
Abstract
Mice exposed to the air pollutant ozone develop eosinophilic rhinitis that is mediated by group 2, GATA-3+, innate lymphoid cells (ILC2s). In the present study, we determined the influx, persistence, and recall of nasal ILC2s and eosinophils in ozone-exposed mice. C57BL/6 (T/B cell sufficient, ILC sufficient), Rag2−/− (T/B cell deficient, ILC sufficient), and Rag2−/−Il2rg−/− (T/B cell deficient, ILC deficient) mice were exposed to 0 or 0.8 ppm ozone for 1 or 9 weekdays and killed 1 or 17 days postexposure. GATA-3+ lymphocytes were sparse in nasal tissue of air-exposed ILC-sufficient mice and absent in ILC-deficient mice. Nine-day, but not 1-day, ozone exposures induced nasal influxes of eosinophils and GATA-3+ lymphocytes in C57BL/6 and Rag2−/− mice but not in Rag2−/−Il2rg−/− mice. Eosinophils waned 17 days postexposure in ILC-sufficient strains of mice. GATA-3+ lymphocytes in C57BL/6 mice also attenuated after exposure but not in ILC-sufficient Rag2−/− mice. Eosinophils, but not GATA-3+ cells, increased rapidly with reexposure in ILC-sufficient mice. Type 2 immune-related messenger RNA expression correlated with cellular responses to ozone. These new findings in mice further elucidate the role of ILC2s in ozone-induced eosinophilic rhinitis and support epidemiologic associations between ozone exposure and eosinophilic inflammation in children.
Introduction
In 2016, our laboratory first reported that healthy, nonatopic, C57BL/6NTac mice repeatedly exposed to a commonly encountered gaseous air pollutant, ozone (O3), without concomitant or previous allergen exposure, develop nasal type 2 immunity and eosinophilic rhinitis with hyperplasia and mucous cell metaplasia of nasal epithelium. 1 Subsequently, we found that these ozone-induced airway alterations are mediated by group 2 innate lymphoid cells (ILC2s) and not by the more classical T and B lymphoid cells that are important in adaptive immune responses typically associated with allergic rhinitis and asthma. 2 In addition, we found that repeated exposures of mice to ozone induce ILC2-mediated airway type 2 immunity, eosinophilic inflammation, and mucous cell metaplasia in the pulmonary airways, similar to our observations in the nasal airways of similarly exposed C57BL/6NTac mice. 3 Together, these animal studies strongly suggest that repeated daily exposures to ambient ozone may induce a nonatopic (nonallergic) asthma phenotype characterized by innate type 2 immunity, eosinophilic inflammation, and mucous cell metaplasia in both the nose and the lung of susceptible individuals (eg, children exposed to air pollution). These research findings in mice have provided for the first time a plausible paradigm for the biological mechanisms underlying the epidemiologically identified associations of airway eosinophilic inflammation and new onset of nonallergic asthma with childhood exposures to elevated ambient concentrations of ozone. 4,5
Although we and others 6,7 have demonstrated in mice that ozone-induced airway inflammation is dependent on ILC2s, the specific airway distribution and density of these lymphoid cells in either the upper or the lower respiratory tract after exposures to this gaseous air pollutant have not been elucidated. In addition, the topographical relationship of eosinophilic inflammation (influx of eosinophils) and ILC2s in ozone-exposed airways has not been determined in mice or any other mammalian species, including humans.
The present study was designed to determine the influx (distribution and density), postexposure persistence, and return upon reexposure (recall) of eosinophils and ILC2s to the nasal mucosa of mice exposed to ozone. We further compared the relative quantitative messenger RNA (mRNA) expression of cytokines and other proteins related to type 2 immunity/inflammation to morphometrically determined densities of eosinophils and GATA-3+ lymphoid cells located in the nasal mucosa of ILC-sufficient and ILC-deficient mice after ozone exposure. Since ILC2s are distinguished from other ILCs by nuclear expression of the GATA-3 transitional factor, 8,9 we were able to immunohistologically identify these group 2 innate lymphoid cells in the nasal mucosa by utilizing immunohistochemistry with a GATA-3-specific antibody and Rag2−/− mice that are ILC sufficient but T cell deficient (including GATA-3+ Th2 cells). Furthermore, we compared the kinetics of ozone-induced mucosal accumulations of GATA-3+ lymphoid cells in a T-cell– and ILC-sufficient mouse strain (C57BL/6 mice) with those of the GATA-3+ ILC2s in Rag2−/− mice and Rag2−/−Il2rg−/− (T/B-cell– and ILC-deficient mice).
Materials and Methods
Animals
Six- to 8-week-old male C57BL/6NTac, Rag2−/−, and Rag2−/−Il2rg−/− mice were obtained from Taconic Farms (Germantown, New York). During inhalation exposures, mice were individually housed in stainless-steel wire cages within whole-body inhalation exposure chambers (H-1000; Lab Products Marywood, New Jersey) with free access to water. During postexposure periods, mice were housed individually in plastic shoe-box cages with Aspen Chips bedding and free access to normal laboratory diet (Harlan Teklad Irradiated 8940, Madison, Wisconsin) and water. Rooms were maintained, during periods of inhalation exposures and no exposures, at a temperature of 23.5°C ± 0.1°C (mean ± standard error of the mean [SEM]), humidity of 41.5% ± 1.2%, and with a 12-hour/12-hour light/dark cycle starting at 6:00
Inhalation Exposures to Ozone
All mice in this study were acclimated for a period of 1 week to the whole-body chambers prior to inhalation exposures. Mice of all strains were exposed to filtered air (0 ppm ozone) or 0.83 ± 0.01 ppm ozone (mean ± SEM), 4 h/d, for 1 day or 9 consecutive weekdays (n = 6 per strain per exposure group). These mice were killed 1 day postexposure (Figure 1). Mice did not receive inhalation exposures over the weekend (no Saturday or Sunday exposures). An additional 12 animals per exposure group of Rag2−/− and C57BL/6 mice were given a 17-day postexposure (recovery) period with no inhalation exposure. Half of these mice (n = 6 per group) were killed at the end of the 17-day recovery period. The other half (n = 6 per exposure group) were reexposed for 1 day (4 hours) to air or ozone (Figure 1).

Experimental study design for inhalation exposures and animal necropsies. All strains of mice were exposed to filtered air (0 ppm ozone) or 0.8 ppm ozone for 4 h/d for 1 day or 9 consecutive weekdays (n = 6 per strain per exposure group). These mice were sacrificed 1-day postexposure (PE). Mice did not receive inhalation exposures over the weekend (no Saturday or Sunday exposures; NE). An additional 12 animals per exposure group of Rag2−/− and C57BL/6 mice were given a 17-day PE (air recovery) period. Half of these mice (n = 6 per group) were sacrificed at the end of the 17-day PE period. The other half (n = 6 per exposure group) were reexposed for 1 day (4 hours) to air or ozone.
Ozone was generated with an OREC Model O3VI-O ozonizer (Ozone Research and Equipment Corp, Phoenix, Arizona), and chamber ozone concentrations were monitored throughout the exposure with a Teledyne T265 Chemiluminescence Ozone Analyzer (Teledyne Advanced Pollution Instrumentation, San Diego, California). It has been previously reported that it takes approximately 4 to 5 times the concentration of ozone to induce pulmonary inflammatory responses in rodents that are comparable to those induced in exercising human participants under controlled acute exposure conditions. 10,11 In other words, 0.8 ppm ozone is (1) equivalent to ozone concentrations of 0.16 to 0.20 ppm that cause pulmonary function impairments in exercising adults receiving short-term exposures 12,13 and (2) approximately 10-fold higher than the 8-hour national ambient air quality standard concentration for ozone (0.070 ppm). 14
Animal Necropsy and Nasal Tissue Selection and Processing
Mice were euthanized by exsanguination under pentobarbital anesthesia 24 hours after the end of the final designated exposure to filtered air or ozone. Immediately after death, the head from each mouse dedicated for light microscopic examination was excised from the carcass, and the lower jaw, skin, and musculature were removed from the head. Nasal passages were flushed retrograde through the nasopharyngeal meatus with 1 mL of 10% neutral-buffered formalin and then the head was immersed in a large volume of the same fixative for at least 24 hours. After fixation, heads were decalcified using 13% formic acid, as previously described in detail. 1 After decalcification, 4 transverse nasal tissue blocks were taken from specific sites, defined by dental and palatine landmarks, of the nasal cavity (proximal T1 to distal T4; Figure 2) by methods previously reported. 15 Tissue blocks were paraffin embedded, and 4- to 5-μm-thick tissue sections from these blocks were further processed and stained with hematoxylin and eosin for routine light microscopic examination. Additional nasal tissues sections (T1-T4) were immunohistochemically stained with antibodies specific for (1) murine major basic protein (MBP; clone MT-14.7, Mayo Clinic, Scottsdale, Arizona) to identify eosinophils 1 and (2) GATA-3+ lymphoid cells (ab199428, rabbit monoclonal, Abcam, Cambridge, Massachusetts). Additional details regarding the immunohistochemistry are available in the Online Supplement.
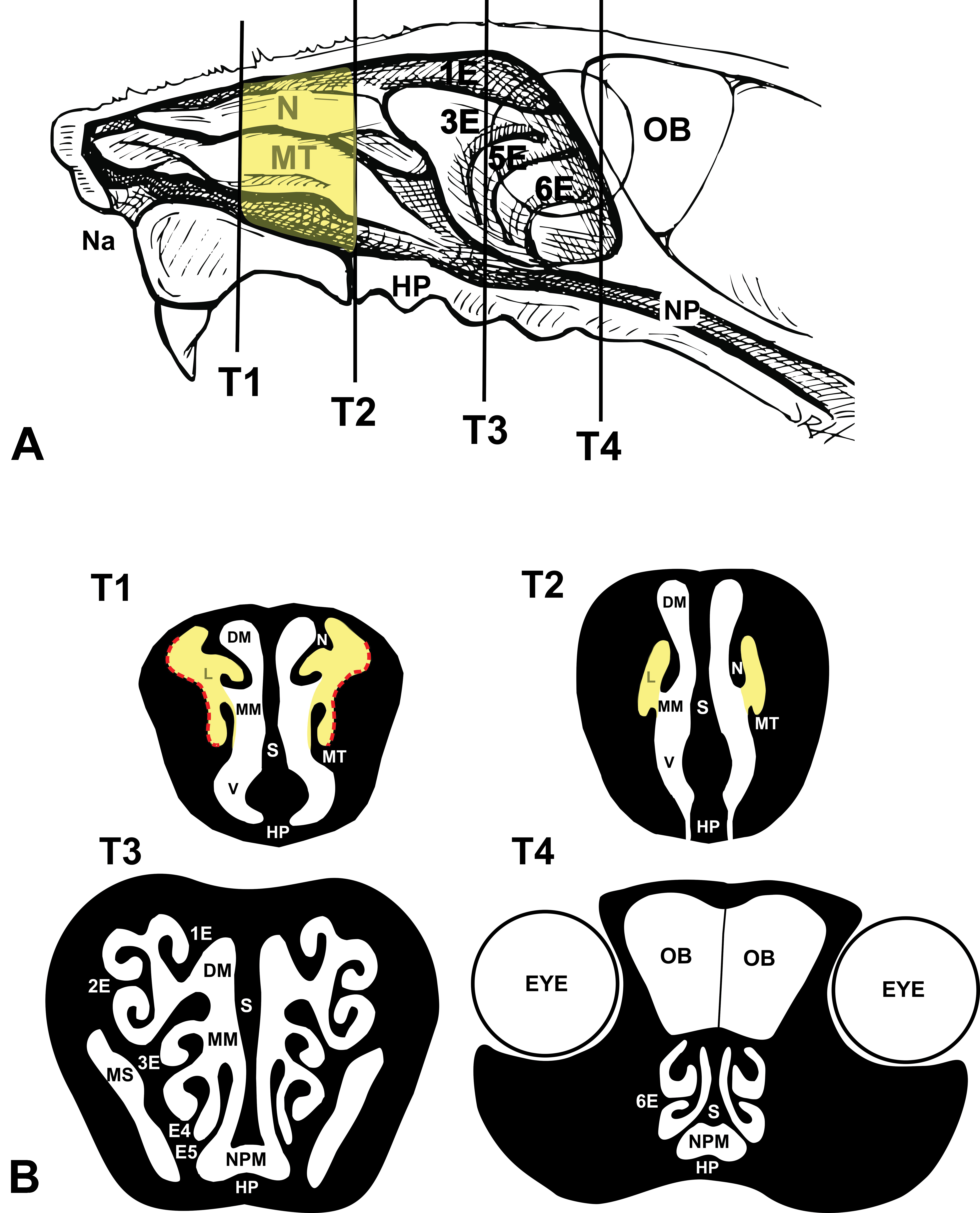
A, Diagrammatic representation of the mouse’s right nasal passage with the septum removed exposing the nasal turbinates. Four transverse tissue blocks were selected for microscopic examination (T1, T2, T3, and T4). Yellow highlight between T1 and T2 indicates the site of mucosal tissues taken for quantitative RT-PCR analysis. B, Rostral face of T1 tissue block that was sectioned for light microscopy and immunohistochemistry. Yellow highlights indicate the bilateral nasal airways that had ozone-induced mucosal injury and inflammation after 9 days of exposure. Red dotted lines indicate the sites of morphometric analyses. HP indicates hard palate; LM, lateral meatus; MT, maxilloturbinate; Na, naris, NP, nasal pharynx; NT, nasoturbinate; OB, olfactory bulb of the brain; RT-PCR, real-time polymerase chain reaction; S, nasal septum.
Nasal Morphometry
Histologic glass slides of nasal tissue sections were digitized with the slide scanner (VS110, Olympus America, Center Valley, Pennsylvania). The nasal mucosa lining the entire lateral wall in both nasal passages in the T1 tissue section, as illustrated in Figure 2B with red stippled lines, was evaluated via morphometric methods using the newCAST software (VisioPharm, Hoersholm, Denmark). Morphometry was used to determine the density of MBP-laden eosinophils and GATA-3+ lymphoid cells in the nasal mucosa by methods previously reported in detail. 1,2 Briefly, the number of points hitting areas positive for MBP or GATA-3 were counted with a point grid in the sampled images of each mouse. The number of points on the reference space (ie, mucosal epithelium and lamina propria) was also counted with a point grid. The total number of points was multiplied by the area/point (a/p) for the MBP+ eosinophil, GATA-3+ lymphoid cell, or reference space to calculate each density. Finally, the percentage of eosinophil density and GATA-3+ lymphoid cell density per reference space was calculated and expressed as the percentage of cells occupying the mucosal tissue.
Quantitative Real-Time Polymerase Chain Reaction of Nasal Mucosal Tissue
For Rag2−/− and C57BL/6 mice designated for real-time polymerase chain reaction (RT-PCR) analyses (n = 6 per exposure group), the entire nasal mucosa lining the nasoturbinates and maxilloturbinates and lateral walls between T1 and T2 were microdissected from both nasal passages (yellow highlighted areas in Figure 2). Total RNA isolation and complementary DNA (cDNA) synthesis were performed as described previously. 16 Total RNA was quantified using a NanoDrop-1000 Spectrophotometer (Thermo Scientific, Wilmington, Delaware). Quantitative RT-PCR was carried out using TaqMan Gene Expression Assays (Applied Biosystems, Waltham, Massachusetts). Selected genes, including housekeeping genes (Actb, Rplp0, Gapd, and Hprt), were analyzed using 0.125 ng cDNA in a 100 nL final reaction volume on a SmartChip RT PCR System (WaferGen, Freemont, California). Replicate ΔCt values of the genes of interest were obtained and normalized by subtracting the mean of Cts from the endogenous controls. Relative gene expression levels were reported as fold change (FC) using the ΔΔCt method, where FC = 2−ΔΔCt. Fold changes in mRNA levels (ozone-exposed mice relative to air-exposed mice) were determined for select genes related to (1) cytokines for eosinophilic inflammation (Ccl11, Ccl24, Il5, and Il13), (2) type 2 cytokines/proteins (Il5, Il13, Chil4, and Retnla), and (3) airway mucus production/secretion (Muc5ac, Clca1, and Clca5).
Statistical Analysis
A Grubbs outlier test was performed on all morphometric data. Multiple comparisons of morphometric data were done using 1-way analysis of variance (ANOVA) followed by a Dunnett post hoc test to detect time-dependent differences, and by t tests to compare effects of exposure (air vs ozone). Data not normally distributed were either transformed or analyzed using ANOVA on Ranks. Molecular data were statistically analyzed using t tests. Significance was assigned to P values ≤ .05 for all statistical analyses.
Results
Intranasal Distribution of Ozone-Induced Eosinophilic Rhinitis and GATA-3+ ILC2s
No exposure-related nasal pathology was present in any of the air-exposed mice (0 ppm ozone, controls) in this study. Marked rhinitis composed of numerous eosinophils interspersed with much fewer lymphoid cells were present in the nasal mucosa of ILC-sufficient mice (C57BL/6 and Rag2−/− strains) exposed for 9 consecutive weekdays to 0.8 ppm ozone but not in ILC-sufficient mice exposed for only 1 day (single 4-hour exposure). This ozone-induced nasal histopathology was restricted to the mucosa lining the proximal lateral meatus in both nasal passages (bilateral lesions) as have been reported in previous studies 1,3,17 (Figure 2A and B). Light photomicrographs at high magnification of the MBP+ eosinophils and GATA-3+ lymphoid cells in the nasal mucosa of ILC-sufficient Rag2−/− mice repeatedly exposed ozone for 9 days and killed 1 day after the last exposure are illustrated in Figure 3A to C. Ozone-induced mucosal eosinophils were approximately 6 to 8 µm in size, spheroid-ovoid in shape, with lobulated nuclei and MBP-laden cytoplasm (Figure 3A and C). In contrast, ovoid-spindle–shaped mucosal lymphoid cells were slightly smaller in size with mononuclear nuclei, scant cytoplasm, and immunohistochemical GATA-3 expression predominately in the nucleus, but some expression was also present in the cytoplasm (Figure 3A and B).
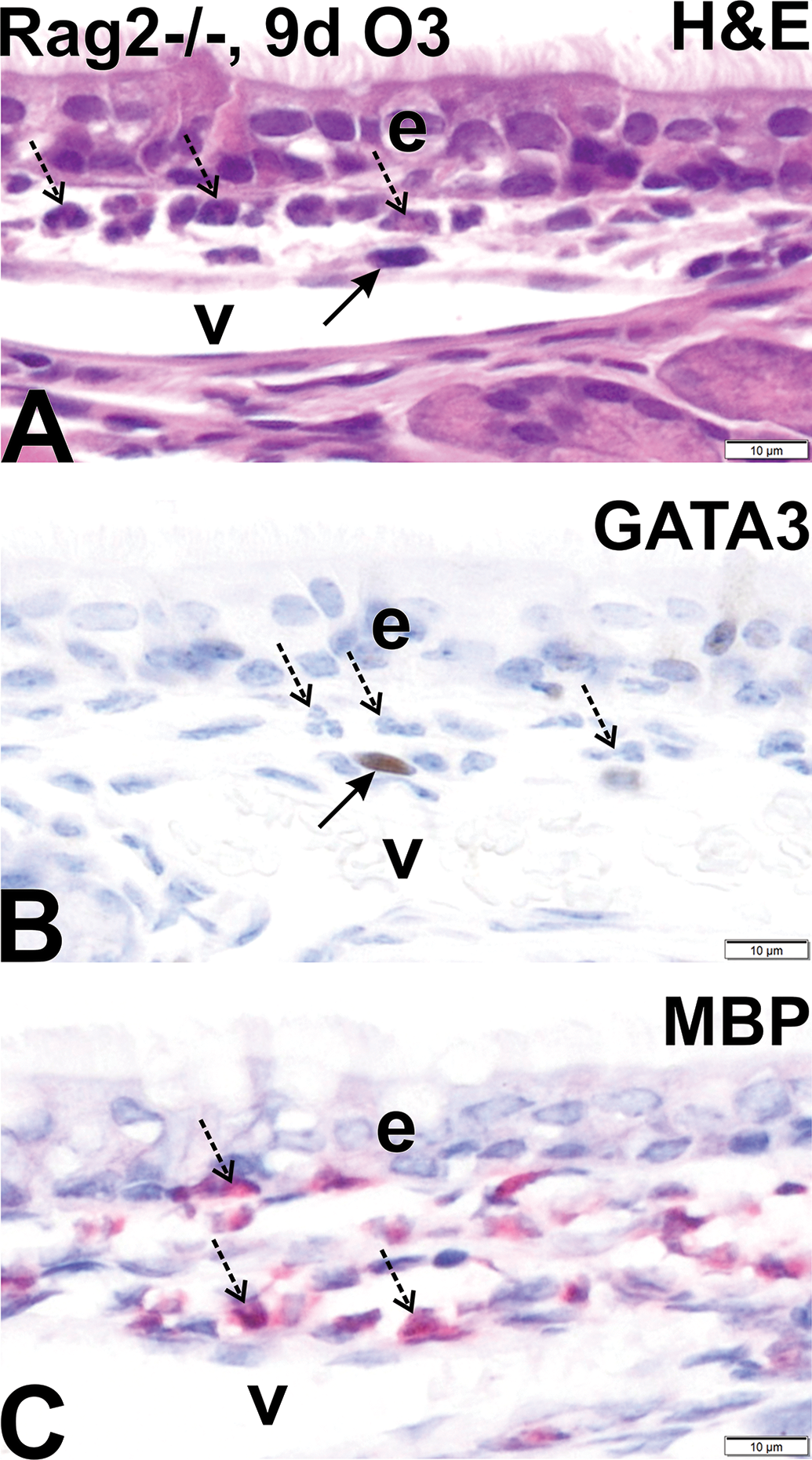
Light photomicrographs at high magnification of digitized images of eosinophils with lobulated nuclei (stippled arrows) and lymphoid cells with ovoid nuclei (solid arrows) in the nasal mucosa lining the proximal lateral wall of Rag2−/− mice repeatedly exposed to ozone for 9 consecutive weekdays and sacrificed 1 day after the last exposure. Tissues were histochemically stained with hematoxylin and eosin (A), immunohistochemically stained for GATA-3 (B; brown chromagen mainly in nucleus), or immunohistochemically stained for murine eosinophil-specific major basic protein (MBP) in cytoplasm (C; red chromagen). e indicates nasal airway epithelium; v, blood vessel in lamina propria of mucosa; scale bar, 10 μm. Tissues in B and C were counterstained with hematoxylin.
Rag2−/−Il2rg−/− mice, deficient in ILCs, T cells, and B cells, which were similarly exposed to ozone for 1 or 9 days had no eosinophilic rhinitis, no GATA-3+ lymphoid cells in the nasal mucosa, and no other associated mucosal lesions (Figure 4B, D, F, and H). In contrast, GATA-3+ lymphoid cells (Figures 3B, 4C, and 7B) were widely interspersed among the greater numbers of eosinophils (Figures 3C, 4G, and 6A) in the nasal mucosa of 9-day ozone-exposed Rag2−/− and C57BL/6 mice. Minimal or no eosinophils or GATA-3+ lymphoid cells were present in the nasal mucosa of air-exposed (Figures 4A, B, E, F, 5A, 6A, 7A, and 8A) or 1-day ozone-exposed mice of all strains (nasal histology similar to air-exposed controls).
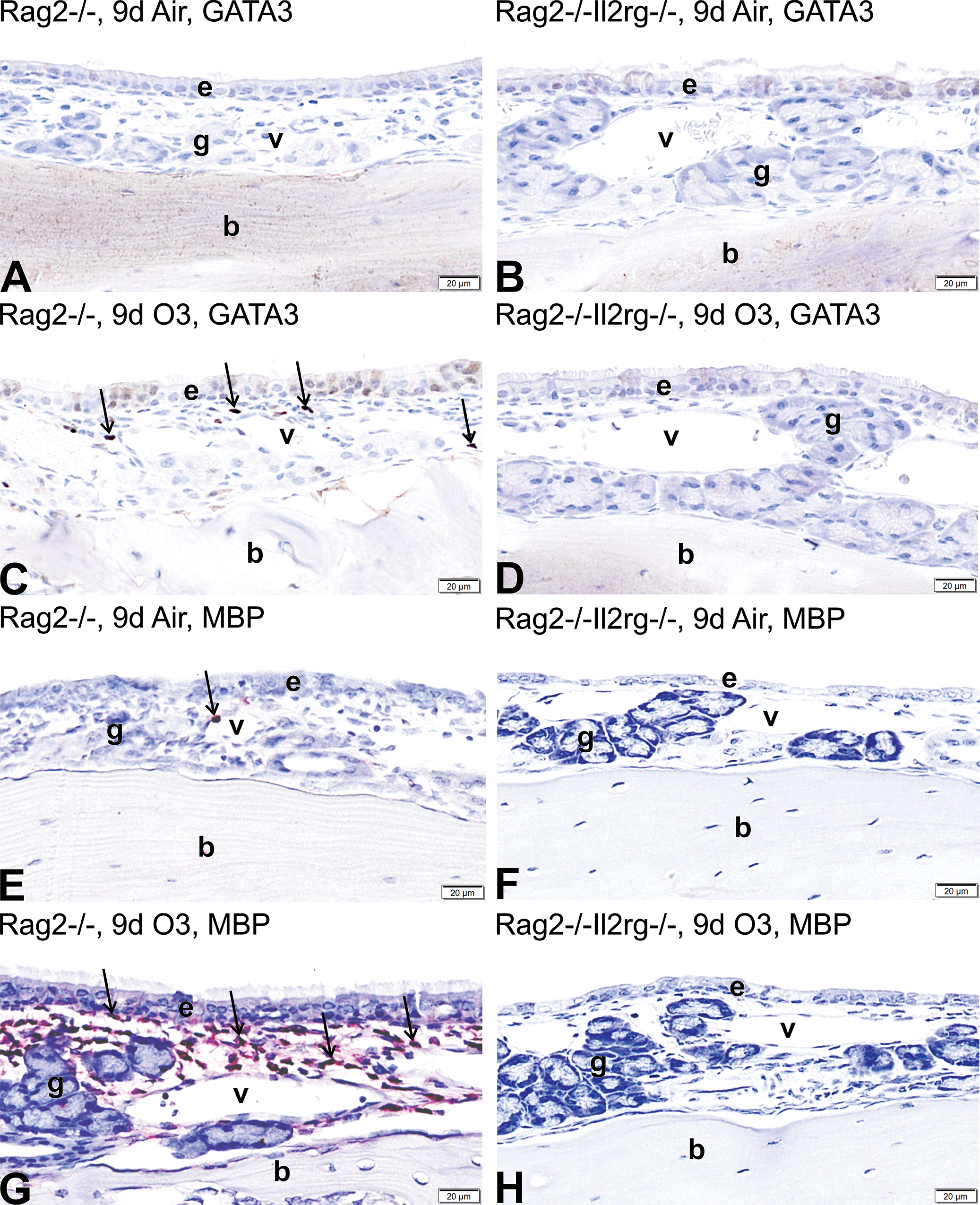
Light photomicrographs of the nasal mucosa lining the proximal lateral wall of Rag2−/− mice (A, C, E, G) and Rag2−/−Il2rg mice (B, D, F, H) exposed for 9 days to air (A, B, E, F) or ozone (C, D, G, H). Mucosal tissues were immunohistochemically stained for GATA-3+ ILC2s (dark brown chromagen, arrows in C) or major basis protein+ eosinophils (red chromagen, arrows in E and G). e denotes nasal epithelium; g, glands in the lamina propria; v, blood vessels in the lamina propria; b, bone in the lateral wall. All tissues were counterstained with hematoxylin.
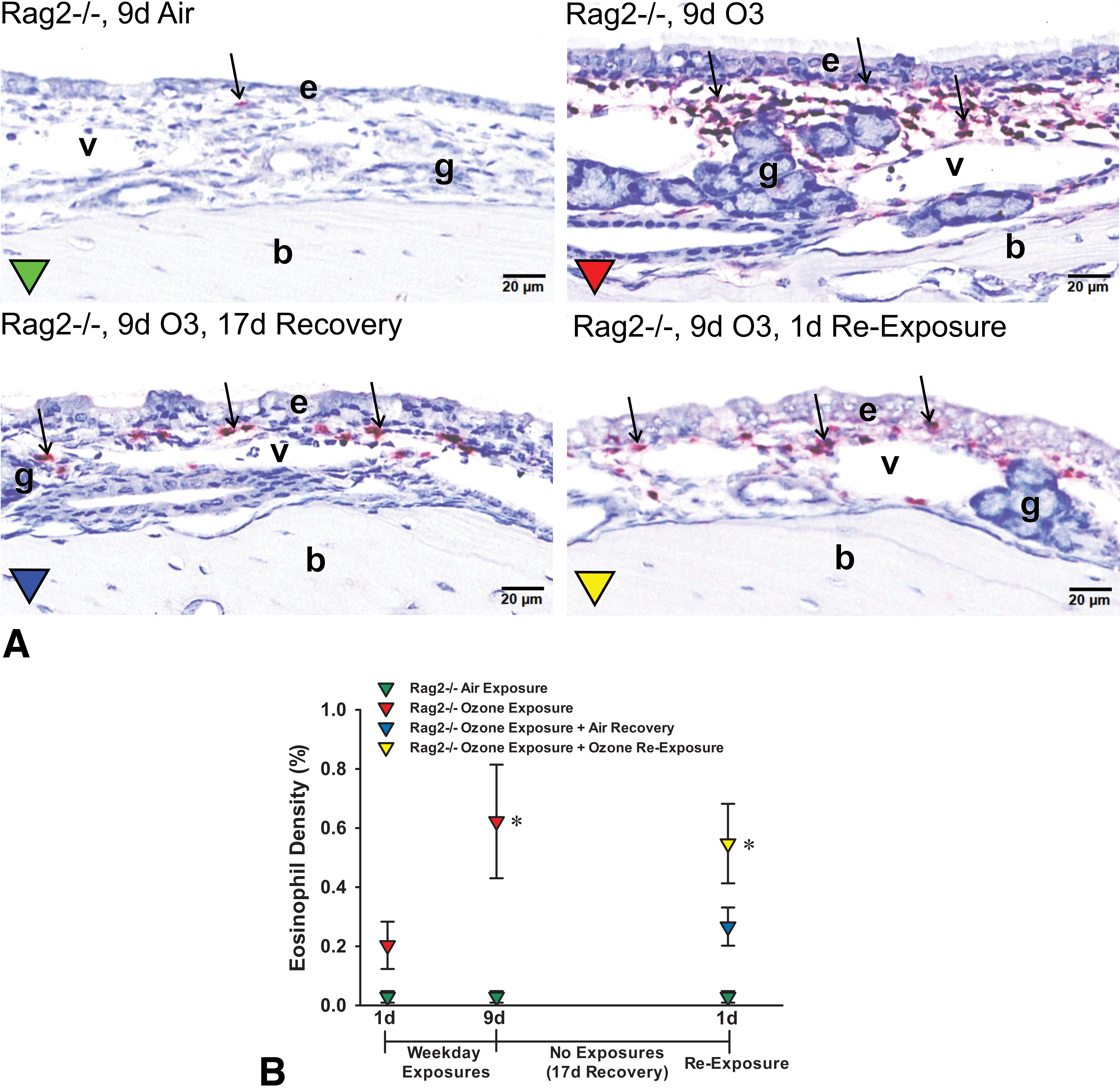
A, Light photomicrographs of the nasal mucosa lining the proximal lateral wall of Rag2−/− mice exposed for 9 days to air, 9 days to ozone, 9 days of ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. Mucosal tissues were immunohistochemically stained for major basis protein+ eosinophils (red chromagen, arrows). e denotes nasal epithelium; g, glands in the lamina propria, v, blood vessels in the lamina propria; b, bone in the lateral wall. All tissues were counterstained with hematoxylin. B, Graphical representation of morphometrically determined eosinophil density (% of mucosal tissue; group means ± standard error of the mean) in Rag2−/− mice exposed for 1 day to air or ozone, 9 days to air, 9 days to air or ozone, 9 days to ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. * Statistically different from respective air control group, P ≤ .05.
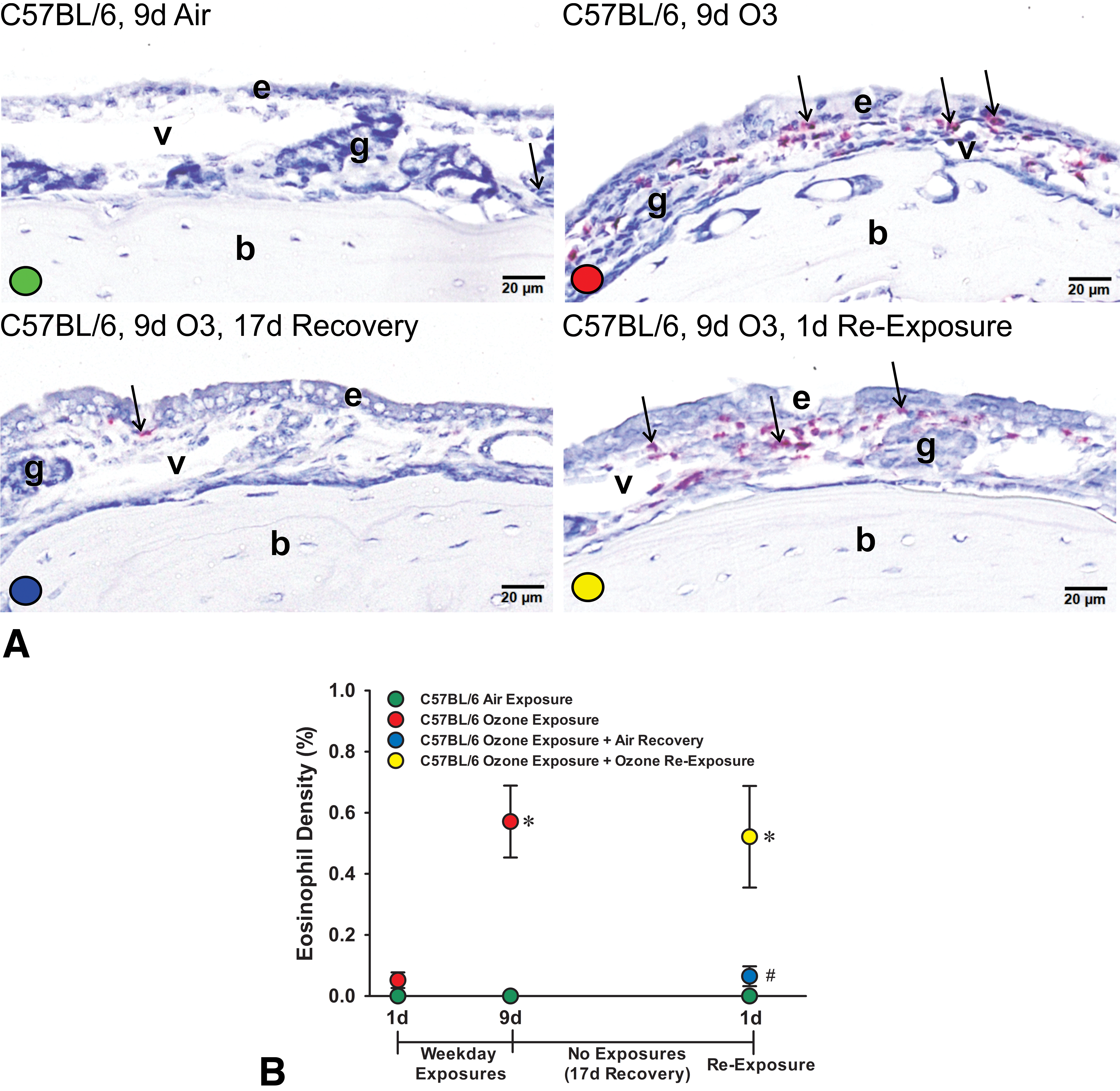
A, Light photomicrographs of the nasal mucosa lining the proximal lateral wall of C57BL/6 mice exposed for 9 days to air, 9 days to ozone, 9 days of ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. Mucosal tissues were immunohistochemically stained for major basis protein+ eosinophils (red chromagen, arrows). e denotes nasal epithelium; g, glands in the lamina propria, v, blood vessels in the lamina propria; b, bone in the lateral wall. All tissues were counterstained with hematoxylin. B, Graphical representation of morphometrically determined eosinophil density (% of mucosal tissue; group means ± standard error of the mean) in Rag2−/− mice exposed for 1 day to air or ozone, 9 days to air, 9 days to air or ozone, 9 days to ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. * Statistically different from respective air control group, P ≤ .05. # Statistically different from 9-day ozone group.
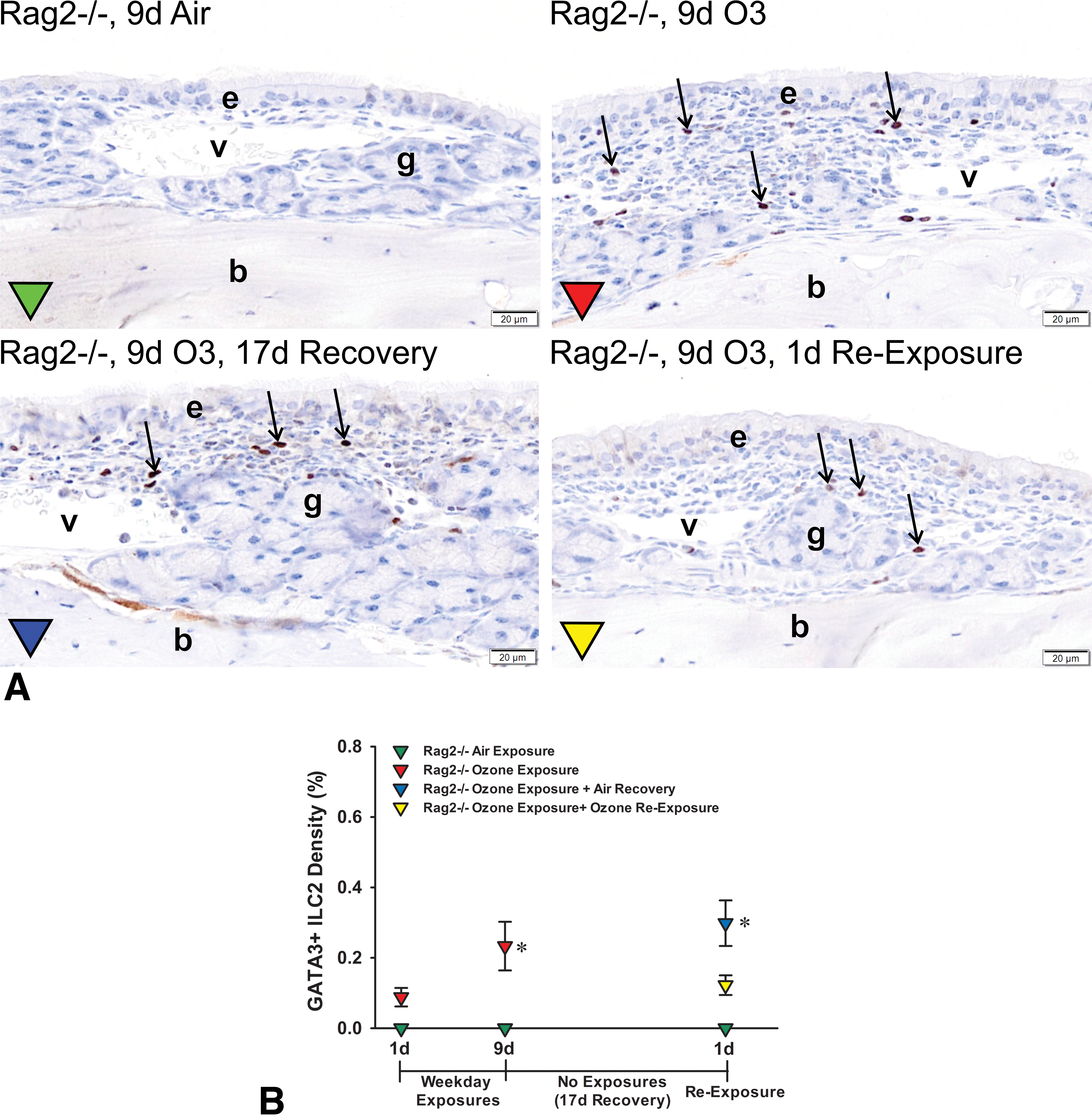
A, Light photomicrographs of the nasal mucosa lining the proximal lateral wall of Rag2−/− mice exposed for 9 days to air, 9 days to ozone, 9 days of ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. Mucosal tissues were immunohistochemically stained for GATA-3+ ILC2s (dark brown chromagen, arrows). e denotes nasal epithelium; g, glands in the lamina propria, v, blood vessels in the lamina propria; b, bone in the lateral wall. All tissues were counterstained with hematoxylin. B, Graphical representation of morphometrically determined GATA-3+ ILC2 density (% of mucosal tissue; group means ± standard error of the mean) in Rag2−/− mice exposed for 1 day to air or ozone, 9 days to air, 9 days to air or ozone, 9 days to ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. * Statistically different from respective air control group, P ≤ .05.
Density of Mucosal Eosinophils After 1- and 9-Day Ozone Exposures, 17-Day Postexposure Period (Air Recovery), and a Single 1-Day Reexposure to Ozone
Morphometrically, <0.03% of the nasal mucosa lining the proximal lateral walls was occupied by MBP+ eosinophils (mucosal density) in air-exposed Rag2−/− or C57BL/6 mice (Figures 5B and 6B). In contrast, Rag2−/−mice and C57BL/6 mice exposed for 9 days to ozone had markedly greater densities of eosinophils in the nasal mucosa as compared to air-exposed controls, 0.62% ± 0.19% and 0.57% ± 0.17% (group means ± SEM), respectively (Figures 5B and 6B).
After a 17-day postexposure period (air recovery), Rag2−/− and C57BL/6 mice previously exposed for 9 days to ozone had decreased mucosal eosinophil densities compared to mice killed 1 day after repeated ozone exposures, 0.27% ± 0.06% and 0.06% ± 0.03%, respectively (Figures 5B and 6B). Interestingly, both 17-day postexposure Rag−/− and C57BL/6 mice that then received a single 4-hour reexposure to ozone had a marked increase in mucosal densities of eosinophils that resembled the 1-day postexposure levels, 0.55% ± 0.13% and 0.52% ± 0.17%, respectively (Figures 5B and 6B).
Density of Mucosal GATA3+ Lymphoid Cells After 1- and 9-Day Ozone Exposures, 17-Day Postexposure Period (Air Recovery), and a Single 1-Day Reexposure to Ozone
Morphometrically, <0.09% and 0.2% of the nasal mucosa lining the proximal lateral walls was occupied by GATA-3+ lymphoid cells in air-exposed Rag2−/− and C57BL/6 mice, respectively (Figures 7B and 8B). In contrast, Rag2−/−mice and C57BL/6 mice exposed for 9 days to ozone had significantly greater densities of GATA-3+ lymphoid cells in the nasal mucosa when compared to air-exposed controls, 0.23% ± 0.07% and 0.41% ± 0.07%, respectively (Figures 7B and 8B).
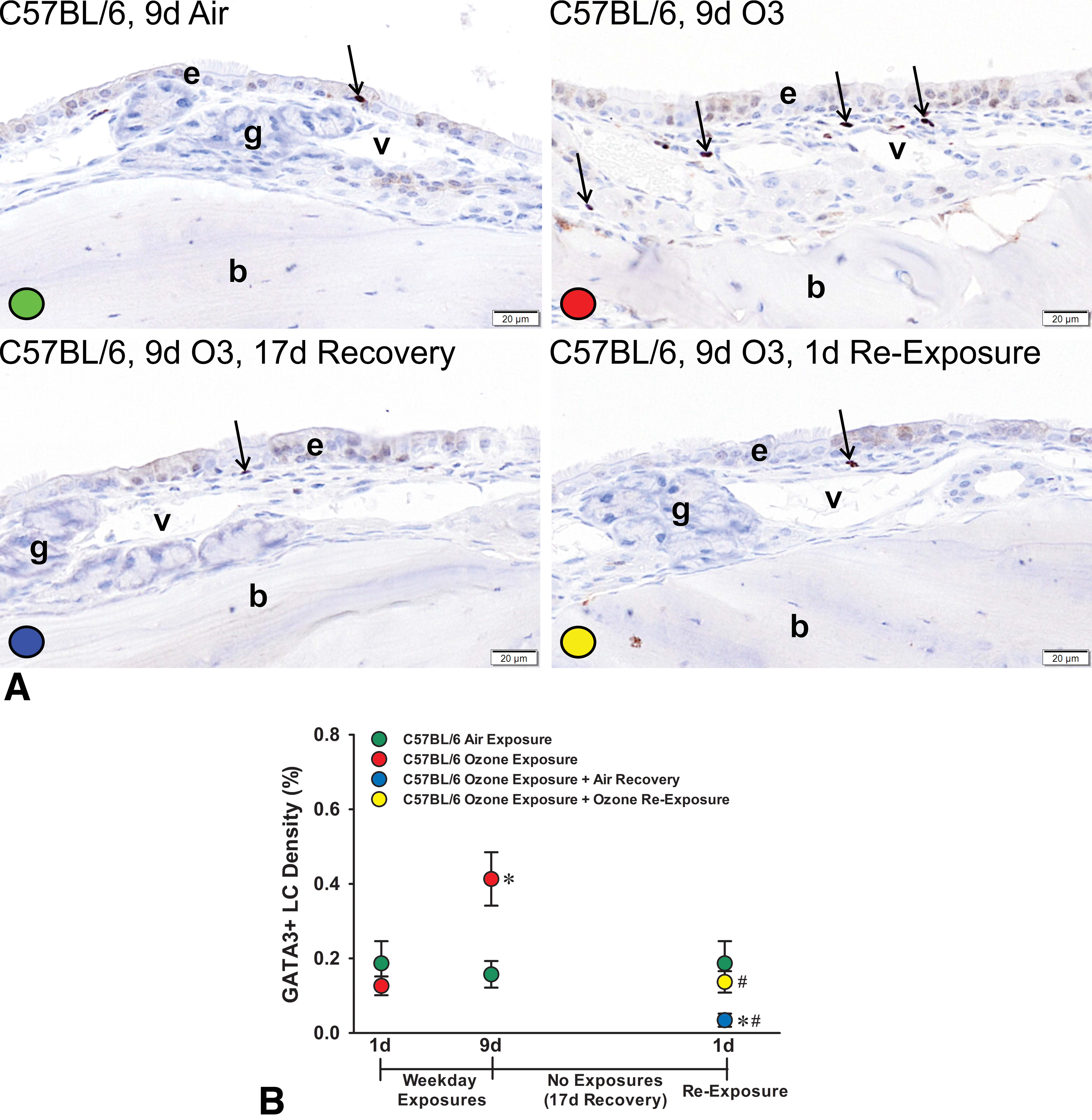
A, Light photomicrographs of the nasal mucosa lining the proximal lateral wall of C57BL/6 mice exposed for 9 days to air, 9 days to ozone, 9 days of ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. Mucosal tissues were immunohistochemically stained for GATA-3+ lymphoid cells (dark brown chromagen, arrows). e denotes nasal epithelium; g, glands in the lamina propria, v, blood vessels in the lamina propria; b, bone in the lateral wall. All tissues were counterstained with hematoxylin. B, Graphical representation of morphometrically determined GATA-3+ lymphoid cell density (% of mucosal tissue; group means ± standard error of the mean) in C57BL/6 mice exposed for 1 day to air or ozone, 9 days to air, 9 days to air or ozone, 9 days to ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. * Statistically different from respective air control group, P ≤ .05. # Statistically different from 9-day ozone group.
After a 17-day postexposure period (air recovery), Rag2−/− previously exposed for 9 days to ozone had a mean mucosal GATA-3 lymphoid cell (ILC2s) density that was similar to Rag2−/−mice killed 1 day after repeated ozone exposures, 0.30% ± 0.65% (Figure 7B). In contrast, after 17 days postexposure, C57BL/6 mice previously exposed for 9 days to ozone had a significant decrease in GATA-3+ lymphoid cell density, 0.03% ± 0.02% (Figure 8B). Unlike the marked increase in mucosal eosinophils after a single 4-hour reexposure to ozone, GATA-3+ lymphoid cell densities in the nasal mucosa of both Rag−/− and C57BL/6 mice did not significantly change after the 1-day ozone reexposure, 0.12% ± 0.03% and 0.14% ± 0.03%, respectively (Figures 7B and 8B).
Messenger RNA Expression of Type 2 Immune-Related Cytokines and Associated Proteins in the Nasal Mucosa in Response to Ozone Exposures Relative to Air Exposures
Rag2−/− and C57BL/6 mice exposed to ozone for 9 days had very similar fold increases (relative to air controls) in mRNA expression for the type 2 cytokine interleukin 5 (IL-5) in the nasal mucosa, 7.63 ± 1.90 and 7.36 ± 1.27 (group means ± SEM), respectively (Figure 9A and B). Likewise, relative fold increases in mRNA expression of Il5 were similarly attenuated 17 days postexposure in both Rag2−/− and C57BL/6 mice, 1.54 ± 0.18 and 2.49 ± 0.61, respectively (Figure 9A and B). After a single 4-hour reexposure to ozone, relative fold increases in the expression of Il5 mRNA in the nasal mucosa were again remarkably similar in both Rag−/− and C57BL/6 mice, 2.57 ± 0.48 and 3.03 ± 0.78, respectively (Figure 9A and B).
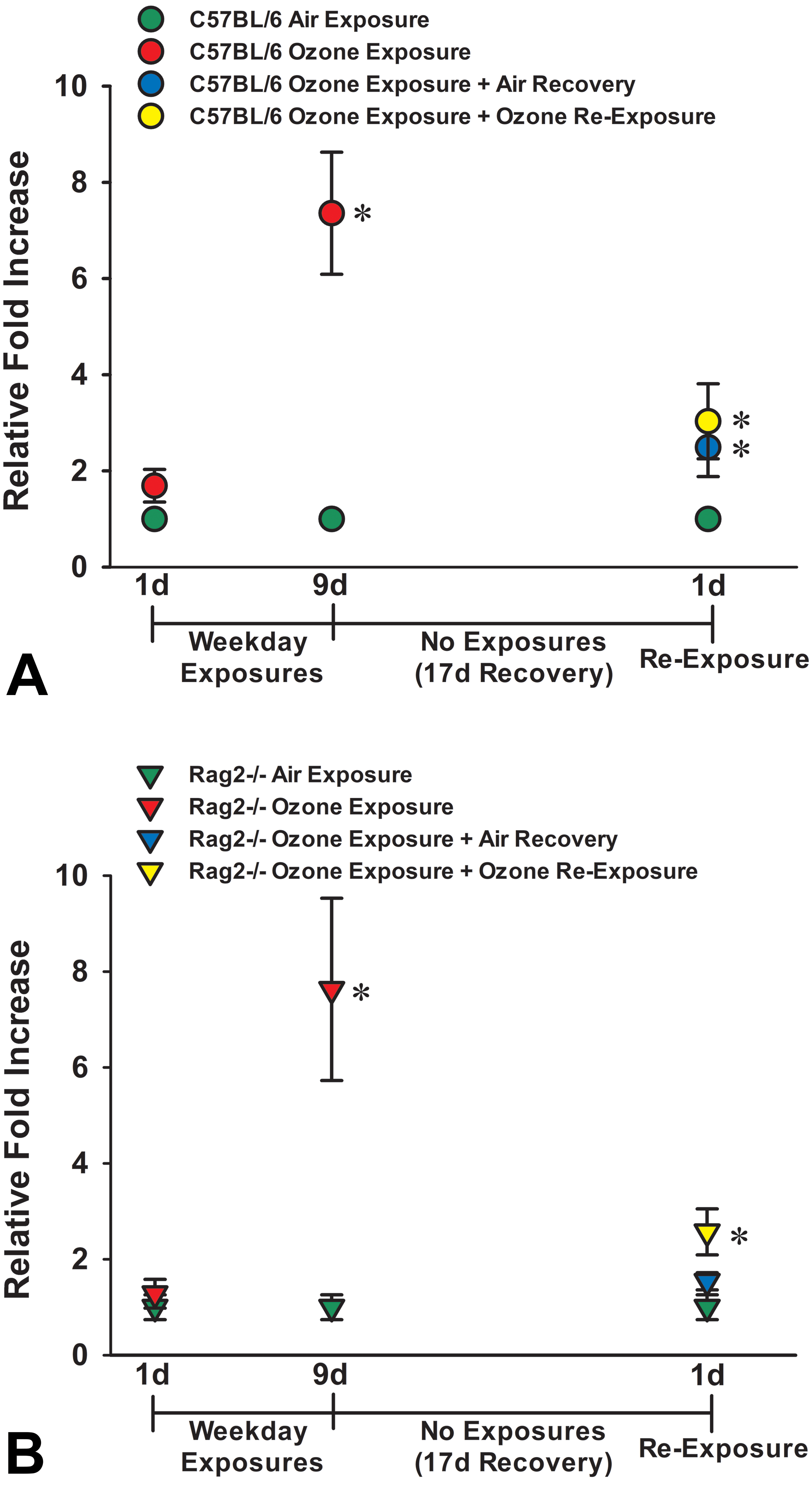
Graphical representation of the fold increases in IL-5 mRNA expression in mucosal tissues of C57BL/6 mice (A) and Rag2−/− mice (B) exposed for 1 day to air or ozone, 9 days to air, 9 days to air or ozone, 9 days to ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. * Statistically different from respective air control group, P ≤ .05. IL-5 indicates interleukin 5; mRNA, messenger RNA.
Like the relative gene expression changes of Il5, relative FCs in mRNA expression of Il13 in the nasal mucosa after 9 days of ozone exposure were also very similar in both Rag2−/− and C57BL/6 mice, 29.12 ± 9.72 and 20.49 ± 5.51 (group means ± SEM), respectively (Figure 10A and B). Seventeen days after the end of the repeated ozone exposures, both Rag2−/− and C57BL/6 mice had marked attenuation of relative Il13 mRNA expression compared to those at 1-day postexposure, 2.71 ± 0.70 and 2.47 ± 0.52, respectively (Figure 10A and B). Furthermore, with a single 4-hour reexposure to ozone after the 17-day air recovery period, both Rag2−/− mice and C57BL/6 mice had a similar relative fold increase of Il13 mRNA in the nasal mucosa, 7.55 ± 1.49 and 6.75 ± 1.50, respectively (Figure 10A and B). Interestingly, a similar pattern of relative FCs of mRNA expression in the nasal mucosa to repeated ozone exposures (increase), postexposure (attenuation), and reexposure (increase) were found in both Rag2−/− and C57BL/6 mice for other selected type 2 immune-associated proteins, that is, eosinophil chemotactic proteins (eotaxin, eotaxin 2), chitinase-like protein (YM2), resistin-like secreted protein (FIZZ1), and proteins related to airway mucus production/secretion (Gob5, Muc5ac; Tables 1 and 2).
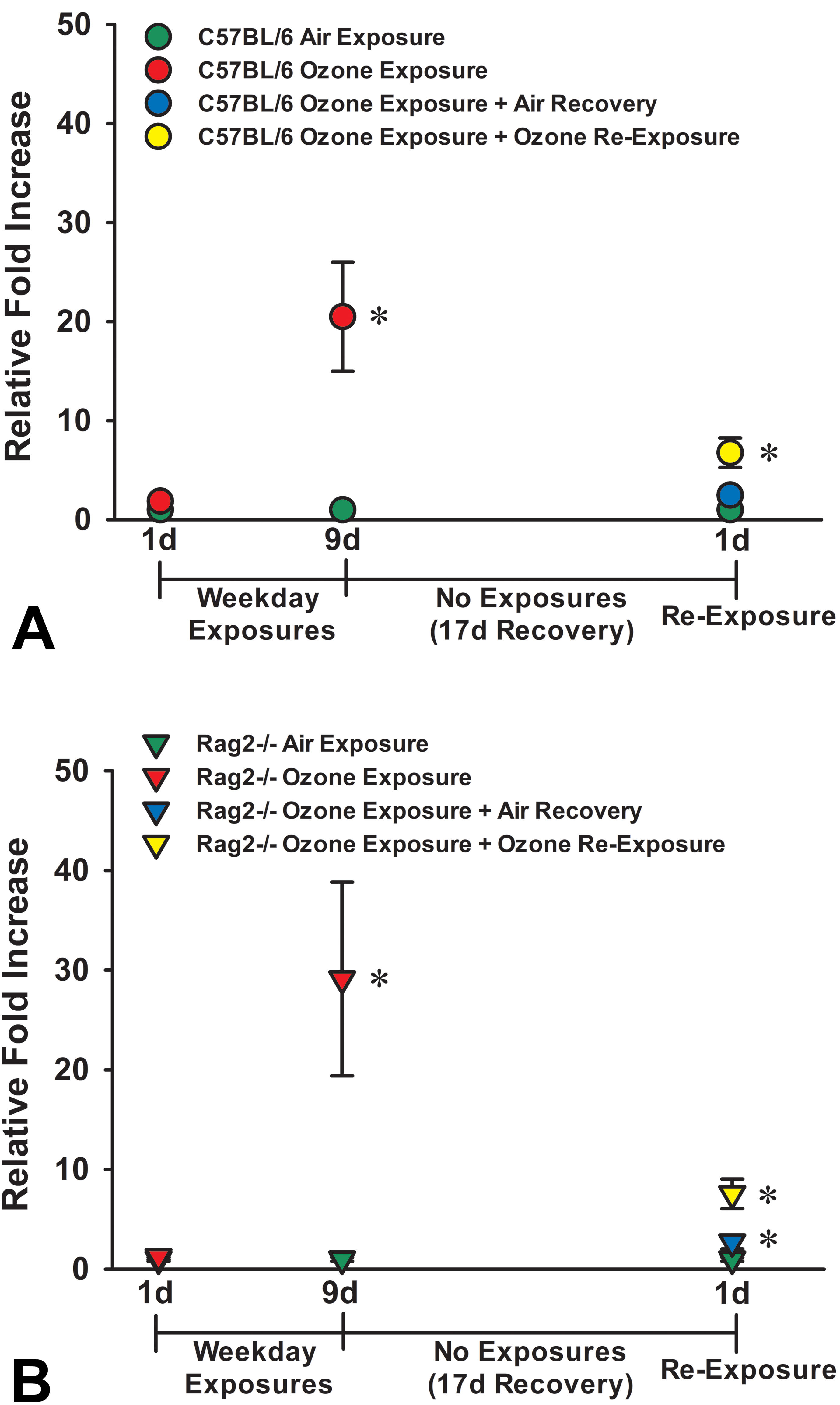
Graphical representation of the fold increases in IL-13 mRNA expression in mucosal tissues of C57BL/6 mice (A) and Rag2−/− mice (B) exposed for 1 day to air or ozone, 9 days to air, 9 days to air or ozone, 9 days to ozone + 17-day air recovery, or 9 days of ozone + 17-day air recovery + 1-day reexposure. * Statistically different from respective air control group, P ≤ .05. IL-3 indicates interleukin 3; mRNA, messenger RNA.
mRNA Expression of Type 2 Immune-Related Cytokines and Associated Proteins in the Nasal Mucosa of C57BL/6 Mice Exposed to Ozone (O3) relative to Air Control Group. n = 6 per group.
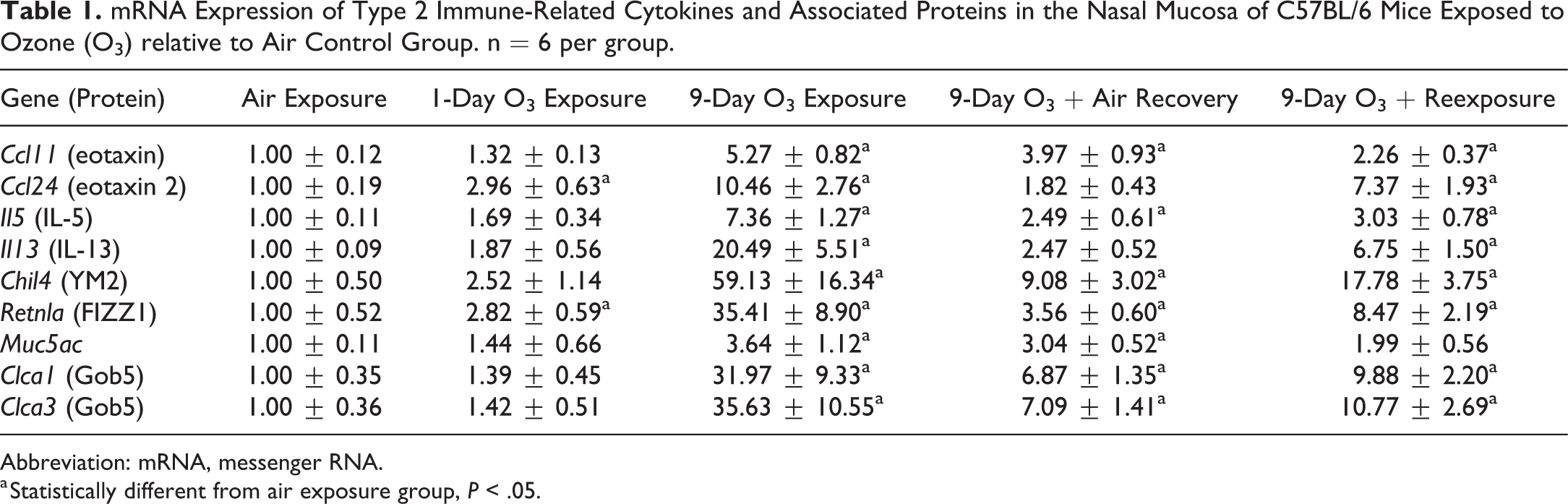
Abbreviation: mRNA, messenger RNA.
a Statistically different from air exposure group, P < .05.
mRNA Expression of Type 2 Immune-Related Cytokines and Associated Proteins in the Nasal Mucosa of Rag2−/− Mice exposed to Ozone (O3) relative to Air Control Group. n = 6 per group.
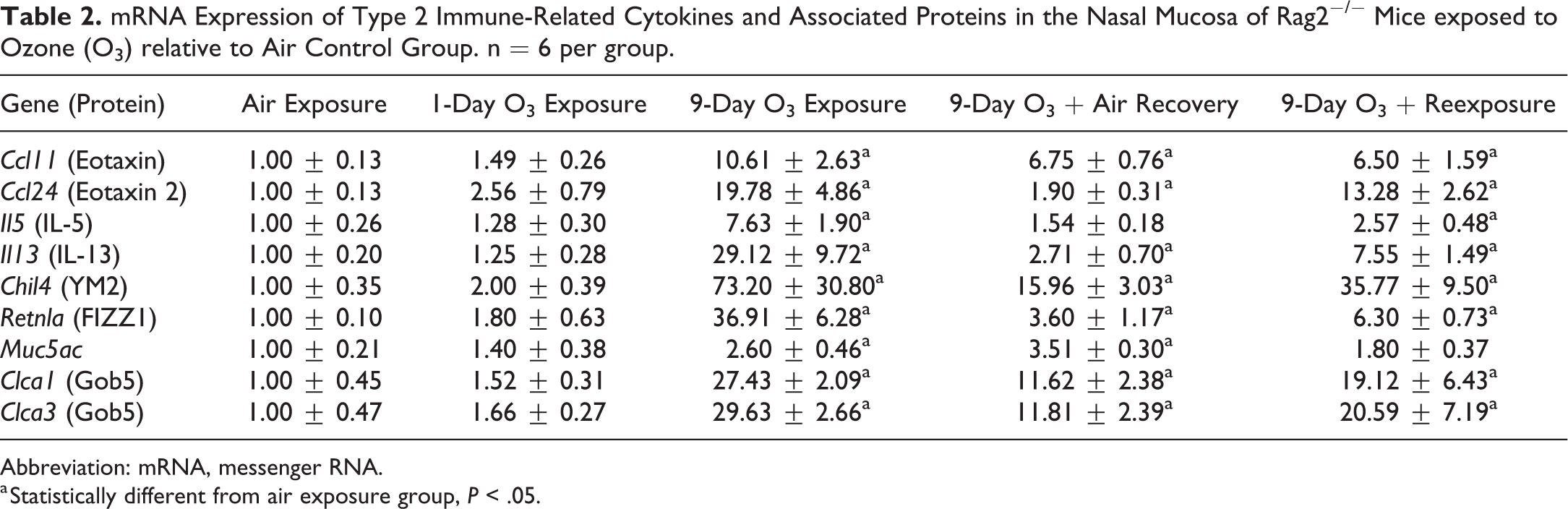
Abbreviation: mRNA, messenger RNA.
a Statistically different from air exposure group, P < .05.
Discussion
The zinc-finger transcriptional factor GATA-3 is a defining feature of both ILC2s and T-helper (Th2) cells along with the production and secretion of their signature type 2 cytokines, for example, IL-5 and IL-13., 8,18 GATA-3 is essential for controlling the lineage, fate, and maintenance of ILC2s and is not found in other groups of ILCs (ILC1 and ILC3) that are defined by other transcriptional factors, such as EOMES, T-bet, and RORγt. 9 Most studies to date have utilized flow cytometry along with a combination of cell surface markers (eg, IL-7Rα, CD25, CD90, SCA-1, ICOS, KLRG1) and the lack of lineage markers (eg, CD3, B220, CD11b, TER119, GR-1) to define, isolate, and quantify ILC2s in mouse tissues. 19 This technique, however, does not allow for the microscopic identification of the exact location where these cells reside in organs and tissues. In the present study, we chose to use a GATA-3 immunohistochemical technique to identify and localize ILC2s (GATA-3+) in mucosal tissues lining the proximal nasal airways of air (controls)- and ozone-exposed Rag2−/− mice that are deficient for all T cells, including Th2 cells, but are ILC sufficient.
GATA-3+ lymphoid cells were also immunohistochemically detected in C57BL/6 mice similarly exposed to air or ozone. Since this wild-type mouse strain (and the background strain for the Rag2−/− and Rag2−/−Il2rg−/− mice) is sufficient for all lymphoid cells (ILCs, T cells, and B cells), we could not distinguish GATA-3+ ILC2s from GATA-3+ Th2s in the nasal mucosa of these mice. A very similar pattern and magnitude of ozone-induced type 2 immunity and eosinophil accumulation were generated in both Rag2−/− and C57BL/6 mice after repeated ozone exposure(s), after a lengthy postexposure period, and following a single-day reexposure to ozone. This strongly suggests that GATA-3+ ILC2s likely played a major role in the C57BL/6 mouse’s response to this inhaled toxicant, though we cannot rule out the possibility of some contribution from Th2 cells in this mouse strain.
Other hematopoietic cell types such as select natural killer cells and basophils, and specific epithelial cells in certain organs (eg, mammary gland, hair follicles, skin, kidney) and at various stages of maturation/speciation, may also immunohistochemically express GATA-3. 18,20 The GATA-3+ cells in the nasal mucosa of mice in the present study, however, had a distinct cellular morphology (small size, an ovoid-spindle shape, mononuclear nucleus, sparse cytoplasm, lack of conspicuous cytoplasmic granules) and intense GATA-3 immunohistochemical staining typical of tissue lymphocytes (ILCs and Th2s). The presence of these GATA-3+ cells in the nasal mucosa (and peribronchiolar interstitial tissue in the lung, data not shown) of T-cell– and B-cell–deficient Rag2−/−mice (ILC sufficient), but not T-cell–, B-cell–, and ILC-deficient Rag2−/−/Il2rg−/− mice, gives further support that these nasal GATA-3+ cells were ILC2s and not another GATA-3+ cell type.
In the present study, repeated daily exposures, but not a single-day exposure, to ozone caused type 2 immune responses with associated eosinophilic inflammation in the nasal airways of ILC-sufficient Rag2−/− and C57BL/6 mice, but not Rag2−/−Il2rg−/− mice. Though we have reported similar findings previously, 1,3,17 our current study is the first to document ozone-induced influx (mucosal distribution and density) and persistence (postexposure) of GATA-3+ lymphoid cells in the nasal airways of ILC-sufficient, but T-cell and B-cell–deficient Rag2−/− mice and ILC-, T-cell– and B-cell–sufficient C57BL/6 mice, as well as the absence of these lymphoid cells in air- or ozone-exposed ILC-, T-cell–, and B-cell–deficient Rag2−/−Il2rg−/− mice.
Our understanding of the pathogenesis of this murine model of ozone-induced, nonallergic eosinophilic rhinitis was furthered by some key findings in the current study. First and foremost, we found that repeated ozone exposures cause an influx of GATA3+ ILC2s in Rag2−/− mice (deficient in T cells) and GATA-3+ lymphoid cells in C57BL/6 mice (most likely including a sizable number of ILC2s in the latter, though not enumerated in this study). These GATA-3+ lymphocytes were few or absent in the mucosa lining the proximal nasal airways of air-exposed control mice in both strains, and only accumulated in mucosal sites of eosinophilic inflammation (interspersed among the eosinophils in the mucosa lining the proximal nasal airways) after 9 days of repeated ozone exposures. Unlike other mucosal surfaces in the body, such as the intestinal mucosa, we found that the nasal mucosa lining the proximal nasal airways of these mice normally contain few (or no) eosinophils and GATA-3+ lymphoid cells.
It is well known that development, recruitment, and survival of eosinophils in mucosal and other tissues after pathogenic exposures (eg, allergens and helminths) are mediated by the local production and secretion of the type 2 cytokine IL-5 and chemokines like eotaxin. 21 In 2013, Nussbaum et al 22 reported that IL-5 and IL-13 from ILC2s during type 2 inflammation results in localized eotaxin production and eosinophilic accumulation in intestinal tissues. It seems plausible that similar controls for recruitment and tissue homeostasis of eosinophils occurred in the nasal mucosa of ozone-exposed mice in our study, though specifically designed studies must yet be conducted to definitively determine the specific cellular and biochemical mechanisms underlying the ozone-induced nasal influx of eosinophils.
It is less clear what mediators promote ILC2 recruitment to tissues. 23 The epithelial cytokines (alarmins) IL-25, IL-33, and TSLP are potent activators of ILC2s eliciting IL-5 and IL-13 production, but their role in migration of ILC2s is still uncertain. In our mouse model, immunohistochemical expression of IL-33, IL-25, and TSLP have all been shown to be elevated in the hyperplastic nasal epithelium of 9-day ozone-exposed ILC-sufficient mice. 2 PGD2 and CysLTs have been found to be more potent chemoattractants of human ILC2s under certain conditions, 24 –26 but their role in the recruitment of ILC2s to nasal or pulmonary airways of mice exposed to ozone has not yet been studied. More studies are needed to fully delineate how, and from where, ILC2s migrate to airway mucosal tissues of mice repeatedly exposed to ozone or other inhaled pollutants.
Another key finding in this study was that mucosal eosinophils (eosinophilic rhinitis) and mRNA overexpression of type 2 immune-related cytokines (and other associated proteins) significantly waned by 17 days postexposure (recovery period) in both ILC-sufficient strains of mice. This suggests that type 2 immunity and nasal airway eosinophilia is not long-lasting once ozone exposure ends. The degree of these postexposure decreases in ozone-induced inflammatory responses after the 17-day recovery period, however, were more marked in C57BL/6 mice than in Rag2−/− mice. Interestingly, GATA-3+ lymphoid cells in C57BL/6 mice also attenuated in the nasal mucosa after this postexposure period, but not in ILC-sufficient Rag2−/− mice that were deficient in both T and B cells. The reason for these strain differences in eosinophil and GATA-3+ lymphoid cell persistence postexposure is not known but does suggest that T (or B) cells directly or indirectly influence the longevity of recruited eosinophils and GATA3+ lymphoid cells in the nasal mucosa after repeated ozone exposures.
Another novel finding of this study was that just a single 4-hour reexposure (“challenge”) to ozone after the postexposure (recovery) period was able to rapidly recall the eosinophil influx and type 2 immune response in the nasal mucosa. This contrasted with no induction of eosinophilic rhinitis or type 2 immunity in mice exposed for only 1 day to ozone without any prior exposure to this inhaled toxicant. This finding suggests that the prior repeated exposures to ozone “primed” the nasal mucosal tissues inducing an “innate memory” or “trained immunity” that was recalled (memory response) upon ozone reexposure resulting in an immediate type 2 immune response with a rapid influx of eosinophils in the mucosal tissue (acute eosinophilic rhinitis). Our laboratory has previously reported 1 that male C57BL/6 mice initially exposed for 1 or 2 days of ozone develop an acute neutrophilic rhinitis and type 1 immunity in response to toxicant-induced nasal epithelial necrosis. In that previous study, these initial inflammatory and epithelial responses to inhaled ozone, however, resolved after continued exposure for 4 days and completely switched to an eosinophilic rhinitis with epithelial hyperplasia, mucous cell metaplasia, and type 2 immunity by 9 days of repeated exposure. In the present study, the rapid recall of eosinophilic rhinitis after a single day of ozone reexposure in ILC-sufficient mice, though not to the same magnitude as after 9 days of initial exposure demonstrates a long-lasting effect of repeated ozone exposure on the nasal mucosa, even after resolution of eosinophilic rhinitis by 17 days postexposure (“recovery”).
Netea et al 27 have implied that “training” of innate immunity due to previous stimulation (ie, priming) would result in an enhanced nonspecific reaction to subsequent challenges. Boraschi and Italiani 28 have recently proposed that “potentiation” is a better term for such memory-induced enhancement of innate immune responses. It is intriguing to speculate that ILC2 memory cells 29 may have played a role in the ozone reexposure-triggered nasal type 2 immune response and acute eosinophilic rhinitis in the Rag2−/− and C57BL/6 mice. More studies, however, are needed to ferret out the specific cellular mechanisms responsible for this recalled immune response to ozone reexposure in the murine nasal airways. We cannot rule out the possibility that ILC1 or ILC3 plasticity (transdifferentiation) toward ILC2s could be an additional pathway in the pathogenesis of ozone-induced type 2 nasal inflammation and immunity. 29,30
Martinez-Gonzalez et al 31 first demonstrated that ILC2s in the murine lung acquire immune memory after exposure to an allergen (papain) or a nonallergen (IL-33, cytokine derived from damaged epithelium/alarmin). The latter is thought to be more critical to ILC2-mediated lung inflammation than other alarmins, such as IL-25 and thymic stromal lymphopoietin (TSLP). 30 In their studies, intranasally administered papain or IL-33 triggered ILC2 expansion and cytokine activation, concurrent with eosinophilic inflammation. This pulmonary expansion of ILCs lasted for a few days and then contracted where numbers declined, and these lymphocytes stopped making type 2 cytokines IL-5 and IL-13. By 2 weeks postexposure, the eosinophilic inflammation in the pulmonary airways had resolved, but the number of ILC2s still remained significantly greater than in naive mice. Upon rechallenge to an unrelated allergen (Aspergillus protease) or IL-33 months after the first exposure, these mice had an enhanced pulmonary inflammation as characterized by increased numbers of eosinophils and a higher number of mucous cells (mucous cell metaplasia) in the airway epithelium as compared to naïve mice receiving the same intranasal challenge. They further found that lung-derived ILC2s that had experienced a previous papain or IL-33 exposure produced higher amounts of IL-5 and IL-13 upon rechallenge as compared to naïve ILC2s.
Likewise, in our study, ILC2-sufficient mice that had experienced ozone (nonallergen)-induced eosinophilic rhinitis had (1) almost full resolution of this inflammatory response by 17 days postexposure and (2) had an immediate recall of a nasal influx of eosinophils upon rechallenge that was not found in naïve mice that received a similar 1-day challenge to ozone. The present study is also the first to demonstrate increased numbers of GATA-3+ ILC2s in the nasal mucosa of air pollutant-exposed mice with concurrent eosinophilic rhinitis. Further studies, however, are needed to elucidate how nasal ILC2s, as well as IL-33–producing nasal epithelial cells, specifically respond to exposure and reexposure to ozone (eg, cytokine activation) to fully delineate the integration of cellular and cytokine pathways underlying this eosinophilic airway disease that is caused by inhalation exposure to this nonallergic air pollutant.
In terms of clinical significance, understanding the potential contribution of commonly encountered air pollutants, such as ozone, to the development or exacerbation of allergic and nonallergic rhinitis is becoming more important in the differential diagnosis, therapeutic management, and prevention of these common upper respiratory diseases. Our finding, in mice, that GATA-3+ ILC2s plays a crucial role in ozone-induced eosinophilic rhinitis suggests a unique innate, rather than adaptive, pathogenic pathway for nasal eosinophilic inflammation recognized in susceptible human populations living in air-polluted environments (eg, children living in communities with photochemical smog). The results of the current study provide additional critical information about the nasal infiltration of eosinophils and GATA-3+ ILC2s and nasal type 2 innate immunity caused by repeated ozone exposures and reexposures in mice, but the translation of these findings to the human condition is yet to be determined.
Although there are numerous reported studies that implicate ILC2s in chronic rhinosinusitis, allergic rhinitis, and allergic asthma, there is a paucity of experimental studies specifically designed to elucidate the role of these unique immune cells in the development of air pollutant-induced nonallergic rhinitis and asthma. Though the present, rather descriptive, study was limited in scope, its hypothesis-driven novel findings are important additions that build upon the results of our earlier reported research and those of others. Together with these previous supportive findings, there is a new paradigm emerging for the pathogenesis of ozone-induced nonallergic airway disease involving ILC2s that may explain the epidemiologic associations of high ambient ozone levels and eosinophilic airway diseases in nonatopic children. 4,5
Supplemental Material
Supplemental Material, Supplemental_Material_Online_Harkema_09172019 - Influx, Persistence, and Recall of Eosinophils and GATA-3+ Innate Lymphoid Cells in the Nasal Mucosa of Mice Exposed and Reexposed to the Gaseous Air Pollutant Ozone
Supplemental Material, Supplemental_Material_Online_Harkema_09172019 for Influx, Persistence, and Recall of Eosinophils and GATA-3+ Innate Lymphoid Cells in the Nasal Mucosa of Mice Exposed and Reexposed to the Gaseous Air Pollutant Ozone by Jack R. Harkema, Elyse A. Eldridge, Ryan P. Lewandowski and James G. Wagner in Toxicologic Pathology
Footnotes
Authors’ Note
Raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Acknowledgments
The authors would like to thank Amy Porter and Kathy Joseph from the Michigan State University Histopathology Laboratory for their excellent technical support in the processing of tissues for light microscopic and immunohistochemical examination.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research was funded in part by USEPA RD83479701 (JRH, JAG), ACC 6168 (JRH), and the Albert C. and Lois E. Dehn Endowment at Michigan State University (JRH).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.