
Abstract
Hemophilia, an X-linked monogenic disorder, arises from mutations in the F8 or F9 genes, which encode clotting factor VIII (FVIII) or clotting factor IX (FIX), respectively. As a prominent hereditary coagulation disorder, hemophilia is clinically manifested by spontaneous hemorrhagic episodes. Severe cases may progress to complications such as stroke and arthropathy, significantly compromising patients’ quality of life. Hemophilia has a monogenic nature, coupled with quantifiable therapeutic endpoints and predictable treatment outcomes. These characteristics render it an ideal candidate for gene therapy studies. Currently, Food and Drug Administration (FDA)-approved gene therapies utilize recombinant adeno-associated virus (AAV) vectors to deliver functional transgene cassettes to hepatocytes. These therapies offer distinct advantages: a single intravenous administration achieves sustained FVIII and FIX activity levels, providing robust hemostatic control while markedly enhancing patients’ quality of life. However, several challenges remain, including immunogenicity, thrombotic risks, potential gene integration, and prohibitive costs. Future endeavors should prioritize expanding patient eligibility and integrating precision gene-editing technologies to mitigate these limitations. In this review, we provide a comprehensive overview of recent advances and emerging strategies in hemophilia gene therapy, with a particular focus on clinical translation and technological innovation. Ongoing research in this field remains pivotal to overcome existing barriers, enhance treatment accessibility, and ultimately realize curative potential for patients with hemophilia.
Introduction
Hemophilia, an inherited coagulation disorder, is defined by compromised activity of either clotting factor VIII (FVIII) or factor IX (FIX), corresponding to hemophilia A and B, respectively. 1 For individuals with severe hemophilia, the current standard of care entails prophylactic replacement of the deficient clotting factor via intravenous infusion. 2 Nevertheless, the inherently short plasma half-life of FVIII and FIX necessitates frequent intravenous administrations (a minimum of 2-3 times weekly), which imposes substantial clinical burdens and economic constraints. Furthermore, patients are at heightened risk of developing neutralizing antibodies (inhibitors), 3 such complications arise in 25% to 30% of hemophilia A cases and 3% to 5% of hemophilia B cases. 4 Consequently, there is an urgent need to develop innovative therapeutic strategies that alleviate these risks while augmenting treatment efficacy.
Gene therapy presents promising therapeutic avenues for diseases refractory to conventional interventions and exhibits substantial efficacy in addressing monogenic disorders such as hemophilia. Concurrent advancements in genetic engineering and materials science have facilitated the expansion of gene therapy applications, extending beyond monogenic conditions to encompass multifactorial diseases. Based on the route of administration, gene therapy can be classified into two primary modalities: ex vivo and in vivo. In the context of hemophilia, ex vivo approaches primarily focus on engineering autologous cells to secrete functional FVIII or FIX. Hematopoietic stem cells (HSCs) 5 and mesenchymal stem cells 6 are among the most studied cell types. Lentiviral vectors are commonly employed for their ability to integrate transgenes into the host genome, ensuring stable expression of FVIII/FIX. For instance, early preclinical studies have demonstrated that lentivirally modified HSCs, when transplanted into hemophilia A mice, can reconstitute long-term FVIII expression, reducing bleeding phenotypes. 7 A key milestone in hemophilia ex vivo gene therapy is the exploration of induced pluripotent stem cells (iPSCs). iPSCs, derived from patient fibroblasts, offer a renewable source of cells that can be genetically edited to correct F8 or F9 mutations and differentiated into hepatocyte-like cells, which naturally secrete coagulation factors. 8 This approach circumvents issues of immune rejection and allows for personalized therapy, particularly valuable for patients with pre-existing antibodies to viral vectors.
In vivo genetic intervention involves targeted delivery of therapeutic transgenes to patients via diverse administration routes. Vectors are typically employed to shield these genes from nuclease-mediated degradation and facilitate their trafficking into recipient target cells. This modality has rapidly emerged as a pioneering therapeutic approach for monogenic disorders. Over the past three decades, numerous gene therapy strategies for hemophilia have been investigated, 9 with adeno-associated virus (AAV)-mediated gene addition emerging as the predominant approach in clinical applications. Notably, Hemgenix (etranacogene dezaparvovec, CSL Behring) and Beqvez (fidanacogene elaparvovec, Pfizer), AAV vector-based gene therapies, were approved by the United States Food and Drug Administration (FDA) for the treatment of adult patients with hemophilia B in 2022 and 2024, respectively.10,11 In 2023, Roctavian (valoctogene roxaparvovec, BioMarin) likewise gained FDA approval for use in adults with severe hemophilia A. 12 These three therapeutics share the characteristic of being one-time, single-dose intravenous infusions of AAV vectors carrying functional genes; upon administration, these genes are expressed in the liver to produce FVIII or FIX. Despite the advancement of multiple gene therapies for hemophilia A and B through various phases of clinical trials, large-scale studies have identified a spectrum of challenges, including immunogenicity, thrombotic risks, potential gene integration, and prohibitive costs. 13 Consequently, gene therapy strategies warrant further refinement by the scientific and clinical communities.
Hemophilia A and Hemophilia B
Characteristic and Pathogenesis of Hemophilia
Hemophilia is frequently dubbed the “royal disease”. In the nineteenth century, Queen Victoria of the United Kingdom transmitted the genetic mutation to her descendants, who subsequently disseminated the disorder to royal families across Europe and Russia via dynastic marriages. Globally, hemophilia ranks among the most prevalent hereditary coagulation disorders. 14 While the majority of cases are hereditary, rare instances of acquired hemophilia have also been documented. 15 Hemophilia A is characterized by deficient production or impaired functionality of coagulation FVIII, whereas hemophilia B is defined by analogous defects in coagulation FIX. Hemophilia A constitutes 80% to 85% of all cases, with an incidence of 1 in 5000 male births. Hemophilia B accounts for approximately 15% of cases, occurring in 1 in 25 000 male births. Based on residual coagulation factor activity levels in the patient, hemophilia severity can be classified as mild (5-40 IU/dL), moderate (1-5 IU/dL), or severe (<1 IU/dL). 16 Patients with mild hemophilia typically experience bleeding episodes following significant trauma, whereas those with moderate hemophilia develop hemorrhage after minor trauma and may suffer sporadic spontaneous bleeding. Individuals with severe hemophilia, by contrast, are prone to frequent spontaneous hemorrhagic events. 17 Pathologically, hemophilia is characterized by recurrent hematoma formation within articular and soft tissue compartments. Persistent intra-articular bleeding can precipitate progressive complications, including muscle atrophy, hemophilic arthropathy and secondary osteoporosis. 18 Life-threatening emergencies may arise from bleeding in critical anatomical sites such as the intracranial spaces, gastrointestinal tract, or pharyngeal region. 19 To mitigate these risks, patients with severe hemophilia require long-term prophylactic administration of clotting factor concentrates. This therapeutic regimen, however, is confronted with three primary challenges: immunogenicity, high treatment burden, and cost constraints. 20
Hemophilia is associated with various types of mutations. Mutations in the F8 gene are categorized as intron 22 inversion, intron 1 inversion, substitution, deletion, duplication, insertion, and polymorphism. 21 The F8 gene encoding FVIII is located on the long arm of the X chromosome (Xq28). It comprises 26 exons and encodes a mature 2332-amino-acid FVIII protein. FVIII is physiologically synthesized in the sinusoidal endothelial cells of the liver, 22 and the majority of circulating FVIII binds to von Willebrand factor (vWF), which attenuates its degradation. The half-life of FVIII is approximately 12 h in adults, with shorter durations observed in children. A multitude of mutations underlying hemophilia A have been identified, among which intron 22 inversion and intron 1 inversion are the most common, affecting roughly 40% to 50% of patients with hemophilia A. 23 Current evidence suggests that these chromosomal rearrangements predominantly occur during male meiosis. 24 The F9 gene exhibits variant types including substitution, deletion, duplication, insertion and polymorphism. The F9 gene is relatively small (approximately 34 kb) and has a simpler genomic structure, consisting of only 8 exons. Compared to FVIII, FIX is synthesized in hepatocytes 25 and forms a 415-amino-acids protein that does not require a chaperone. Missense mutations are the predominant variant in the F9 gene, accounting for approximately 58% of all reported variants. Hemophilia B Leyden, 26 a rare yet severe subtype of hemophilia B, is characterized by a unique molecular mechanism involving the disruption of specific transcription factor binding sites within the proximal promoter of F9 gene. Under these conditions, abnormal bleeding typically manifests postnatally but resolves spontaneously during adolescence, with circulating FIX levels gradually normalizing by adulthood. 27
Current Therapies for Hemophilia
Coagulation Factor Replacement Therapy
The mainstay of hemophilia therapy lies in the replacement of the deficient coagulation factor to achieve the target peak in factor activity. Factor replacement therapy is administered either as prophylaxis to prevent hemorrhagic episodes or as on-demand intervention for acute bleeding management. 28 However, the requirement for frequent intravenous administration, typically 2–3 times weekly, significantly escalates both economic and temporal burdens on patients while necessitating strict adherence. To address these limitations, extended half-life (EHL) factor formulations have been developed to sustain therapeutic levels of FVIII or FIX over prolonged periods, thereby reducing infusion frequency. Strategies to extend the plasma half-life of coagulation factors include polyethylene glycol (PEG) conjugation, albumin fusion, incorporation of immunoglobulin fragment crystallizable (Fc) domains, 29 and engineering of single-chain FVIII variants. These modifications can prolong the half-life of FVIII by 1.2- to 2-fold, whereas a more pronounced 4- to 6-fold extension is achieved for FIX. 30 The development of inhibitors represents the most severe complication of hemophilia replacement therapy, occurring in approximately 30% of patients with hemophilia A and 3% of those with hemophilia B. 20 This immunological complication significantly compromises clinical management by rendering standard factor replacement therapies ineffective.
Non-Factor Therapy
Non-factor therapies include bispecific antibodies that mimic activated FVIII, small interfering RNA (siRNA) targeting antithrombin (AT), and anti-tissue factor pathway inhibitor (TFPI) antibodies. 20 Emicizumab, a bispecific antibody targeting both factor X (FX) and activated factor IX (FIXa), effectively mimics FVIII function. Since 2017, emicizumab (Hemlibra) has been approved for clinical use in the United States to reduce bleeding frequency in patients with hemophilia A, regardless of inhibitor status. 31 Clinical evidence, such as from the HAVEN 1-4 trial, demonstrates a significantly lower annualized bleeding rate in patients receiving emicizumab prophylaxis compared to non-prophylaxis groups. 32 Furthermore, in inhibitor-negative patients, emicizumab reduces bleeding rates more effectively than traditional FVIII prophylaxis. Emicizumab is generally well-tolerated, with mild injection-site reactions as the most common adverse event and no serious thrombotic complications observed in most studies. Long-term safety data from real-world experience and ongoing trials further support its favorable safety profile, and owing to its half-life of approximately one month, it is administered subcutaneously weekly, biweekly, or monthly.
Fitusiran, an siRNA targeting AT, reduces circulating AT levels by inhibiting hepatic AT synthesis. 33 Marstacimab (Hympavzi) and concizumab (Alhemo) are monoclonal antibodies that antagonize TFPI-mediated anticoagulant pathways. In October 2024, marstacimab received its first approval in the United States for prophylaxis in adolescents and adults with hemophilia A or B who are inhibitor-negative, 34 whereas concizumab was approved by the FDA in December 2024 for patients aged ≥12 years with hemophilia A or B. 35 These novel therapeutic approaches exhibit efficacy across patient populations regardless of inhibitor status while reducing administration frequency. Nevertheless, such therapies may raise safety concerns, most notably the risk of thrombosis. 36
Gene Therapy
Historically, the management of hemophilia has evolved from replacement therapy to gene therapy (as depicted in Figure 1). Hemophilia, caused by a monogenic defect in F8 or F9, presents an ideal candidate for gene therapy, as the underlying genetic aberration can be corrected through gene delivery to achieve sustained and stable clotting factor activity. AAV-based gene therapies, which effectively express clotting factors, have been approved by the FDA for the chronic treatment of hemophilia. To date, three therapeutic products, including Roctavian (for hemophilia A), 37 Hemgenix, and Beqvez (both for hemophilia B), have entered clinical practice.38,39 Table 1 summarizes FDA-approved gene therapies in terms of vector type, efficacy, safety, and cost. Despite these advancements, several critical challenges must be addressed during the rapid progression of gene therapy. The presence of pre-existing neutralizing antibodies directed against AAV capsids in treatment-naive individuals may significantly diminish therapeutic efficacy. Moreover, all three FDA-approved gene therapies have exhibited elevated alanine aminotransferase (ALT) levels in clinical trials. 40 Additionally, prohibitive costs remain a major barrier to widespread adoption. Further research is required to comprehensively characterize and optimize these promising therapeutic modalities.

Key Milestones in the Therapy of Hemophilia.
Comparison of FDA-Approved Gene Therapies.

Gene Vectors
Gene vectors, alternatively termed as gene delivery systems, enable the introduction of exogenous target genes into host cells, where functional expression occurs to modulate gene activity or treat diseases. Gene vectors are broadly classified into two primary types: viral vectors and non-viral vectors (the latter encompassing physical and chemical delivery approaches), as elaborated in subsequent sections.
Viral Vectors
Viral vectors are recombinant constructs derived from parental viruses, which have been genetically modified to abrogate pathogenicity while retaining their capacity to deliver therapeutic genes. The primary viral vector systems include AAVs, adenoviral vectors, retroviral vectors, and lentiviral vectors. These engineered vectors enable the targeted delivery of therapeutic genetic material into host cells for disease intervention.
AAV Vectors
The majority of developed AAV vectors are utilized for the treatment of monogenic diseases, which are classified as rare diseases. 41 Notably, AAV does not integrate into the host genome and exhibits the most favorable safety profile among viral vectors. To date, numerous AAV-based gene therapeutics have obtained regulatory approval. Prominent examples include FDA's authorization of Hemgenix and Beqvez, two therapies targeting hemophilia B caused by congenital FIX deficiency. For the treatment of hemophilia A, the FDA has approved Roctavian, an AAV5 vector engineered to deliver the B-domain-deleted FVIII (BDD-FVIII) transgene that is under the control of a liver-specific promoter. 42 Additionally, the FDA has approved Zolgensma, 43 Luxturna, 44 and Elevidys 45 as AAV-based gene therapies.
Adenovirus (AdV) Vectors
Human AdV serotypes 2 and 5 are predominantly utilized in gene therapy applications. Like AAV, AdV genomes persist as episomal elements following infection, with no integration into the host genome. AdV vectors-mediated gene therapy has been primarily employed for cancer treatment and vaccination against infectious diseases. In 2003, Gendicine® was approved by China Food and Drug Administration (CFDA) as the first globally authorized gene therapeutic agent for managing squamous cell carcinoma of the head and neck. 46 Beyond oncology, AdV vectors have shown considerable potential in combating infectious diseases such as coronavirus disease 2019 (COVID-19) pandemic.
Lentiviral Vectors
The lentiviral vector systems have been predominantly utilized in ex vivo gene therapies. Chimeric antigen receptor T cell (CAR-T) therapy, which involves genetically engineering autologous T cells to target and eliminate tumor cells, has demonstrated remarkable efficacy in treating specific hematologic malignancies across multiple clinical trials. Current clinical protocols primarily utilize lentiviral or retroviral transduction as the principal approach for genetically engineering T lymphocyte. Several CAR-T products, including Kymriah, Breyanzi, Abecma, and Carvykti, have been developed via lentiviral vector-mediated T-cell modification and have obtained regulatory approval. 47
Physical Non-Viral Gene Delivery Systems
Non-viral gene delivery platforms are classified into physical and chemical approaches. Physical methods involve the introduction of naked nucleic acids into target cells via techniques such as microinjection, gene gun delivery, electroporation, and ultrasound-mediated transfection.
Microinjection
As a gene delivery method, microinjection constitutes a critical technique in biomedical research, owing to its ability to support diverse genetic applications. Key advantages of microinjection include the absence of gene length limitations and broad applicability across cell types. However, microinjection requires specialized expertise and involves time-consuming, labor-intensive protocols. 48 To address these limitations, Andrew D. Alegria and colleagues developed a fully automated, versatile robotic platform designed to standardize the microinjection process. This automated system holds the potential to enable high-throughput microinjection, thereby significantly enhancing scalability and reproducibility in genome-wide functional genomics research. 49
Gene Gun
Particle bombardment technology, utilizing a gene gun, was initially developed for gene delivery in plant cell transformation. Recent studies have extended its application to the mammalian systems, including both in vitro and in vivo contexts. This technique is particularly valuable for transfecting cell lines refractory to conventional methods, such as dendritic cells and 3T3-L1 adipocytes. 50 A key limitation, however, is its shallow penetration depth (0.5-1 mm), which restricts its primary use to superficial tissues such as skin and muscle. 51 Additional drawbacks include high costs and transient gene expression profiles.
Electroporation
Electroporation is a biophysical process wherein controlled electrical pulses induce transient pores in the plasma membrane, enabling the entry of nucleic acids into the cellular cytoplasm. Successful transfection requires rigorous optimization of the electrical parameters used in the experiment. 52 Critical factors influencing transfection efficiency include electric field strength, pulse waveform, number of applied pulses, and buffer ionic composition. A primary limitation of this method is potential cytotoxicity arising from improper pulse parameters; excessive voltage can induce up to 50% cell mortality in sensitive cell lines.
Ultrasound-Targeted Microbubble Destruction (UTMD)
UTMD technology involves encapsulating therapeutic agents or genetic material within microbubble carriers. Upon exposure to ultrasound waves, these microbubbles undergo cavitation, a process in which their rupture generates radiation pressure, microjets, and shockwaves. These phenomena induce shear stresses on cell membranes, creating small pores that enhance membrane permeability. 53 While UTMD provides a simple and efficient method for cellular gene transfection, prolonged ultrasound exposure may induce cytotoxicity in target cells.
Chemical Non-Viral Gene Vectors
Recently, several types of chemical non-viral vectors, including cationic polymers, cationic liposomes, inorganic nanoparticles and nanomicelles, have garnered significant attention as gene transfection agents (Figure 2). 54
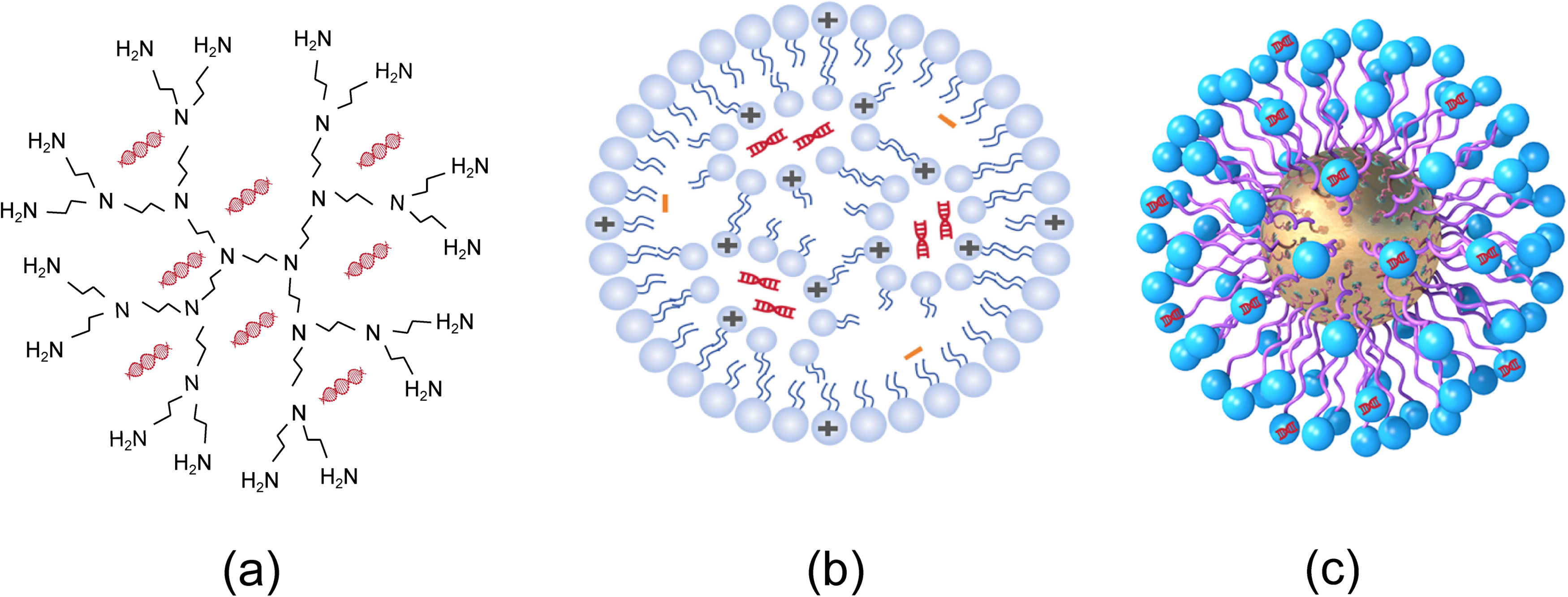
Common Chemical Non-viral Gene Delivery Vectors. (a) Cationic Polymers; (b) Cationic Liposomes; (c) Various Forms of Inorganic Gene Vectors, Take Gold Nanoparticles for Example.
Cationic Polymers
Cationic polymers (Figure 2a) exhibit either linear or branched architectures, with molecular weights ranging from 800 to 1 000 000 Daltons. Polyethyleneimine (PEI) is among the most commonly used cationic polymers; notably, the 25 000-Dalton variant achieves high ex vivo transfection efficiency. For in vivo applications, the commercially available low-molecular-weight PEI formulation (in vivo-jetPEI) has been designed to effectively deliver nucleic acids to targeted organs or tissues. Another widely studied class of cationic polymers, poly β-amino esters (PBAEs), have attracted significant attention for both in vivo and in vitro gene delivery. Our previous study demonstrated that PEI-capped PBAEs mediate effective and safe intraperitoneal transfection of minicircle DNA in mice, highlighting their potential for clinical translation. 55
Cationic Liposome and Lipid Nanoparticles (LNPs)
Another category of chemical gene vectors encompasses cationic liposomes. As illustrated in Figure 2b, these structures characteristically consist of a hydrophilic head group (either mono- or polycationic) covalently linked to an extended lipophilic hydrocarbon tail. LNPs are an advanced formulation of cationic liposomes, comprising ionizable lipid species, zwitterionic phospholipids, cholesterol derivatives, and PEGylated lipids. 56 With the authorization of COVID-19 vaccines such as Comirnaty and Spikevax, LNPs are recognized as the most successful mRNA delivery vehicles. 57 Jeong Pil Han and colleagues. 58 demonstrated that co-delivery of LNP-encapsulated Cas9 mRNA and antithrombin-targeting guide RNA to the mouse liver significantly reduced spontaneous bleeding frequency in both hemophilia A and B mouse models. Furthermore, the administration of FVIII mRNA-LNPs successfully restored hemostatic function and sustained therapeutic FVIII protein levels in Hemophilia A mice. 59
Inorganic Nanoparticles
In addition to organic agents, inorganic nanoparticles (Figure 2c), including calcium phosphates and silicon-based materials, are also used for gene delivery. Recent studies have shown that modified calcium phosphate nanocomposites, such as erythrocyte membrane-coated or polymer-encapsulated calcium phosphate nanoparticles, exhibit significantly improved transfection efficiency. 60 Furthermore, a calcium phosphate nanoneedle-based gene delivery system yielded remarkable anticancer therapeutic effects in a human ovarian cancer xenograft mouse model. 61 Zhang and colleagues developed PEI-functionalized silicon nanoparticles for in vivo siRNA delivery, which demonstrated significant gene silencing efficacy. 62
The transfection process for DNA delivery involves five sequential steps: DNA loading, endocytosis, endosomal escape, nuclear entry and protein expression (Figure 3a). In contrast, the siRNA delivery follows a distinct pathway: following siRNA loading and endocytosis, successful endosomal escape facilitates siRNA release into the cytoplasm, where it is incorporated into the RNA-induced silencing complex (RISC) to mediate target mRNA degradation and gene silencing (Figure 3b). Compared to viral vectors, non-viral vectors exhibit superior stability, cost-effectiveness and operational flexibility. To enhance their transfection efficiency, researchers have focused on three key strategies: optimizing receptor-mediated endocytosis, enhancing endosomal escape mechanisms, and integrating physical delivery methods. It is anticipated that further research will yield efficient and clinically viable non-viral gene vectors.

Intracellular Processes of Exogenous Nucleic Acid Delivery via non-Viral Vectors (Take Cationic Liposomes for Instance). (a) DNA; (b) siRNA.
Gene Therapy Research in Hemophilia
Given the monogenic nature of hemophilia, even modest elevations in coagulation factor activity (≥5% of physiological levels) can markedly ameliorate hemorrhagic phenotypes in severe cases. This unique characteristic renders hemophilia particularly suitable for genetic intervention strategies. 14 Gene therapy represents a transformative approach by correcting disease-causing gene defects, enabling hemophilia patients to restore normal physiological levels of clotting factors through in vivo or ex vivo strategies (Figure 4). This biological restoration not only mitigates bleeding risks but also attenuates long-term joint impairments, thereby establishing gene therapy as the first definitive one-time curative approach for hemophilia. Moreover, it significantly reduces the frequency of prophylactic injections and lowers the infectious risk associated with repeated exposure to blood-derived products. Emerging clinical evidence supports that this approach exhibits robust safety and efficacy profiles, facilitating not only patients’ reintegration into social life but also meaningful improvements in their quality of life.
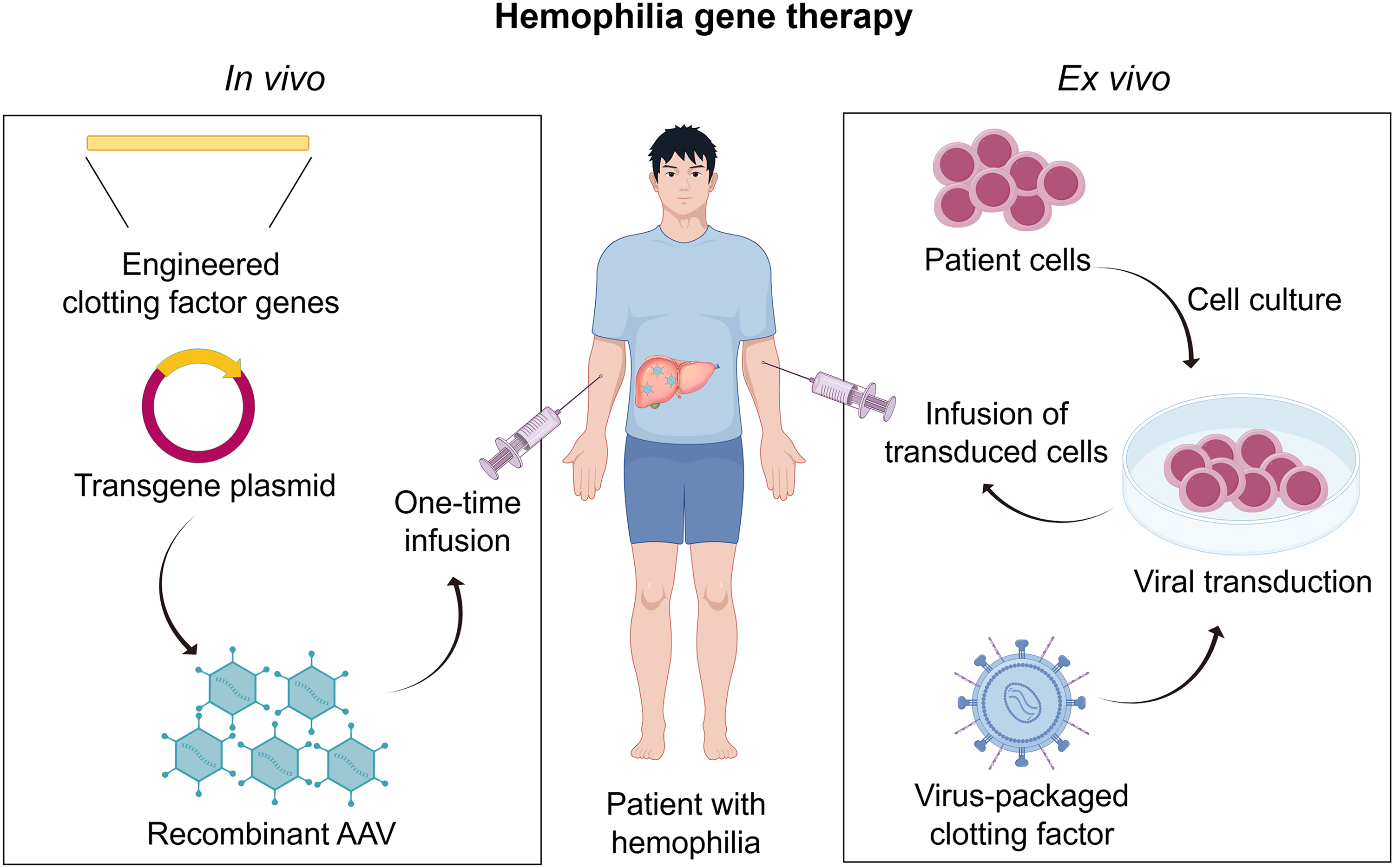
Gene Therapy Strategies for Hemophilia. (Left Panel) In Vivo; (Right Panel) Ex vivo.
Hemophilia A Gene Therapy
AAV vectors are the most widely adopted delivery system, although a small number of studies have employed alternative systems such as retroviral vectors 63 or autologous cells-based approaches. 64 The AAV vector has a packaging capacity of approximately 5 kb, posing a considerable challenge for hemophilia A gene therapy due to the 7 kb length of the FVIII coding sequence. 19 To address this limitation, the majority of hemophilia A gene therapy strategies utilize a transgene encoding B-domain-deleted (BDD) form of human FVIII. The B-domain, which is unnecessary for the procoagulant activity of this cofactor, has been replaced with a 14-amino-acid sequence to promote efficient intracellular cleavage. 65 In current clinical trials, the predominant administration route involves intravenous delivery of vectors carrying the transgene. 19 Despite the outstanding therapeutic efficacy, adverse effects associated with these therapies remain a significant concern. Table 2 and Table S1 summarize the pivotal clinical studies on hemophilia A gene therapy reported in recent years.
Hemophilia A Gene Therapy.

N/A: Not Applicable
On June 29, 2023, the FDA granted regulatory approval to Roctavian, an innovative gene therapeutic product developed by BioMarin Pharmaceuticals for managing severe hemophilia A. This therapeutic modality employs AAV5 as a vector and utilizes the human liver-specific promoter (HLP) to drive the expression of BDD-hFVIII expression. The approval of Roctavian was primarily supported by data from the phase 3 GENEr8-1 trial, 68 which is one of the largest phase 3 trials conducted in the field of hemophilia gene therapy. Updated long-term safety and clinical efficacy profiles of Roctavian were presented at the 2024 Congress of the International Society on Thrombosis and Haemostasis. Results from the phase 3 GENEr8-1 study revealed that durable bleed control and sustained FVIII expression were maintained for 4 years following Roctavian administration. Compared with previously reported outcomes,42,68 FVIII activity remained stable, without additional safety concerns identified. Among the 134 subjects who received a single dose of Roctavian, 112 (83.6%) had completed a minimum 6-month FVIII prophylactic regimen prior to treatment initiation. Baseline annualized bleeding rate (ABR) data were prospectively collected during the use of FVIII concentrates. Of the initial 112 participants, 2 discontinued the study. In the remaining cohort evaluated at the 4-year follow-up (n = 110), 81 (73.6%) achieved complete bleeding cessation. The mean FVIII activity, quantified via one-stage clotting and chromogenic substrate assays, was 27.1 IU/dL and 16.1 IU/dL, respectively. These levels remained nearly stable compared with the previously reported 3-year data. 77 Over the 4-year study period, the rollover population exhibited a mean ABR of 0.8 treated bleeding events per year, versus 1.3 bleeding events per year for all reported bleeding episodes.
Hemophilia B Gene Therapy
Hemophilia B has emerged as a prominent focus of numerous clinical investigations, with gene therapy approaches predominantly leveraging AAV vectors of various serotypes. These therapeutic strategies have centered largely on the delivery of a variant of F9 gene harboring the gain-of-function R338L mutation (designated FIX Padua). This specific mutation endows the encoded FIX protein with approximately 8-fold higher coagulant activity compared to its wild-type counterpart. Despite the demonstrated therapeutic efficacy of these interventions, immune-mediated responses and hepatotoxicity remain the most clinically impactful adverse events. A summary of key clinical trials of evaluating gene therapy for hemophilia B is provided in Table 3 and Table S1.
Hemophilia B Gene Therapy.
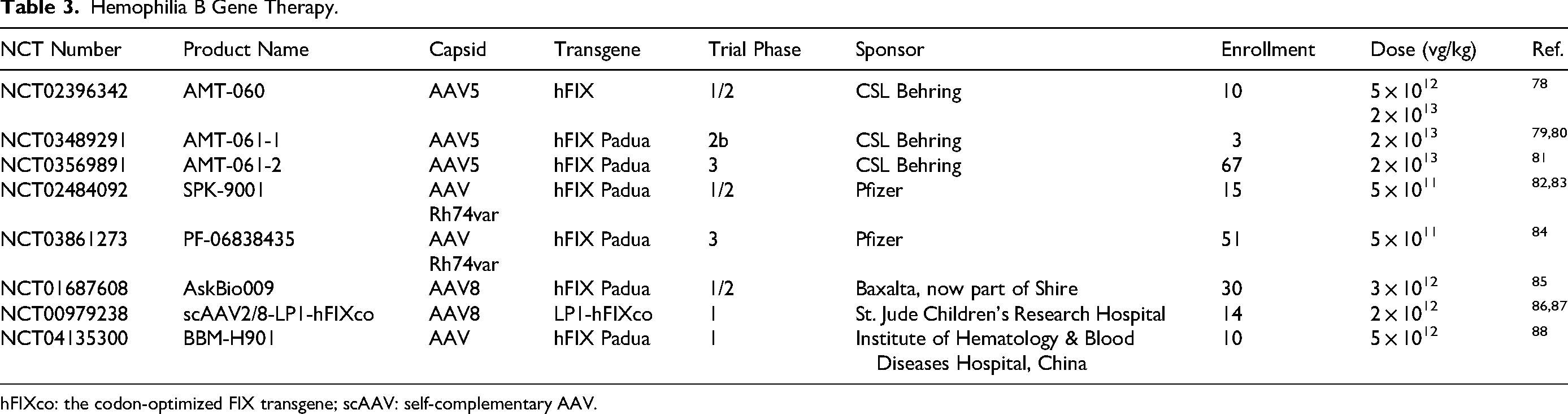
hFIXco: the codon-optimized FIX transgene; scAAV: self-complementary AAV.
In November 2022, Hemgenix received FDA authorization as the first gene therapy product for hemophilia B, with subsequent European Medicines Agency (EMA) approval in February 2023. This therapy utilizes an AAV5 vector, which demonstrates the lowest immunogenicity among viral serotypes and is generally unaffected by pre-existing anti-AAV antibodies in the majority of patients. The therapeutic construct features a codon-optimized Padua variant of human F9 gene, which is regulated by a liver-specific promoter. During the 2025 European Hematology Association Congress, CSL Behring presented the longitudinal outcomes of the phase 3 HOPE-B trial (NCT04645148). The results confirmed the durable therapeutic efficacy of Hemgenix in adults with hemophilia B up to 48 months post-infusion. This AAV5-based therapy demonstrated sustained FIX activity. Specifically, 93% of the participants (n = 57) maintained at least a 90% reduction in ABR relative to baseline.
Beqvez (fidanacogene elaparvovec) is an AAV gene therapy vector for hemophilia B, containing a high-activity human FIX variant (FIX-R338L). The activity of FIX-R338L can be measured in clinical laboratories using one-stage clotting and chromogenic substrate assays, which provide insights into assay variability when assessing endogenously produced FIX-R338L. 89 In a phase 1/2 study (NCT03587116), sustained therapeutic expression of FIX-R338L activity was observed in 15 participants with moderately severe to severe hemophilia B following infusion of Beqvez. These participants were able to discontinue prophylactic treatment with minimal bleeding episodes and reduced factor use. 90 In a follow-up study spanning up to 5 years, Beqvez remained well-tolerated. 83 Developed by Pfizer, Beqvez received FDA approval in April 2024 for the treatment of adult patients with moderate to severe hemophilia B. This approval was based on the positive results of the phase 3 BENEGENE-2 trial (NCT03861273), which demonstrated a 71% reduction in the ABR from 4.42 to 1.28 episodes per year. The mean FIX activity at 15 months was 26.9 IU/dL measured by the one-stage SynthASil assay. At 24 months after therapy, 82% of participants maintained factor IX levels above 5 IU/dL. No infusion-related serious adverse events, thrombotic events, development of FIX inhibitors, or malignant conditions were observed. 84
Clinical Case Studies in Gene Therapy
Case study results in the literature highlight the transformative efficacy and durability of gene therapy for hemophilia, particularly in hemophilia B. Hemgenix was approved by the FDA in November 2022 based on pivotal data from the phase 3 HOPE-B trial, a single-arm, open-label study evaluating its safety and efficacy in 54 adult males with severe or moderately severe hemophilia B. The trial demonstrated that the ABR decreased from 4.19 during the lead-in period to 1.51 during months 7 through 18 after a single intravenous infusion of Hemgenix. Mean FIX activity attained 36.9 IU/dL by 18 months and remained stable at 36.7 IU/dL through 24 months, with 94% of patients achieving a FIX activity level exceeding 5 IU/dL. 91 The usage of FIX concentrate decreased by mean of 248 825 IU per year per participant in the post-treatment period. Hemgenix has good tolerance, and no serious adverse events related to treatment have been reported. The treatment-related adverse events mostly occurred in the first six months after treatment, and the most common adverse event was alanine aminotransferase elevation. 92 In clinical trials for HEMGENIX, the most common side effects reported in more than 5% of patients were liver enzyme elevations, headache, elevated levels of a certain blood enzyme, flu-like symptoms, infusion-related reactions, fatigue, nausea, and feeling unwell. 93
The gene therapy BBM-H901 for hemophilia B, developed by Belief BioMed, has shown excellent efficacy in case studies. 88 In a phase 3, multi-center, single-arm trial, 26 adult patients with severe or moderately severe hemophilia B received a single intravenous infusion of BBM-H901. At 52 weeks post-treatment, the mean ABR dropped to 0.6, a 96% reduction compared to pre-treatment ABR. Among the 26 subjects, 21 (80.8%) had no bleeding events after treatment. FIX activity rapidly increased post-infusion, reaching 49.7 IU/dL within 3 days and stabilizing at 55.08 IU/dL at 52 weeks. During the 52-week follow-up, the most common grade 1-2 adverse events related to BBM-H901 included elevation of alanine aminotransferase (26.9%), elevation of aspartate aminotransferase (7.7%), decrease of fibrinogen (11.5%). 94 No serious adverse events, thromboembolic incidents, or FIX inhibitor formation were reported. Based on the above clinical trial data, BBM-H901 was approved by China's National Medical Products Administration in April 2025, it marks a milestone as Asia's first and globally the third hemophilia B gene therapy.
Adult and Pediatric Hemophilia Gene Therapy
Gene therapy outcomes for hemophilia diverge significantly between pediatric and adult populations due to physiological variations and developmental factors. In adults, AAV-mediated gene therapy achieves stable, long-term clotting factor production. Clinical data reveal persistent therapeutic levels of FVIII/FIX, correlating with marked ABR reductions and diminished need for prophylaxis. The FDA-approved Roctavian has demonstrated sustained efficacy in long-term follow-up. At 7 years post-treatment, the high-dose cohort maintained a mean FVIII activity of 16.2 IU/dL, which is indicative of mild hemophilia levels. 95 For hemophilia B, therapies like Hemgenix have shown even more durable FIX activity (>30 IU/dL) with minimal bleeding events over 5 years. 96
Pediatric hemophilia gene therapy remains at an earlier developmental stage, with particular emphasis on longitudinal safety monitoring of growth parameters, organ maturation, and age-specific immune reactivity. Current protocols employ conservative vector dosing and enhanced safety surveillance. The landmark SPK-8011 trial, a multicenter study coordinated by Children's Hospital of Philadelphia, evaluated this AAV-LK03 vector in males aged 18–52 years (n = 12, including post-pubertal adolescents). At 48-month follow-up, 83% maintained FVIII >12 IU/dL (mean 22.4 ± 8.7 IU/dL), correlating with 91.5% ABR reduction. 71 While primarily an adult study, its demonstration of sustained hepatocyte transduction informs pediatric applications, though developmental considerations warrant separate dose-escalation studies in prepubertal cohorts.
In summary, while gene therapy for adult hemophilia patients has achieved commercialization, pediatric applications demand customized approaches to address unique developmental, physiological and immunological challenges. Critical advancements in vector engineering and immunomodulation strategies will be essential to realize the one-time cure vision for all age groups. Regulatory agencies like the FDA particularly stress the importance of stringent long-term monitoring for off-target effects and immune responses in pediatric populations, considering their extended lifespan and potential risks of delayed adverse effects.
Continuing Challenges for Gene Therapy
Despite significant advancements in the development of novel gene vectors and therapeutic strategies, gene therapy for hemophilia has yet to achieve widespread clinical implementation. A multitude of challenges need to be addressed prior to its establishment as a standard-of-care intervention.
Immuogenicity and Hepatotoxicity
AAV gene therapy elicits diverse immune responses that compromise therapeutic efficacy and result in adverse effects, including systemic inflammation, hepatotoxicity, dorsal root ganglion toxicity, and myocarditis. 97 Clinical evidences consistently identify three principal immune mechanisms: pre-existing humoral immunity targeting viral capsids, innate immune activation triggered by pathogen-associated molecular patterns, and adaptive immune responses directed against transgene products. 98 In the context of AAV-mediated gene therapy for hemophilia, immune responses are primarily driven by two clinically relevant factors: (1) pre-existing anti-AAV neutralizing antibodies, and (2) the development of inhibitory antibodies against coagulation factors FVIII and FIX.
Neutralizing antibodies against AAV arise from environmental exposure to wild-type AAV in the general population. Notably, 20% to 70% of patients with hemophilia exhibit pre-existing anti-AAV immunity, which significantly compromises the efficacy of gene therapy delivery. 99 Current strategies to mitigate anti-AAV neutralization include immunomodulation, physical removal, vector engineering, and dosage escalation.40,100 The fundamental pathogenesis of hemophilia is attributed to deficiencies in functional coagulation factors. Therapeutic administration of these factors triggers immune recognition as non-self antigens, leading to the development of inhibitors that compromise treatment efficacy. Clinically significant inhibitors emerge in 25% to 30% of patients with hemophilia A, compared to only 3% to 5% of those with hemophilia B. 101 Immune tolerance induction remains the gold-standard approach for eradicating inhibitors in hemophilia. This protocol involves repeated high-dose administration of FVIII/FIX concentrates over 12 to 24 months, achieving immune tolerance through antigen-specific desensitization and clonal deletion of inhibitor-producing B lymphocytes. 102 Clinical outcomes demonstrate success rates of 60% to 80% in inhibitor eradication, with superior efficacy observed in patients with low-titer inhibitors. 103
Although gene therapy demonstrates favorable tolerability profiles, elevations in alanine aminotransferase (ALT) levels and transient reductions in coagulation factor activity are commonly observed. Administration of AAV vectors activates both innate and adaptive immune pathways, leading to the persistent formation of neutralizing antibodies. This immunogenic cascade is strongly associated with hepatic impairment, 13 and the risk of such immune responses exhibits a clear dose-dependent relationship with vector exposure. High doses of vectors may directly induce hepatocyte damage or elicit hepatic inflammation. Furthermore, the overexpression and aberrant deposition of coagulation factors within hepatocytes can disrupt cellular metabolic processes and impair hepatic function. Current strategies to mitigate hepatotoxicity include optimizing vector design to reduce immunogenicity, adjusting vector dosage according to patient's clinical status, and administering immunomodulatory agents to suppress excessive immune activation.19,40
Thrombotic Risk and Cancerogenesis
Overexpression of coagulation factors subsequent to gene therapy may result in abnormal systemic accumulation, thereby increasing the risk of thrombosis. Clinical evidence indicates that sustained FIX levels exceeding 150 IU/dL and FVIII levels exceeding 100 IU/dL are associated with an increased incidence of thrombotic complications. 104 Genetic susceptibility to thrombosis also plays a critical role, as highlighted by a recent study on Han Chinese patients with familial venous thromboembolism. Whole genome sequencing identified novel pathogenic variants in genes such as GP6, TET2, and JAK2, which significantly increased venous thromboembolism risk independent of coagulation factor levels. 105 The phase 1/2 B-AMAZE trial (NCT03369444 and NCT03641703) evaluated FLT180a, an AAV3 capsid vector engineering to encode the F9 gene variant harboring the gain-of-function Padua (R338L) mutation, and enrolled a cohort of 10 participants. Notably, one patient with supraphysiological FIX activity developed right-arm arteriovenous fistula thrombosis, requiring hospitalization and anticoagulant therapy with dalteparin. 106 Similarly, transient elevations in FVIII levels have been documented in recipients of hemophilia A gene therapy, with 7 of 134 patients (5.2%) exceeding the upper normal limit (150 IU/dL). 71 Therefore, vigilant monitoring is essential for patients with a history of thrombosis undergoing gene therapy.
Gene therapy harbors a potential oncogenic risk owing to random integration of viral vectors into the host genome, which may disrupt tumor suppressor genes or activate proto-oncogenes. Although the risk of insertional mutagenesis in AAV-mediated gene therapy has traditionally been regarded as minimal, given the predominantly episomal persistence of the delivered transgene, emerging evidence challenges this assumption. Long-term animal studies, 107 ultra-deep sequencing analyses, 108 and clinical case reports 109 collectively indicate that AAV exhibits a low yet non-negligible oncogenic risk. The possibility of insertional oncogenesis associated with gene therapy cannot be definitively ruled out, which underscores the ongoing necessity of long-term safety monitoring in clinical applications.
Affordability of AAV Gene Therapy
The research and development of gene therapies for hemophilia require significant capital investment, with considerable costs incurred in basic research, clinical trials and manufacturing technologies. 110 Additionally, the production of recombinant AAV vectors requires advanced technical expertise and specialized equipment. These factors collectively contribute to the high pricing of three FDA-approved hemophilia gene therapies. Roctavian, indicated for hemophilia A, is priced at $2.9 million, 37 while Hemgenix 111 and Beqvez, 96 both indicated for hemophilia B, are each priced at $3.5 million. The high costs associated with gene therapy impose significant economic burdens on both patients and healthcare system.
To address this challenge, three key measures should be implemented to enhance the affordability of treatment. First, prioritizing cost reduction in raw materials and production processes through technological innovation is crucial, particularly via advancements in gene-editing platforms and optimized viral vector systems. Second, establishing strategic pricing negotiations between national health authorities and biopharmaceutical manufacturers is essential. This process should involve two key components: inclusion of hemophilia gene therapies in national reimbursement formularies, and development of progressive tiered reimbursement frameworks to ensure at least 80% coverage for low-income populations. The National Institute for Health and Care Excellence evaluated Hemgenix and found it cost-effectiveness via manufacturer rebates, considering long-term savings from fewer hospitalizations and reduced infusions. 112 Hemgenix met the £20k to £30k per quality-adjusted life-year threshold within 10 years, with performance-based rebates offsetting high upfront costs, leading to the National Health Service reimbursement approval in July 2024. Finally, the implementation of innovative payment models such as value-based reimbursement frameworks, structured installment plans, and risk-sharing agreements should be prioritized to alleviate the financial burdens on patients confronting high upfront treatment costs. For instance, Germany included Hemgenix in its national health insurance in 2024 based on 4-year HOPE-B trial data showing 94% discontinued FIX therapy, 90% bleeding rate drop, and stable FIX activity. This milestone decision established Hemgenix as the first gene therapy in Germany to adopt a nationwide efficacy-linked reimbursement model. Similarly, France has implemented a risk-sharing agreement for Roctavian, where payments are adjusted based on the actual reduction in bleeding events experienced by patients.
Transfection Efficiency of Non-Viral Vectors
The clinical translation of non-viral gene delivery technologies is hindered by significant challenges related to inferior transfection performance, as demonstrated by empirical studies showing 2–3 log reductions relative to viral vector systems. 113 Physical delivery modalities (eg, electroporation, sonoporation) transiently enhance membrane permeability via controlled energy deposition, achieving in vitro transfection efficiencies of up to 40% by facilitating cytosolic nucleic acid entry. 114 However, inefficiencies in intracellular trafficking and barriers to nuclear membrane translocation impede clinical translation, with fewer than 5% of preclinical candidates progressing to phase 1 trials.
Conclusions and Outlook
This article presents a systematic review of the principles and technological advancements in gene therapy development, with particular focus on therapeutic strategies for hemophilia. Among existing gene delivery vectors, viral vectors predominate in current hemophilia clinical trials owing to their superior transfection efficiency and precise tissue-targeting capabilities. In contrast, non-viral vectors encounter significant challenges, including reduced in vivo transfection efficiency and limited capacity to sustain long-term transgene expression. However, these non-viral systems offer distinct advantages over their viral counterparts, such as scalability for industrial-scale production, extended shelf stability, enhanced payload capacity, and superior safety profiles. 113 Currently, physical non-viral gene delivery methods remain largely restricted to laboratory settings rather than clinical applications. Accordingly, we propose that future vector development strategies should prioritize chemically engineered delivery systems augmented by physical enhancement techniques.
Viral vectors have emerged as the cornerstone of clinical gene therapy development, as evidenced by the FDA approval of multiple therapeutic products. The commercialization of three gene therapies (Roctavian, Hemgenix, and Beqvez) has accelerated their clinical adoption, with these interventions gaining widespread recognition among hematologists and hemophilia patients. Advancements in in vivo delivery systems are anticipated to enhance bleeding prophylaxis and improve quality-of-life outcomes for patients. Despite the remarkable progress in gene therapy for hemophilia, it is important to emphasize that, as noted by the World Federation of Hemophilia (WFH) in its 2024 guidelines, 115 gene therapy does not yet constitute a standard-of-care treatment for either hemophilia A or B. The WFH emphasizes that gene therapy remains investigational and should be administered under strict regulatory frameworks, with long-term safety monitoring for liver toxicity, immune responses, and durability of factor expression. WFH guidelines also highlight the need for equitable access and cost-effectiveness evaluations, as current therapies may exclude patients with preexisting anti-AAV antibodies or advanced liver disease. The selection of patients for gene therapy requires a comprehensive assessment protocol that encompassing anti-AAV antibody titers, coagulation factor inhibitor profiles, hepatic function evaluation, and detailed clinical history. 116 These clinical imperatives underscore the critical need for parallel development of non-viral vector-mediated hemophilia therapies, which theoretically circumvent both preexisting AAV5-neutralizing antibodies and hepatic dependency. Furthermore, current mainstream gene therapies for hemophilia exhibit persistent limitations, including prohibitive cost structures, eligibility constraints confined to adult populations, and unresolved challenges pertaining to treatment durability. 117
To further promote the development of gene therapy for hemophilia, future research should focus on the following aspects: first of all, gene therapy primarily targets adult patients with hemophilia, but hemophilia is a hereditary disease that manifests from birth. Therefore, research on pediatric formulations and treatment for children is particularly important. However, pediatric cohorts face unique challenges: ongoing growth and liver maturation can cause vector dilution over time, potentially reducing long-term factor expression and necessitating adjusted dosing strategies. 8 The development of strategies for repeated dosing is also crucial for patients who require additional treatment due to immune reactions or reduced gene expression. Future studies should explore how to optimize gene therapy regimens to make them applicable to a broader patient population. In addition, CRISPR-Cas9 and its variants have shown great potential in gene editing. Studies have demonstrated that CRISPR-Cas9 can effectively repair point mutations in hemophilia B, 118 while base-editing technology exhibits higher efficiency and lower off-target effects in repairing specific mutations. 119 Future research should further optimize these gene editing technologies to enhance their application in hemophilia treatment. Furthermore, although current clinical trials have shown promising short-term results, long-term efficacy and safety remain areas requiring further investigation. Finally, future research should explore how to tailor treatment strategies to individual patient characteristics and develop efficient delivery methods to enhance the precision and safety of treatment.
Supplemental Material
sj-docx-1-cat-10.1177_10760296251378455 - Supplemental material for Recent Advances in Gene Therapy for Hemophilia
Supplemental material, sj-docx-1-cat-10.1177_10760296251378455 for Recent Advances in Gene Therapy for Hemophilia by Xiaojuan Pang, Jinxian Fu, Zhongqi Zhou, Nannan Tang, Cia-Hin Lau, Yingfei Wen, Ping Chen, Jing Zhao and Hongman Xue in Clinical and Applied Thrombosis/Hemostasis
Footnotes
List of Abbreviations
Author Contributions
XP and ZZ reviewed the literature and wrote the original draft. NT and XP prepared the figures and edited the manuscript. CL, YW and PC made the charts and organized related literature. JZ and HX conceptualized the framework of the article, supervised the completion and retrieval of the manuscript, and provided funding support.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Shenzhen Medical Research Fund (Grant No. D2301010), Shenzhen Science and Technology Program (Grant No. RCYX20231211090346060), Shenzhen Key Laboratory of Chinese Medicine Active substance and Translational Research (Grant No. ZDSYS20220606100801003), Science and Technology Planning Project of Shenzhen Municipality (Grant No. KJZD20230923115002005 and JCYJ20220530144613030).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.