
Abstract
Epidemiological associations have been made between the new onset of childhood rhinitis/asthma and exposures to elevated ambient levels of ozone, a commonly encountered gaseous air pollutant. Our laboratory was the first to find that mice repeatedly exposed to ozone develop nasal type 2 immunity and eosinophilic rhinitis with mucous cell metaplasia. More recently, we have found that these ozone-induced upper airway alterations are mediated by group 2 innate lymphoid cells (ILC2s) and not by T and B cells that are important in adaptive immune responses typically associated with allergic rhinitis and asthma. Furthermore, repeated exposures of mice to ozone cause ILC2-mediated type 2 immunity and airway pathology in the lungs, like those found in the nasal airways. Our recent findings in ozone-exposed mice complement and extend previous reports of nonallergic nasal airway disease in ozone-exposed rats and nonhuman primates. Overall, these experimental results in laboratory animals suggest a plausible ILC2-dependent paradigm for the toxicologic pathobiology that underlies the development of nonallergic rhinitis/asthma in children who live in environments with repeated occurrences of high ambient concentrations of ozone.
Keywords
Introduction
Asthma and rhinitis are heterogeneous and recurring inflammatory airway diseases with many different phenotypes and endotypes that are defined by clinical, cellular, and molecular manifestations. 1 –3 Asthma is often separated into 2 large categories, recurrent airway diseases with allergic sensitization and those without allergic sensitization (atopic and nonatopic asthma), as well as those with or without type 2 immunity. To better understand the pathobiology of specific subtypes within these 2 broad classifications, clinical, cellular, and molecular phenotypes must be linked to other factors, such as genetics, age, obesity, respiratory infections, psychosocial stressors, and environmental triggers (eg, air pollution). Complex animal models of asthmatic phenotypes are often used to better define the underlying pathobiologic pathways for various allergic and nonallergic airway diseases. 3 –5 This review article predominantly highlights research from our laboratory that was designed to (1) explore if repeated exposures to airborne ozone cause epithelial and inflammatory airway pathology that resembles nonallergic asthma/rhinitis and (2) elucidate the immune cellular and cytokine responses underlying the development of this ozone-induced airway disease (a possible endotype of asthma and rhinitis).
Human Health Effects of High Ambient Levels of Ozone
Ozone (O3) is a gaseous, secondary (formed in the troposphere) air pollutant found in photochemical smog that is commonly encountered on hot sunny summer days in urban environments experiencing high motor vehicle traffic (eg, Los Angeles, CA; Houston, TX; and Mexico City, Mexico). Short-term exposure (minutes to hours) of this irritating oxidant gas at high ambient concentrations can trigger acute health effects often manifested with narrowing of conducting airways (hyperreactivity), mucus hypersecretion, and acute inflammation that together may result in difficulty breathing (dyspnea) and increased hospital emergency department visits of asthmatics. 6 –15 Clinically relevant decrements in pulmonary function with short-term exposures had also been well documented in controlled human inhalation studies of exercising patients exposed to high ambient ozone concentrations. 16 –21
Chronic respiratory health effects of repeated, long-term ozone exposures (weeks, months, and years) are somewhat less well defined. Research on the human health effects of long-term ozone exposure, and that of other outdoor air pollutants, are largely dependent on (1) epidemiologic cohort/population studies 22 –25 that are designed to identify associations between elevated ambient concentrations of an air pollutant and indices of adverse health effects (eg, hospital visits, gene/protein biomarkers, onset of respiratory diseases) and (2) controlled chronic inhalation studies of laboratory animals (eg, rats, mice, and nonhuman primates) that provide toxicologic end points (eg, histopathology, relative fold changes in gene/protein expression, measurements of pulmonary dysfunction). 26 –34
Inhalation toxicology studies of ozone are specifically designed to discover dose-dependent, cellular, and molecular mechanisms underlying the pathogenesis of airway toxicity, as well as providing plausibility for epidemiologic associations between ambient ozone exposure and specific health-related effects (eg, incidence and severity of asthma). Epidemiologically, associations continue to be reported between increased ambient airborne concentrations of ozone and increased incidences of chronic respiratory diseases (morbidity) and mortality. 23 Many uncertainties, however, remain concerning toxicologic mechanisms (or modes of action) underlying ozone-related health effects and confounding factors such as concurrent exposures to other gaseous and particulate air pollutants (eg, oxides of nitrogen, particulate matter) that may also contribute to respiratory or other diseases (eg, cardiovascular, metabolic). It is predicted there will be increases in the occurrences of high ambient levels of ozone due to climate change. Future increases in ozone that are derived from various chemistry-climate forecast models project up to a 14% increase in ozone-related mortality by 2030. 35,36
As mentioned previously, more targeted research is needed to identify environmental factors, such as air pollution and obesity, that may cause specific endotypes and phenotypes of chronic diseases such as asthma and rhinitis (eg, allergic vs nonallergic, type 1 vs type 2 inflammation/immunity). Linking specific clinical manifestations and cellular/molecular biomarkers to subtypes of chronic airway diseases will markedly advance population-based and precision medicine approaches to prevent and treat chronic respiratory diseases.
Ozone-Induced Mucous Cell Metaplasia/Hyperplasia in the Nasal Airways of Laboratory Monkeys and Rats
In 1987, Harkema et al first reported that repeated inhalation exposures to high ambient concentrations of ozone induce mucous cell metaplasia (microscopic appearance of mucous goblet cells in an airway surface epithelium normally containing few or no mucus-secreting cells) and mucous cell hyperplasia (increase in the number of mucus-secreting cells in airway epithelium that normally contains these secretory cells) in the nasal mucosa of nonhuman primates. 30,31 Mucous cell metaplasia and hyperplasia are key morphologic changes in the airway epithelium of many chronic respiratory diseases, such as asthma, rhinitis, bronchitis, and cystic fibrosis. 37 These phenotypic changes of airway epithelium are often associated with excessive accumulation of viscous mucus that may result in dysfunction of mucociliary clearance, airway obstruction, and compromised gas exchange in the deep lung. The reader is referred to some excellent recent reviews on this topic that describe in more detail current understanding of the biology and pathobiology of airway mucus and mucous cells in health and disease. 37 –44
In that early nonhuman primate study, macaque monkeys exposed to 0.15 ppm ozone for 6 or 90 days (6 h/d) had marked epithelial changes in the proximal nasal passages 30,31 and centriacinar regions of the lung. 32 Pulmonary centriacinar lesions in terminal and respiratory bronchioles were similar to previously reported ozone-induced alterations (eg, epithelial hyperplasia, macrophage accumulations) in this region of the lung. This animal toxicology study was the first, however, to describe the effects of inhaled ozone on nasal epithelium. Nasal lesions were confined to the surface epithelium in the proximal nasal passages and were characterized by increased mucous cells in the transitional epithelium (TE; nonciliated, stratified, cuboidal epithelium with few mucous goblet cells) and respiratory epithelium (ciliated, pseudostratified respiratory epithelium normally containing mucous cells). Interestingly, the degree of these changes was not dependent on the concentration of the ozone (ie, 0.15 ppm vs 0.30 ppm), but on the duration of exposure (ie, more mucous goblet cells after 90 days of exposure than after 6 days of exposure). In the TE of ozone-exposed monkeys, the numbers of nonciliated cells with secretory granules were 15% to 20% greater than in air-exposed controls. In addition, the amount of epithelial mucosubstances (quantitative indicator of mucous cell metaplasia) in TE increased dramatically (300% greater than air-control monkeys) after ozone exposure. Accompanying the increase in mucous cells in the nasal passages of ozone-exposed monkeys were ciliated cell necrosis (6 and 90 days of exposure), attenuation of ciliary length (6 and 90 days of exposure), and inflammatory cell influx (only after 6 days of exposure).
Many inhalation studies in laboratory rats (eg, F344/N and Sprague-Dawley) have been conducted that demonstrate the development of ozone-induced nasal pathology similar to that in monkeys. In 1989, it was first reported that F344/N rats exposed for 7 days to 0.8 ppm ozone, 6 h/d, develop mucous cell metaplasia in the TE lining the maxilloturbinates, lateral wall, and lateral aspects of the nasal turbinates in the proximal nasal passages. 45 There were, however, some ozone-induced changes in the nasal epithelium of rats that differed from that in nonhuman primates.
Ozone-induced increases in mucous cells in rat nasal airways were primarily restricted to the TE. Transitional epithelium in rats is only 1 to 2 cells thick and normally contains no secretory cells and only a few widely scattered ciliated cells. 46 Transitional epithelium in the monkey, however, is a stratified epithelium (4-6 cells thick) with few ciliated cells and widely scattered mucous cells and other secretory cells. Therefore, the ozone-induced, nasal epithelial alteration in the rat was a true metaplastic response that differed from the hyperplastic response (mucous cell hyperplasia) that occurred in ozone-exposed monkeys. In addition, no epithelial alterations were evident in the respiratory epithelium in the rat, in contrast to the development of mucous cell hyperplasia in the nasal respiratory epithelium that occurs in ozone-exposed monkeys. In that initial rat study, no metaplastic alterations were evident in the TE of rats exposed to 0.12 ppm, 6 h/d for 7 days. This contrasted with the conspicuous mucous cell hyperplasia that was induced in monkeys after a 6-day (8 h/d) exposure to 0.15 ppm ozone. These later results suggest that the rat nasal epithelium may not be as sensitive to ozone-induced injury as the nonhuman primate TE. 47
Cho et al 48 demonstrated that ozone-induced mucous cell metaplasia in the TE of rats could be induced with only a 3-day exposure of 0.5 ppm, 6 h/d, and the amount of epithelial mucosubstances at 7 days after the start of this 3-day exposure was similar to the amount found in TE after a 7-day consecutive exposure to the same concentration of ozone. The fact that the character and severity of these alterations induced by 3 and 7 days of acute ozone exposure were similar suggested that, once initiated, the development of this acute ozone-induced phenotypic change in the nasal epithelium was not dependent on further ozone exposures. Four additional days of exposure to 0.8 ppm ozone had no significant effect on the location or magnitude of the ozone-induced mucous metaplastic response over that observed in rats exposed to 0.8 ppm ozone for 3 days followed by 4 days of exposure to air.
Our laboratory has also examined the effects of chronic ozone exposure on the structure and function of the nasal epithelium in rats. In our first chronic inhalation exposure study, F344/N rats were exposed to 0, 0.25, 0.5, or 1 ppm ozone for 20 months, 6 h/d, 5 d/wk. 29,49 Animals were killed 1 week after the end of the exposure. Immediately after death, mucous flow rates throughout the nasal passages were determined using in vitro video motion analysis. 49 Following assessment of mucociliary function, nasal tissues were processed for light microscopy and histochemically stained for detection of epithelial mucosubstances. Rats exposed to 0.5 and 1 ppm ozone had markedly slower mucous flow rates over the lateral walls and turbinates of the proximal third of the nasal passages as compared to no changes in rats exposed to 0 and 0.12 ppm ozone. Nasal tissues from areas of decreased mucous flow rates (rats exposed to 0.5 and 1 ppm ozone) had marked mucous cell metaplasia with 20 and 40 times greater amounts of epithelial mucosubstances than controls (0 ppm ozone), respectively. There were no significant differences in the mucous flow rates between 0 and 0.12 ppm exposed rats, and no ozone-induced morphologic alterations were present in the nasal epithelium of rats exposed to 0.12 ppm ozone.
This was the first study to demonstrate that ozone-induced mucous cell metaplasia in rat nasal epithelium is associated with a functional alteration, that is, decreased nasal mucous flow. This finding suggested that ozone exposures may impair mucociliary clearance, an important respiratory defense mechanism of the upper airways that protects not only the nose but also the lung from potentially injurious concentrations of subsequent or concurrent inhaled xenobiotics (eg, bacteria, viruses, particulate matter).
In a subsequent chronic inhalation study, 28 we determined the persistence of ozone-induced mucous cell metaplasia in the nasal epithelium after the end of a 3-month exposure. Male F344/N rats were exposed to 0, 0.25, or 0.5 ppm ozone, for 8 h/d, 7 d/wk for 13 weeks. Animals were killed 8 hours, 4 weeks, or 13 weeks after the end of this chronic exposure. Ozone-related alterations in the nasal epithelium were qualitatively and quantitatively characterized through histochemistry, image analysis, and morphometric techniques. Some rats were exposed for an additional 8 hours to 0.5 ppm ozone after the 13 week-recovery period to determine whether chronic ozone exposure results in persistent changes in the sensitivity of nasal epithelium to subsequent acute toxicant exposure.
At the end of the chronic exposure, hyperplasia was present in the nasal epithelium (ie, increase in total epithelial cells) of rats exposed to 0.25 and 0.5 ppm ozone. By 13 weeks postexposure, this proliferative change in the nasal epithelium was still evident only in the rats exposed to 0.5 ppm ozone. Ozone-induced mucous cell metaplasia, associated with morphometrically determined increases in the amount of epithelial mucosubstances and overexpression of the airway mucin gene Muc5AC, was evident only in the nasal tissues of rats exposed to 0.5 ppm ozone. Although attenuated, these alterations in the nasal mucous apparatus were still detectable at 13 weeks after the end of the exposure. At this same time after the chronic exposure, an acute (8 hours) exposure to 0.5 ppm ozone induced an additional increase in mucosubstances and mucin gene expression in the nasal epithelium of rats previously exposed to 0.5 ppm ozone, but not in rats chronically exposed to 0 or 2.5 ppm ozone. The persistent nature of ozone-induced mucous cell metaplasia in rats documented in this study suggests that ozone exposure may have the potential to induce similar long-lasting effects in human nasal airways.
Ozone-Induced Mucous Cell Metaplasia, Eosinophilic Inflammation, and Type 2 Immunity in the Airway Mucosa of Mice: Innate Lymphoid Cell Dependence
Even though important discoveries were made concerning the comparative pathology and toxicology of ozone-induced airway epithelial remodeling in monkeys and rats, this earlier research was primarily descriptive in nature and limited in mechanistic design. In these animal species, we had insufficient data regarding (1) the inflammatory and immune reactions (eg, types of lymphoid cells and cytokines) associated with the airway epithelial changes caused by single or repeated ozone exposures, (2) the gene expression changes in nasal and pulmonary tissues after single or repeated exposures to ozone, and (3) the cellular and molecular mechanisms underlying the epithelial, inflammatory, and immune reactions to inhaled ozone.
With subsequent technological advances in immunohistochemistry, molecular biology, and laboratory animal science and technology, we have subsequently conducted mechanistic studies using wild-type and gene-specific knockout mice. In these murine studies, we tested a novel hypothesis that ozone-induced mucous cell metaplasia in airway epithelium and a concurrent eosinophilic inflammation were lymphoid cell dependent and, more specifically, were dependent on innate lymphoid cells (ILCs). The discovery of ILCs and their functions has only been made in the last decade, but their wide-ranging importance in health and disease has rapidly emerged through elegant and escalating research. 48 Few studies, however, have been specifically designed to elucidate the role of these distinct lymphoid cells in the pathogenesis of respiratory diseases caused by the inhalation of airborne toxicants, such as ozone, found in air pollution.
Innate lymphoid cells are a specific family of innate immune cells that lack specific antigen receptors, but produce a broad range of effector cytokines, and have many diverse roles at barrier surfaces located between the body and the environment, such as airway and gut mucosa and skin. 50 –54 These unique lymphocytes, though relatively small in number within barrier tissues, have been found to be extremely important in the early stages of immune protection from bacterial and viral infections, as well as in the mediation of acute responses to allergen or toxicant exposures. Innate lymphoid cells also play significant roles in tissue homeostasis, repair of tissue injury, and the formation of lymphoid structures. 55
Innate lymphoid cells are broadly classified into 3 subsets or groups based on surface markers, cytokine production, and transcriptional factors needed for development and function. Group 1 ILCs (ILC1s) are most often associated with immune responses to viral infections or tumors, produce interferon γ (IFN-γ) and require Tbet, a Tbox transcription factor family member. Group 2 ILCs (ILC2s) are classically associated with immune reactions to allergens or helminths, produce interleukin 4 (IL-4), IL-5, and IL-13 and require the transcriptional factor GATA-3. Group 3 ILCs (ILC3s) have been found to be important in infectious and autoimmune diseases, defined by the retinoid-related orphan receptor γt, and produce IL-17, IL-22, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor α (TNF-α). Cytokines or chemokines secreted by epithelial cells or other immune cells activate ILCs (eg, alarmins IL-33, IL-25, and thymic stromal lymphopoietin [TSLP] that activate ILC2s). Other studies have recently established that ILCs are also highly plastic, allowing them to adapt to their microenvironment (eg, stromal niches) by changing their cytokine production profiles. 56 As an example, ILC3s and ILC2s can transdifferentiate into IFN-γ-producing ILC1s in the presence of IL-12.
Since mucous cell metaplasia of airway epithelium is a common feature of both allergic and nonallergic rhinitis/asthma and ILC2s are potent producers of IL-13, a well-documented cytokine mediator of airway mucus production, 57 we speculated that these or other lymphoid cells (eg, Th2 cells) play a role in airway epithelial and inflammatory responses to inhaled ozone. Described below are the results of our recent studies that suggest that ILCs, and especially ILC2s, are important in the airway epithelial, inflammatory, and immune responses in the nose and lung of mice repeatedly exposed to ozone.
To better elucidate the development of inflammatory/immune responses to repeated exposures to ozone, investigators in our laboratory first designed a study to examine the nasal airways of mice that received daily ozone exposures for 1, 2, 4, or 9 consecutive weekdays. In this study, Ong et al 58 determined the quantitative pathology that occurs in nasal airways with increasing daily exposures to ozone, along with exposure-related messenger RNA (mRNA) expression changes in nasal mucosa. Figure 1 illustrates the site-specific locations of ozone-induced nasal injury (epithelial necrosis), inflammation (rhinitis), and epithelial remodeling (mucous cell metaplasia) in mice that closely resembled the intranasal location of ozone-induced rhinitis and remodeling that occurs in ozone-exposed rats and monkeys. Mice repeatedly exposed to ozone (0.5 ppm, 4 h/d, for 9 days) had eosinophilic rhinitis and nasal type 2 immunity without any previous or concurrent inhalation exposures to an allergen. Based on these initial findings, these investigators further explored the role of lymphoid cells in the development of the eosinophilic rhinitis and nasal epithelial remodeling. Most importantly, they found that the nasal lesions (mucous cell metaplasia and eosinophilic inflammation) caused by 9 days of ozone exposure were indeed lymphoid cell dependent.
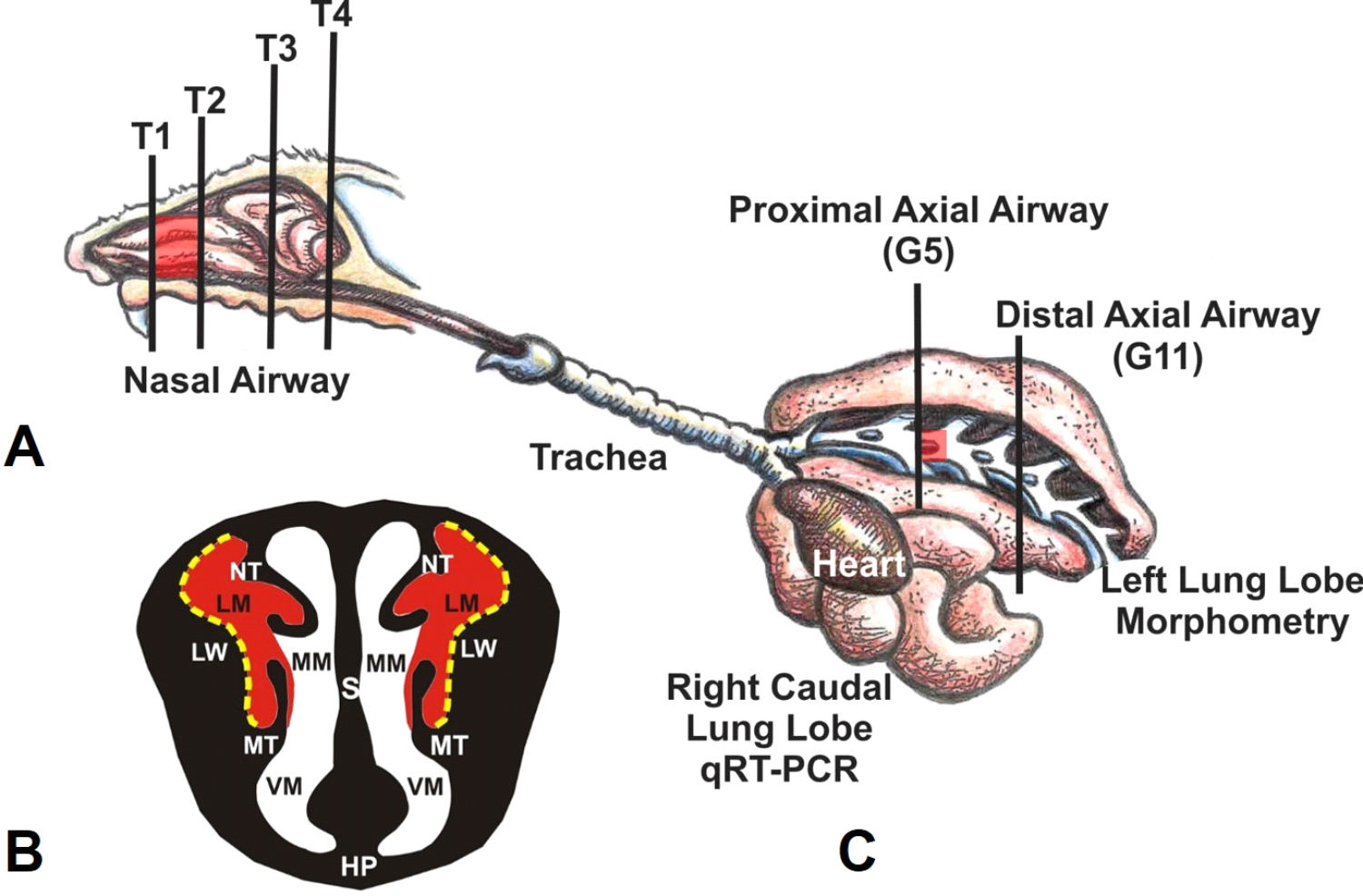
A, Diagrammatic representation of the right nasal passage, with the septum removed, in the mouse. The vertical lines (T1-T4) indicate the locations of the transverse tissue blocks that were selected for light microscopic examination. Red highlights indicate the proximal airway location of ozone-induced nasal lesions. B, Illustration of the anterior face of T1 section. The lateral meatus is highlighted in red, indicating the bilateral location of ozone-induced nasal mucosal histopathology. Stippled yellow lines indicate the lateral wall locations that were selected for morphometric analyses. C, Illustration of the murine lung with the exposed main axial airway in the left lung lobe. Vertical lines indicate the location of transverse tissue blocks that were taken at the level of axial airway generation 5 (G5; proximal) and generation 11 (G11; distal) for microscopic examination and morphometry of airway epithelium. HP indicates hard palate; LM, lateral meatus; LW, lateral wall; MM, medial meatus; MT, maxilloturbinate; NT, nasoturbinate; S, septum; VM, ventral meatus. From Harkema et al. Toxicol. Pathol. 2017;45(1):161-171.
Lymphoid cell–sufficient C57BL/6 mice were exposed to 0 or 0.5 ppm ozone for 1, 2, 4, or 9 consecutive weekdays (4 h/d). Lymphoid cell–deficient, Rag2−/−Il2rg−/− double knockout mice (on a C57BL/6 background) were similarly exposed for 9 weekdays. Nasal tissues were taken at 2 or 24 hours after exposure for morphometric and gene expression analyses. C57BL/6 mice exposed to ozone for 1 day had acute neutrophilic rhinitis, with airway epithelial necrosis and overexpression of mucosal Ccl2 (MCP-1), Ccl11 (eotaxin), Cxcl1 (KC), Cxcl2 (MIP-2), Hmox1, Il1b, Il5, Il6, Il13, and Tnf mRNA. In contrast, 9-day ozone exposure elicited type 2 immune responses in C57BL/6 mice, with mucosal mRNA overexpression of Arg1, Ccl8 (MCP-2), Ccl11, Chil4 (Ym2), Clca1 (Gob5), Il5, Il10, and Il13; increased density of mucosal eosinophils; and nasal epithelial remodeling (eg, hyperplasia/hypertrophy, mucous cell metaplasia). Rag2−/−Il2rg−/− mice exposed to ozone for 9 days, however, had no nasal pathology or overexpression of transcripts related to type 2 immunity, suggesting that some type of lymphoid cell (T lymphocyte, B lymphocyte, or ILCs) are necessary for the manifestation of type 2 immune responses after repeated ozone exposures.
Epidemiological cohort studies have reported associations of elevated ambient ozone concentrations with increases in eosinophil-derived proteins in nasal lavage or urine of atopic and nonatopic children, supporting the hypothesis that elevated ambient ozone exposures may be directly inducing eosinophilic inflammation in the airways. 59,60 Therefore, the results of our initial mouse study provided a plausible new paradigm for the activation of eosinophilic inflammation and type 2 immunity found in the nasal airways of nonatopic individuals subjected to episodic exposures to high ambient ozone. Understanding the contribution of air pollutants, like ozone, to the development and exacerbation of allergic and nonallergic rhinitis is important for the differential diagnosis, therapeutic management, and prevention of this and related chronic respiratory diseases, such as rhinitis and asthma.
Kumagai et al 61 in a subsequent study determined the role of ILCs in the pathogenesis of ozone-induced eosinophilic rhinitis using lymphoid-sufficient C57BL/6 mice; Rag2−/− mice, which are devoid of T cells and B cells; and Rag2−/−Il2rg−/− mice, which are depleted of all lymphoid cells including ILCs. Similar to the study by Ong et al, 58 mice were exposed to 0 or 0.8 ppm ozone for 9 consecutive weekdays (4 h/d). Mice were killed 24 hours postexposure and nasal tissues were selected for histopathology and gene expression analysis. The ILC-sufficient C57BL/6 and Rag2−/− mice exposed to ozone developed marked eosinophilic rhinitis and mucous cell metaplasia (Figure 2).
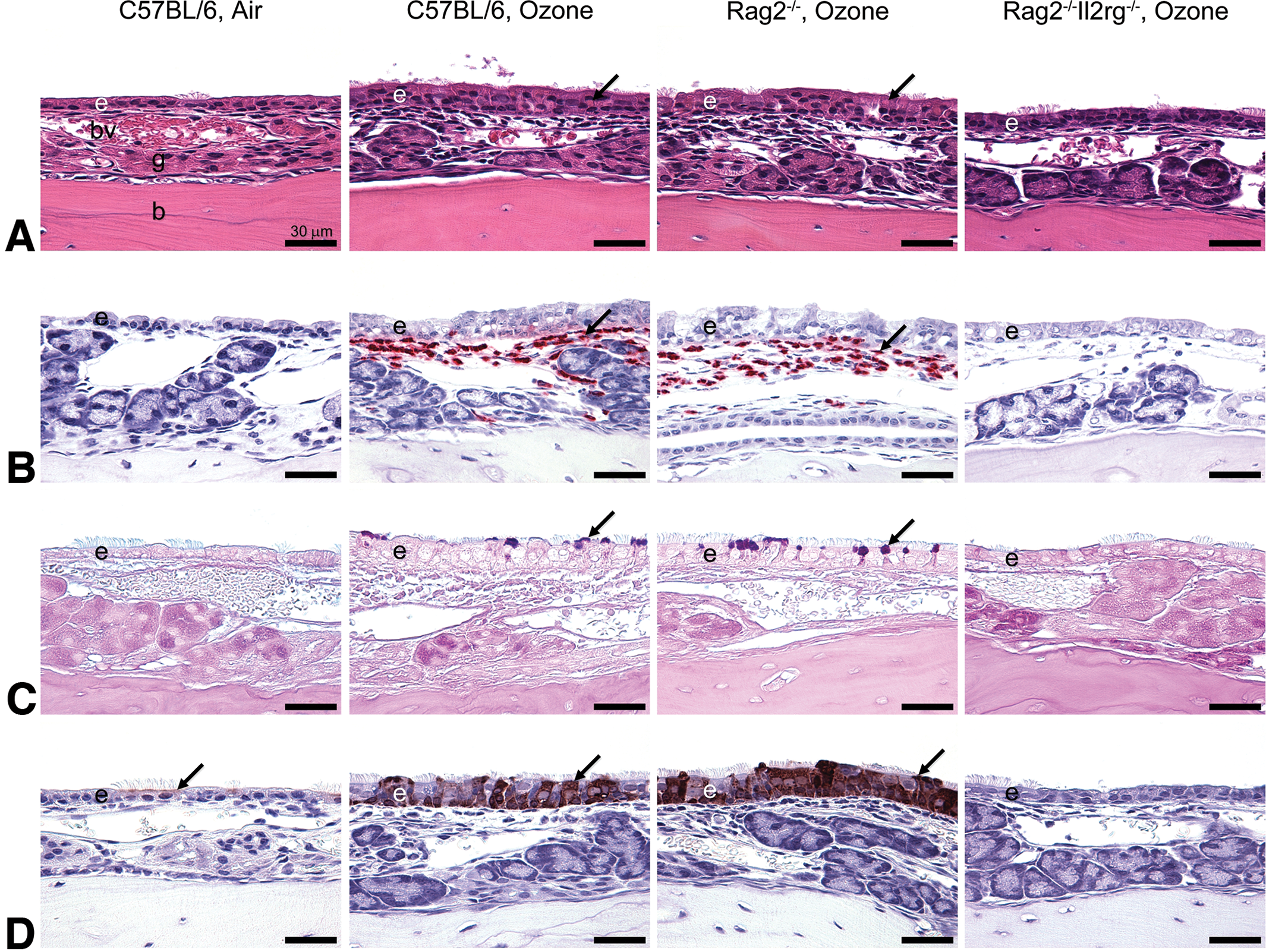
Light photomicrographs of eosinophilic inflammation and epithelial remodeling in the nasal mucosa lining the lateral wall of the proximal nasal passages after repeated ozone exposure. A, Hematoxylin and eosin stain. Exposure of 0.8 ppm ozone for 9 days caused marked rhinitis and epithelial remodeling (ie, hyperplasia, mucous cell metaplasia, and hyalinosis) in innate lymphoid cell–sufficient C57BL/6 mice and Rag2−/− mice. By contrast, innate lymphoid cell–deficient Rag2−/−Il2rg−/− mice exposed to ozone had no nasal lesions. Arrows indicate eosinophilic globules (hyalinosis) in the transitional epithelium. B, Immunohistochemical stain for major basic protein in murine eosinophils. Large numbers of eosinophils (arrows) were detected in the nasal mucosa of C57BL/6 mice and Rag2−/− mice after 9-day exposure to ozone. C, Alcian blue (pH 2.5)/periodic acid–Schiff stain for acidic and neutral mucosubstances. C57BL/6 and Rag2−/− mice exposed to ozone for 9 days stored Alcian blue (pH 2.5)/periodic acid–Schiff–stained intraepithelial mucosubstances in the apical aspect of nonciliated epithelial cells (arrows). D, Immunohistochemical stain for chitinase-like 3 (YM1) and chitinase-like 4 (YM2) proteins. Air-exposed mice contained small amounts of YM1/YM2 proteins in the apical aspect of ciliated epithelial cells (arrows). YM1/YM2 proteins (arrows) were found throughout the full thickness of the nasal transitional epithelium of C57BL/6 and Rag2−/− mice exposed to ozone for 9 days. Scale bars, 30 mm. e, nasal epithelium; g, gland in lamina propria; bv, blood vessel in lamina propria; b, bone. From Kumagai et al. Am J Respir Cell Mol Biol. 2016;54(6):782-791.
Epithelial cytokines (alarmins; IL-33, IL-25, and TSLP) were also morphometrically increased in the nasal epithelium of ozone-exposed C57BL/6 and Rag2−/− mice. Ozone exposure elicited increased expression of Th2- or ILC2-related transcripts, including Il4, Il5, Il13, St2, eotaxin, MCP-2, Gob5, Arg1, Fizz1, and Ym2 mRNA in C57BL/6 and Rag2−/− mice. In contrast, ozone-exposed ILC-deficient Rag2−/−Il2rg−/− mice had no nasal lesions or overexpression of Th2- or ILC2-related transcripts. These results indicated that ozone-induced eosinophilic rhinitis, epithelial mucous cell metaplasia, and type 2 immunity are dependent on ILCs (most likely ILC2s) and not on T- or B-lymphoid cells. This was the first study to demonstrate that ILCs play an important role in the nasal pathology induced by repeated ozone exposure.
Kumagai et al 62 further investigated the role of ILCs in the pulmonary pathology of ozone-exposed C57BL/6, ILC-sufficient Rag2−/−, and Rag2−/−Il2rg−/− mice. As in our previous murine studies, animals were exposed to 0 or 0.8 parts per million ozone for 1 day or 9 consecutive weekdays (4 h/d). A single exposure to ozone caused neutrophilic inflammation, airway epithelial injury, and reparative DNA synthesis in the lungs of all strains of mice, irrespective of the presence or absence of ILCs. In contrast, 9-day exposures induced eosinophilic inflammation and mucous cell metaplasia (Figure 3) only in the lungs of ILC-sufficient mice.
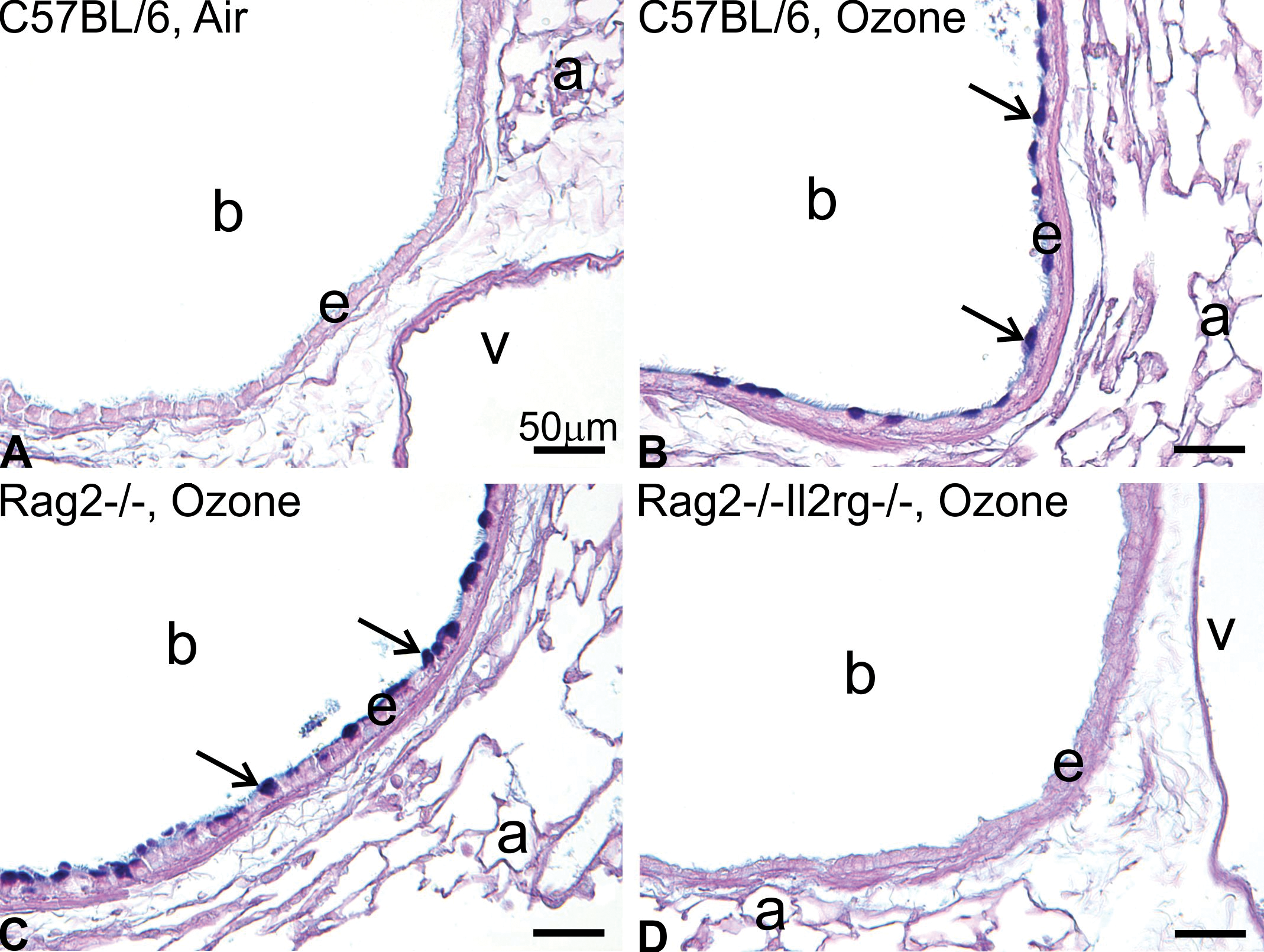
Mucous cell metaplasia in the bronchiolar epithelium after repeated exposure to ozone for 9 days. Light photomicrographs of AB/PAS-positive mucosubstances in the left lung microdissected at the level of the fifth axial airway generation. A, C57BL/6 mouse exposed to filtered air. B, C57BL/6 mouse exposed to 0.8 ppm ozone. C, Rag2−/− mouse exposed to 0.8 ppm. D, Rag2−/−Il2rg−/− mouse exposed to 0.8 ppm. E, Morphometric analysis for AB/PAS-positive mucosubstances in the bronchiolar epithelium at the fifth and 11th airway generation. * Significantly different from respective mice exposed to filtered air at the same region, P < .05. Data are expressed as mean ± standard error of the mean (n = 6 per group). a indicates alveolus; b, terminal bronchiole; v, blood vessel. Scale bars = 50 µm. From Kumagai et al. Toxicol Pathol. 2017;45(6):692-704.
Nine days of repeated ozone exposures also elicited increased mRNA expression of transcripts associated with type 2 immunity and airway mucus production in the lungs of ILC-sufficient mice similar to that previously found in the nasal airways of ozone-exposed mice. Likewise, ILC-deficient mice repeatedly exposed to ozone had no pulmonary pathology or increased gene expression related to type 2 immunity (Figure 4). In addition, these investigators used an anti-Thy1.2 antibody to deplete ILCs in the lungs of ozone-exposed Rag2−/− mice and measured pulmonary ILC2s by flow cytometry. These mice, pharmacologically depleted of ILCs, and without T or B cells, also did not develop type 2 immune-related mucous cell metaplasia in bronchiolar airways or an influx of eosinophils in the lung, further suggesting that ozone induces nonallergic asthma-like features by way of ILC2s and their cytokines (eg, IL-5, IL-13).
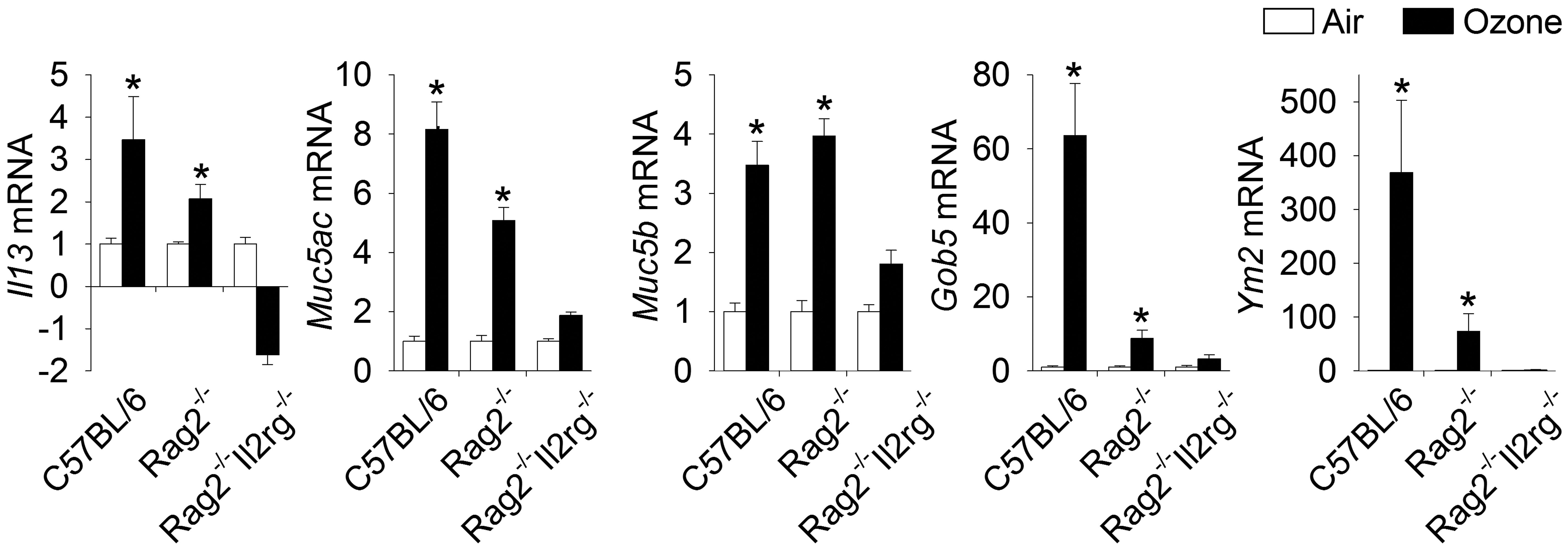
Gene expression in 9-day ozone-exposed lungs. Relative fold increases in mRNA transcripts were analyzed for Muc5ac, Muc5b, Gob5, Il13, and Ym2. * Genes showing statistical significances (air vs ozone/same mouse strain, P values <.05). Data are expressed as fold changes relative to their respective air-exposed controls ± standard error of the mean (n = 6 per group). mRNA indicates messenger RNA. From Kumagai et al. Toxicol Pathol. 2017;45(6):692-704.
Summary, Conclusions, and Future Studies
In early studies, we found that repeated inhalation exposures to high ambient concentrations of ozone caused conspicuous mucous cell metaplasia/hyperplasia in the nasal airway epithelium of laboratory monkeys and rats. In rats chronically exposed to ozone, we found that this striking histologic appearance of mucous cell metaplasia in airway epithelium was correlated with increased mRNA expression of the airway mucin gene Muc5ac and decreases in nasal mucus flow rates, indicating mucociliary dysfunction. Mucous cell metaplasia and Muc5ac overexpression in chronically exposed rats were long-lasting nasal airway alterations that persisted for many weeks postexposure.
Airway mucous cell metaplasia/hyperplasia is a key histopathologic feature of allergic and nonallergic rhinitis and asthma. Therefore, in more recent studies, we used wild-type and gene-specific knockout mice to test the hypothesis that mucous cell metaplasia and concurrent eosinophilic inflammation, which we found in both the nose and lung of ozone-exposed mice, are dependent on ILCs. The results of our murine studies indicate that ozone-induced mucous cell metaplasia, eosinophilic inflammation, and type 2 immunity are indeed dependent on ILCs, and more specifically ILC2s. Reports from other investigators using single exposures to higher concentrations of ozone have demonstrated that ILC2s and their cytokines are also important mediators of ozone-induced pulmonary inflammation and airway hyperreactivity in mice. 63 -65
Although associations between human exposures to elevated levels of ambient ozone and the new onset of chronic airway diseases such as asthma have been epidemiologically identified, 66 few toxicology-based models have been proposed for the toxicologic pathobiology responsible for air pollutant-driven respiratory diseases. Figure 5 summarizes our proposed ILC2-dependent paradigm for the development of airway mucous cell metaplasia, eosinophilic inflammation, and type 2 immunity in mice repeatedly exposed to ozone—a novel model for the new onset of nonallergic rhinitis and asthma associated with exposures to high ambient concentrations of ozone in children living in areas with repeated occurrences of photochemical air pollution (smog).
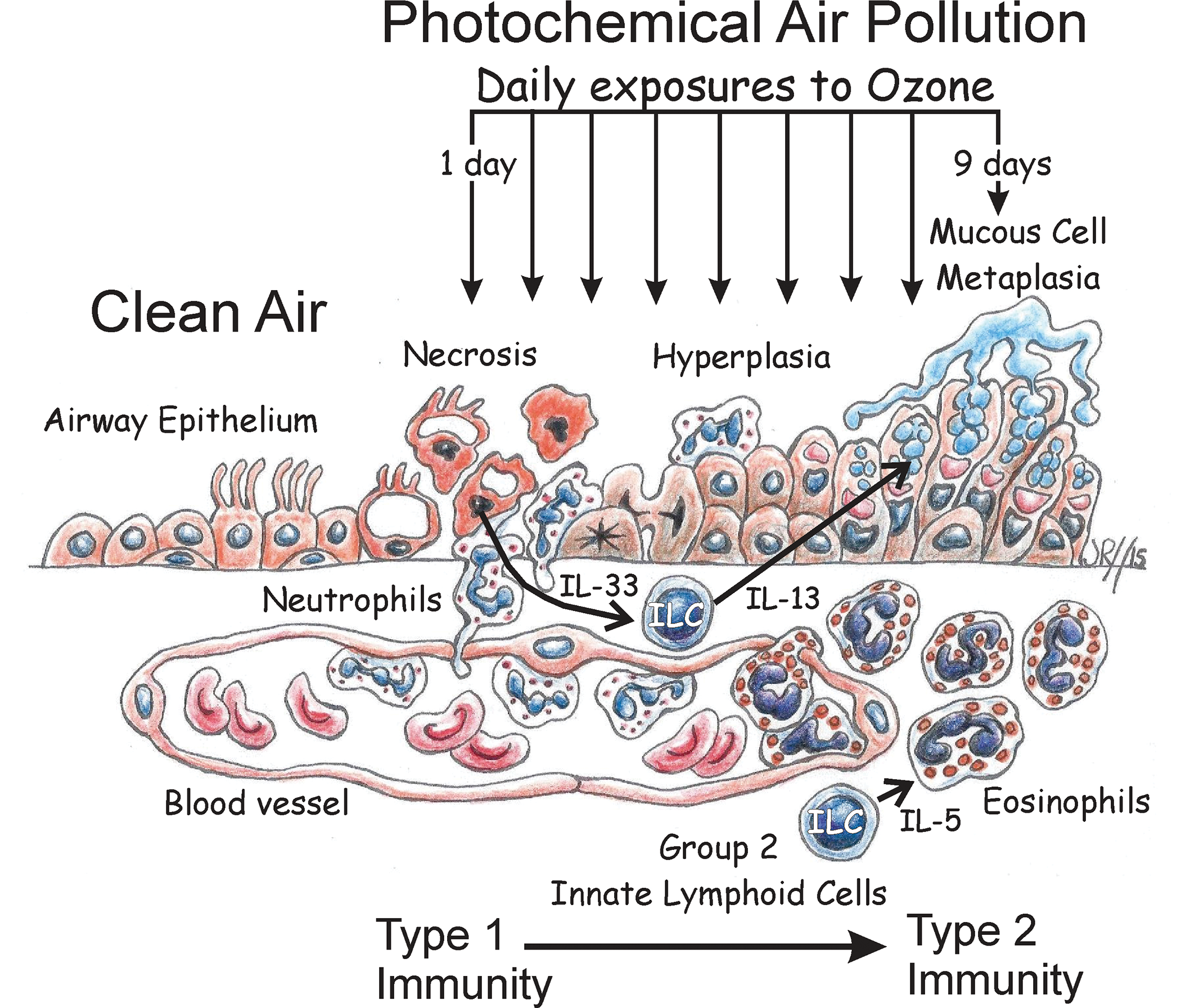
Cartoon depiction of an ILC2-dependent paradigm for the pathogenesis of epithelial remodeling (hyperplasia and mucous cell metaplasia) and eosinophilic inflammation in the nasal and pulmonary airways of mice after repeated daily exposures to ozone. ILC2 indicates group 2 innate lymphoid cell.
There are, however, many data gaps that still need to be filled to confirm this pathogenesis model. Though we recently have immunohistochemically demonstrated that GATA-3–positive ILC2s infiltrate the nasal mucosa along with eosinophils in Rag2−/− mice repeatedly exposed to ozone (currently unpublished data), the source of these nasal ILC2s (eg, bone marrow, gut, proliferation, and/or differentiation of nasal ILCs) 67 is yet unknown. In addition, further studies are needed to confirm the role of proposed ILC2–cytokines responsible for mediating epithelial mucous cell metaplasia (eg, IL-13) 57 and the inflammatory influx of eosinophils (eg, IL-5) 68 in the nose and lung of ozone-exposed mice. Furthermore, the role of epithelial cytokines (eg, IL-33, IL25, TSLP) 69 in the initiation and maintenance of the ILC2-mediated immune responses in this murine model needs to be further elucidated. Since ILCs could also interact with other adaptive or innate immune/inflammatory cells (eg, T lymphocytes, eosinophils, dendritic cells) 70 and the airway microbiome, 71 research in these areas needs to be conducted with carefully designed approaches using state-of-art technologies (eg, single-cell RNA sequencing). 72,73
Footnotes
Acknowledgments
The authors thank Amy Porter and Kathy Joseph of the Michigan State University Histopathology Laboratory for their excellent histology slide preparation; Ryan Lewandowski for his technical assistance in the inhalation ozone exposures and RT-PCR analyses; and the many graduate, undergraduate students, and collaborating faculty for their contributions to the design and execution of our studies reviewed in this article.
Declaration of Conflicting Interests
The author(s) declared no potential, real or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The most recent research was supported with funds in part from the US EPA’s Clean Air Research Center grant RD 83479701, the NIH grant T35OD016477, and the Albert C. and Lois E. Dehn Endowment to Michigan State University for Veterinary Medicine (Pathobiology and Diagnostic Investigation).