
Abstract
Ozone is an irritating gas found in photochemical smog. Epidemiological associations have been made between the onset of asthma and childhood exposures to increasing levels of ambient ozone (i.e., air pollutant–induced nonatopic asthma). Individuals, however, vary in their susceptibility to this outdoor air pollutant, which may be due, in part, to their genetic makeup. The present study was designed to test the hypothesis that there are murine strain-dependent differences in pulmonary and nasal pathologic responses to repeated ozone exposures. C57BL/6NTac and BALB/cNTac mice were exposed to 0 or 0.8 ppm ozone, 4 hr/day, for 9 consecutive weekdays. In both strains of mice, ozone induced eosinophilic inflammation and mucous cell metaplasia in the nasal and pulmonary airways. Lungs of ozone-exposed C57BL/6NTac mice, however, had greater eosinophilic inflammation, mucous cell metaplasia, and expression of genes related to type 2 immunity and airway mucus hypersecretion, as compared to similarly exposed BALB/cNTac mice. Ozone-exposed C57BL/6NTac mice also had greater eosinophilic rhinitis but a similar degree of mucous cell metaplasia in nasal epithelium, as ozone-exposed BALB/cNTac mice. These findings suggest that nonatopic individuals may differ in their inflammatory and epithelial responses to repeated ozone exposures that are due, in part, to genetic factors.
Asthma (and rhinitis) is no longer viewed as one single respiratory disease that is solely allergen dependent but as a heterogeneous entity with a growing number of specific subtypes, or phenotypes, that are defined by common clinical, inflammatory, and molecular characteristics (Gauthier, Ray, and Wenzel 2015; Ilmarinen, Tuomisto, and Kankaanranta 2015; Wenzel 2016). This evolving concept of asthmatic phenotypes has provided a broad differentiation of the asthmatic condition into those with and without allergic sensitization (atopic and nonatopic asthma) as well as type 2 immunity and inflammation. To foster the development of new and better therapeutics and strategies for prevention, asthmatic clinical and molecular phenotypes must be linked to factors underlying the variability in asthma such as genetics, age, obesity, respiratory infections, psychosocial stressors, and environmental exposures (e.g., air pollution). Complex animal models of novel asthmatic phenotypes are needed to better define the biomolecular pathways underlying the onset of these specific subtypes of asthma (Wenzel 2016).
Recently our laboratory has reported that healthy (nonatopic) C57BL/6NTac mice repeatedly exposed to ozone, without concomitant or previous allergen exposure, develop nasal type 2 immunity and eosinophilic rhinitis with mucous cell metaplasia (Ong et al. 2016). We have also found that these ozone-induced airway alterations are mediated by group 2 innate lymphoid cells (ILC2s) and not by the more classical T and B lymphoid cells that are important in adaptive immune responses typically associated with allergic rhinitis and asthma (Kumagai et al. 2016). Furthermore, preliminary findings (unpublished data) indicate that similar repeated exposures of mice to ozone induce ILC2-mediated airway type 2 immunity, eosinophilic inflammation, and mucous cell metaplasia in the pulmonary airways, like what we observed in the nasal airways of similarly exposed C57BL/6NTac mice. Together these studies in mice strongly suggest that repeated daily exposures to ambient ozone may induce a nonatopic asthma phenotype characterized by innate type 2 immunity, eosinophilic inflammation, and mucous cell metaplasia in both nose and lung of susceptible individuals. These research findings in mice have provided for the first time a plausible paradigm for the biological mechanisms (or mode of action) underlying the epidemiologically identified associations of airway eosinophilic inflammation, and new onset of nonatopic asthma, with childhood exposures to increasing ambient concentrations of ozone (Frischer et al. 2001; Nishimura et al. 2016).
Investigators using controlled human exposure studies have found that individuals vary in their respiratory response to a single, short-term (1 to 8 hr) exposure to a high ambient ozone exposure (Blomberg et al. 1999; McDonnell et al. 1985; McDonnell 1996; Holz et al. 1999). Some exposed individuals develop marked deficits in pulmonary function and/or airway inflammation after short-term ozone exposure, while others have little or no changes in lung function and develop little or no pulmonary inflammation. Similarly, different strains of mice have been found to vary in their acute inflammatory and pulmonary function responses to single or short-term exposure (1 to 3 days) to ozone (Bauer, Malkinson, and Kleeberger 2004; Goldstein et al. 1973; Kleeberger et al. 1990, 1997, 2000; Prows and Leikauf 2001; Vancza et al. 2009; Yang et al. 2016). In contrast, little is known about genetic variability in airway responses to repeated, longer-term exposures to air pollutants (i.e., subacute or chronic inhalation exposures). The present study was designed to determine if strain is a factor in the inflammatory, epithelial, and molecular manifestations in pulmonary and nasal airways of mice repeatedly exposed to ozone (i.e., model of new-onset, nonatopic asthma, and rhinitis with pathologic signatures of eosinophilic inflammation and mucous cell metaplasia).
Materials and Methods
Animals
Six- to eight-week-old male C57BL/6NTac and BALB/cNTac mice were obtained from Taconic Biosciences, Inc. (Germantown, NY). Mice were individually housed in stainless steel wire cages within whole-body inhalation exposure chambers (H-1000; Lab Products Marywood, NJ). All animal procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee at Michigan State University.
Inhalation Exposures
After an acclimatization period of 1 week, mice were exposed to filtered air (0 ppm ozone; air controls) or 0.8 ppm ozone for 9 consecutive weekdays (4 hr/day). All mice were exposed for 5 days (Monday to Friday) in the first week and 4 days the following week (Monday to Thursday). Animals were exposed to the same daily inhalation air or ozone exposures (i.e., a single experiment of repeated daily exposures). Mice did not receive inhalation exposures over the weekend of the first week (Saturday and Sunday).
Ozone was generated with an Ozone Research and Equipment Corp. (OREC) Model O3VI-O ozonizer (Phoenix, AZ) and chamber ozone concentrations were monitored throughout the exposure with a Teledyne T265 Chemiluminescence Ozone Analyzer (Teledyne Advanced Pollution Instrumentation, San Diego, CA). It has been previously reported that it takes approximately 4 to 5 times the concentration of ozone to induce pulmonary inflammatory responses in rodents that are comparable to those induced in exercising human subjects under controlled acute exposure conditions (Hatch et al. 2013, 1994). In other words, 0.8 ppm ozone is (1) equivalent to ozone concentrations of 0.16 to 0.20 ppm that cause pulmonary function impairments in exercising adults receiving short-term exposures (Avol et al. 1984; Folinsbee, Bedi, and Horvath 1984) and (2) approximately 10-fold higher than the 8-hr national ambient air quality standard concentration for ozone (0.070 ppm; US Environmental Protection Agency 2015).
Necropsy and Bronchoalveolar Lavage
Mice were anesthetized with pentobarbital and euthanized by exsanguination 24 hr after the end of the 9-day chamber exposure to filtered air or ozone. Immediately after death, the trachea was exposed and cannulated, and the heart and lungs were excised en bloc. A volume of 0.8 mL saline was instilled through the tracheal cannula and withdrawn to recover bronchoalveolar lavage fluid (BALF). A second intratracheal saline lavage was performed and the collected BALF was combined with the first sample for cellular analysis (i.e., single analysis per mouse with no replicates).
BALF Cytology
Total number of inflammatory cells in the collected BALF was estimated using a hemocytometer. Cytological slides were prepared by centrifugation at 400g for 10 min and stained with Diff-Quick (Siemens Healthcare Diagnostics, Inc., Newark, DE). Differential cell counts for macrophages, neutrophils, eosinophils, and lymphocytes were assessed from a total of 200 counted cells.
Pulmonary Histology
After lavage collection, the left lung lobe was intratracheally infused with 10% neutral-buffered formalin at a constant pressure (30 cm H2O) for approximately 2 hr and then stored in a large volume of the same fixative until further processing. After fixation, the lung lobe was microdissected along the main axial airway and two transverse tissue blocks were taken at the level of the 5th (proximal) and 11th (distal) airway generation along the main axial airway (G5 and G11, respectively; Figure 1C), as previously described in detail (Farraj et al. 2003). Five-micron thick, paraffin-embedded lung tissue sections from mice exposed to filtered air or ozone for 9 days were stained with either Alcian Blue (pH 2.5)/Periodic Acid Schiff (AB/PAS) to detect acidic and neutral mucosubstances in bronchiolar airway epithelium or hematoxylin and eosin (H&E) for routine histopathology.
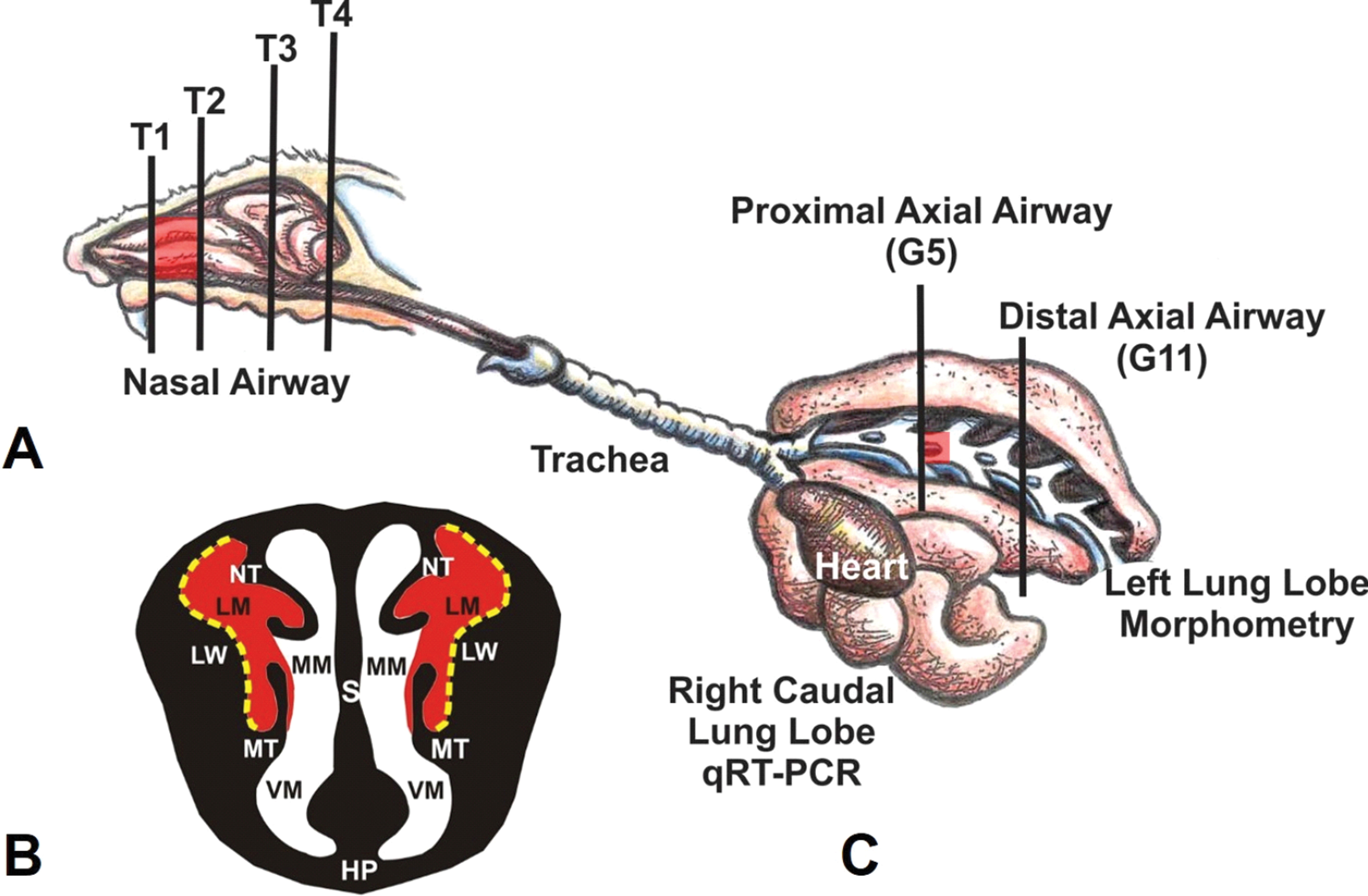
(A) Diagrammatic representation of the right nasal passage, with the septum removed, in the mouse. The vertical lines (T1–T4) indicate the locations of the transverse or coronal tissue blocks that were selected for light microscopic examination. Red highlights indicate the proximal airway location of ozone-induced nasal lesions. (B) Illustration of the anterior face of T1 section. The lateral meatus is highlighted in red indicating the bilateral location of ozone-induced nasal mucosal histopathology. S, Septum; LW, lateral wall; NT, nasoturbinate; MT, maxilloturbinate; HP, hard palate; LM, lateral meatus; MM, medial meatus; VM, ventral meatus. Stippled yellow lines indicate the lateral wall locations that were selected for morphometric analyses. (C) Illustration of the murine lung with the exposed main axial airway in the left lung lobe. Vertical lines indicate the location of transverse tissue blocks that were taken at the level of axial airway generation 5 (G5; proximal) and generation 11 (G11; distal) for microscopic examination and morphometry of airway epithelium.
Pulmonary Morphometry
Histologic slides containing the lung tissue sections were scanned and digitized with a slide scanner (VS110; Olympus America, Center Valley, PA) and evaluated via stereological methods with newCAST software (VIS version 5.2.1.1485; VisioPharm, Hoersholm, Denmark). For quantification of AB/PAS-positive, intraepithelial mucosubstances, all bronchiolar epithelium lining G5 along the main axial airway were selected and captured at 400× magnification (one lung tissue section per animal). A point intercept grid was placed over the sampled images to estimate density of mucosubstances per basal lamina. The number of points hitting AB/PAS-positive mucosubstances (Pm
) was counted. The density of AB/PAS-positive mucosubstances
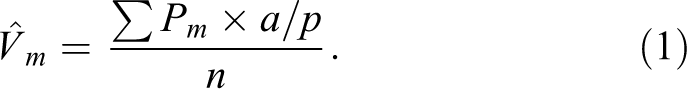
The surface density of the basal lamina
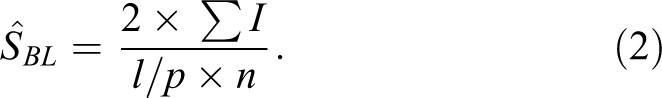
The positive density per basal lamina of the bronchiolar epithelium was then estimated by dividing
Nasal Histology
The heads from all mice were also collected at necropsy, formalin fixed, and decalcified using 13% formic acid, as previously described in detail (Ong et al. 2016). After decalcification, 4 transverse nasal tissue blocks were taken from specific sites, defined by dental and palatine landmarks, of the nasal cavity (proximal T1–distal T4; Figure 1A) by methods previously reported (Harkema, Carey, and Wagner 2006). Like the lung tissue sections, 5 μm thick, paraffin-embedded nasal tissue sections were histochemically stained with H&E and AB/PAS. Additional nasal sections were immunohistochemically stained with an antibody specific for murine major basic protein (MBP; clone MT-14.7; Mayo Clinic, Scottsdale, AZ) to identify eosinophils in the nasal mucosa (Ong et al. 2016).
Nasal Morphometry
Glass slides of nasal tissue sections were also digitized with the slide scanner (VS110). Similar to the pulmonary airways, the nasal mucosa lining the lateral wall in the T1 section (Figure 1B) was evaluated via morphometric methods using the newCAST software. The volume density of AB/PAS-stained mucosubstances in the nasal epithelium lining the lateral wall of the proximal nasal passages (T1) was morphometrically measured using the same methodology described above for the epithelium lining the proximal main axial airway in the left lung lobe.
In addition, morphometry was used to determine the density of MBP-laden eosinophils in the mucosa lining the proximal lateral wall of each nasal passage (T1), by methods previously reported in detail (Kumagai et al. 2016; Ong et al. 2016). Briefly, the number of points hitting areas positive for MBP antibody was counted with a point grid in the sampled images of each mouse. The number of points on the reference space (i.e., mucosal epithelium and lamina propria) was also counted with a point grid. The total number of points was multiplied by the area/point (a/p) for the eosinophil or reference space to calculate each density. Finally, the percentage of eosinophil density per the reference space was calculated and expressed as the percentage of eosinophilic influx in the mucosal tissue.
Quantitative Real-time Polymerase Chain Reaction (RT-PCR) of Lung Tissue
After lavage collection, the right lung lobes were stored at −20°C until further use. Total RNA isolation and complementary DNA (cDNA) synthesis were performed as described previously (Brandenberger et al. 2013). Total RNA was quantified using a NanoDrop-1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). Quantitative RT-PCR was carried out using TaqMan Gene Expression Assays (Applied Biosystems, Waltham, MA). Selected genes, including housekeeping genes (Gusb, Hprt, and Rplp0 messenger RNA [mRNA] for proteins beta-glucuronidase, hypoxanthine-guanine phosphoribosyltransferase, and 60S acidic ribosomal protein P0, respectively), were analyzed using 0.125 ng cDNA in a 100 nL final reaction volume on a SmartChip Real-Time PCR System (WaferGen, Freemont, CA) or using 10 ng cDNA in a 10 µL final reaction volume on the ABI PRISM 7900HT platform (Applied Biosystems). Replicate ΔCt values of the genes of interest were obtained and normalized by subtracting the geometric mean of Cts from the endogenous controls. Relative gene expression levels were reported as fold change using the ΔΔCt method where FC = 2−ΔΔCt. Fold changes in mRNA levels (ozone-exposed mice relative to air-exposed mice) were determined for select genes related to (1) airway mucus production/secretion (Muc5ac, Muc5b, and Clca1/Gob5), (2) epithelial cell-derived cytokines (Il33, Il25, Tslp), and (3) type 2 cytokines/proteins (Il5, Il13, Chia, and Chil4/Ym2).
Statistical Analysis
A Grubbs outlier test was performed on all data. Multiple comparisons of cytologic, morphometric, and molecular data were done by two-way analysis of variance (ANOVA; exposure and strain factors) and Student–Newman–Keuls’ post hoc test. Significance was assigned to p values <.05 for all statistical analyses.
Results
Strain Differences in Pulmonary Inflammatory Response to Ozone
Figures 2 and 3 graphically illustrate the number of total and inflammatory cells, respectively, in the recovered BALF of filtered air- and ozone-exposed mice. Repeated ozone exposures induced a marked increase in total inflammatory cells, macrophages/monocytes, neutrophils, and eosinophils in the BALF of both strains of mice. Ozone-exposed C57BL/6 mice, however, had 4× more eosinophils in BALF, as compared to similarly exposed BALB/c mice (Figure 3). In contrast, ozone-exposed BALB/c mice had approximately twice as many macrophages and 2.5× more neutrophils in BALF, as compared to ozone-exposed C57BL/6 mice. Interestingly, filtered air (0 ppm ozone)–exposed BALB/c mice had significantly more macrophages and neutrophils in BALF, as compared to filtered air–exposed C57BL/6 mice. Filtered air controls of both mouse strains had no detectable eosinophils in their BALF.
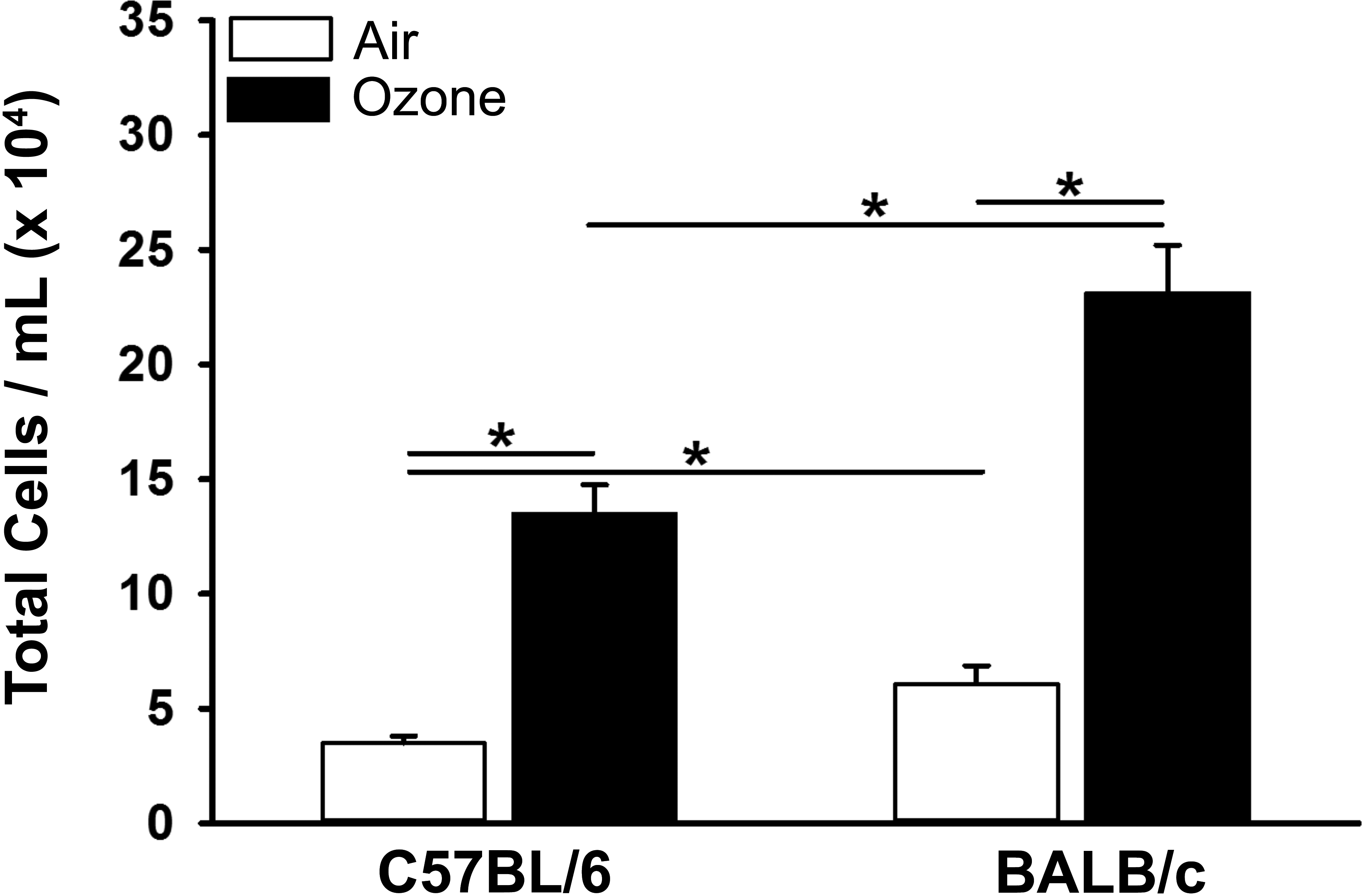
Graphical representation of total cells in the bronchoalveolar lavage fluid taken from the lungs of C57BL/6 and BALB/c mice exposed to filtered air or ozone. Bars represent group means with standard error of the means. Horizontal lines (with asterisks) indicate statistically significant differences between groups, p ≤ .05 (n = 6 mice/group).
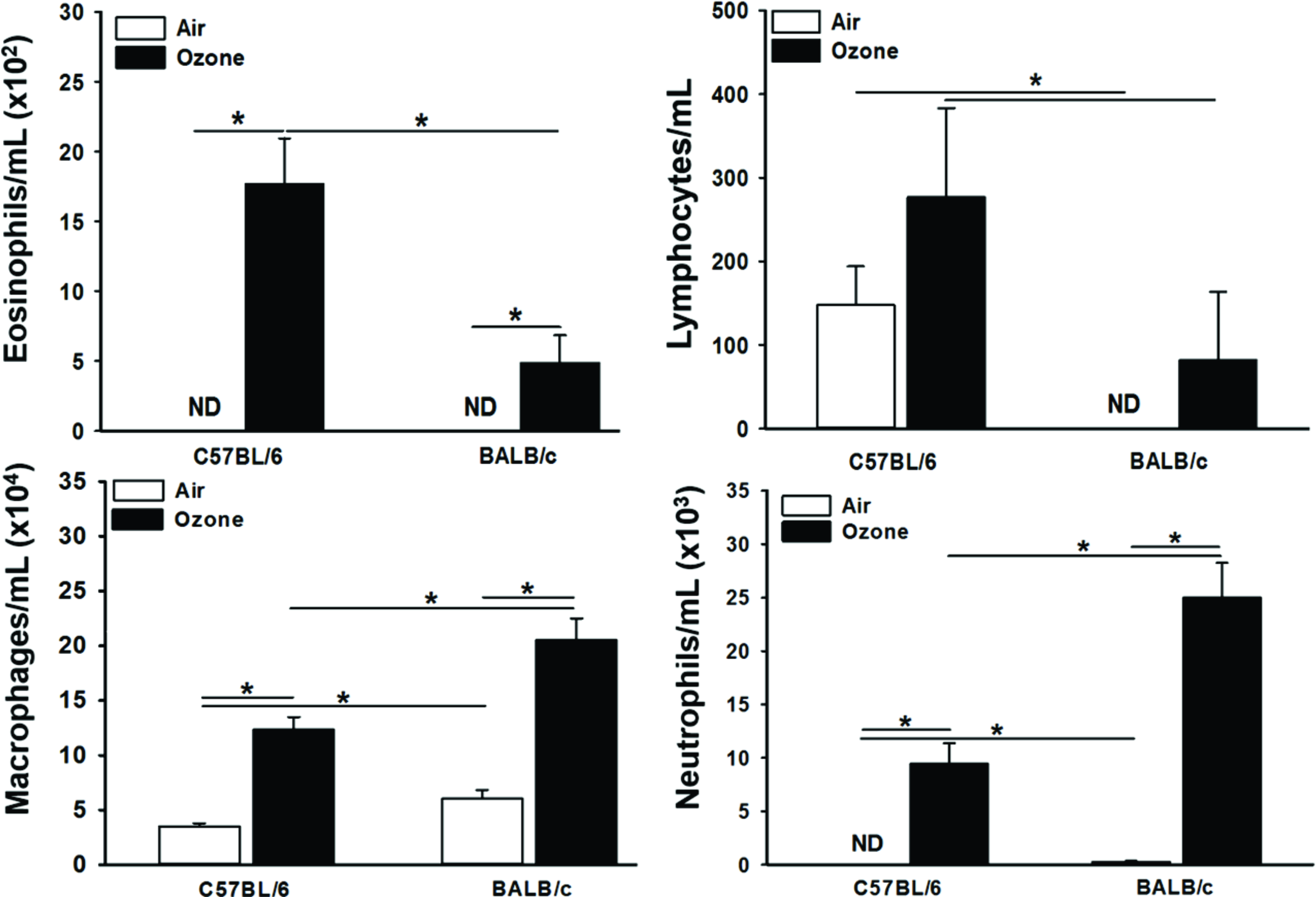
Graphical representation of differential inflammatory cells (macrophages/monocytes, eosinophils, neutrophils, and lymphocytes) in bronchoalveolar lavage fluid taken from the lungs of C57BL/6 and BALB/c mice exposed to filtered air or ozone. Bars represent group means with standard error of the means. Horizontal lines (with asterisks) indicate statistically significant differences between groups, p ≤.05 (n = 6 mice/group). ND, not detected.
Greater Ozone-induced Mucous Cell Metaplasia in Lungs of C57BL/6 Mice
Nine-day exposures to ozone induced marked mucous cell metaplasia in the luminal epithelium lining the proximal (G5), but not the distal (G11), aspect of the main axial airway in the left lung lobe of both strains of mice (Figure 4A). The amount of intraepithelial AB/PAS-stained mucosubstances (i.e., volume density) in proximal pulmonary airway of ozone-exposed C57BL/6 mice was approximately 5× greater than ozone-exposed BALB/c mice (Figure 4B). Little or no mucosubstances were detected in the epithelium lining the proximal or distal airways of filtered air–exposed mice of both strains.
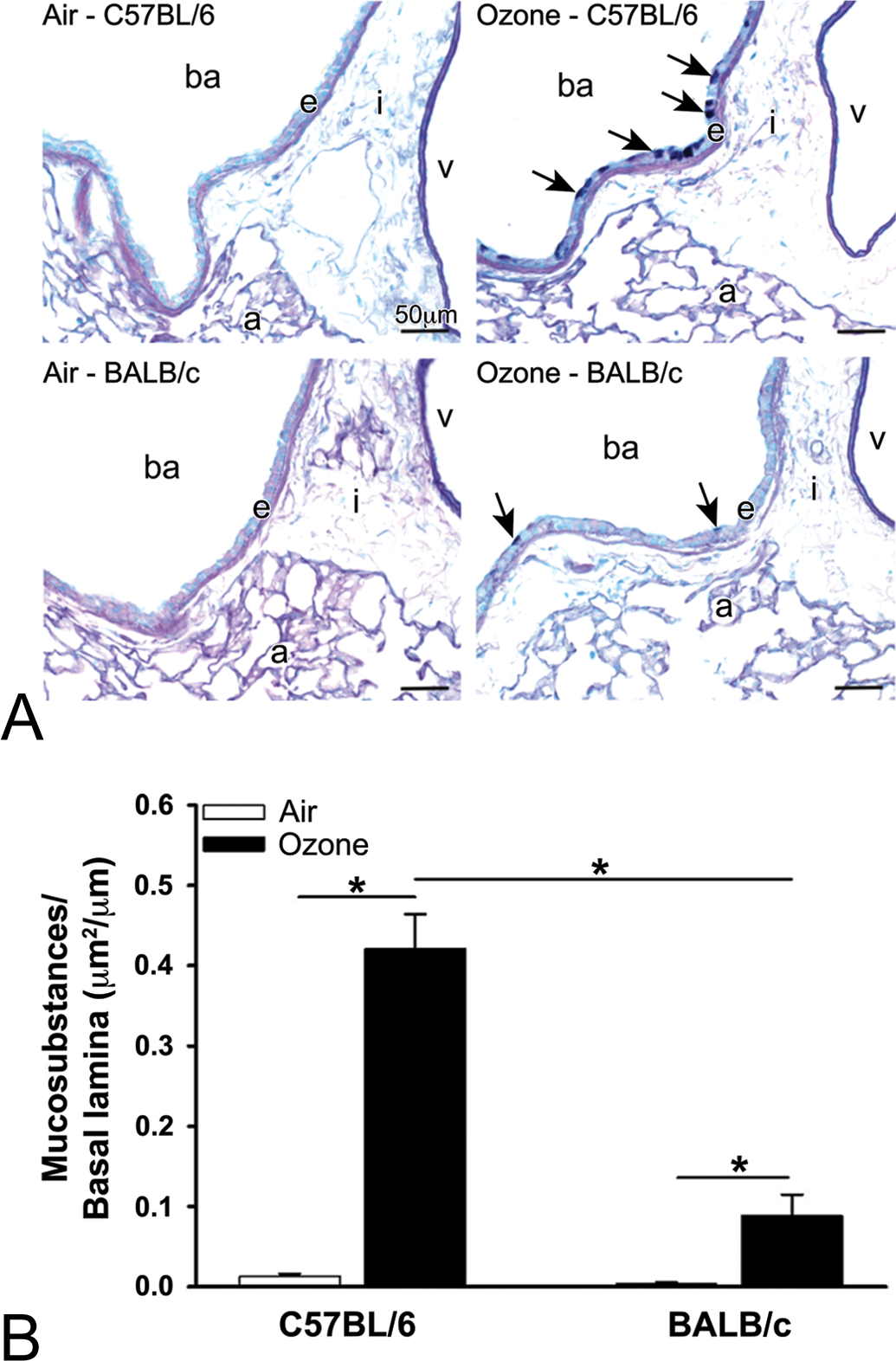
(A) Light photomicrographs of the respiratory epithelium (e) lining the proximal main axial airway (G5) in the left lung lobe of C57BL/6 and BALB/c mice exposed to filtered air or ozone. Tissue sections were histochemically stained with Alcian Blue/Periodic Acid Schiff (AB/PAS) to identify intraepithelial mucosubstances (arrows). ba, lumen of bronchiolar airway; a, alveolus; i, peribronchiolar/perivascular interstitial tissue; and v, lumen of blood vessel. (B) Graphical illustration of the volume density of intraepithelial AB/PAS-stained mucosubstances lining the proximal axial airway of the left lung lobe in C57BL/6 and BALB/c mice exposed to filtered air or ozone. Bars represent group means with standard error of the means. Horizontal lines (with asterisks) indicate statistically significant differences between groups, p ≤.05 (n = 6 mice/group).
Strain Differences in Ozone-induced Pulmonary Gene Expression
Both ozone-exposed C57BL/6 and BALB/c mice had statistically significant relative fold increases, as compared to filtered air–exposed controls, in the pulmonary mRNA levels of mucin-5AC (Muc5ac), mucin-5B (Muc5b), chloride channel accessary-1 protein (Clcal/Gob5), acidic mammalian chitinase (Chia), chitinase-like-4 protein (Chil4/Ym2), Interleukin-33 (Il33), and Interleukin-13 (Il13) (Figure 5; Table S1 in online supplemental data). Relative fold increases in Il33 mRNA was found in the lungs of ozone-exposed C57BL/6 mice but not in the lungs of similarly exposed BALB/c mice (Figure 5). The magnitude of ozone-induced fold changes in the mRNA for Muc5ac, Muc5b, Clca1/Gob5, Chil4/Ym2, Il33, and Il13 were greater (approximately 6×, 2×, 43×, 18×, 1.5×, and 5×, respectively) in the lungs of C57BL/6 mice compared to BALB/c mice. There was no ozone-induced fold change in the pulmonary mRNA for IL5, Il25, or thymic stromal lymphopoietin (Tslp) in either strain of mouse.
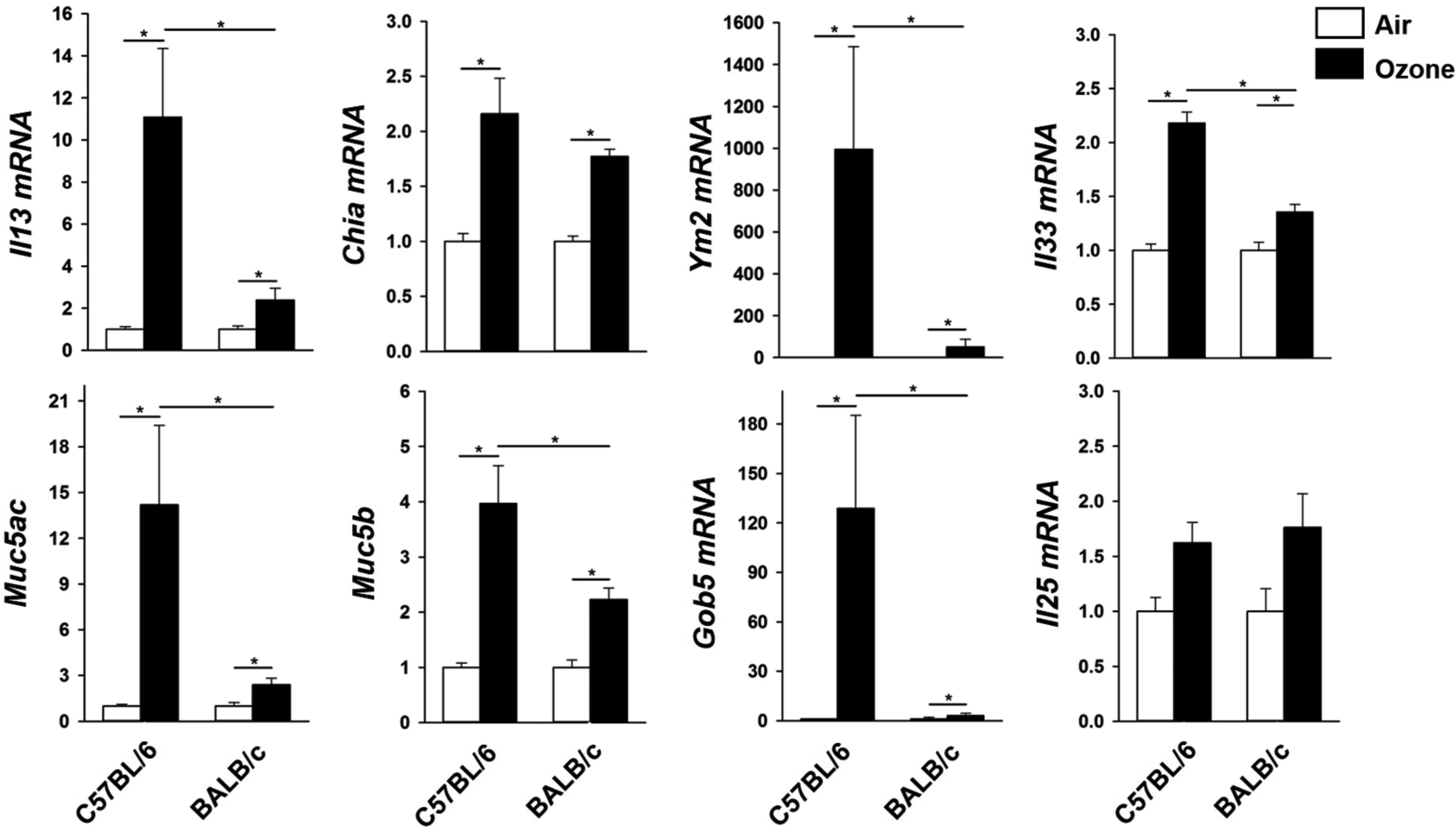
Graphical representation of ozone-induced fold changes in messenger RNA (mRNA; % relative to filtered air-exposed) for genes in the lungs of C57BL/6 and BALB/c mice. Selected genes are related to (1) type 2 immunity/inflammation (Il13, Chia, Ym2), (2) airway mucus production/secretion (Muc5ac, Muc5b, Gob5), or (3) epithelial cytokines (alarmins; Il33, Il25). Horizontal lines (with asterisks) indicate statistically significant differences between groups, p ≤.05 (n = 6 mice/group).
Greater Ozone-induced Eosinophilic Rhinitis in C57BL/6 Mice
Ozone-induced eosinophil infiltration of the nasal mucosa was restricted to the nasal mucosa lining the proximal lateral meatus of both nasal passages (i.e., T1 section; bilateral eosinophilic rhinitis; Figure 6A) in both strains of mice and similar to what we have previously reported (Ong et al. 2016; Kumagai et al. 2016). Interestingly, the mucosal density of the eosinophilic influx in ozone-exposed C57BL/6 mice was approximately twice as great as that in ozone-exposed BALB/c mice (Figure 6B).
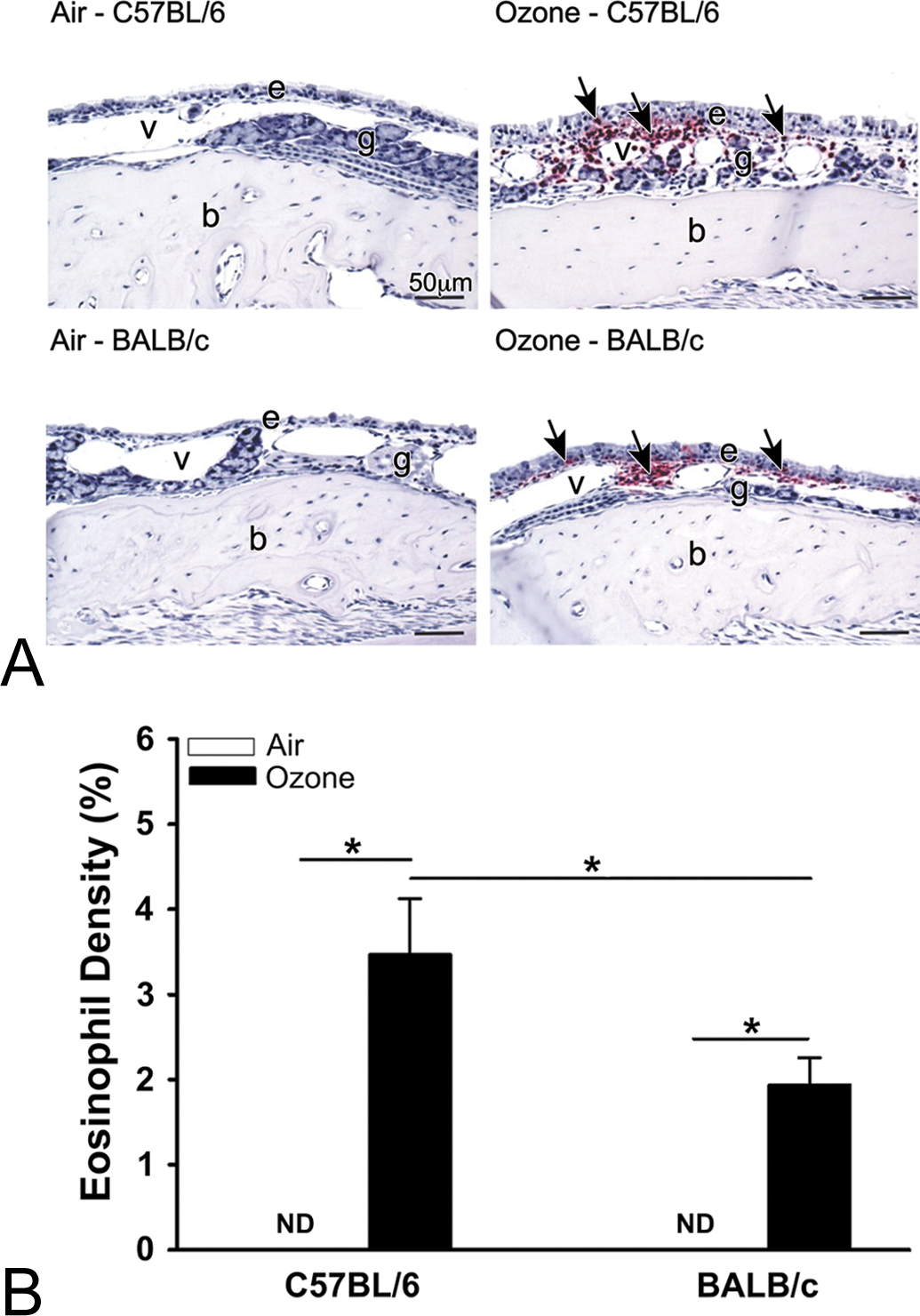
(A) Light photomicrographs of the nasal epithelium (e) lining the lateral wall in the proximal nasal airway (T1) of C57BL/6 and BALB/c mice exposed to filtered air or ozone. Tissue sections were immunohistochemically stained to identify major basic protein-laden eosinophils (arrows; red chromagen), and counterstained with hematoxylin; v, lumen of blood vessel in subepithelial lamina propria; g, gland in lamina propria; b, bone. (B) Graphical representation of the numeric cell density of eosinophils in the nasal mucosa lining the lateral wall of the proximal nasal airways in C57BL/6 and BALB/c mice exposed to filtered air or ozone. Bars represent group means with standard error of the means. Horizontal lines (with asterisks) indicate statistically significant differences between groups, p ≤.05 (n = 6 mice/group).
Similar Ozone-induced Mucous Cell Metaplasia in Nasal Epithelium of Mice Strains
Mucous cell metaplasia of nasal epithelium accompanied the eosinophilic inflammation in the proximal nasal passages of both strains of ozone-exposed mice (Figure 7A). In contrast to the strain-related difference in the magnitude of ozone-induced eosinophilic rhinitis mentioned above, the volume densities of intraepithelial mucosubstances (i.e., quantitative marker of mucous cell metaplasia) were not statistically different between ozone-exposed C57BL/6 and BALB/c mice (Figure 7B).
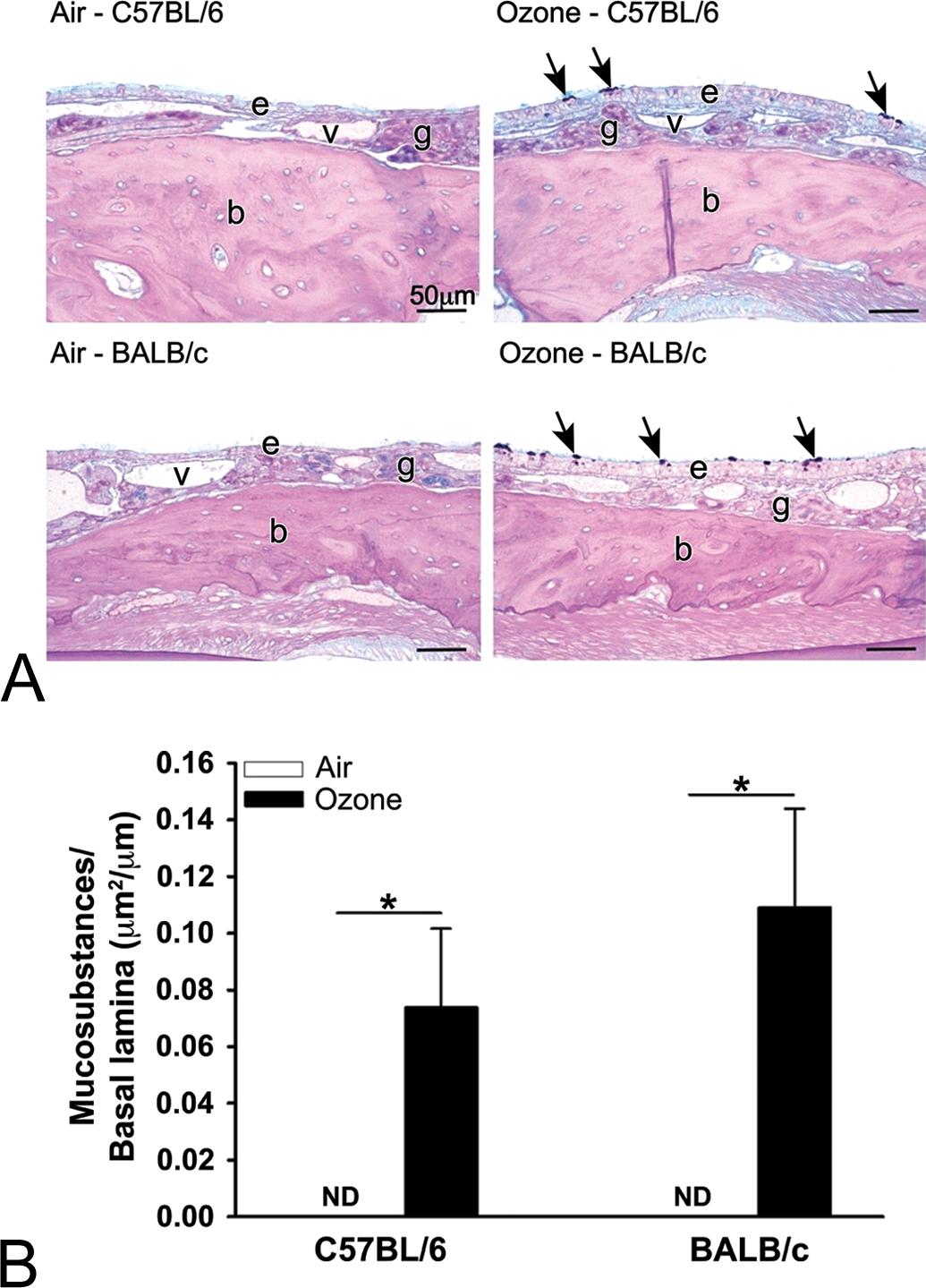
(A) Light photomicrographs of the nasal epithelium (e) lining the lateral wall in the proximal nasal airway (T1) of C57BL/6 and BALB/c mice exposed to filtered air or ozone. Tissue sections were histochemically stained with Alcian Blue/Periodic Acid Schiff to identify intraepithelial mucosubstances (arrows); v, lumen of blood vessel in subepithelial lamina propria; g, gland in lamina propria; b, bone. (B) Graphical representation of the volume density of intraepithelial mucosubstances in the nasal mucosa lining the lateral wall of the proximal nasal airways in C57BL/6 and BALB/c mice exposed to filtered air or ozone. Bars represent group means with standard error of the means. Horizontal lines (with asterisks) indicate statistically significant differences between groups, p ≤.05 (n = 6 mice/group).
Discussion
In the present study, repeated exposures to ozone caused type 2 immune responses with associated eosinophilic inflammation and epithelial mucous cell metaplasia (key pathologic and molecular markers of asthma and rhinitis) in both pulmonary and nasal airways of C57BL/6 and BALB/c mice. However, the magnitude of these ozone-induced, type 2 immune-associated airway inflammatory/epithelial changes, and pulmonary gene expression increases were consistently greater in C57BL/6 compared to BALB/c mice. These strain differences in airway responses to repeated ozone exposures indicate that genotype is an important factor in this murine model of nonatopic asthma and rhinitis. It further suggests that an individual’s genetic background may determine the clinical manifestation of ozone-induced new-onset asthma (and rhinitis) in nonatopic children or adults (i.e., without allergic sensitization). Interestingly, in a recent epidemiological study of a very large and diverse population of Latino children recruited from multiple urban regions across the United States, investigators found that children with asthma without allergic sensitization (nonatopic asthma) were more likely to have been exposed to higher levels of ozone pollution during their first year of life compared with children with atopic asthma (Nishimura et al. 2016). They reported that for every 5-ppb increase in average ozone exposure during the child’s first year of life, the odds for having asthma without sensitization increased by 32%. This epidemiological study, however, was not designed to decipher the variability in asthmatic responses to ozone in these nonatopic Latino children.
It has been long recognized that allergic rhinitis often accompanies or precedes the onset of asthma (Bousquet et al. 2001, 2008; Cingi et al. 2015). The pathology of allergic rhinitis is so similar to the airway pathology of asthma that the two respiratory maladies are commonly referred to as one-airway disease or united airway disease (Jeffery and Haahtela 2006; Giavina-Bianchi et al. 2016). Likewise, we found with the present and past studies, that our murine model of nonatopic asthma and rhinitis has similar pulmonary and nasal pathology that are associated with type 2 immunity and dependent on ILCs. Ong et al. (2016) conducted a study in which lymphoid cell-sufficient, C57BL/6 mice were exposed to 0 or 0.5 ppm ozone for 1, 2, 4, or 9 consecutive weekdays (4 hr/day). Lymphoid cell-deficient (no ILCs, T or B cells), Rag2−/−Il2rg−/− mice were similarly exposed for 9 weekdays. Nasal tissues were taken at 2 hr or 24 hr postexposure for morphometric and gene expression analyses. C57BL/6 mice exposed to ozone for 1 day had acute neutrophilic rhinitis with airway epithelial necrosis and overexpression of mucosal Ccl2 (monocyte chemoattractant protein-1 [MCP-1]), Ccl11 (eotaxin), Cxcl1 (CXC-1 chemokine [KC]), Cxcl2 (CXC chemokine-2 [MIP-2]), heme oxygenase 1 (Hmox1), Il1b, Il5, Il6, Il13, and tumor necrosis factor (Tnf) mRNA. In contrast, 9-day ozone exposure elicited type 2 immune/inflammatory responses in C57BL/6 mice with mucosal mRNA overexpression of arginase-1 (Arg1), chemokine ligand 8 (monocyte chemoattractant protein-2) [Ccl8 (MCP-2)], Ccl11, Chil4 (Ym2), Clca1 (Gob5), Il5, Il10, and Il13, increased the density of mucosal eosinophils, and nasal epithelial remodeling (i.e., mucous cell metaplasia). Importantly, Rag2−/−Il2rg−/− mice exposed to ozone for 9 days, however, had no nasal pathology or overexpression of transcripts related to type 2 immunity.
In a subsequent study, Kumagai et al. (2016) determined the role of ILCs in the pathogenesis of ozone-induced eosinophilic rhinitis by using lymphoid-sufficient C57BL/6 mice, Rag2−/− mice that are devoid of T cells and B cells, and Rag2−/−Il2rg−/− mice that are depleted of all lymphoid cells including ILCs. Animals were exposed to 0 or 0.8 ppm ozone for 9 consecutive weekdays (4 hr/day). Mice were sacrificed 24 hr postexposure and nasal tissues were selected for histopathology and gene expression analysis. ILC-sufficient C57BL/6 and Rag2−/− mice exposed to ozone developed marked eosinophilic rhinitis and epithelial remodeling (i.e., mucous cell metaplasia). Alarmins (Interleukin [IL]-33, IL-25, and thymic stromal lymphopoietin [TSLP]) were also morphometrically increased in the nasal epithelium of ozone-exposed C57BL/6 and Rag2−/− mice. Ozone exposure elicited increased expression of Il4, Il5, Il13, St2, eotaxin, MCP-2, Gob5, Arg1, small cysteine-rich secreted protein associated with type 2 inflammation (FIZZ1), and Ym2 mRNA in C57BL/6 and Rag2−/− mice. In contrast, ozone-exposed ILC-deficient Rag2−/−Il2rg−/− mice had no nasal lesions or overexpression of Th2- or ILC2-related transcripts. These results indicated that ozone-induced eosinophilic rhinitis, epithelial mucous cell metaplasia, and type 2 immune activation are dependent on ILCs (most likely group 2 ILCs). This was the first study to demonstrate that ILCs play an important role in the nasal pathology induced by repeated ozone exposure.
ILCs are recently discovered non-T or non-B lymphocytes (lineage negative) that are essential for (1) the innate mucosal immunity generated in response to a number of pathogens (e.g., viruses, fungi, and helminths) and allergens (e.g., house dust mite antigen), (2) promoting wound healing, and (3) maintaining tissue homeostasis (Artis and Spits 2015; Sonnenberg and Artis 2015). Mature ILCs have been broadly classified into 3 groups, based primarily on their specific surface markers and cytokine production profiles: group 1 (ILC1s), group 2 (ILC2s), and group 3 (ILC3s). ILC2s lack lineage markers but express several surface cell antigens such as CD25, c-kit, Sca-1, T1/ST2, and Thy1.2 (CD90.2). This group of ILCs is the innate lymphocyte equivalents to T-helper type 2 (Th2) cells and produce large amounts of type 2 cytokines including IL-5 and IL-13 in response to initial exposures to allergens (e.g., house dust mite) and pathogens (e.g., viruses and parasites; Klein Wolterink et al. 2012). Although the roles of ILC2s in the early onset and persistence of human allergic airway diseases are not fully understood, increased numbers of ILC2s have been detected in peripheral blood, sputum, and BALF of asthmatic patients (McKenzie 2014; Christianson et al. 2015; Kim, Umetsu, and Dekruyff 2016; Smith et al. 2016).
Using various murine models of allergic airway diseases, ILC2s have been shown to contribute to type 2 immune-related pathologic airway responses including IL-5-mediated recruitment, and activation of eosinophils and IL-13 induction of mucus hyperproduction and airway hyperresponsiveness (AHR) to methacholine challenge (Doherty 2015). It has been suggested that there may be a common pathway of ILC2-mediated pulmonary allergic immunity and inflammation that is initiated by injury to airway epithelium caused by allergen or pathogen exposure (Hammad and Lambrecht 2015; Karta, Broide, and Doherty 2016). Injured or necrotic airway, or alveolar, epithelial cells release specific cytokines (alarmins), including IL-33, IL-25, and/or TSLP, that either individually or in combination activate ILC2s through specific ligand/receptor binding (e.g., IL-33/ST2). Activated ILC2s then produce IL-5 that induces lung eosinophilia, and IL-13 that promotes mucous cell metaplasia/mucus hypersecretion and AHR. In addition, it is thought that ILC2s play a major role in the initiation of adaptive Th2 responses to allergens and thus link the innate and adaptive responses caused by inhaled allergens (van Rijt, von Richthofen, and van Ree 2016).
In the present study, we found that 9 consecutive weekday exposures to ozone also caused type 2 associated gene expression in the lung that is reminiscent of the ozone-induced gene expression increases and histopathology that we previously observed in the nasal mucosa of ILC-sufficient C57BL/6 and Rag2−/− mice but not in the ILC-deficient Rag2−/−Il2rg−/−. Like in the nasal mucosal tissues of ILC-sufficient mice from our previous ozone studies, we found in the present study that repeated ozone exposures caused, in lung tissues, relative fold increases in mRNA related to mucous cell metaplasia and mucus production/secretion (i.e., Il13. Muc5ac, Muc5b, and Clca1/Gob5) and type 2 immunity (i.e., Il13, Chia, and Chil4/Ym2). The overexpression of these pulmonary genes in ozone-exposed mice correlated with type 2 associated pulmonary histopathology of mucous cell metaplasia in bronchiolar epithelium and the increase of eosinophils in BALF.
With this new lung data, we suspect that ozone also activates pulmonary ILC2s, possibly through alveolar or airway epithelial cell–derived IL-33, to produce IL-13 and possibly other type 2 cytokines (e.g., IL-5) that drive airway epithelial and inflammatory changes in mucous cell metaplasia and eosinophilic inflammation. However, with design limitations of our present study, further investigations are needed to determine if ILC2s really do mediate ozone-induced type 2 immunity, mucous cell metaplasia, and eosinophilic inflammation in the lung, like these lymphoid cells do in the nose. Since the numbers of ILCs in the murine lung and nose are extremely small and microscopic markers for these lymphoid cells have not yet been developed, future studies will have to rely on flow cytometric techniques for ILC identification (Moro and Koyasu 2015) along with the use of ILC-sufficient and ILC-deficient C57BL/6 or BALB/c mice.
Another limitation of the current study was the absence of pulmonary function testing to determine if ozone-exposed mice had AHR, a key functional feature of asthmatics that is clinically manifested as wheeze. Others, however, have recently demonstrated that ILC2s mediate AHR, release of type 2 cytokines, and eosinophil influx in the lungs of BALB/c mice after a single, 2 hr-exposure to a very high concentration (3.0 ppm) of ozone (Yang et al. 2016). Additional studies are needed to determine if repeated, longer-term ozone exposures at lower, more environmentally relevant, concentrations will also cause AHR in BALB/c or C57BL/6 mice.
Others have investigated strain differences in pulmonary epithelial, inflammatory, and functional responses to allergen sensitization and challenge (i.e., model of allergic airway disease or atopic asthma). In a study by Kelada et al. (2011), BALB/cJ and C57BL/6J mice were intraperitoneally sensitized with house dust mite allergen, without an exogenous adjuvant, and then given a single airway challenge to this same antigen. They found that the allergen-treated BALB/cJ mice developed all the key features of allergic airway disease. In comparison, similarly treated C57BL/6J mice had exaggerated Th2-biased responses and eosinophilic inflammation but had an unexpected decrease in AHR compared with control mice. Their study demonstrated that inflammation and AHR can be decoupled, and the authors suggested that a down modulation of expression of G-protein-coupled receptors involved in regulating airway smooth muscle contraction may have contributed to this dissociation.
It is interesting that in both the murine model of atopic asthma (with allergen sensitization; Kelada et al. 2011) and our murine model of ozone-induced nonatopic asthma (without allergen sensitization), C57BL/6 mice exhibited a greater eosinophilic inflammatory response in the lung compared to BALB/c mice. In contrast to our findings, however, they found that the magnitude of allergen-induced mucous cell metaplasia in bronchiolar epithelium did not differ between these 2 strains. Further in-depth comparisons of similar key pathologic, immunologic, and molecular end points among different murine models of asthma and rhinitis are needed to better understand the genetic and environmental factors underlying the growing number of asthmatic phenotypes. In addition, the impact of other factors, such as substrain, gender, and obesity, on these murine models of rhinitis and asthma needs to be explored and elucidated in future studies.
In summary, the results of our study confirm our hypothesis that murine airway epithelial, inflammatory, and immunologic responses to repeated ozone exposures, in the nose and lung, are strain dependent. The variability in responses between the C57BL/6 and BALB/c mice was primarily in the magnitude, rather than the character, of the ozone-induced type 2 immunity, eosinophilic inflammation, and mucous cell metaplasia (i.e., murine model of airpollutant–induced nonatopic asthma and rhinitis). Specific genomic factors responsible for these strain differences are yet to be determined.
Footnotes
Acknowledgments
The authors thank Ms. Amy Porter and Ms. Kathy Joseph of Michigan State University Histopathology Laboratory for the histology slide preparation. We also thank Dr. Jeff Landgraf for conducting the qRT-PCR analysis at Research Technology Support Facility in Michigan State University.
Author Contribution
All authors (JRH, LAH, NAV, DNJ-H, RPL, JGW) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported with funds from the USEPA’s Clean Air Research Center grant RD 83479701 and from NIH grant T35OD016477.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.