
Abstract
Progress in understanding the molecular bases of human health and disease in recent decades has flourished making it possible for the field of gene therapy (GT) to offer new possibilities for treating, and even curing, a plethora of medical conditions such as monogenic disorders and metabolic diseases. GT is a therapeutic intervention to genetically alter or modify living cells by means of gene delivery achieved using either viral vectors or nonviral vectors, with adeno-associated virus (AAV) vectors constituting market-share majority. Although GT is conceptually attractive, adverse and even fatal iatrogenic complications have marred the initial enthusiasm of clinical successes. The properties of investigational AAV-based GT may pose safety concerns unique from those of small molecule drugs and other macromolecular biologics, such as ectopic or unregulated expression of the transgene, long-term persistence, and off-target distribution. Herein, we discuss considerations in the design of a comprehensive preclinical safety program for AAV-based GT prior to administration in humans.
Gene therapy (GT) is a therapeutic intervention to genetically alter or modify living cells using either viral or nonviral vectors to replace a missing or defective gene in order to correct a disease of genetic origin or a chronic pathologic process (e.g. heart failure). The delivered DNA may be inserted into the host genome (e.g., retroviral vectors) or remain episomal (e.g., adeno-associated virus [AAV] vectors). The delivery of the gene to a host cell may take place ex vivo and the altered cell delivered to the patient, or by a vector containing the new genetic material injected directly into the patient and the host cell is altered in vivo. Some of the viruses that have been modified to serve as vectors include retrovirus, AAV, adenovirus (Ad), and herpesvirus. Nonviral vectors include liposomes or other types of nanoparticles (Keeler, ElMallah, and Flotte 2017).
AAV is a single-stranded DNA parvovirus discovered as “viruslike” particles in an Ad preparation in 1965 (Hastie and Samulski 2015). AAV is replication defective by nature and incapable of productive replication without the help of Ad or herpesviruses (Carter 2004). AAV is not associated with causing disease in human and animals. The absence of pathogenicity and benign persistence of AAVs within the host cell established the foundation for using AAV as a GT vector.
AAV vectors are made up of two components: a protein capsid and an internal DNA expression construct. The protein capsid is made up of three proteins (VP-1, 2, and 3) and is responsible for the tropism of the vector for different cell types (Carter 2004). There are numerous naturally occurring AAV vector serotypes with different capsid peptide sequences typically designated with a number (e.g., AAV-1, 2). The DNA in AAV vectors has been modified from wild-type AAV by removing the genes responsible for replication (Rep) and capsid (Cap) formation and replacing them with a promoter that drives the expression of a gene of interest. The only components retained from the wild-type virus are the inverted terminal repeats (ITRs) found at each end of the DNA construct, which are important for packaging of the DNA into the capsid during vector production and genome replication. In addition to the vector capsid and ITRs, key components of the DNA expression construct for AAV vectors are the promoter, the transgene coding the messenger RNA (mRNA) of interest, and the polyA termination sequence (Hastie and Samulski 2015). The AAV genome is 4.7 kb, which poses a key limitation in the size of the transgene that can be packaged in the vector. Another form of AAV vector known as self-complementary AAV (scAAV) does not require second strand DNA synthesis and hence is capable of more efficient and earlier gene expression. However, the packaging capacity of scAAV is reduced in half.
Although AAV does not cause disease in the natural setting, their use as GT vectors may carry risks associated with ectopic or unregulated expression of the transgene, immunogenicity including inflammatory host tissue responses, long-term persistence, and off-target distribution.
Regulatory Framework for AAV-based GT
There are several key regulatory guidance documents released by U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) that discuss the considerations expected in the development of GT products including those related to the preclinical package, the design of clinical trials, and the manufacturing of GT products. These guidance documents are continuously evolving, and the FDA has recently committed to accelerating the approval process for GT products and consequently released six additional guidance documents, some of which are tailored toward specific disease entities that have demonstrated promising results with GT.
While GT products carry features unique to this biological therapeutic modality, the general regulatory framework has elements that still overlap with other large molecule biologics, such as therapeutic monoclonal antibodies. Preclinical study design needs to mimic the proposed clinical trial design in the target population and the route of administration (ROA) as well as build an understanding of the persistence of the vector DNA and transgene expression in both the target tissue and off-target vector biodistribution. As with other biologics, the immune response to the vector capsid and transgene protein product is another key criterion for consideration while taking into account the caveats in translatability of that immune response into the clinical target population. Other elements that are considered, but with less emphasis, in AAV-based GT are potential consequences of insertional mutagenesis (i.e., the vector genome is unintentionally integrated into host cell DNA), germline transmission, and environmental shedding.
The preclinical safety and efficacy programs for GT are generally case-by-case with regulatory agencies encouraging early interaction and innovative risk-based approaches. The FDA has recently created a new process that replaces the pre-pre-investigational new drug (pre-preIND) interaction called Initial Targeted Engagement for Regulatory Advice on CBER products Meetings (INTERACT) that is intended to facilitate early interactions on innovative programs.
The following is a nonexhaustive list of regulatory guidance documents related to the development of GT products: EMA (http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000410.jsp&mid=WC0b01ac058002958d) Guideline on the quality, preclinical, and clinical aspects of GT products. Preclinical studies required before first clinical use of GT products. Reflection paper on quality, preclinical, and clinical issues related to the development of recombinant adeno-associated viral vectors. Guideline on preclinical testing for inadvertent germ-line transmission of gene transfer vectors. FDA (https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/) Guidance for industry: Preclinical assessment of investigational cellular and GT products. Design and analysis of shedding studies for virus- or bacteria-based GT and oncolytic products. Long-term follow-up after administration of human GT products (Draft 2018). Chemistry, manufacturing, and control information for human GT IND applications (Draft 2018).
General Considerations of Preclinical Study Design
According to the IND regulations in 21 CFR 312.23 (a)(8)—Pharmacology and Toxicology, the ultimate goal of any preclinical program is to generate “…adequate information about the pharmacological and toxicological studies…on the basis of which the sponsor has concluded that it is reasonably safe to conduct the proposed clinical investigations. The kind, duration, and scope of animal and other tests required varies with the duration and nature of the proposed clinical investigations” (Government 2017).
As with other modalities, the objective of the preclinical program for a GT is to predict the human efficacious dose range, establish a safe starting dose, and characterize a dose-related toxicity profile. Because a systemically administered GT can only be administered once, due to a neutralizing antibody response to the vector capsid, a key difference with other therapeutic modalities is that the starting clinical dose should have the potential to provide clinical benefit (Manno et al. 2006). The design of a preclinical safety package should mimic the clinical scenario by using the intended final test article construct and ROA. Because the final test article construct expresses a transgene protein product foreign to the preclinical animal species, it may induce an immunogenic response. This immune reaction is most likely unique to the animal and may not affect the intended human patient population. To appropriately study the biology and safety of the GT in the animal, consideration should be given to the possibility of using immunosuppressed animals or a surrogate homologous transgene that is autologous and nonimmunogenic to the animal. For genetic diseases that have significant progressive degenerative changes, early intervention in the disease has a higher likelihood of a successful outcome for a GT. This usually involves treating a pediatric population and consequently the preclinical program should consider the use of juvenile animals that are age-matched for the intended clinical population.
The study duration is another important consideration since a GT is typically only administered as a single administration and studies intended to characterize the toxicity for first-in-human trials may be the only studies done to support registration of the product. The study should be designed to assess acute effects of the viral vector interacting with the animal test system, changes associated with initial peak expression of the transgene, and effects associated with prolonged expression of the transgene. These studies can be utilized to assess the biodistribution of the vector within tissues, its persistence, and the onset and stability of transgene expression. The study duration, number of necropsy time points, and data end points are all key discussion points with regulatory agencies. These end points may vary based on prior experience with the vector capsid, expression construct, and the biology of the transgene. Studies may require two necropsy time points; for example, at 4 and 13 weeks to allow the assessment of the persistence of the vector DNA and transgene expression and effects associated with the onset of peak (4 weeks) and steady-state expression exposure to the transgene (13 weeks; Zincarelli et al. 2008). Early interaction of the transgene with the animal test system can typically be assessed by clinical pathology end points and in-life clinical assessments that may include safety pharmacology evaluation of the cardiovascular, nervous, and respiratory systems.
A key component of GT preclinical studies is assessing the biodistribution of the vector DNA and transgene expression (mRNA or protein) in tissues. Typically, blood, major organs, and gonads are evaluated by polymerase chain reaction (PCR), in situ hybridization (ISH), and immunohistochemistry (IHC) for the presence and persistence of vector DNA and transgene expression. Preclinical assays demonstrating shedding (or lack of shedding) of vector into biofluids (semen, urine, feces, saliva, and tears) may also be included. EMA guidance requires that preclinical studies assess shedding, while the FDA guidance does not (when using replication defective vectors such as AAV). However, assays evaluating shedding into biofluids are required by both FDA and EMA during the clinical development program. Evaluation of the relationship between toxicity findings and the presence of the vector DNA or transgene expression is important. For example, while persistence of vector DNA in gonads may raise concern for germline transmission of vector DNA, there are many cell types in the testis and ovary other than spermatocytes and ova that can be transduced by a viral vector. In that case, appropriate interrogation of the cell type harboring the vector DNA may mitigate the need for breeding studies to assess germline transmission. In addition to the biodistribution assessment, the preclinical toxicology program is otherwise similar to other modalities and includes standard clinical pathology, in-life, safety pharmacology, ophthalmology, organ weight, and tissue histopathology that are required of other modalities.
Species Selection
Comparable physiology and anatomy is a primary consideration for selection of species in the efficacy and safety assessment of any therapeutic modality. However, additional criteria may significantly influence the choice of the preclinical animal species for GT preclinical safety evaluations. Both the FDA and EMA guidance documents indicate two species are not required for preclinical toxicology programs and nonrodent species are not necessary; therefore, a single relevant rodent species may suffice. Additionally, the incorporation of safety end points into disease model studies may substitute for toxicology studies in normal animals and may be important to understanding the safety profile of the GT in the disease setting. The inclusion of safety end point(s) in a disease model study may substitute for one of the species used if a two species toxicology program is required. The choice of species and use of disease models should be scientifically justified and agreed to with the appropriate regulatory agencies.
Additional points to consider in the selection of the preclinical species include: If a delivery system will be used, is it feasible to use it in the animal? If a device is required for delivery, then the preclinical species needs to accommodate the use of the device and mimic the anatomy that the device will interact with in humans. Is the selected species susceptible to vector transduction? Translation of tissue tropism for a given vector between animals and human may be unknown. There is no stipulation as to what species should be used. However, if there are significant differences in transduction efficiency for the therapeutic target organ or off-target tissue distribution between species, then consideration should be given as to which species is most representative to human. What is the potential in animals of mounting an immune response to the human therapeutic transgene? If the therapeutic transgene product is immunogenic in an animal species and that leads to neutralization of the transgene’s pharmacologic activity or toxicity, then either immune suppression or a species-specific transgene should be considered. The transgene should be pharmacologically active in the preclinical species used for safety evaluation. If not, then an animal equivalent transgene should be used as a surrogate. What is the prevalence of neutralizing antibodies in the nonrodent species? Preexisting antibodies to AAV will prevent transduction of tissues, and as a consequence, nonrodents are typically screened for preexisting antibodies prior to being placed on study. The prevalence of preexisting neutralizing antibodies in a large animal species needs to be considered, as a high prevalence may make the selection of sufficient number of negative animals for study inclusion difficult. Prevalence of antibodies may vary between different large animal species, vendors of large animals, and between species of nonhuman primates (NHPs).
If safety is evaluated in a disease model, there should be adequate characterization of that model to enable distinguishing low incidence background lesions from test article–induced lesions. Disease models can be very specialized and conducted in academic laboratories where good laboratory practices (GLP) are not strictly followed. Nonetheless, with a robust study design and improved recordkeeping, these studies can be transformed into GLP-like studies by paying attention to prospective adequate planning, completing reports with detailed documentation of procedures, recruitment of independent study monitors, and the use of contract research organizations that perform necropsy, histopathological evaluation, and clinical pathology in GLP-compliant manner.
Species-specific differences in susceptibility to toxicity may exist and should be considered in the design of the preclinical safety assessment strategy. In a recent report addressing species-dependent differences, direct intracranial injection of AAVrh8 vectors encoding species-specific hexosaminidase in mice, cats, and sheep (disease models) didn’t result in any evidence of toxicity. However, when the same vector expressing a species-specific transgene was tested in NHPs, animals developed neurological signs associated with overexpression of the protein in the form of eosinophilic aggregates in neurons despite the lack of any evidence of humoral and cell-mediated immune responses (Golebiowski et al. 2017).
Similarly, juvenile pigs and NHPs treated with the same AAVhu68 vector demonstrated a remarkable difference in the amount of viral genome in the liver of these two species, highlighting the species-specific effect on vector transduction efficiency, that were associated with a different toxicologic response (Hinderer et al. 2018). When the same AAV-PHP.B vector was administered in mice, the vector exhibited enhanced neurotropism uniquely in strain C57BL/6J mice, but not in strain BALB/cJ mice or NHPs, highlighting mouse strain-specific as well as species-specific differences (Hordeaux et al. 2018).
DNA Integration
Unlike retroviral vectors, AAV vectors do not need to integrate into the host cell genome to produce their transgene protein product. However, AAV vectors do integrate into the host cell genome at a very low frequency (Deyle and Russell 2009). While considered to be a nonintegrating vector, data emerged that AAV can integrate and result in possible genotoxic effects leading to neoplastic transformation in certain animal models that are prone to tumor development (Donsante et al. 2007; Chandler et al. 2015). Nonetheless, the oncogenic potential of recombinant AAV vectors is still controversial, and the translatability of current mouse data to nonrodent animal species and humans needs to be established (Gil-Farina et al. 2016). The need to assess DNA integration may vary between regulatory agencies in different geographies and in the same geography (Aiuti et al. 2013). Consequently, this topic should be a discussion point with regulatory agencies, and considerations should be given to collecting appropriate samples from toxicology studies so that DNA integration can be assessed, at the request of a regulatory authority, as a program develops over time or if an observation of concern arises in a preclinical study. The proactive collection of tissues will avoid the need to repeat studies to address this issue, thus minimizing animal use. The approval of Glybera, the first EMA-approved AAV-based GT product, involved extensive assessment of AAV integration and demonstrated that very low frequency of integration occurs but with no association with proliferative or preneoplastic changes (Kaeppel et al. 2013).
Reproductive Toxicology
Other than assessing the clearance of AAV vectors from semen in programs supporting clinical trials in hemophilia, there has been little experience with reproductive toxicology (Manno et al. 2006; Arruda et al. 2001; Schuettrumpf et al. 2006). However, assessment of potential reproductive toxicology may be warranted as more AAV-based therapeutics target women of childbearing age. Additionally, embryo-fetal development studies may be required in the future to investigate the ability of AAV to pass the placental barrier, stage-dependent susceptibility of placental transfer, and potential serotype differences in mediating such effects (Karda et al. 2014).
Preclinical Assessment of Immune Responses
AAV GT vectors are derived from naturally occurring viruses that are widespread in humans and animals. As discussed above, this requires prescreening animals to be used in studies and selecting animals that are seronegative for neutralizing antibodies that will prevent tissue transduction (Masat, Pavani, and Mingozzi 2013).
Following vector administration, a humoral immune response will develop to the AAV vector capsid protein. The resulting antibodies will prevent readministration of the vector, but they have not been associated with toxicity. Humoral immune responses can also occur in response to the transgene protein that can neutralize the pharmacology of the transgene or lead to antigen-antibody-mediated adverse effects (Masat, Pavani, and Mingozzi 2013). Cell-mediated immune responses can occur to both the transgene and capsid proteins that can lead to T cell–mediated destruction of transduced cells (Mingozzi and High 2017). This T-cell-mediated destruction may only be an efficacy concern as it results in elimination of transduced cells expressing the intended transgene protein product. However, adverse toxicities may develop if the target cells are within a vital organ that has minimal reserve or regenerative capacity. Similar to other biologic modalities, preclinical studies do not consistently predict the type or magnitude of the immune response in humans. Of note, the T-cell immune response to AAV vector capsid observed in certain individuals in hemophilia clinical trials was not observed in the preclinical safety studies in rodents or NHP (Mingozzi and High 2013). There are complex, poorly understood interactions between the vector DNA, transgene (mRNA and protein), and the vector capsid as well as host genetics (e.g., human leukocyte antigen [HLA] haplotype) that influence the development of the immune response (Mingozzi and High 2017).
Impact of Preexisting Anti-AAV Antibodies
Preexisting antibodies, particularly neutralizing antibodies, may play a significant role in the success or failure of an AAV-based GT product (Masat, Pavani, and Mingozzi 2013). Not only may it affect the safety profile of the test article, but it significantly impacts the efficacy of the test article, the availability of appropriate preclinical animal species, and the commercial feasibility of the entire program. High titers of preexisting neutralizing antibodies to vector capsid proteins neutralize the vector and preclude efficient transduction. Additionally and similar to other protein-based therapeutics, neutralizing antibodies to the transgene protein product may limit adequate and accurate assessment of the pharmacology of that transgene by neutralizing its function in vivo.
Similar to humans, large animals (particularly NHPs and to a lesser extent dogs) have widespread exposure to AAV and hence may have preexisting antibodies that cross-react with AAV serotypes used for therapeutic development (Colella, Ronzitti, and Mingozzi 2018). Accordingly, screening animals for lack of neutralizing antibodies is required for both species selection and animal selection within the species. Similarly, in a clinical setting, high prevalence of seropositive individuals for a given AAV serotype within the intended target population may limit the economic viability of such a vector. While certain therapeutic AAV vectors might prove to be safe and efficacious in the preclinical animal species lacking preexisting antibodies, developing such therapeutic vectors becomes uneconomical if a high prevalence of preexisting antibodies excludes a large percentage of the target patient population.
Neutralizing antibodies assays are most widely used to assess antibody neutralizing activity in vitro by defining the serum dilution required to inhibit 50% or less of in vitro cell transduction (Falese et al. 2017; Meliani et al. 2015). Regardless of the assay ultimately used, establishing the biological relevance of the in vitro assay measurement relative to the impact of such inhibition on vector transduction ability in vivo is vital to the utility of such measurement and the success of the entire program. Typically, animals and patients that have titers greater than 1:5 of neutralizing antibodies will have marked inhibition of tissue transduction and are typically excluded from studies.
The impact of binding, nonneutralizing antibodies is evolving and poorly understood, particularly in animals and patients that are neutralizing negative but positive for binding antibodies. Emerging data suggest that nonneutralizing binding antibodies may enhance the uptake of vector by hepatocytes (Fitzpatrick et al. 2018).
GT Product Characteristics
A key consideration in the preclinical program design is the development of assays to assess the titer and potency of test material and show comparability between lots of material produced. Assays should be in place to document the amount of test material administered to animals in safety and efficacy studies. The comparability of these assays to those used to determine the dose given to humans needs to be established.
Assessment of potency may be completed by either in vivo or cell-based in vitro assays. PCR, which quantitates vector genome components, improves the ability to properly characterize the active and inactive products, but it has limitations. The application of alternative or multiple orthogonal assays should be considered to assess potency such as delineating particle numbers, UV 260/280 protein to nucleic acid ratio, and assessment of empty versus full capsids.
A variety of process-related impurities needs to be characterized and quantified. Similar to other modalities, test material used in preclinical studies should have either equivalent or higher concentration of impurities than those going into the clinical studies. Examples of types of process-related impurities that should be characterized include surfactants, nucleases, antibiotics, purification reagents, residual plasmid DNA, and host cell DNA and proteins.
There are 4 different production methods for AAV detailed by Penaud-Budloo et al. (2018). If the production method changes during the program, understanding the difference in these production methods as far as quality of the vector, the different potential contaminants or process-related impurities, and the ratio of infective to noninfective particles is needed to establish product comparability. Bridging safety and efficacy studies may need to be conducted if the production process is changed.
Dose Selection
Dose extrapolation and designing the preclinical studies to define a dose escalation scheme that is safe and biologically plausible are key considerations. An ethical consideration unique to gene and cell therapy products dictates that the starting dose in clinical studies needs to have the potential to provide therapeutic benefit as subtherapeutic dose levels will result in development of neutralizing antibodies that will preclude future redosing. Therefore, the initial clinical dose will have some potential benefit to the patient, and then dose escalation schema will be designed based on maximum efficacious dose and the safety profile characterized in preclinical studies.
Dose selection in preclinical toxicology will typically bracket the minimally efficacious and maximum efficacious doses identified in preclinical efficacy models. The high dose in the toxicology study will be a multiple of the anticipated maximum efficacious dose. Typically, a 10× multiple is desired or a maximum feasible dose if 10× is not achievable (EMA guidance). The maximum feasible dose will be defined by the concentration at which the vector can be formulated and the volume that can be delivered to the test system.
Dose extrapolation may differ depending on the target organ and ROA. Doses of products administered via intravenous ROA are typically calculated and extrapolated based on viral genome per kilogram body weight. Targeting the eye on the other hand may require a fixed amount considering the restricted defined size and capacity of the eye. In the case of the CNS, doses are extrapolated based on tissue or CSF volume.
Biodistribution
Due to poor in vitro to in vivo and animal to human translatability associated with developing novel AAV vector capsids and transgene expression constructs (Lisowski et al. 2014; Naso et al. 2017), it is recommended that smaller exploratory phase studies be conducted to assess the broad biodistribution of that vector in two preclinical animal species; typically, utilizing the preclinical pharmacology animal model (e.g., mouse model of disease) and normal NHPs to understand the nonrodent AAV vector biodistribution. If nonrodent species other than NHP are considered for the preclinical program, understanding tropism differences between the species should be factored into the strategy for dose extrapolation and therapeutic margin considerations.
While the use of a tissue-specific promotor mimicking the intended clinical test article is intuitive and rational, the use of a separate, ubiquitous promoter capable of unrestricted transgene expression may also be helpful at an early stage of vector characterization. Broad tissue evaluation with such a construct may help investigators fully understand the maximum potential biodistribution of the vector DNA. Outcome from the exploratory studies then guides the design of regulatory phase studies in terms of triaging tissue collection and selecting interim and terminal necropsy time points adequate to evaluating maximum vector biodistribution, maximum transgene expression, and vector DNA and transgene expression persistence. This is critical since tissue collection criteria are not well defined in the regulatory guidance documents, and consequently, empirical evidence is required to identify and justify the list of target and potential off-target tissues that may carry safety concerns beyond the expected list of major organs. Additionally, lesions observed macroscopically at the time of necropsy should be collected for histopathology assessment as well as DNA assessment of the vector. Evaluation of vector DNA and transgene (mRNA or protein or both) product are critical for the assessment of efficacy and safety of a particular test article. Regulatory guidance requires that a GLP-validated quantitative PCR (qPCR) assay with sensitivity able to detect <50 copies of vector/1-μg genomic DNA be used for the assessment of vector DNA biodistribution. A reverse-transcription PCR (RT-PCR) assay that is scientifically qualified is sufficient for the assessment of transgene mRNA from a regulatory perspective.
Morphology-based Biodistribution
The addition of morphology-based methods such as ISH for the assessment of test article–related nucleic acids (DNA and RNA) and IHC for the assessment of transgene protein product may be critical to understand the cellular distribution within a tissue and provide unique insight that can influence safety considerations of the expression of a given transgene in a cell type that may have unique physiologic function.
There may be a disconnect in the presence of AAV vector DNA and transgene expression (mRNA or protein) in cells with fewer cells expressing the transgene (authors’ unpublished data; Figure 1). While qPCR and RT-PCR provide accurate quantitation of vector DNA and transgene mRNA, respectively, these assays lack the cellular context necessary to understanding vector tropism and the assessment of DNA and mRNA on the single-cell level. Morphology-based assessment of biodistribution, as carried out by fluorescent reporter or IHC for reporter protein or tagged transgene protein product and ISH for DNA and mRNA, complements and adds a spatial context to the biodistribution assessment necessary to associate certain study outcomes with cell-specific and tissue-specific vector spread. Demonstrating transduction of nongerm interstitial cells in the male and female gonads (testes and ovaries) will mitigate the need to do extensive reproductive toxicology studies. Morphology-based assessment may provide biological relevance of low vector DNA and transgene mRNA copy numbers that is restricted to a few cells with important physiologic function.
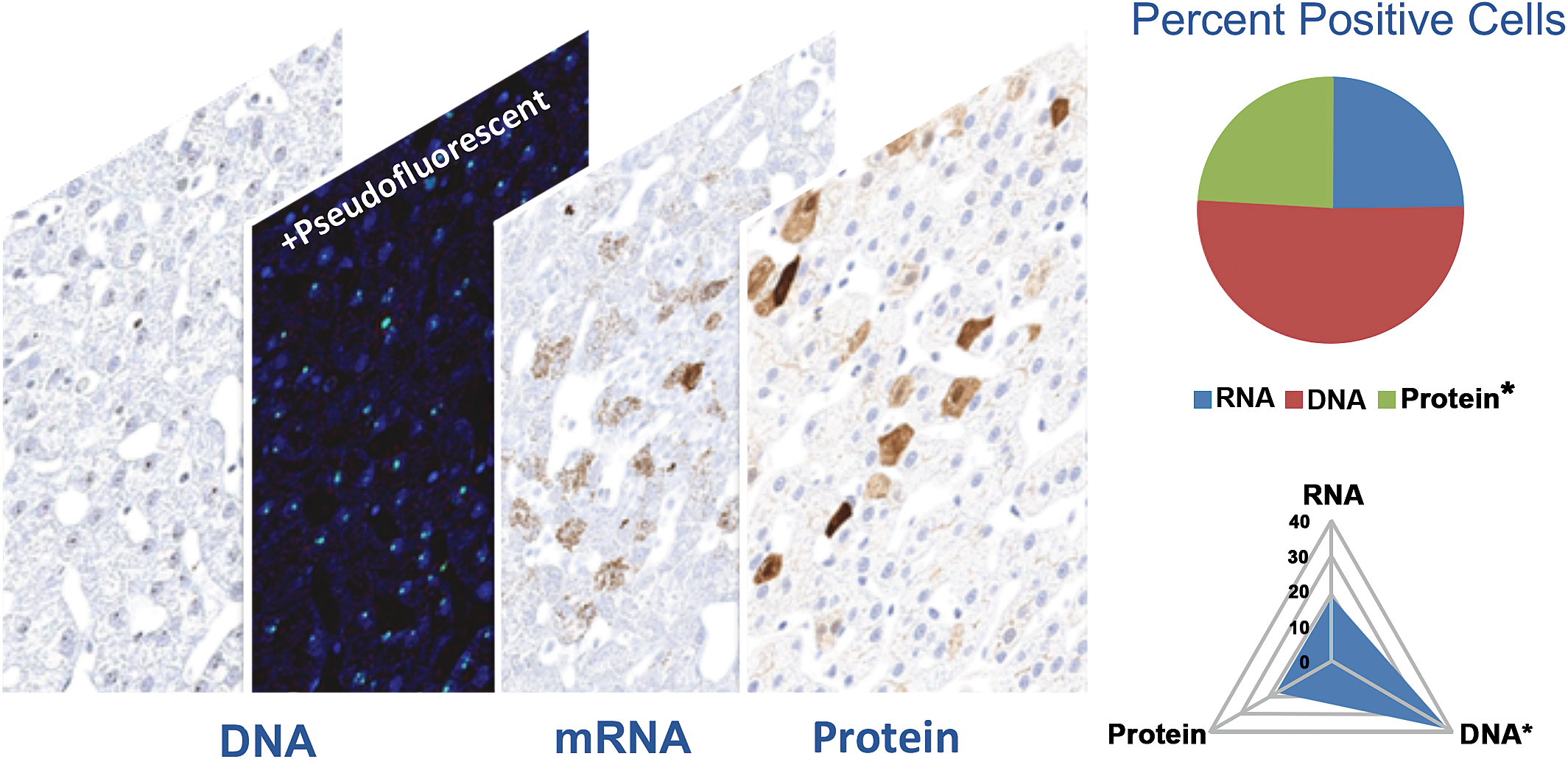
A disconnect in the number of vector DNA-positive cells compared with transgene mRNA-positive cells and transgene protein product in the liver of a cynomolgus macaque–administered adeno-associated virus (AAV) expressing a transgene. AAV vector DNA and transgene mRNA signals were assessed by RNAScope assay with protocols optimized to detect DNA or RNA only. Transgene protein is assessed by immunohistochemistry using antibody against transgene protein product. Pseudofluorescent image of DNA signal was generated to enhance visualization. Quantitation of signal distribution was digitally analyzed using Visiopharm newCAST software version 2017.7.3.4069.
Summary and Future Directions
As summarized in Figure 2, there are several important points to consider during the design of GT preclinical safety packages and interactions with regulatory agencies, including understanding the vector capsid biology across different species, optimizing transgene expression constructs, using tissue-specific or ubiquitous promotors, selecting doses for toxicology studies based on an understanding of the predicted minimum and optimal efficacious dose, and finally having key discussion points with regulatory agencies.
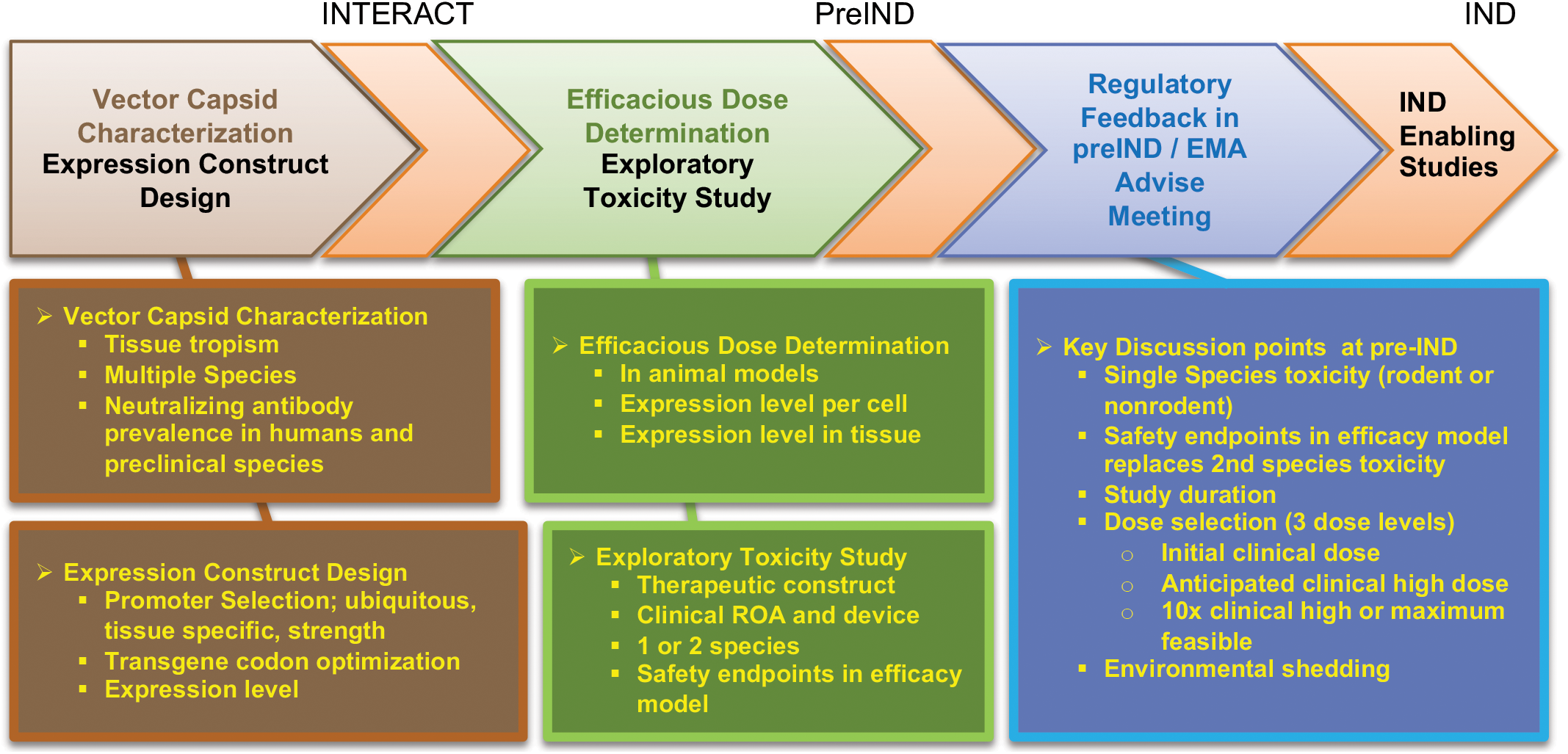
General summary and outline of points to consider during the design of gene therapy preclinical safety package and the interaction with regulatory agencies.
To develop GT products in a more feasible and cost-effective way, there may be opportunities to simplify and accelerate a preclinical toxicology program. Once a vector serotype, expression construct, promoter, delivery route, and dose levels have been assessed, the delivery of additional transgenes targeting the same target tissue(s) for the same or different disease may enable a reduction in the extent of the preclinical safety package that is focused on assessing specific risks only associated with the transgene and disease condition. Specific aspects of a safety program that may be shortened include study duration, fewer interim time points in studies, use of 1 species (e.g., rodent only), and reduced number of end points.
Footnotes
Authors’ Note
This article contains only public information and has been prepared solely for educational purposes to contribute to the understanding of preclinical safety assessment of adeno-associated virus (AAV) gene therapy (GT) product. These materials reflect only the personal views of the authors and may not be the views of their employer. The reader should understand that the development of each AAV GT product is fact-specific and that the appropriate solution or approach to assessing preclinical safety may vary. Therefore, these materials may or may not be relevant to any particular assessment of preclinical safety of AAV GT products.
Acknowledgment
The authors would like to thank Karrie Brenneman for the critical review of this article and Beth Mahler for the generation of figures.
Author Contributions
Authors contributed to conception or design (BA, LW); data acquisition, analysis, or interpretation (BA, LW); drafting the manuscript (BA); and critically revising the manuscript (BA, LW). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.