
Abstract
Exposure to elevated levels of ambient ozone in photochemical smog is associated with eosinophilic airway inflammation and nonatopic asthma in children. In the present study, we determined the role of innate lymphoid cells (ILCs) in the pathogenesis of ozone-induced nonatopic asthma by using lymphoid cell-sufficient C57BL/6 mice, ILC-sufficient Rag2−/− mice (devoid of T and B cells), and ILC-deficient Rag2−/−Il2rg−/− mice (depleted of all lymphoid cells including ILCs). Mice were exposed to 0 or 0.8 parts per million ozone for 1 day or 9 consecutive weekdays (4 hr/day). A single exposure to ozone caused neutrophilic inflammation, airway epithelial injury, and reparative DNA synthesis in all strains of mice, irrespective of the presence or absence of ILCs. In contrast, 9-day exposures induced eosinophilic inflammation and mucous cell metaplasia only in the lungs of ILC-sufficient mice. Repeated ozone exposures also elicited increased messenger RNA expression of transcripts associated with type 2 immunity and airway mucus production in ILC-sufficient mice. ILC-deficient mice repeatedly exposed to ozone had no pulmonary pathology or increased gene expression related to type 2 immunity. These results suggest a new paradigm for the biologic mechanisms underlying the development of a phenotype of childhood nonatopic asthma that has been linked to ambient ozone exposures.
Keywords
Ozone, a commonly encountered gaseous air pollutant in photochemical smog, is often found in urban communities with large amounts of sunlight and motor vehicle traffic. High concentrations of ambient ozone are associated with increases in morbidity and mortality (Bell et al. 2004; Jerrett et al. 2009), decrements in pulmonary function (Brown, Bateson, and McDonnell 2008; Kahle et al. 2015), and exacerbation of asthma and allergic rhinitis (Peden 2001; Guarnieri and Balmes 2014). The US Environmental Protection Agency (USEPA) has recently revised the 8-hr national ambient air quality standard concentration (NAAQS) for ozone from 0.075 to 0.070 parts per million (ppm) in consideration of ozone-related health effects including those for “at-risk” populations such as children with asthma (USEPA 2015). It has been predicted that ambient ground levels of ozone are likely to increase and cause more exposure-related health effects in the future based on expected climate changes, such as annual elevations in outdoor temperatures (D’Amato et al. 2015). It has been estimated that higher ozone concentrations due to rising temperatures in 2020 will lead to an average of 2.8 million more occurrences of acute respiratory symptoms, such as asthma attacks (Perera and Sanford 2011). Although many harmful health effects of short-term ozone exposure have been identified in asthmatics, the biological mechanisms responsible for the epidemiologically associated early onset (development) of asthma caused by this and other air pollutants (e.g., particulate matter) have not been fully elucidated and need further exploration.
Asthma is no longer viewed as a single lung disease that is solely allergen dependent but as a heterogeneous entity with a growing number of specific subtypes, or phenotypes that are defined by common clinical, inflammatory, and molecular characteristics (Gauthier, Ray, and Wenzel 2015; Ilmarinen, Tuomisto, and Kankaanranta 2015; Wenzel 2016). This evolving concept of asthmatic phenotypes has provided a broad differentiation of the asthmatic condition into those with atopic and nonatopic asthma (with and without preexisting allergic sensitization) as well as those with and without type 2 immunity and inflammation. In order to develop better therapeutics and strategies for prevention, asthmatic clinical and molecular phenotypes must be linked to factors responsible for the variability in asthma, such as genetics, age, respiratory infections, psychosocial stressors, obesity, and environmental exposures (e.g., air pollution). Animal models of asthmatic phenotypes are needed to better define the biomolecular pathways underlying the onset of these specific subtypes of asthma (Wenzel 2016).
Our laboratory has recently reported that repeated daily exposures of mice to ozone induce eosinophilic rhinitis, airway epithelial remodeling (e.g., epithelial hyperplasia and mucous cell metaplasia), and increased expression of type 2 immune-related transcripts in the nasal airways. Interestingly, we further found that ozone-induced nasal type 2 immunity was dependent on innate lymphoid cells (ILCs). Both ILC-sufficient Rag2−/− mice and C57BL/6 mice (genetic background animals) developed ozone-induced nasal type 2 immunity and associated pathology (Kumagai et al. 2016; Ong et al. 2016). Congenic Rag2−/− mice are incapable of developing adaptive immunity but sustain the ability for innate immunity by way of ILCs like that of C57BL/6 mice (Yasuda et al. 2012; Doherty et al. 2013). In contrast, we found that nasal type 2 immune responses were completely absent in ozone-exposed Rag2−/−Il2rg−/− mice that are deficient in both recombinase activating gene (Rag) 2 and IL-2 receptor γ chain (Il2rg) and therefore incapable of producing T cells, B cells, and ILCs (Colucci et al. 1999; Moro et al. 2010; Vonarbourg et al. 2010).
ILCs are recently discovered non-T and -B lymphocytes that are essential for innate mucosal immunity generated in response to a number of pathogens (e.g., viruses, fungi, and helminths) and allergens (e.g., house dust mite), for promoting wound healing, and for maintaining tissue homeostasis (Sonnenberg and Artis 2015). Although these unique lymphocytes lack antigen receptors, they do express several surface cell antigens, such as CD25, c-kit, Sca-1, T1/ST2, and Thy1.2 (CD90.2; Moro et al. 2015). Mature ILCs have been broadly classified into 3 subtypes, based primarily on their specific surface markers and cytokine production profiles, namely, group 1 ILCs (ILC1s), group 2 ILCs (ILC2s), and group 3 ILCs (ILC3s). ILC2s, the focus of the present study, are the innate lymphocyte equivalents to T-helper type 2 (Th2) cells and produce large amounts of IL-4, IL-5, IL-9, IL-13, and amphiregulin (AREG) in response to tissue injury/stress caused by xenobiotic agents, such as allergens, pathogens, or toxicants.
ILC2s contribute to airway type 2 immune-related responses (e.g., mucus hypersecretion, eosinophilic inflammation, and airway hyperresponsiveness [AHR]) in various murine models of allergic airway disease (Doherty 2015). Interestingly, it has recently been reported that ILC2s mediate AHR, release of type 2 cytokines, and eosinophil influx in the lungs of BALB/c mice after a single, 2-hr exposure to a high concentration (3.0 ppm) of ozone (Yang et al. 2016). Although the roles of ILC2s in the early onset and persistence of allergic airway diseases are not fully understood, increased numbers of ILC2s have been detected in peripheral blood, sputum, and bronchoalveolar lavage fluid (BALF) of asthmatic patients (Christianson et al. 2015; Smith et al. 2015).
ILC populations are known to vary in number (and possibly in functional activity) in various organs of the body (Moro et al. 2010; Kim, Hashimoto-Hill, and Kim 2016). In addition, ozone-induced inflammatory and epithelial lesions are very site specific (both in character and severity) along the respiratory tract of laboratory animals. Therefore, the present study was designed to determine whether repeated ozone exposure in mice would result in ILC-dependent type 2 immunity in the lung like that previously observed in the nasal airways of similarly exposed mice (Kumagai et al. 2016). We tested the hypotheses (1) that repeated ozone exposures induce pulmonary histopathologic hallmarks of asthma (i.e., eosinophilic inflammation, mucous cell metaplasia, and type 2 inflammatory cytokine gene overexpression) and (2) that these type 2 immune responses to ozone are mediated by ILC2s. C57BL/6, Rag2−/−, and Rag2−/−Il2rg−/− mice were exposed to 0 or 0.8 ppm of ozone for 1 day or 9 consecutive weekdays and their lungs were analyzed by cytological, morphometric, and molecular methods. In addition, we used an anti-Thy1.2 antibody to deplete ILCs in the lungs of ozone-exposed Rag2−/−mice and measured pulmonary ILC2s by flow cytometry. Here we report for the first time that ozone-induced eosinophilic inflammation, mucous cell metaplasia, and type 2 immunity in the lungs of mice are mediated by ILCs (most likely ILC2s) and not by T or B lymphoid cells (nonatopic asthma). The results of this study in mice suggest a new paradigm for the early onset and pathogenesis of pulmonary eosinophilic inflammation and other type 2 immune-related responses epidemiologically associated with high ambient ozone exposure in nonatopic healthy children.
Material and Method
Animals
Six- to 8-week-old male C57BL/6, Rag2−/−, and Rag2−/−Il2rg−/− mice were obtained from Taconic Farms (Germantown, NY). Mice were individually housed in stainless steel wire cages within whole body inhalation exposure chambers (H-1000; Lab Products Maywood, NJ). All animal procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee of Michigan State University.
Inhalation Exposures
After an acclimatization period of 1 week, mice were exposed to filtered air (0 ppm ozone; air controls) or 0.8 ppm ozone for 1 day or 9 consecutive weekdays (4 hr/day). Ozone was generated with an Ozone Research and Equipment Corp. (OREC) Model O3VI-O ozonizer (OREC Phoenix, AZ) and chamber ozone concentrations were monitored throughout the exposure with Dasibi 1003 AH ozone monitors (Dasibi Environment Corp., Glendale, CA).
It has been previously reported that it takes approximately 4 to 5 times the concentration of ozone to induce pulmonary inflammatory responses in sedentary laboratory rats that are comparable to those induced in exercising human subjects under controlled acute exposure conditions (Hatch, Slade, et al. 1994; Hatch, McKee, et al. 2013). In other words, rodents exposed to 0.8 ppm ozone is (1) roughly equivalent to ozone concentrations of 0.16 to 0.20 ppm that cause pulmonary function impairments in exercising adults receiving short-term exposures (Avol et al. 1984; Folinsbee, Bedi, and Horvath 1984) and (2) approximately 10-fold higher than the 8-hr NAAQS for ozone (0.070 ppm; USEPA 2015).
Anti-Thy1.2 Antibody Injection
Male Rag2−/− mice were injected intraperitoneally with vehicle (saline) or 500 µg/body of anti-Thy1.2 antibody (clone 30H12, Bio X Cell, West Lebanon, NH) every other day beginning 5 days before the first inhalation exposures (3 doses) and during the repeated ozone exposures for 9 consecutive weekdays (5 doses).
Animal Necropsy and Lung Lavage
Mice were euthanized by exsanguination under pentobarbital anesthesia 24 hr or 2 weeks (15 days) after the end of the 1- or 9-day chamber exposure(s) to air or ozone. Mice that were acutely exposed to filtered air or ozone for 1 day and designated for lung morphometry were injected intraperitoneally with bromodeoxyuridine (BrdU, Sigma Aldrich, St. Louis, MO) 2 hr prior to sacrifice.
Immediately after death, the trachea was exposed and cannulated, and the heart and lungs were excised en bloc. A volume of 0.8 ml saline was instilled through the tracheal cannula and withdrawn to recover BALF. A second intratracheal saline lavage was performed and the collected BALF was combined with the first sample for cellular analysis.
Inflammatory Cells in BALF
Total number of leukocytes in the collected BALF was estimated using a hemocytometer. Cytological slides were prepared by centrifugation at 40 × g for 10 min and stained with Diff-Quick. Differential cell counts for macrophages/monocytes, neutrophils, eosinophils, and lymphocytes were assessed from a total of 200 counted cells.
Tissue Selection and Processing for Lung Histology
After lavage collection, the left lung lobe was intratracheally fixed with 10% neutral buffered formalin at a constant pressure (30 cm H2O) for approximately 2 hr and then stored in a large volume of the same fixative until further processing. After fixation, the lung lobe was microdissected along the main axial airway, and 2 transverse tissue blocks were taken at the level of the 5th (proximal) and 11th (distal) airway generation along the main axial airway (G5 and G11, respectively). These specific airways were selected in order to obtain a representative proximal, large-diameter airway and a distal, small-diameter axial airway for morphologic examination and morphometric analysis. Our laboratory has previously conducted similar morphometric analyses of airway lining epithelial cells from these airway generations in murine studies of allergen-induced airway disease (e.g., Farraj et al. 2003; Brandenberger et al. 2013).
Paraffin-embedded lung tissue sections from mice exposed to filtered air or ozone for 9 days were stained with Alcian Blue (pH 2.5)/periodic acid-Schiff (AB/PAS) to detect acidic and neutral mucosubstances in bronchiolar airway epithelium or hematoxylin and eosin for routine histopathology. For BrdU immunohistochemistry, a mouse anti-BrdU antibody (0.625 µg/ml in final volume, clone B44, BD Biosciences, San Jose, CA) was added to 3% normal horse serum. The anti-BrdU antibody was preincubated with a horse antimouse IgG (1:1,000 dilution in final volume) for 30 min at room temperature. Normal mouse serum (1:50 dilution in final volume) was added to the antibody mixture for eliminating the excess of antimouse IgG and incubated for 30 min at room temperature. Slides were subsequently reacted with the precomplex antibodies, horseradish peroxidase, and Nova Red (Vector Laboratories, Burlingame, CA). Sections were then counterstained with Mayer’s hematoxylin.
Lung Morphometry
Histologic slides containing the lung G5 and G11 tissue sections were scanned and digitized with a slide scanner (VS110, Olympus America, Center Valley, PA) and evaluated via stereological methods with newCAST software (version 5.2.1.1485; VisioPharm, Hoersholm, Denmark). For quantification of BrdU-positive nuclei in bronchiolar epithelium, digitized images of the lung G5 and G11 lung sections were selected as regions of interest and 40% of the lung tissue was captured at 400× magnifications by systematic random sampling. Percentage of BrdU-positive nuclei in the bronchiolar epithelium was estimated with a point grid by dividing number of points hitting areas positive for BrdU by the total number of points falling on all epithelial nuclei (BrdU positive and negative).
For quantification of AB/PAS-positive mucosubstances in the bronchiolar epithelium, all bronchiolar epithelium lining G5 of the main axial airway were selected and captured at 400× magnifications. Morphometric analyses for the volume density of intraepithelial AB/PAS-stained mucosubstances were restricted to this proximal axial airway (G5) since morphologic examination indicated that this ozone-induced airway epithelial response was restricted to this large-diameter axial airway and was not present in the epithelial cells lining the more distal, small-diameter G11 axial airway of ILC-sufficient mice exposed to ozone (see Results section). A point intercept grid was placed over the sampled images to estimate density of mucosubstances per basal lamina. The number of points hitting AB/PAS-positive mucosubstances (Pm
) was counted. The density of AB/PAS-positive mucosubstances (
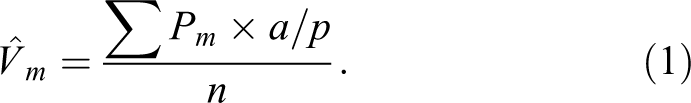
The surface density of the basal lamina (

The positive density per basal lamina of the bronchiolar epithelium was then estimated by dividing
Quantitative Reverse Transcription-Polymerase Chain Reaction
After lavage collection, the right lung lobes were stored in RNAlater (Qiagen Inc., Valencia, CA) at −20°C until further use. Total RNA isolation and cDNA synthesis were performed as described previously (Brandenberger et al. 2013). Total RNA was quantified using a NanoDrop-1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). Quantitative reverse transcription-polymerase chain reaction (RT-PCR) was carried out using TaqMan Gene Expression Assays (Applied Biosystems, Waltham, MA). Selected genes, including housekeeping genes (Gusb, Hprt, and Rplp0 messenger RNA [mRNA]), were analyzed using 0.125 ng cDNA in a 100 nl final reaction volume on a SmartChip Real-Time PCR System (WaferGen, Fremont, CA) or using 10 ng cDNA in a 10 µl final reaction volume on the ABI PRISM 7900HT platform (Applied Biosystems). ΔCt values of the genes of interest were obtained and normalized by subtracting the geometric mean of Cts from the endogenous controls. Relative gene expression levels were reported as fold change using the ΔΔCt method, where Fold Change = 2−ΔΔCt.
Flow Cytometry for ILC2s
Dissection of the lungs and preparation of whole lung cell suspensions were performed, according to the method of Moro and coworkers (2015) with minor modifications. Lungs were minced in a 70-μm nylon cell strainer in a 6-well plate containing 7 ml of RPMI 1640 (Life Technologies, Grand Island, NY) with 100 μg/ml Liberase and 1 μg/ml DNase. The lung segments were subsequently digested for 45 min at 37°C. The whole lung cell suspension was obtained by manually homogenizing the lung tissues over the span of 3 rinses of Hank’s balanced salt solution (HBSS) containing 5% fetal bovine serum (FBS-HBSS, Life Technologies).
Red blood cells were removed by BD Pharm Lyse™ lysis buffer (BD Biosciences, San Jose, CA) for 2 min at room temperature. Remaining cells were then washed with FBS-HBSS (500 g, 5 min at 4°C), resuspended in PBS containing 2% FBS, and blocked with rat antimouse CD16/CD32 (0.5 μg/1 × 106 cells, clone 2.4G2, BD Biosciences) for 5 min at 4°C. Lung cells (1 × 106) were stained with lineage cell detection cocktail biotin, which included monoclonal antibodies against CD5, CD11b, CD45R, anti-7-4, anti-Gr-1, and anti-Ter-119, and anti-CD11c-biotin. This was followed by antibiotin-phycoerythrin staining according to manufacturer’s instructions (Miltenyi Biotec, San Diego, CA).
Cells were also stained with rat anti-T1/ST2-FITC (fluorescein isothiocyanate; 1 μg/1 × 106 cells, MD Bioproducts, St Paul, MN) and rat anti-Thy-1.2-APC (allophycocyanin; 0.12 μg/1 × 106 cells, BioLegend, San Diego, CA) for 20 min at 4°C and resuspended in washing buffer for flow cytometric analysis. 4′,6-diamidino-2-phenylindole (1 μg/ml, BD Biosciences) was added immediately before the analysis. Flow cytometry and data analysis were performed using LSR II and FACSDiva software (version 8.0.1; BD Biosciences). Viable ILC2s were identified as DAPI−/lineage (PE)−/T1/ST2 (FITC)+/Thy-1.2 (APC)+ cells.
Flow cytometric analyses were performed only on 9-day air- or ozone-exposed Rag2−/− mice and 9-day air-exposed Rag2−/−Il2rg−/− mice to specifically determine the effects of ozone exposure and anti-Thy1.2 antibody treatment on the number of lung ILC2s.
Statistical Analysis
A Grubbs outlier test was performed on data of BALF cytology and lung morphometry. Two group comparisons were performed by a Student’s or Welch’s t-test. Multiple comparisons were done by one-way analysis of variance (ANOVA) with post hoc Student–Newman–Keuls’s test or ANOVA on ranks with post hoc Dunn’s test. Significance was assigned to p values < 0.05 for all statistical analyses.
Results
Disparate Ozone-induced Airway Inflammation after Single and Repeated Exposure(s)
BALF neutrophils were significantly increased in all 3 strains of mice exposed to ozone after 1 day and 9 days as compared with their respective air-exposed controls, though the numbers of these inflammatory granulocytes after 9-day exposure were conspicuously less than those after 1-day exposure in each strain (Figure 1). BALF macrophages in all mouse strains were also significantly elevated after 9-day exposure to ozone as compared to air-exposed controls. After 9 days, C57BL/6 and Rag2−/− mice exposed to ozone had significantly greater BALF eosinophils and lymphocytes compared to their respective air-control groups.
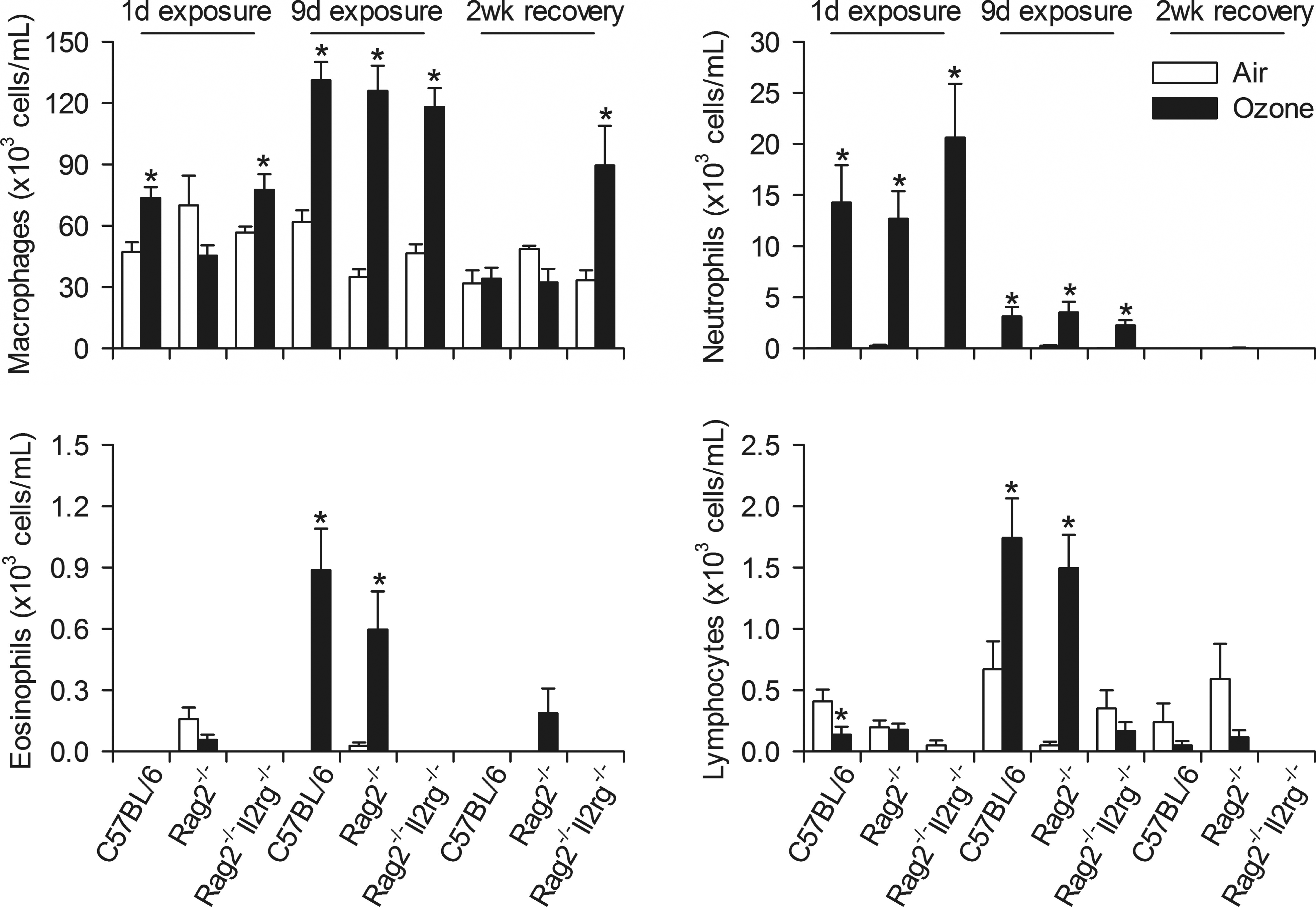
Inflammatory cells in bronchoalveolar lavage fluid (BALF) after air or ozone exposure for 1 day or 9 days and after a postexposure period of 2 weeks. Amount of macrophages/monocytes, neutrophils, eosinophils, and lymphocytes in BALF is graphically illustrated. Data are expressed as mean ± standard error of the mean (n = 12/group for 1-day and 9-day exposure, n = 6/group for 2-week recovery). *Significantly different from the respective control group at the same time point, p < 0.05.
In contrast, Rag2−/−Il2rg−/− mice exposed for 9 days to ozone had no increases in BALF eosinophils or lymphocytes as compared to their respective air-exposed controls. After a 2-week postexposure period in filtered air, 9-day, ozone-induced increases in BALF cell numbers did not persist, except for a persistent increase of macrophages in ozone-exposed Rag2−/−Il2rg−/− mice (Figure 1).
Bronchiolar Epithelial Injury Caused by Single Exposure to Ozone
BrdU-positive epithelial nuclei lining bronchiolar airways were frequently observed in all 3 strains after 1-day exposure to ozone, suggesting acute epithelial cell injury/death followed by regenerative DNA synthesis/cell proliferation (Figure 2A–D). Morphometrically, BrdU-positive density of epithelial nuclei was significantly greater (compared to air-exposed controls) in the bronchiolar airways of the G5 lung tissue section in all 3 strains of mice and in the bronchioles of G11 sections in Rag2−/− and Rag2−/−Il2rg−/− mice (Figure 2E). BrdU-positive nuclear density of the bronchiolar epithelium in the more proximal lung section (G5) of ozone-exposed mice was approximately twice as high as those in the more distal lung section (G11).
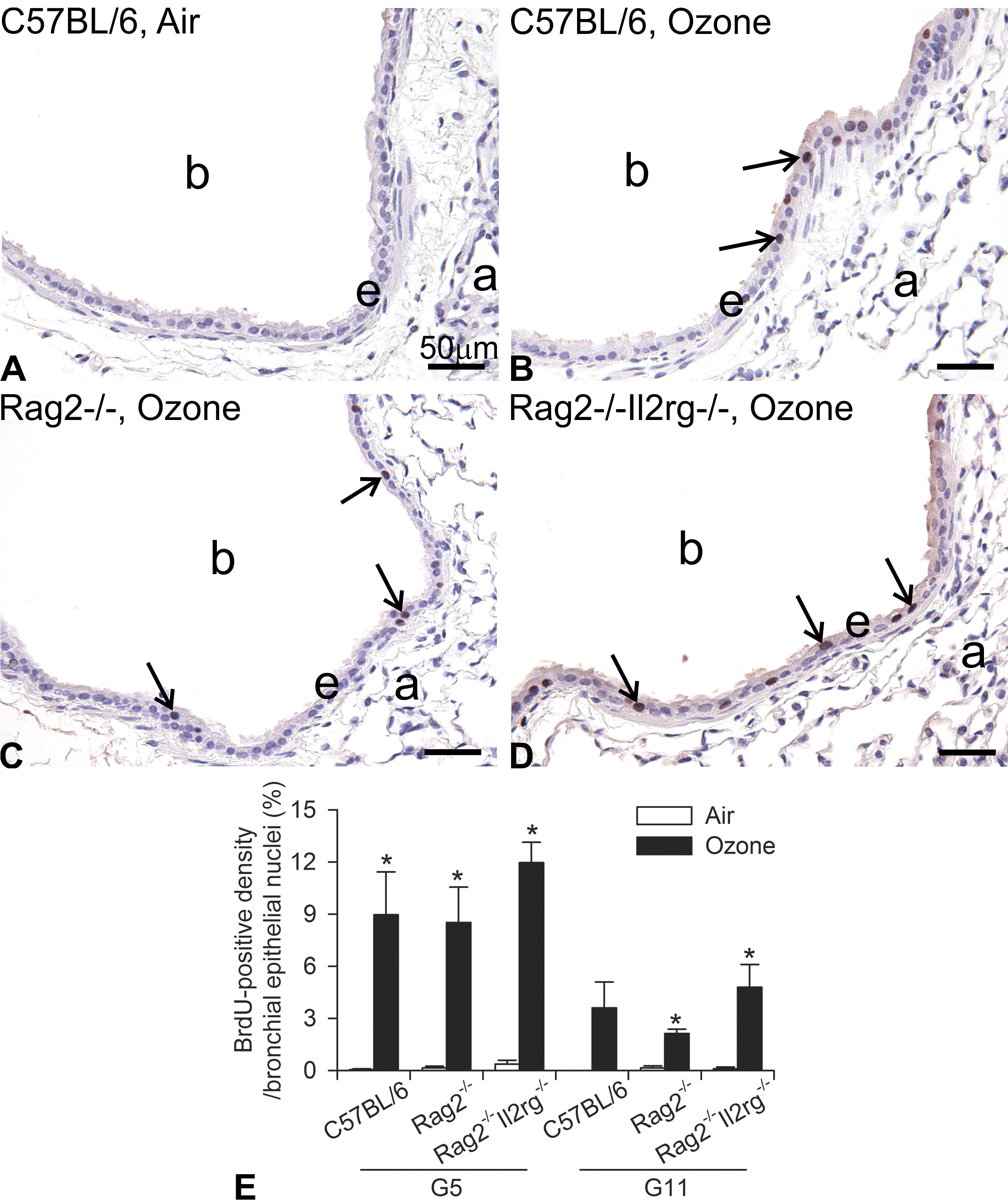
Incorporation of bromodeoxyuridine (BrdU) in the nucleus of bronchiolar epithelial cells after a single 1-day exposure to ozone (induced regenerative DNA synthesis/cell proliferation). Light photomicrographs of BrdU immunostaining in the left lung microdissected at the level of the 5th axial airway generation (G5). (A) C57BL/6 mouse exposed to filtered air. (B) C57BL/6 mouse exposed to 0.8 ppm ozone. (C) Rag2−/− mouse exposed to 0.8 ppm ozone. (D) Rag2−/−Il2rg−/− mouse exposed to 0.8 ppm ozone. a = alveolus; b = terminal bronchiole; e = airway epithelium. Scale bars = 50 µm. (E) Morphometric determinations of BrdU-positive nuclear density in bronchiolar epithelium at the 5th and 11th airway generation. Data are expressed as mean ± standard error of the mean (n = 6/group). *Significantly different from respective mice exposed to filtered air at the same region, p < 0.05.
Mucous Cell Metaplasia in ILC-sufficient Mice, but Not in ILC-deficient Mice, that Were Repeatedly Exposed to Ozone
There were little or no AB/PAS-stained mucosubstances in the epithelium lining the main axial airways (G5 and G11) in any of the air-exposed mice (Figure 3A). C57BL/6 and Rag2−/− mice exposed to ozone for 9 days had conspicuous mucous cell metaplasia in the proximal aspect of the main axial airway (G5, Figure 3B and C). Morphometric analyses demonstrated greater volume densities of mucosubstances in airway epithelium lining this proximal large-diameter bronchiole of these ozone-exposed ILC-sufficient mice as compared with their respective air-exposed mice (Figure 3E).
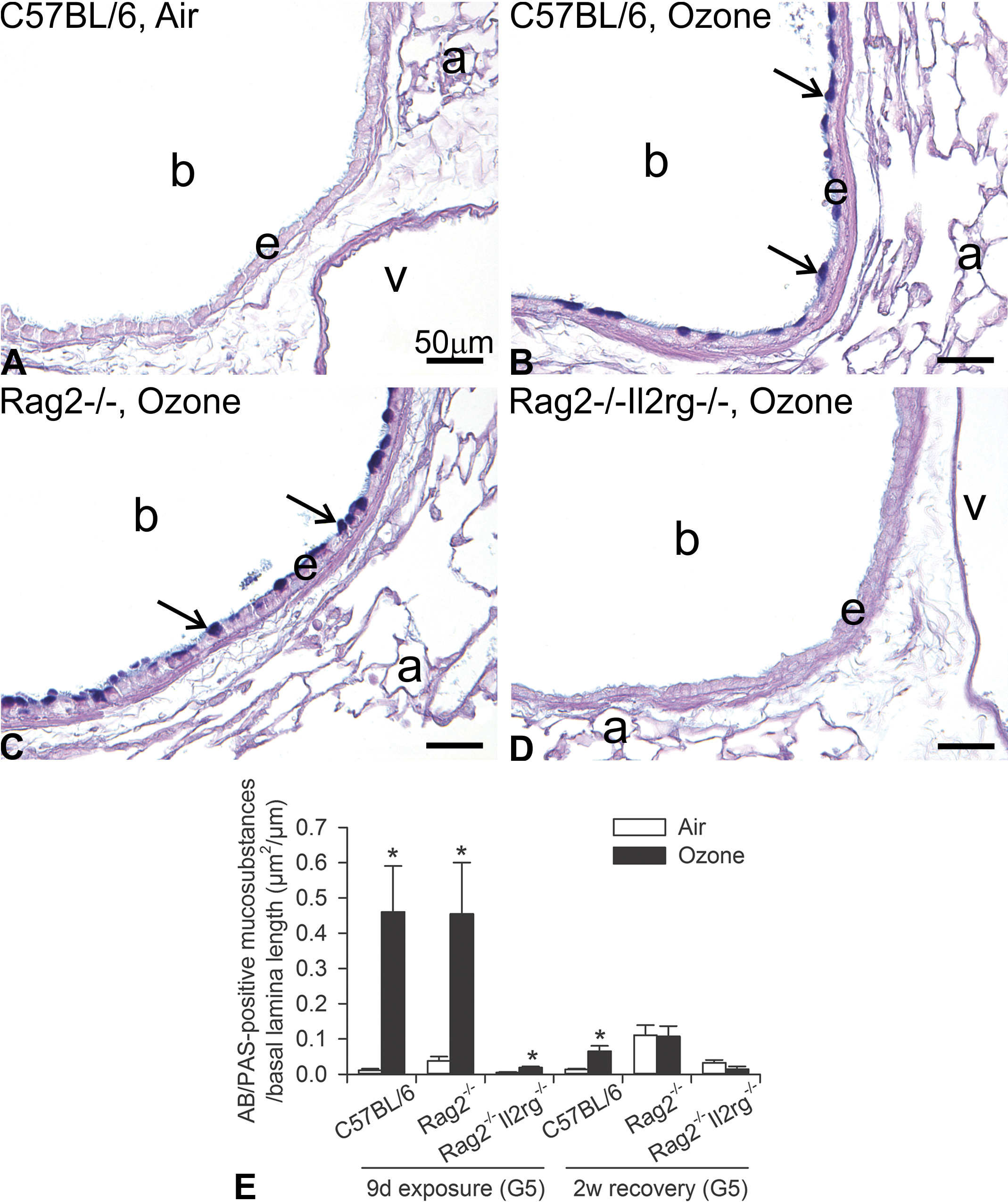
Mucous cell metaplasia in bronchiolar epithelium after repeated daily exposure to ozone for 9 days. Light photomicrographs of Alcian Blue (pH 2.5)/periodic acid-Schiff (AB/PAS)-positive mucosubstances (arrows) in the axial airway of the left lung lobe at the level of the 5th axial airway generation (G5 proximal airway). (A) C57BL/6 mouse exposed to filtered air. (B) C57BL/6 mouse exposed to 0.8 ppm ozone. (C) Rag2−/− mouse exposed to 0.8 ppm ozone. (D) Rag2−/−Il2rg−/− mouse exposed to 0.8 ppm ozone. a = alveolus; b = terminal bronchiole; e = airway epithelium; v = blood vessel. Scale bars = 50 µm. (E) Morphometric analysis for AB/PAS-positive mucosubstances in bronchiolar epithelium at the 5th and 11th airway generation (G5 proximal airway and G11 distal airway). Data are expressed as mean ± standard error of the mean (n = 6/group). *Significantly different from respective mice exposed to filtered air at the same airway generation, p < 0.05.
In contrast, 9-day ozone-exposed, ILC-deficient, Rag2−/−Il2rg−/− mice had minimal mucous cell metaplasia with approximately 24 times less AB/PAS-stained mucosubstances than that in the ozone-exposed ILC-sufficient C57BL/6 and Rag2−/− mice. Mucous cell metaplasia in the proximal main axial airway of ozone-exposed C57BL/6 and Rag2−/− mice was markedly attenuated after 2 weeks postexposure in filtered air.
Increased Expression of Genes Involved in Type 2 Immune Responses in ILC-sufficient, but Not ILC-deficient, Mice after Repeated Ozone Exposure
Among the 40 selected transcripts evaluated in the lung tissues by quantitative RT-PCR, statistically significant changes (p < .05) related to ozone exposure (>2-fold increase) versus the corresponding air control group were detected in the Il13, Muc5ac, Muc5b, Gob5 (Clca1), and Ym2 (Chil4) mRNA of the ILC-sufficient C57BL/6 and Rag2−/− mice exposed to ozone for 9 days (Online Tables S1 and S2, Figure 4). In addition, 9-day ozone exposure induced significant increases in Fizz1 (Retnla) and MCP-2 (Ccl8) mRNA in the lungs of all 3 strains of mice. After the 2-week postexposure period in filtered air, there were no significant exposure-related differences in mRNA expression of these selected transcripts among all strains of mice (data not shown).
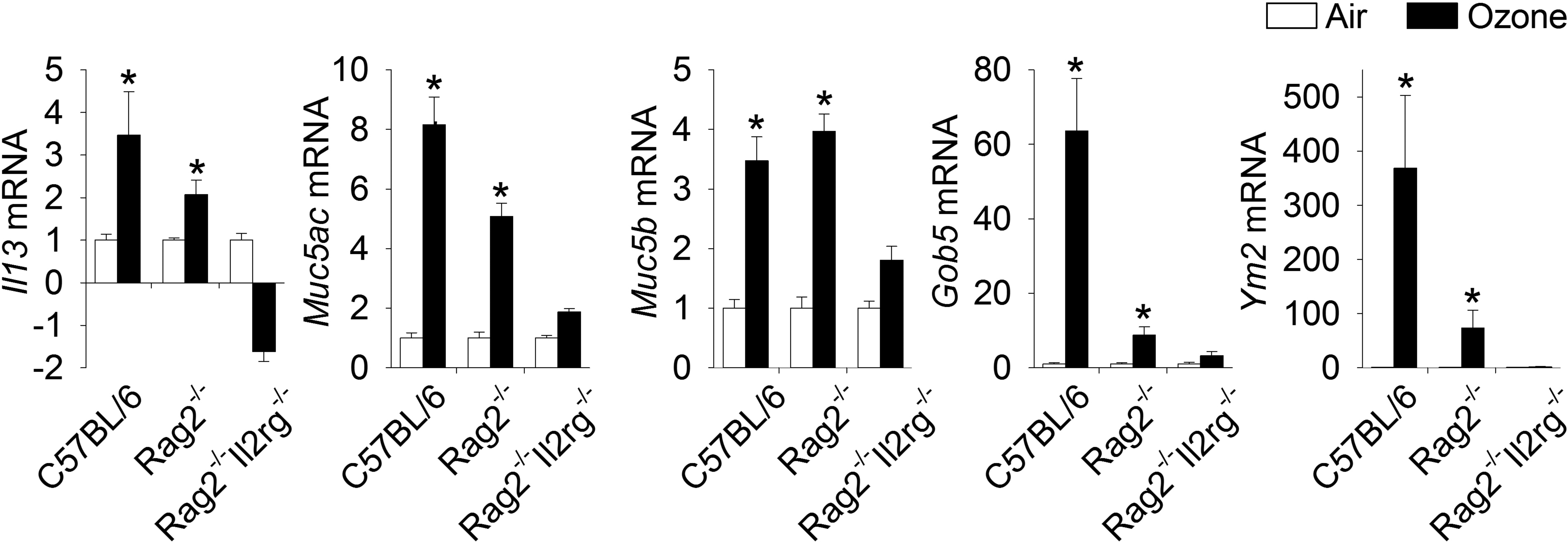
Gene expression in 9-day ozone-exposed lungs. Relative fold increases in mRNA transcripts were analyzed for Muc5ac and Muc5b on SmartChip Real-Time PCR System and Gob5, Il13, and Ym2 on ABI PRISM 7900HT Sequence Detection System. *Genes showing statistical significances (air vs. ozone/same mouse strain, p values < 0.05). Data are expressed as fold changes relative to their respective air-exposed controls ± standard error of the mean (n = 6/group).
Flow Cytometric Identification of Lung ILC2s in Rag2−/− but Not in Rag2−/−Il2rg−/− Mice after Repeated Exposed to Ozone
We determined the presence of ILC2s in the lungs of Rag2−/− and Rag2−/−IL2rg−/− mice by using flow cytometry. A small number of ILCs were present in ozone-exposed Rag2−/− but not in similarly exposed Rag2−/−IL2−/− mice. Air- and ozone-exposed Rag2−/−mice had statistically similar numbers of lung ILC2s (Figure 5). This later finding, however, was not definitive because of the small number of animals analyzed and the individual variation in the number of these ILCs (n = 3/group).
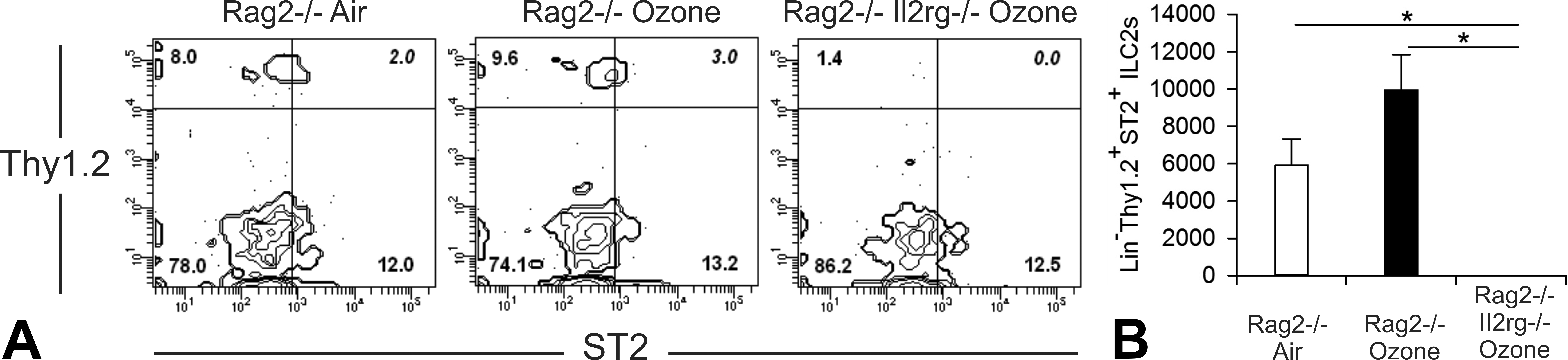
Flow cytometric analysis of innate lymphoid cells (ILCs) in air-exposed or ozone-exposed ILC-sufficient Rag2−/− mice and ILC-deficient Rag2−/−Il2rg−/− mouse. Exposure to ozone (0.8 ppm) for 9 days did not have a statistically significant impact on the number of ILC2s in the lungs of mice. (A) Representative contour plots for Thy1.2 and ST2 expression gated on the live, lineage-negative cell population. Thy1.2 and ST2 double-positive cells are identified as ILC2s, with the percentage shown in the upper-right quadrant. (B) Number of ILC2s per mouse lung. Numbers represent means ± standard error of the means (n = 3). *p < 0.05 compared to the Rag2−/−Il2rg−/− ozone group.
Depletion of Lung ILC2s and Attenuation of Ozone-induced Mucous Cell Metaplasia and Eosinophilic Inflammation in Anti-Thy1.2 Antibody-treated Rag2−/− Mice
Anti-Thy1.2 antibody treatment successfully depleted ILC2s in the lungs of ozone-exposed Rag2−/− mice (Figure 6). Furthermore, these treated mice had significantly less eosinophils in BALF (2.9×) and AB/PAS-positive mucosubstances in bronchiolar epithelium (1.8×) as compared with saline-treated control Rag2−/− mice that were similarly exposed to ozone for 9 days (Figure 7).
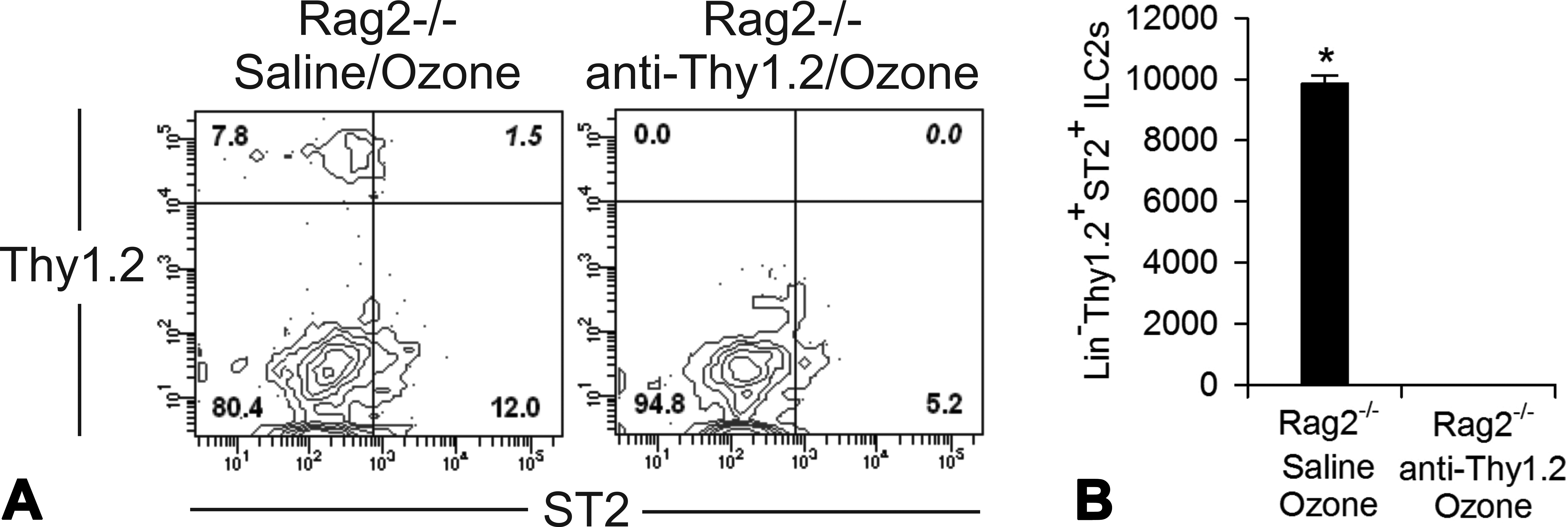
Anti-Thy1.2 antibody (Ab)-induced depletion of innate lymphoid cells (ILCs) in ozone-exposed Rag2−/− mice. Intraperitoneal injection of anti-Thy1.2 antibody depleted ILC2s in the lungs of Rag2−/− mice. Mice were injected with saline or antibody prior to and during the 9-day ozone exposure (0.8 ppm). (A) Representative contour plots for Thy1.2 and ST2 expression gated on the live, lineage-negative cell population. Thy1.2 and ST2 double-positive cells are identified as ILC2s, with the percentage shown in the upper-right quadrant. (B) Number of ILC2s per mouse lung. Numbers represent means ± standard error of the means (n = 3). *p < 0.05 compared to the anti-Thy1.2 Ab injection group.
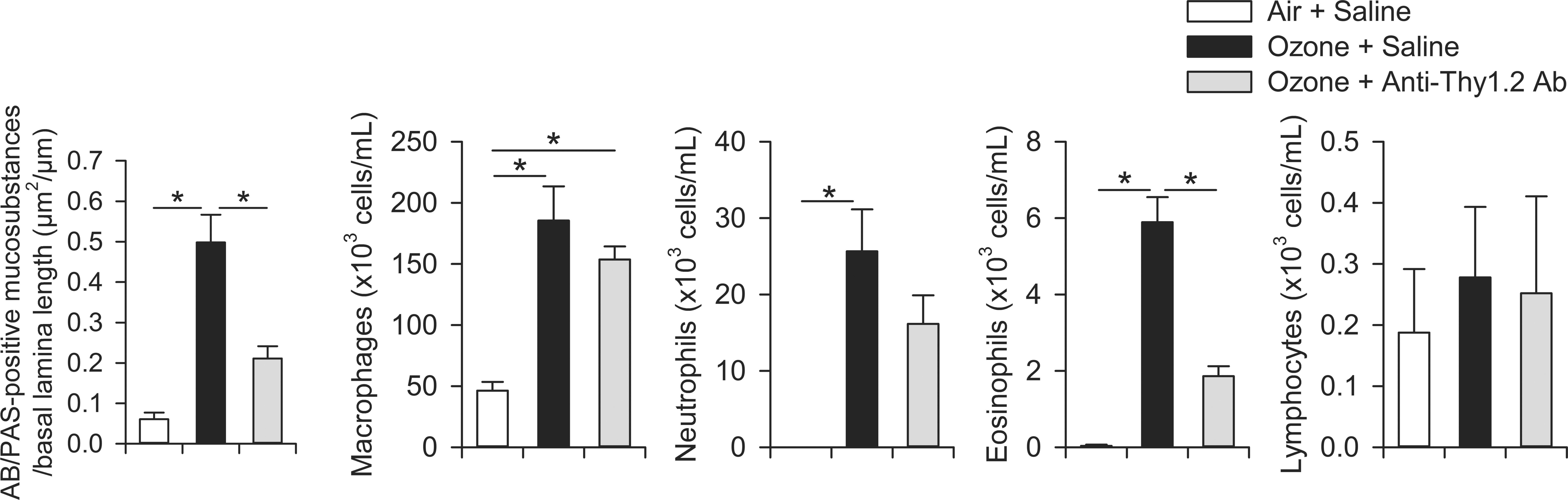
Effects of anti-Thy1.2 antibody (Ab) treatment on airway mucosubstances and inflammatory cells in bronchoalveolar lavage fluid (BALF) of Rag2−/− mice. Anti-Thy1.2 antibody treatment resulted in a loss of type 2 immune responses to 9-day ozone exposure, confirming that ILC2s mediate these effects. Ozone-induced increase in Alcian Blue (pH 2.5)/Periodic Acid-Schiff–positive mucosubstances (mucous cell metaplasia) and BALF eosinophils were lost with antibody treatment. Bars represent means ± standard error of the means (n = 3). *Significantly different group means, p < 0.05.
Discussion
In this animal toxicology study, we found that repeated exposures to ozone in ILC-sufficient C57BL/6 and Rag2−/− mice, but not ILC-deficient Rag2−/−Il2rg−/− mice, induced pulmonary eosinophilic inflammation, mucous cell metaplasia in proximal airway epithelium, and increased expression of lung mRNA transcripts associated with type 2 immunity. In addition, pharmacological depletion of lung ILCs in Rag2−/− mice with anti-Thy1.2 antibody treatment resulted in a dramatic loss of ozone-induced histopathologic hallmarks of pulmonary type 2 inflammation and asthma-like disease (i.e., influx of eosinophils and mucous cells metaplasia). Together, the results of this study strongly suggest that ILCs (most likely ILC2s), and not T or B cells, play a major role as mediators of ozone-induced type 2 immunity in the murine lung.
In many respects, our findings in the lung closely mirrored those previously described in the nasal airways of ILC-sufficient and ILC-deficient mice exposed to 0 or 0.8 ppm ozone for 1 day or 9 consecutive weekdays (Kumagai et al. 2016), with only a few exceptions. In both the lung and the nose of ILC-sufficient mice, but not ILC-deficient mice, repeated ozone-exposure mounted type 2 immunity, eosinophilic inflammation (rhinitis and pneumonitis), mucous cell metaplasia of airway epithelium, and increased mRNA expression of type 2 immune-related transcripts (relative to respective air-exposed controls) that included transcripts for Il13, Muc5ac, Muc5b, Gob5 (Clca1), and Ym2 (Chi3/l4).
Pulmonary responses to the repeated ozone exposures in the ILC2 sufficient mice (C57BL/6 and Rag2−/−), though similar in character, appeared to be of a lesser magnitude than those in the nasal mucosa as we previously reported (Kumagai et al. 2016). As compared to our findings in the lung, the nasal mucosa from ILC-sufficient mice had an overall greater ozone-induced type 2 immune response including a stronger influx of eosinophils and a greater number of type-2 signature transcripts exhibiting ozone-induced increased expression (e.g., Il4, Il5, eotaxin/Ccl11, MCP-2, and Arg 1 in nasal tissue but not in lung tissue). The reasons for these regional airway differences in response to ozone exposure are not known, but may be due, in part, to differences in ozone dosimetry in the respiratory tract, in cell/tissue sensitivity to ozone toxicity, and/or differences in the time course of nasal and pulmonary cellular and cytokine responses. Nevertheless, the results of our study do confirm our primary hypothesis that the type 2 immunity/inflammation caused by repeated ozone exposure in the lung is dependent on ILCs, and not T or B cells, like that previously observed in the mouse nasal mucosa (Kumagai et al. 2016).
In contrast to the differences in airway epithelial response observed in the ozone-exposed lungs of ILC-sufficient and ILC-deficient mice after 9-day exposures to ozone, no differences in airway epithelial response after a single, 4 hr, exposure to ozone were observed among the ILC-sufficient and ILC-deficient groups of mice. In all of the single day ozone-exposed mice, there was a clear increase in the number of BrdU-labeled epithelial cells in both the proximal and distal axial airways (G5 and G11) at 1-day postexposure. This increase in epithelial DNA synthesis was similar to what we had previously observed in the nasal epithelium of mice (Kumagai et al. 2016) and in the nasal and bronchiolar epithelium of rats (Johnson et al. 1990; Hotchkiss, Harkema, and Johnson 1997; Vesely et al. 1999), when exposed to only a single acute ozone exposure. This finding demonstrates that the acute ozone-induced epithelial cell injury/death and the subsequent rapid reparative increase in DNA synthesis/cell proliferation in both the nasal and bronchiolar airways are independent of ILCs, unlike the ILC-dependency of the type 2 immune responses in the nose and lung after repeated 9 days of ozone exposure.
The mucous cell metaplasia observed in the surface epithelium lining the proximal axial airway (G5) in the ILC-sufficient mice after repeated 9-day exposure was also similar in character and magnitude to the ozone-induced metaplastic responses that we have previously observed in the nasal (Kumagai et al. 2016; Ong et al. 2016) and pulmonary airways (Harkema et al. 2017) of similarly exposed C57BL/6 mice. Mucous cell metaplasia is a commonly reported nasal epithelial response to repeated daily ozone exposures in rats (Cho, Hotchkiss, and Harkema 1999; Harkema et al. 1999) as well as in nonhuman primates (Harkema, Plopper, Hyde, St George, et al. 1987; Harkema, Plopper, Hyde, St George, Wilson, et al. 1987). This airway epithelial change after repeated exposures increases in magnitude with increased ozone concentrations and exposure duration (Harkema, Plopper, Hyde, St George, Wilson, et al. 1987; Harkema, Hotchkiss, and Griffith, 1997; Harkema et al. 1999). Ozone-induced mucous cell metaplasia in rats has also been shown to correlate well with marked reductions in flow rate of nasal mucus, suggesting a dysfunction in mucociliary clearance, a major defense mechanism of the upper airways in the respiratory tract (Harkema and Mauderly 1994).
Mucous cell metaplasia is also associated with mucus hypersecretion in the airway lumen that may lead to airflow obstruction, reduced pulmonary function, heightened risk of microbial infections, and increased mortality in patients suffering from obstructive lung diseases like asthma. In our current study, ozone-induced mucous cell metaplasia in ILC-sufficient mice was restricted to the proximal main axial airway (G5; large-diameter bronchiole). It was not found in epithelium lining the distal axial airway (G11; small diameter bronchiole) of these animals. Others have previously demonstrated that airway Club cells (formerly known as Clara cells) in proximal bronchioles transform into mucous cells after sensitization and challenge to aeroallergens-like ovalbumin (OVA). Both Reader et al. (2003) and Evans et al. (2004) have demonstrated that Club cells in proximal airway generations of the murine lung are more prone to differentiate into mucous cells than those in the more distal airways of mice exposed to OVA. Likewise, in the present study, we found that ozone-induced mucous cell metaplasia was limited to proximal and not distal bronchioles. This differential response in airway epithelium may have been due to regional airway differences in ozone dosimetry and/or disparities in epithelial cell sensitivity to this oxidant gas.
We have recently reported that the magnitude of ozone-induced mucous cell metaplasia in the proximal pulmonary airway is strain dependant (Harkema et al. 2017). In this previous study, we found that C57BL/6 male mice exhibit greater metaplasia than BALB/c male mice, even though both strains of mice were of similar age and exposed to the same exposure regimen (0.8 ppm ozone, 4 hr/day, for 9 consecutive weekdays, as in the present study). Lungs of ozone-exposed C57BL/6 mice, as compared to the lungs of BALB/c mice, also had greater eosinophilic inflammation and greater expression of genes related to airway mucus production/secretion (e.g., Clca, Muc5ac, and IL-13) and type 2 immunity/inflammation (IL-13, Chil4/Ym2).
Although our data from the ILC-sufficient C57BL/6 and Rag2− /−and ILC-deficient Rag2−/−Il2rg−/− mice exposed to ozone for 9 days clearly suggested that ozone-induced type 2 immune, inflammatory, and epithelial responses in the lung are dependent on ILCs, we further investigated the role ILCs in Rag2−/− mice. We intranasally instilled anti-Thy1.2 antibody in the airways of ozone-exposed Rag2−/− mice and found that these animals did not have the increases in BALF eosinophils or epithelial mucosubstances (mucous cell metaplasia) like that in the nontreated ozone-exposed Rag2−/− mice. These results complemented our results in the ozone-exposed ILC-deficient Rag2−/−Il2rg−/− mice and add support to our conclusion that ILCs (and most likely ILC2s), and not T or B cells, mediate ozone-induced type 2 immunity, inflammation, and epithelial mucous cell metaplasia.
We cannot, however, totally rule out the possibility that other ILCs, such as ILC1s or ILC3s, or other not yet identified immune cells may be playing a role, since the anti-Thy1.2 antibody could have also altered the number (or function) of these cells in the Rag2−/− mice. Therefore, further in vivo studies such as the reconstitution of ILC2s in immunodeficient mice as has been used by other investigators in single acute ozone exposure studies (Yang et al. 2016; Mathews et al. 2017) could be conducted in the future to better define the role of ILC2s in our model. Still this technique also has its limitations. Since only small numbers of ILC2s can be isolated from the mouse lung (Kim, Hashimoto-Hill, and Kim 2016), it is necessary to expand these cells in vitro with type 2 cytokines in order to have enough ILC2s for adoptive transfer. This in vitro treatment may affect ILC2s function before exposure to ozone, making the interpretation of results difficult. The development of a compound, or other technique, that specifically targets and removes ILC2s from the lung is needed to better understand the role of ILC2s in murine models of asthma and other chronic respiratory diseases dependent on type 2 immunity.
Others have also found a role of ILC2s in ozone-induced toxicity of mice, but after single acute, rather than multiple repeated, exposures and at much higher concentrations of ozone than we used in the present study. Yang et al. (2016) have reported that ILC2s play an important role in ozone-induced AHR, airway inflammation, and gene expression of type 2 cytokines in the lungs of BALB/c mice after an acute 2 hr exposure to 3 ppm ozone. Mathews et al. (2017) have more recently found that IL-13-positive ILC2s play an important role, along with IL-13-positive γδ T cells, in the IL-33-driven augmentation of ozone-induced acute responses (e.g., AHR, neutrophilic inflammation) in obese mice. The increased gene expression of IL-13 and AREG in the lungs of ILC-sufficient, but not in ILC-insufficient, mice in our present study suggests that these animals exhibiting mucous cell metaplasia in pulmonary airway epithelium may have also had AHR, although we did not measure this functional end point. Carefully designed airway function studies in mice using aerosol challenges to increasing doses of a bronchoconstrictor (e.g., methacholine) are needed to determine whether ILC2-dependent AHR is a feature of mice repeatedly exposed to ozone like in mice that received a single acute exposure (Yang et al. 2016; Mathews et al. 2017).
Taken together with our findings, there is growing evidence from animal toxicology studies that the health effects of this commonly encountered gaseous air pollutant is mediated, at least in part, by ILC2s. These findings have yet to be substantiated in humans exposed to elevated ambient concentrations of ozone. In addition, the role of ILCs in the toxicity of other air pollutants, like particulate matter, is yet to be explored and elucidated.
As we have observed in the nasal mucosa of ILC-sufficient mice, repeated ozone exposure also caused a profound increase in the relative gene expression of Ym2/Chi3l3 in the ILC-sufficient murine lung. Ym2 is a chitinase-like protein that is highly homologous to another chitinase-like protein Ym1 (Ym2/Chi3l4), both of which are specific to rodents (Lee et al. 2011). Although the biological roles of Ym1 and Ym2 have not been fully elucidated, results from several studies have suggested that these proteins may play important roles in the development of type 2 immune-inflammatory responses in allergic airway diseases of mice (Webb, McKenzie, and Foster 2001; Cai et al. 2009; Lee et al. 2011). In our study, relative fold increases in the Ym2 mRNA transcript, but not in the Ym1, transcript was dramatically increased in C57BL/6 and Rag2−/− mice, but not in Rag2−/−Il2rg−/− mice, after a repeated 9-day exposure to ozone. We have previously reported that ozone causes immunohistochemically detectable increases in Ym1/Ym2 proteins in the nasal epithelium (as well as increased relative expression of Ym2 mRNA) in the nasal mucosa of ILC-sufficient, but not ILC-deficient, mice (Kumagai et al. 2016; Ong et al. 2016). Therefore, Ym2 may play an important role in type 2 immune responses in ILC-sufficient mice after repeated exposure to ozone. Immunohistochemical identification of the precise cellular location (e.g., macrophages, airway epithelium) of Ym1/Ym2 proteins in the lungs of ILC-sufficient and ILC-deficient mice exposed to ozone are yet to be done.
Activation of ILC2s and other type 2 immune cells that drive type 2 immunity in chronic respiratory diseases, like asthma, are triggered by epithelial cytokines (alarmins), such as IL-33, IL-25, and TSLP, that are released upon cell injury or death caused by environmental pathogens (e.g., viruses), allergens (e.g., house dust mite), or toxicants (e.g., cigarette smoke, air pollutants; Holtzman et al. 2014). Increased relative fold expression of IL-33 mRNA, but not IL-25 or TSLP, was present in the lung tissues of all our mice repeatedly exposed to ozone, irrespective of ILC sufficiency or deficiency. This suggests that IL-33, an IL-1 family cytokine that signals through a receptor complex composed of ST2 and IL-1RAcP, may be an important initiator of the ILC2-dependant type 2 immunity in the murine lung after repeated ozone exposure, as others have demonstrated after a single acute exposure (Mathews et al. 2017). We suspect that ozone activates lung ILC2s, either through alveolar or airway epithelial cell-derived IL-33, to produce IL-13 and other mediators of type 2 immune responses like IL-5, Ym1/2 proteins and AREG. With the design limitations of the present study, further studies are needed to elucidate the role of IL-33 in this mouse model of ozone-induced nonatopic asthma.
In conclusion, the results of this study demonstrate for the first time that murine ILCs, but not T or B cells, play a crucial rule in ozone-induced mucous cell metaplasia, eosinophilic inflammation, and type 2 immunity in the lungs of mice. This suggests a new and plausible paradigm for the early onset and pathogenesis of nonatopic asthma epidemiologically associated with children exposed to elevated ambient concentrations of ozone.
Footnotes
Acknowledgments
The authors thank Ms. Amy Porter and Ms. Kathy Joseph of the Michigan State University Histopathology Laboratory for their preparation of the histology slides.
Author Contribution
All authors (KK, RL, DJ, NB, KW, SV, JW, JH) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported with funds from the US Environmental Protection Agency’s Clean Air Research Center grant no. RD 83479701.
Supplementary Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.