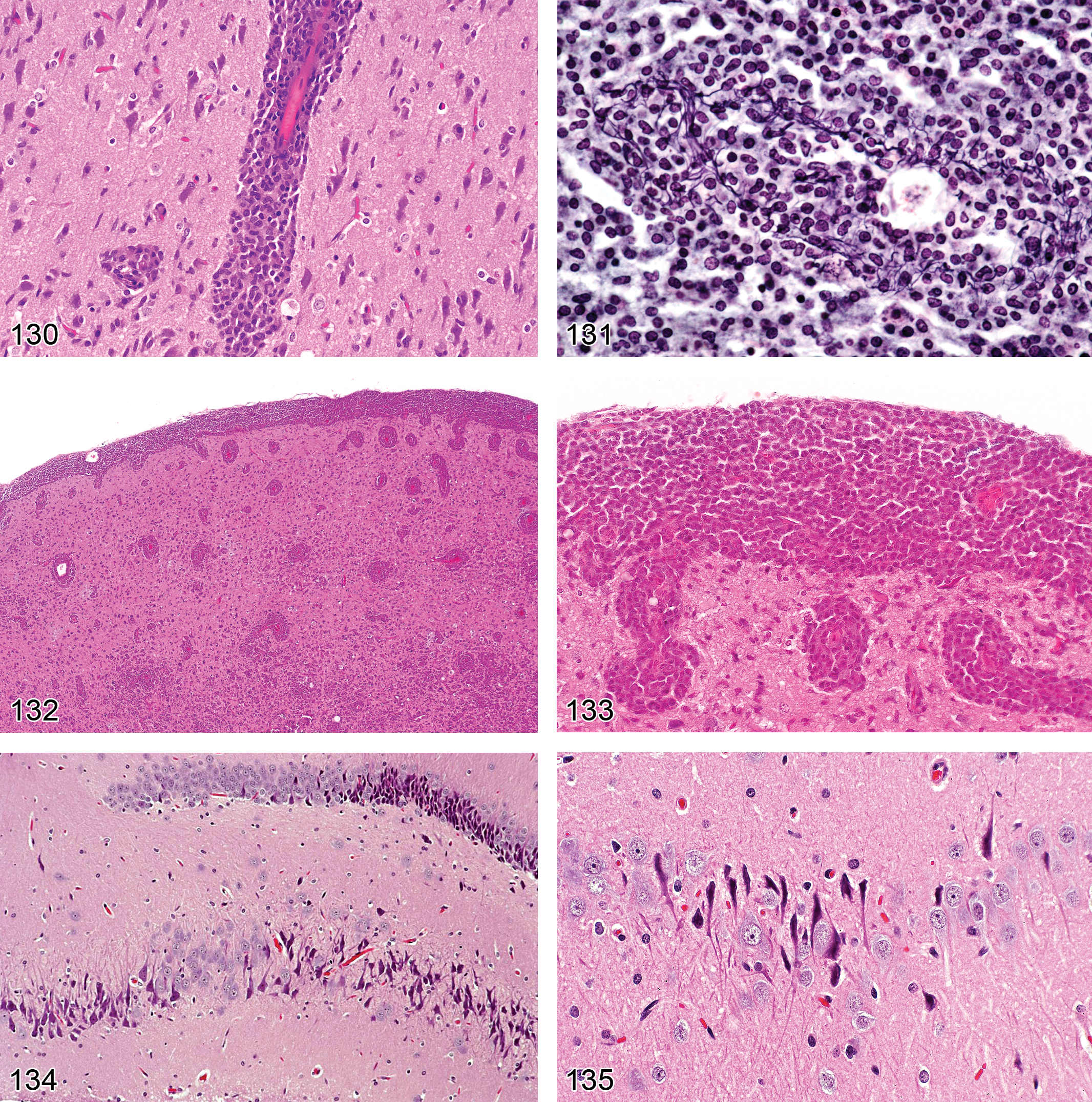
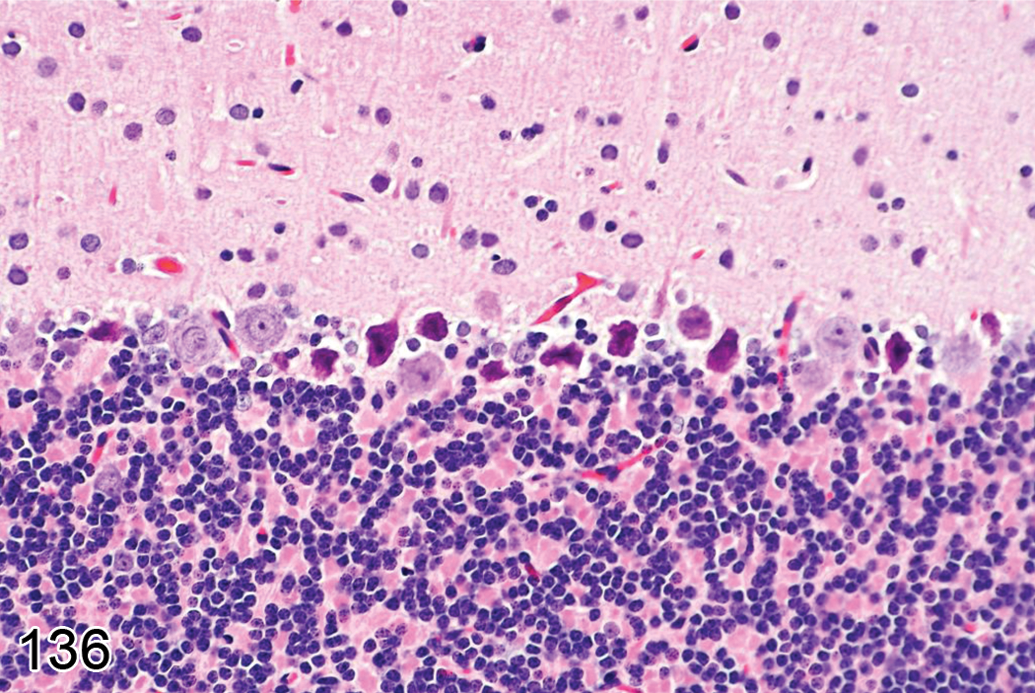
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
Acknowledgments
The authors wish to express their thanks for excellent editorial support and suggestions provided by members of the STP, ESTP, BSTP, and JSTP. Figure numbers 19 through 22 were provided by Dr. J. M. Ward; figure numbers 36 through 39 by Dr. M. D. Norenberg. Special thanks go to Beth Mahler for her support to prepare all the photography provided by different companies, NTP (National Toxicology Program) and RITA database sources.