
Abstract
Prevalence of immune-mediated glomerulonephritis has increased in preclinical toxicity studies, with more frequent use of biotherapeutic agents (especially antigenic humanized molecules) and antisense oligonucleotide (ASO) therapies. Immune complex disease affects a small number of study monkeys, often correlates with antidrug antibody (ADA) titers, and occurs at a dose that favors immune complex formation or impedes clearance. While preclinical glomerulonephritis often fails to correlate with evidence of glomerular or vascular injury in human clinical trials and is not considered predictive, additional animal investigative immunohistochemical work may be performed to substantiate evidence for immune complex pathogenesis. While ADA is most commonly encountered as a predisposing factor with biotherapeutic agents, complement activation may occur without circulating complexes, and other mechanisms of non-ADA immune-mediated glomerulonephritis have been observed including nonendogenous immune aggregates and immunoregulatory pharmacology. Although glomerulonephritis associated with oligonucleotide therapies has been noted occasionally in preclinical studies and more rarely with human patients, pathophysiologic mechanisms involved appear to be different between species and preclinical cases are not considered predictive for humans. ADA is not involved in oligonucleotide-associated cases, and complement fixation plays a more important role in monkeys. Recent screening of ASOs for proinflammatory activity appears to have decreased glomerulonephritis incidence preclinically.
Introduction
In the practice of toxicologic pathology of the kidney, the most frequently encountered lesions in historical terms have been localized to the tubules, and the most significant impediments to drug development in regard to nephrotoxicity have been related to compounds that induce tubule degeneration and/or necrosis. However, in recent years, the paradigm has begun to shift with the increasing importance of biotherapeutic (antibody and peptide) drugs and antisense oligonucleotide (ASO) therapies that have the potential to affect the glomerulus. Due to the recent industry practice using humanized antibodies and multimeric complex biologics as therapeutic modalities, glomerular lesions have been noted with much greater frequency in preclinical toxicity studies (Rojko et al. 2014). In the case of antibody and peptide therapies, the close relationship between the use of these agents and preclinical glomerular injury is generally based on immunogenicity of these new modalities and the development of antidrug antibody (ADA) in nonhuman primates (Rojko et al. 2014). ASOs have recently been implicated in a few cases of preclinical and more rarely, clinical glomerulonephritis, but the mechanism appears to be slightly different than those associated with biotherapeutics.
There are four major categories of preclinical glomerular toxicity based on mechanism, including (1) cytotoxic or charge-related injury to the filtration barrier, (2) toxicity related to accumulation of a xenobiotic within the glomerular compartment based on pharmacologic or off-target activity, (3) glomerulosclerosis, and (4) immune-mediated injury (Figure 1A–D). The histologic lesions associated with these four types of injury are quite different and the distinguishing characteristics relatively easy to discern for most pathologists making differentiation based on mechanism possible by histology alone. In the first type, typified by exemplar compounds such as puromycin or adriamycin, direct injury to podocytes by cytolytic agents results in blunting and retraction of foot processes, but uniquely by degenerative changes directly to the podocytes, mesangial cells, or glomerular capillary endothelium (Morgan, Kaler, and Bach 1998). Conversely, cationic agents such as gentamicin can disrupt or even destroy pedicel slit diaphragms and injure the overall filtration apparatus (Martínez-Salgado et al. 2007). Podocyte membranes are composed of anionic sialoglycoproteins, which can be disrupted by highly positively charged molecules (Martínez-Salgado et al. 2007). Morphologic changes can be observed soon after high-dose administration and are accompanied by rapid and significant declines in glomerular filtration rate. Rare biotherapeutic agents can induce this kind of lesion, usually as a result of pharmacologic activation of direct injury pathways in endothelium or podocytes. For example, interferon therapy causes glomerular injury resulting in collapsing focal segmental glomerular sclerosis by poorly defined mechanisms that include both direct and indirect effects on podocytes. (Markowitz et al. 2010). Long-term interferon therapy has also been associated with thrombotic microangiopathy by a suspected mechanism causing injury to capillary endothelial cells through induction of apoptosis (Zuber et al. 2002).
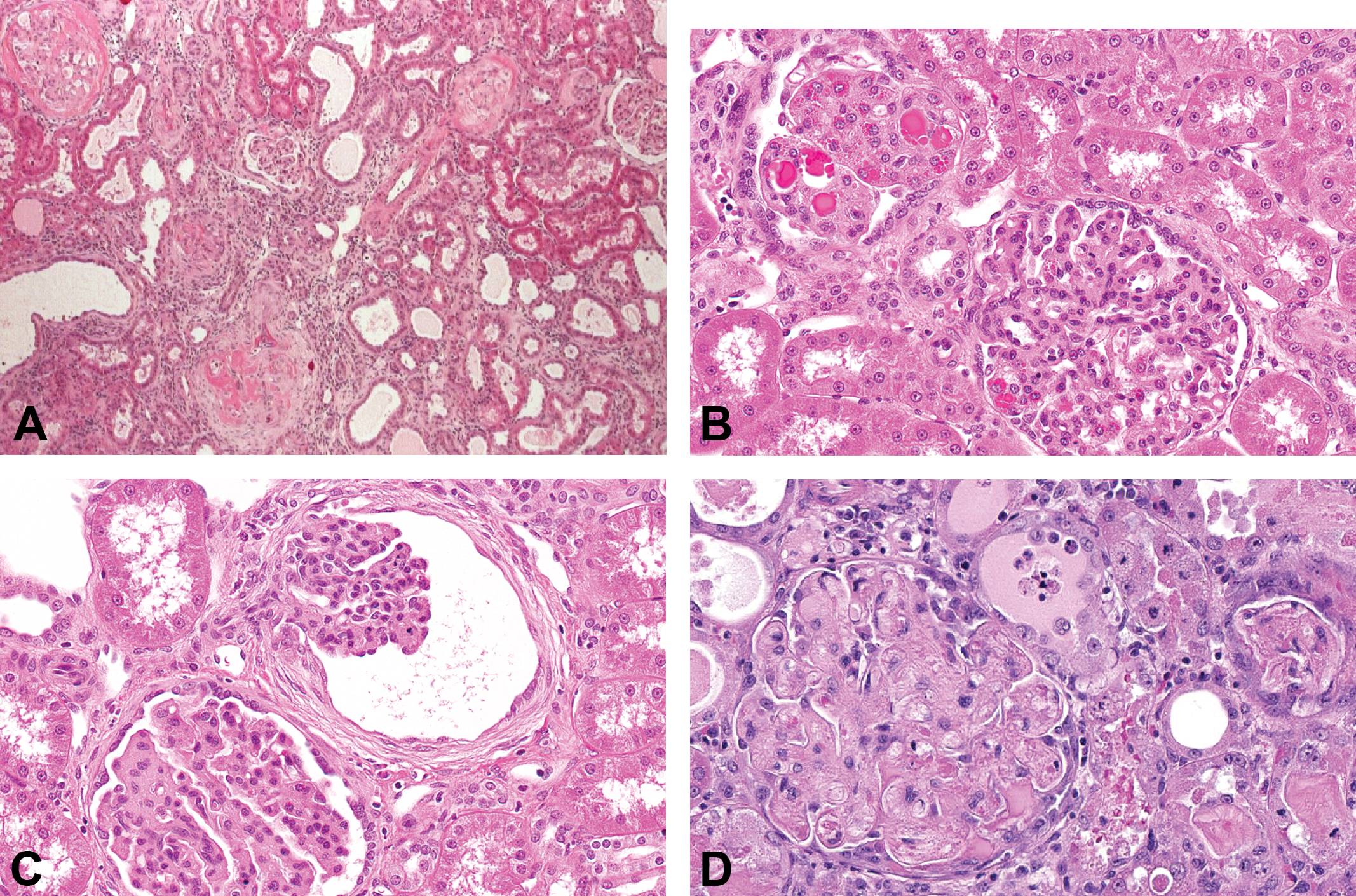
Four major types of preclinical glomerular injury noted in toxicity studies. (A) Rat kidney: administration of an anticancer agent resulted in direct cytotoxic injury, with hyalinized membranes and necrosis of glomerular cells. (B) Monkey kidney: accumulation of a vascular endothelial growth factor receptor inhibitor resulted in protein droplets and mesangial expansion. (C) Monkey kidney: age-related (spontaneous) glomerulosclerosis characterized by mesangial expansion and pericapsular fibrosis. (D) Monkey kidney: glomerulonephritis due to antidrug antibody-related immune complex disease. Note that fibrin, fibrocellular crescents (synechia), increased mesangial matrix and thickened basement membranes at the podocyte–endothelial interface due to immune deposits.
Similarly, the second mechanism may involve direct cellular injury. Degeneration or necrosis of glomerular cellular elements can arise from accumulation of a drug or chemical within podocytes, endothelial cells, or mesangial cells (Frazier et al. 2012). Absorption from the capillaries into endothelium may occur even with agents that are not rapidly passed into urinary filtrate, while xenobiotics enter mesangial cells after filtration due to phagocytic properties of this cell population. Accumulation of agents into podocytes may occur through either active or passive transport at the site of the filtration barrier within Bowman’s space. This accumulation of drug or its metabolite into the endolysosomal compartment can then initiate degenerative changes. The hallmark histopathologic lesion for this type of mechanism is the presence of inclusion bodies in one or more cell types. Ultrastructurally, variably sized, dark, homogenous osmiophilic bodies or even vacuoles may be present. Examples include the vascular endothelial growth factor (VEGF) inhibitors (Abbas et al. 2015; Frazier et al. 2015). In the case of VEGF inhibitors, however, the mechanism of toxicity is pharmacologic rather than through direct cytotoxicity. After endothelial uptake, thrombotic microangiopathy induces systemic and intrarenal endothelial injury as a result of disturbed angiogenesis through inhibition of the VEGF pathway (George and Nester 2014; Izzedine et al. 2010) and has been noted with both circulating VEGF decoy-receptor molecules (aflibercept) and VEGF receptor tyrosine-kinase inhibitors such as sunitinib, sorafenib, axitinib, and pazopanib (Frazier et al. 2015; Gurevich and Perazella 2009). Interestingly, glomerular lesions including evidence of podocyte injury (podocytouria and apoptosis by electron microscopy [EM]) can also be induced by a biotherapeutic anti-VEGF therapy, bevacizumab, providing proof of pharmacologic basis (Muller-Deile et al. 2010; Izzedine et al. 2014); however, inclusion bodies are not present within the cells.
The third mechanistic category of drug- or chemical-mediated glomerular toxicity is easiest to differentiate based on histopathology. Glomerulosclerosis refers to the replacement of glomerular matrix with fibrosis. It often has been associated with hemodynamic alterations and is a frequent accompaniment to chronic progressive nephropathy (CPN) in rats and hypertension in aged monkeys (Frazier et al. 2012; Yamada et al. 2013). With rodent CPN, glomerular lesions parallel effects in tubules and can be noted initially between 6 and 9 months of age with increased collagen deposition within mesangium and surrounding Bowman’s capsule. In cynomolgus monkeys, glomerular changes can be noted by 6 to 10 years of age but have been occasionally noted in even younger monkeys. While early findings are characterized by increased numbers of mesangial cells and mesangial expansion, the most consistent lesion involves a thin band of fibrosis around Bowman’s capsule, which becomes thickened with collagen given time. Not all cases of glomerulosclerosis are spontaneous background changes. A chemical that selectively induces glomerulosclerosis in rats is N-nitrosomorpholine (Romen, Bannasch, and Aterman 1975). Many other agents induce glomerular fibrosis indirectly such as toxicity by cisplatin through ill-defined mechanisms that likely involve primary mesangial injury and replacement of normal mesangial matrix over time (Frazier et al. 2000; Lee and Song 2009; Pabla and Dong 2008).
With a few exceptions, the three previous mechanistic classes of glomerular injury have largely been associated with small molecule drugs and chemicals. However, as noted in the beginning of the Introduction section, the increasing use of biotherapeutic agents and ASO therapies have resulted in a surge in preclinical cases of glomerulonephritis that are mechanistically linked to the fourth category: immune-mediated glomerular damage. The vast majority of drug- and chemical-related glomerulopathies in preclinical studies today are recognized to be immune mediated and driven by immune complex formation involving antibody and/or complement (Engelhardt 2016). Fortunately, clinical cases of drug-induced glomerular injury in humans occur only rarely. Immune-mediated injury from drug-induced autoimmunity occurs in less than 1% of patients exposed to drugs and of these patients only 5% develop lupus-like lesions or vasculitis in the kidney (Hogan, Markowitz, and Radhakrishnan 2015). The Spanish Study Group of Biological Agents in Autoimmune Disease (BIOGEAS) Registry reviewed approximately 13,000 reported cases of autoimmune diseases that developed in patients exposed to biotherapeutic agents, and only 50 cases of glomerulonephritis were noted. Of these, 50% were associated with tumor necrosis factor (TNF)-targeted therapies (Pérez-De-Lis et al. 2017). While antibody therapies and other peptide-based biologics represent most cases in laboratory animal toxicity studies and account for a few cases clinically, some small molecules such as penicillin, D-penicillamine, or cephalosporins have also been responsible for some cases of immune-mediated glomerulonephritis in humans, usually while acting as haptens or partial antigens to endogenous antibodies (Adams et al. 1993; Remuzzi and Perico 1995). Drug-induced lupus (DIL) is one such type of clinical adverse glomerular reaction. DIL is uncommon, but drugs most commonly associated with DIL are hydralazine and procainamide, with incidences as high as 8% to 20% during the first year of therapy (Rubin 2015). DIL has also been rarely reported with biotherapeutic therapies (e.g., anti-TNFα therapy) but at a much lower incidence (0.1%; De Bandt et al. 2015).
From a pathology perspective, glomerulonephritis can be subcategorized into membranoproliferative and crescentic forms in rodents, dogs, and monkeys (Frazier and Seely 2012). Both forms are associated with inflammatory cytokine involvement and potential complement activation within the glomerular capillaries. As the name suggests, membranoproliferative forms are centered on capillary basement membranes and tend to be chronic in progression. Crescentic forms may be either acute or chronic in their presentation and involve adhesions of visceral podocytes to Bowman’s capsule. In humans, crescentic glomerulonephritis often presents clinically in the form of rapidly progressive glomerulonephritis (RPGN) and is one of the most common presentations of drug-induced forms of glomerular injury (Zhou and Silva 2007). It can lead to renal failure and dialysis dependency. Both membranoproliferative and crescentic forms of glomerulonephritis have been associated with drug administration in laboratory animals (particularly monkeys) in toxicity studies (Frazier and Seely 2012), but there is poor concordance with human susceptibility and clinical translation is often of uncertain or questionable validity. This is largely because ADA formation is species-dependent, and ADA is by far the most common factor resulting in drug-induced glomerulonephritis in monkey studies. In humans, immune-mediated reactions to biologic agents also depend on the foreignness of the molecule, such that murine and chimeric antibodies have a greater potential to cause adverse clinical reactions than humanized antibodies. Therefore, preclinical ADA-related glomerulonephritis invariably fails to correlate with any incidence of renal or vascular injury in clinical trials in humans and is generally not a translatable toxicity (Rojko and Price-Schiavi 2008; Rojko et al. 2014; Van Meer et al. 2013). The regulatory agencies have agreed, as the International Conference on Harmonization S6R1 stipulates, “the demonstration of ADAs in any nonclinical species is not considered predictive for ADA response in humans” (ICH 2011).
Even with small molecules, the idiosyncratic pattern of drug-related glomerulonephritis demonstrates profound species sensitivity. For instance, while DIL-inducing drugs such as procainamide and hydralazine occur clinically, this type of reaction is incredibly rare in rodents, dogs, or monkeys, and only specific animal models (e.g., rabbits) have been demonstrated to be susceptible (Adams et al. 1993). Therefore, clinical cases of hapten-like glomerulonephritis with small molecule drugs have been unfortunately very difficult to predict using routine preclinical toxicity studies. Given the idiosyncratic nature of immune reactions in general and marked differences between species in susceptibility based on autoantibody formation, clinical translation and risk assessment can be challenging with any type of immune-mediated glomerulonephritis and especially those related to biotherapeutic administration. While drug-induced glomerulonephritis only represents a small fraction of clinical cases of glomerular injury, it is important for those working in preclinical drug development to understand the implications and ramifications of preclinical glomerular toxicity to put animal lesions in appropriate context and properly inform clinical risk assessment and decision-making and help prevent future clinical cases of drug-induced glomerulonephritis. This review will focus on the idiosyncrasies of biotherapeutic and ASO-related glomerulonephritis separately and provide some guidelines for development when investigators are faced with each of these types of reactions.
Biotherapeutic-associated Glomerulonephritis
Increasing numbers of biologic modalities are being investigated by drug companies, especially for inflammatory and oncology indications. Monkeys are often the pharmacologically relevant species for recombinant human therapeutic proteins and monoclonal antibodies (mAbs). Human proteins may differ enough from the native monkey proteins to be immunogenic resulting in an ADA response, but so-called humanized antibodies are much more likely to be immunogenic in monkeys (Brinks, Jirkoot, and Schellekens 2011; Leach et al. 2014; Ponce et al. 2009; Rojas et al. 2005). Surprisingly, less than 60% correlation has been demonstrated between ADA development in monkeys and ADA development in humans (Van Meer et al. 2013), and this has led to the established dogma that immunogenicity in animals is not considered predictive of immunogenicity in humans (Brinks, Jirkoot, and Schellekens 2011; Leach 2013; Leach et al. 2014). Beyond the simplest explanation that a foreign protein is being introduced into a heterologous species, there are additional reasons for the large discrepancy in prevalence between autoantibody formation in monkeys and humans with current biologic modalities. Cynomolgus and rhesus macaque monkeys seem to be prone toward immunoreactivity. In one report, 44% of monkeys previously treated with a human mAb developed ADAs that cross reacted with another test mAb in subsequent studies, indicating how common and cross-reactive ADAs can be in nonhuman primates (Han et al. 2015).
In the case of antibody/antibody-like drugs, cell surface targets have a greater potential for inducing immunogenicity than other targets (Leach et al. 2014). While preexisting circulating antibodies to the test article are rarely present prior to dosing, immune effects can develop rapidly after administration of the first drug dose (Leach et al. 2014). As far as glomerulonephritis development, monkeys are not only prone to immunoreactivity, they also appear to be more susceptible to the development or persistence of circulating immune complexes (CICs). Naturally occurring CICs are frequently encountered in cynomolgus monkeys and they appear to have higher levels of spontaneously occurring CICs than humans (Alexander, Clarkson, and Fulgham 1985; Hebert 1991; Hebert, Birmingham, Shen, et al. 1994). There are strain and age differences, as macaques originating from Indonesia have higher CIC levels than those from Malaya or the Philippines and adults have higher CIC levels than juveniles (Alexander, Clarkson, and Fulgham 1985). Mauritius-origin macaques appear to be more susceptible to immune-related phenomenon than other strains (Engelhardt et al. 2015). Likewise, Immune complex (IC) formation and deposition have been observed in glomeruli of undosed monkeys, representing the normally high levels of biodistribution and transport of CICs in macaques (Rojko et al. 2014). In addition, the clearance of these immune complexes appears to be slower than in humans (Cornacoff et al. 1983; Davies et al. 1990; Rojas et al. 2005).
Immune complex disease in monkeys treated with biotherapeutics is commonly associated with a high incidence of clinical signs, detectable ADA, and accelerated drug clearance (Kronenberg et al. 2017). ADAs typically occur within 10 to 14 days after initiation of dosing (Clarke et al. 2010; Leach 2013). IgG2 antibody isotype is most commonly associated with ADA-related immune reactions, but other isotypes may be involved (Leach et al. 2014). The larger and more complex the antibody protein (for instance, conjugation with albumin or other motifs), the greater the propensity for immunogenicity. Immune complexes can either form directly within the tissues or within the circulation. Antibody to antigen ratio is the most important factor determining whether a reaction will occur, but quantity, size, charge, and binding strength are also important factors (McGavin and Zachary 2005). The formation and deposition of IC depends on the balance between drug and ADA response and is not necessarily dose proportional (Rojko et al. 2014). Higher concentrations of antigen and antibody increase the risk for IC formation, but the lattice size and amount depend on the antigen to antibody ratio and in many cases very high doses inhibit IC persistence (Ponce et al. 2009). An excess of either antigen or antibodies usually results in smaller complexes, which can remain in circulation or are rapidly cleared. As the ratio of antibody to antigen increases, larger soluble immune complexes form which may persist in the circulation and deposit in the vascular wall activating complement causing vascular injury in tissues/organs. When an equal molar equivalence of antigen and antibody exists in circulation, large insoluble complexes can form which quickly precipitate and are cleared by phagocytes unless the clearance mechanisms are impeded or saturated. Under those conditions, the larger complexes can deposit in the vascular wall, activate complement, and cause tissue damage (Rojko et al. 2014).
When glomerulonephritis or vasculitis is encountered in a preclinical study involving the administration of a biologic, it has been common practice within the pharmaceutical industry to try to evaluate and attempt to correlate both ADA titers in individual monkeys and, in some cases, to perform immunohistochemistry (IHC) for IgG (both autogenous and exogenous), IgM, and complement in affected tissues (Abbas et al. 2015; Rojko et al. 2014; Frazier et al. 2015). In monkeys with drug-induced ADA-ICs in the kidney, mesangial IgM granular deposits are the most frequent finding (Rojko et al. 2014), but they can also be observed in monkeys with spontaneous glomerular diseases including control monkeys in toxicity studies (Rojko et al. 2014; Poskitt et al. 1974). When antigen has been demonstrated in glomerular granular deposits in experimental/toxicity studies involving administration of foreign proteins, it may be found in mesangial, subendothelial, and/or subepithelial locations (Rojko et al. 2014; Cosio, Hebert, et al. 1987; Cosio, Birmingham, et al. 1987; Hebert, Allhiser, and Koethe 1978; Hebert, 1991; Hebert, Birmingham, Mahan, et al. 1994; Hebert, Birmingham, Shen, et al. 1994).
IHC is not without procedural difficulties and should be undertaken with the understanding that data interpretation is often difficult. Importantly, IC granular deposits may only contain drug half of the time and can vary between individuals on a study or between studies (Rojko et al. 2014). In addition, some monkeys have increased IgM staining of the extraglomerular mesangium and distal tubule/macula densa regions (Rojko et al. 2014). While in many cases, the immunohistochemical approach definitively demonstrates a direct correlation in an individual animal, it is actually quite common for IC deposition to be seen in a study animal without evidence of an ADA response in that individual or vice versa. There are multiple reasons including ADA and IHC assay sensitivity issues, timing of the clearance of circulating ICs, and simply sampling discrepancies (especially in the case of vasculitis lesions, as glomerular immune complexes are much easier to detect). Different animals can have different outcomes based on their propensity to form and deposit ICs in tissue. Sometimes, the drug is not evident by IHC in the glomerular IC granular deposits. This can result from clearance of the drug during the recovery period, masking/steric hindrance by ADA, quantities below the limit of IHC detection, or just from not being present (Rojko et al. 2014). Increased glomerular granular deposits that do not contain drug may also result from other circulating autoantibodies that are induced by or from immunomodulation of other immune factors (e.g., complement) by the drug (Rojko et al. 2014). Different individuals have different immune responses which can vary over time and different IC structures can be present within the same individual (Frazier et al. 2015; Wener 2007). Additional factors that influence the development of ADA in an individual animal include the dose level, frequency of dosing, route of administration, genetic predisposition (human leukocyte antigen class and presence of genetic defects), age, immune status and competence, and presence of other diseases (Leach et al. 2014). For these reasons, it must be stressed that finding ADA in a monkey from the same dose group should be a strong suggestion that ADA may have also occurred in the individual in question regardless of individual assay results. With the concurrent presence of glomerulonephritis or vasculitis, immune complex pathogenesis should be highly suspected. Detectable/significant levels of ADA in any monkey from a preclinical study regardless of the presence or absence of vascular/glomerular lesions is considered evidence for an ADA response for that biotherapeutic. The same logic applies for detecting CICs. Sampling only the area of injury in only the individual monkey lessens the chances that IHC for CICs will be effective, and it is the authors’ suggestion (based on experience) that tissues from multiple animals from the same dosing group should be examined histologically by IHC. If no detectable ADA or CICs are noted in a preclinical study where vascular and/or glomerular lesions are found in multiple animals, the clinical risk should be considered increased as an ADA-independent mechanism must be considered.
If only one monkey is involved, conclusions may be difficult to draw if ADA or CICs are not present. Is it a spontaneous glomerular lesion or instead a case of non-ADA related, but still drug-induced glomerulonephritis? Additional procedures such as EM or special stains may be necessary to better characterize the glomerular lesions and help determine mechanism. Given only a limited amount of tissue can be examined by EM, it is best used in conjunction with IHC to support an overall interpretation (Leach et al. 2014). Matrix-assisted laser desorption/ionization-mass spectrometry imaging is an additional or alternative technique that can be utilized to visualize biotherapeutic drug, native immunoglobulin (Ig), and/or ICs directly within a tissue section (Lalowski et al. 2013; Groseclose et al. 2015). By using such techniques, other mechanisms of injury or even nondrug-related lesions may be identified/characterized, such as inclusion body glomerulopathy or glomerulosclerosis, respectively, to better inform clinical risk.
Rapid infusion of the biotherapeutic can result in greater immune complex formation and the potential for more severe immune reactions (Frazier et al. 2015). Generalized drug-ADA IC formation has also been associated with acute infusion reactions that can be severe enough to result in death due to systemic complement activation (Rojko et al. 2014). The complement, coagulation, and kinin systems are closely intertwined, and complement activation can promote activation of other inflammatory pathways. Bradykinin activation results in multiple systemic effects including increased vascular permeability, venular dilation, coronary and pulmonary vascular constriction, uterine and gastrointestinal smooth muscle contraction, bronchoconstriction, increased arachidonic acid metabolism, and hypotension (Joseph and Kaplan 2005; Robbins and Cotran 1999). Thus, triggering of the kinin pathway may explain the unresponsiveness to antihistamine pretreatment in some instances of acute infusion reactions (Rojko et al. 2014). Immune complexes can also cause platelet activation in primates by cross-linking of FcgRIIa resulting in pulmonary thromboemboli, complement activation, release of vasoactive amines, and possibly even disseminated intravascular coagulation (Del Conde et al. 2005; Hedin and Smedegard 1979; Radegran and McAslan 1972; Revenas, Smedegard, and Saldeen 1980; Smedegard, Revenas, and Arfors 1979; Frazier et al. 2015).
In the authors’ experience at GlaxoSmithKline over the last decade, glomerulonephritis occurs in two-thirds of cases of drug-induced CIC disease in monkeys preclinically, but vascular injury is commonly encountered in the same animal, and in one-third of cases, drug-induced vascular injury is present without glomerular involvement. Other institutions appear to have similar relative incidences (Leach 2013). Immune complex deposition may be more frequently observed in the glomerulus than in peripheral vessels as a consequence of the tissue sampling requirements for preclinical studies. Only a small percentage of vascular area are ever examined in a typical toxicity study. In monkeys with vascular/perivascular inflammation, the densely IHC-positive staining granular deposits can be discrete or aggregated and are found within the intima and/or media of affected small arteries/arterioles and less often small veins/venules or as globular intravascular microthrombi. They are most prominent at vascular branch points and/or at the internal elastic lamina consistent with the initial sites of IC deposition in vascular walls at points of turbulent flow/shear stress (Rojko et al. 2014; Cochrane and Hawkins 1968; Germuth 1953; Guilpain et al. 2005; Kauffmann et al. 1980; Kniker and Cochrane 1968; Sams 1985). More often the ICs contain IgM with lesser amounts of C3, drug, IgG, and rarely sC5b-9 (Rojko et al. 2014). The reason for the different phenotypic presentations (glomerular vs. vascular and intimal vs. medial) may be related to IC physiochemical characteristics which influence the tissue deposition location, ability to activate complement, and IC lattice formation (Nangaku and Couser 2005; Gauthier and Mannik 1990; Michelin et al. 2002; Theofilopoulos and Dixon 1979; Wener 2007; Mannik 1980). Larger-sized immune complexes are more likely to deposit in or around the intima, whereas smaller immune fragments can traverse the vascular wall, especially with endothelial compromise. Large ICs also can deposit in subendothelial and/or mesangial locations in kidney glomerulus (Mannik 1980), whereas small ICs are more likely to be retained in circulation, undergo dissociation, or cleared (Wener 2007). Circulating antigens can diffuse into the vessel wall at various levels. In situ immune complex formation can occur if circulating anti-antigen antibodies find their way to the antigen. Cationic antigens have a predilection for anionic sites in glomerular basement membrane, knee joint, or dermoepidermal junction. ICs containing cationic antibodies may deposit in glomerular and/or small myocardial blood vessel walls, whereas ICs containing neutrally charged antibodies often remain in the circulation (Krant, Gauthier, and Mannik 1989).
It is not uncommon for thrombosis, thrombocytopenia (from platelet consumption), hemorrhages, or other manifestations of vascular disease to occur systemically. Intratubule hemorrhage is occasionally encountered within the kidney of these cases. In the past, this has been attributed to glomerular filtration effects, but the lack of evidence for glomerular damage in many or most cases (especially locally in associated nephron segments) has led the authors to conclude that it more likely represents vascular damage in the peritubule interstitium from immune activation of the endothelium and leakage into adjacent tubules after secondary inflammatory effects on the basement membranes. Different Ig classes (IgG1, IgG3, IgM, IgA, etc.) have varying capacities for activating the classical or alternative complement pathways which can affect immune complex deposition within organs and/or vessels. Large ICs activate the classical complement pathway resulting in the attachment of C3b to the Fc region of the IC antibody component which mediates the binding of IC to the complement receptor 1 (CR1) on primate erythrocytes. Erythrocyte-bound ICs are cleared by liver Kupffer cells or spleen macrophages via FcR and/or CR1. If erythrocyte CR1 mechanisms in humans and monkeys are saturated, IC clearance can occur in liver and spleen macrophages via binding to platelet FcgRII (Mahan et al. 1993). When FcR- or complement-mediated clearance is saturated, large ICs can deposit in tissues or bind to FcgR on circulating or tissue neutrophils (Cornacoff et al. 1983; Kavai et al. 1988; Medgyesi et al. 1981; Ratnoff, Fearon, and Austen 1983; Waller et al. 1981; Wener 2007). In monkeys, ICs that bind to circulating neutrophils and monocytes may sequester in tissue capillaries causing vascular damage due to local cytokine release (Hart, Alexander, and Dransfield 2004). In addition, antibody strength may contribute to delayed IC clearance and tissue deposition; ICs formed from low-affinity or low-avidity antibodies are cleared more slowly than those having high-affinity and high-avidity antibodies and are more likely to deposit in the kidney glomerulus (Hebert et al. 1991; Herbert, Birmingham, Shen et al., 1994; Theofilopoulos and Dixon 1979). Thus, the diverse localization of biotherapeutic-induced injury and different individual animal phenotypic responses can be attributed to the variability associated with IC composition, sequestration, and immune cell activation.
The predilection sites for immune complexes deposition in tissues are small postcapillary venules where there is loss of laminar blood flow, sites of ultrafiltration where there is high pressure and fenestrated endothelium (e.g., choroid plexus, ciliary body, synovium, and glomeruli), and sites of turbulent blood flow (e.g., coronary artery branches off aorta, aortic bifurcations, cardiac valve leaflets, and glomerular tufts). In monkeys, the glomerular change is usually diagnosed as membranous/membranoproliferative glomerulopathy or glomerulonephritis (Rojko et al. 2014). For toxicologic pathologists, these terms should not be used interchangeably, and the difference in terminology should be based on whether or not there is evidence of capillary tuft inflammatory infiltrates, degeneration or whether there is evidence of synechia. Synechia (presence of adhesions of visceral podocytes to Bowman’s capsule) occur only with the presence of fibrin within the urinary space and, by definition, should be indicative of glomerulonephritis. Glomerulopathy is reserved for lesions which primarily involve mesangial hyperplasia and podocyte/filtration barrier degenerative effects without significant basement membrane changes (Frazier et al. 2012; Frazier and Seely 2012).
Biotherapeutics may also result in other types of immune-mediated glomerular disease which is unrelated to ADA formation. Occasionally, the drug can form large complexes or aggregates with itself or protein within the dosing solution that can result in immune recognition of a substance as “foreign” (Rosenberg 2006; Leach 2013). Nonendogenous IgG aggregates (i.e., mAb) can simulate immune complexes within the bloodstream causing local hypersensitivity reactions especially if they retain the capacity to fix complement (Vazquez-Rey and Lang 2011). Aggregates can form during the production, formulation, or repackaging stages that can be further enhanced by heat, shear stress, or agitation (Leach et al. 2014; Liu et al. 2011; Rojko et al. 2014; Vazquez-Rey and Lang 2011, Kahook et al. 2010). As noted in the Introduction section, the most commonly encountered human cases of drug-induced glomerulonephritis involve anti-TNFα antibody therapies. DIL disease may be limited to the skin and resemble lupus; however, glomerulonephritis has been reported in 7% of patients receiving anti-TNFα therapy (Stokes et al. 2005; Ramos-Casals et al. 2007). The lesion is not one involving ADA as it occurs preclinically in monkeys with most biologics, but one of other immunomodulatory pharmacologic activity (Rihova et al. 2005). It has been proposed that anti-TNFα agents can cause autoantibody production via a cytokine switch paradigm that results in a shift from Th1 to Th2 cytokine production (Rubin 2015; Singh, Mehrotra, and Agarwal 1999). In addition, anti-TNFα drugs may cause apoptosis in inflammatory cells, releasing antigens that trigger autoantibody production (Gonnet-Gracia et al. 2008). As more and more immunomodulatory biotherapeutics enter the drug discovery realm, particularly in immuno-oncology, there are an increasing number of these biologic drugs harkening to the anti-TNFα experience and resulting in glomerulonephritis in monkeys that seems unrelated to ADA development. These drugs pose a larger dilemma for the toxicologist, as the clinical risk increases and the preclinical findings cannot be so easily written off as the ADA cases. For these programs, mechanistic data, IHC results, and knowledge of pharmacology are necessary to properly frame clinical risk. Immune complex–mediated glomerulonephritis can lead to secondary renal changes in the tubules from protein cast–induced damage, tubuloglomerular feedback to the nephron, or from ischemia associated with immune complex–related vascular injury or even thrombosis. Tubules in the corticomedullary junction, and particularly thick ascending limb, pars recta (S3 segment of proximal tubules) and distal convoluted tubules are particularly susceptible to hypoxic injury (Frazier et al. 2012). However, the presence of significant diffuse cortical renal tubule toxicity (that does not involve a traditional ischemic pattern of toxicity) in addition to glomerular injury could suggest a mechanism involving pharmacologic activity of a particular biologic agent in addition to renal effects solely attributable to ADA and immune complexes.
Standard urinary biomarkers such as urine protein, urine albumin, urine protein: creatinine ratio, and/or urine albumin: creatinine ratio are considered the most useful analytes for detecting glomerulonephritis (Frazier and Seely 2012). Large increases (e.g., >1.5 g/L) or the presence of large molecular weight proteins in urine may aid in identifying glomerular origin and signal a risk for clinical glomerulonephritis. Small or trace amounts of protein and a predominance of albumin would be consistent with tubular effects and of considerably less clinical concern.
While the clinical risk for most cases of biotherapeutic-associated preclinical glomerulonephritis remains small, regulatory agency reviewers may want additional clinical biomarkers above urine protein analytes when monitoring subsequent clinical trials. Since the immune mechanisms and pathologic responses to a protein therapy are similar in humans and monkeys, immune analytes that translate across species including C-reactive protein (CRP) and monocyte chemoattractant protein-1 (MCP-1) might also be helpful for clinical (and preclinical) monitoring (Brinks, Jiskoot, and Schellekens 2011; Monach et al. 2011; Matsuyama et al. 2003; Egashira et al. 2002; Pai et al. 2016; Gonzalez-Quesada and Frangogiannis 2009; Frazier et al. 2015; Tarrant 2010; Demeule, Gurny, and Arvinte 2006). Radiolabeled imaging techniques have been recently utilized to demonstrate the formation, distribution, and clearance of ADA complexes in patients and cynomolgus monkeys, and the utility of this technology for imaging immune complex development in preclinical studies may be practical in the immediate future (van der Laken et al. 2007; Rojas et al. 2005).
ADA alone may not be a useful biomarker since it can be present in animals without any vascular injury (Alpers 2009; Nangaku and Couser 2005). However, reduction in plasma concentrations of the therapeutic protein or antibody along with detection of ADA prior to the onset of immune complex disease may be considered an early marker for its potential development (Heyen et al. 2014). There are other methods beyond determination of ADAs that can be used to detect evidence of an immune response. Patients in early clinical trials are often monitored using serum biomarkers for complement components (C3a, SC-5b9), cytokines (IL-6), coagulation parameters (activated partial thromboplastin time [APTT], partial thromboplastin time [PTT], and platelets), and serum albumin (Heyen et al. 2014). Similarly, in addition to routine hematology, clinical chemistry, and urinalysis evaluations, some possible assays for investigating immune-related toxicity study findings may include measurement of CICs, CR1, complement split products (C3a and C5b-9), CH50, IgE levels, cytokines, autoantibodies, and mast and basophil activation assays (Leach et al. 2014). Decreased CR1 expression on erythrocytes may be associated with chronic increased clearance of immune complexes (Cosio et al. 1990) and may be suggestive of ongoing immune complex formation. In addition, complement-mediated decreases in neutrophils and monocytes can occur (Birmingham et al. 1999; Davies et al. 1990; Smedegard, Revenas, and Saldeen 1980; Kinsell et al. 1941; Kopeloff and Kopeloff 1941). In monkeys with glomerular disease, proteinuria often coincides with reduced CR1 expression and reduced IC clearance (Cosio, Hebert, et al. 1987; Cosio, Birmingham, et al. 1987; Hebert 1991; Hebert, Birmingham, Shen, et al. 1994). In some cases, the deposition of immune complexes in glomeruli does not result in clinicopathologic alterations despite the presence of morphologic changes and determination of whether an immune response is affecting individual animals or a group is often based on a weight of evidence approach (Leach 2013). Even in such rare cases, glomerulonephritis is virtually always considered adverse.
ASO-associated Glomerulonephritis
ASOs are single stranded, synthetic deoxynucleotide sequences designed to hybridize to specific and complimentary mRNA sequences, which have been developed as therapeutic drugs for a variety of diseases. Renal tubular changes associated with ASO administration are considered class effects and have been well characterized (Henry et al. 1999, 2008, 2012; Marquis and Grindel 2000; Rao et al. 2004). Glomerular toxicity in laboratory animals has also been rarely noted with ASO administration. We previously reported the potential for some proinflammatory ASOs to induce renal glomerular pathology in mice and monkey following chronic administration (Frazier et al. 2014; Frazier 2015). Changes originally described as glomerulopathy were noted in preclinical toxicity studies of at least three months duration, involving drisapersen, a 2′-oMe phosphorothiorate (PS) ASO (Frazier et al. 2014). Enlarged, hypercellular glomeruli with increased mesangium and occasional inflammatory cells were noted in the kidneys of monkeys. Immune-mediated dense deposits, characterized by strong C3 complement fragments, were demonstrated by immunofluorescence with endothelial cell complement damage considered as the probable initial site and mechanism of injury. Glomerular changes were described in mice, given the same 2′-oMe PS ASO. Glomerular lesions in mice were characterized by increased mesangial matrix, increased cellularity, with or without inflammatory cells in glomerular tufts. With prolonged administration of greater than six months in mice, marked mesangial accumulation of Ig fragments mixed with amyloid developed, consistent with the murine syndromes of hyaline glomerulopathy and contributory amyloidosis (Frazier et al. 2014; Hoane et al. 2016; Linder, Pasternack, and Edgington 1972; Wojcinsky et al. 1991). Fibrillary deposits corresponded to immune fragments devoid of complement mixed with smaller deposits of amyloid. The mechanism of the changes was related to innate immune activation involving murine-specific toll-like receptor (TLR) activity as well as upregulation of serum amyloid A protein and was not considered necessarily relevant to humans (Frazier et al. 2014; Frazier 2015).
Subsequently, similar findings were noted in a few preclinical studies in monkeys and/or mice with other ASOs, but notably, these were primarily associated with PS ASOs of either 2′-oMe or 2′-MOE backbones (Frazier 2015; Engelhardt 2016). This should not be construed as a class effect of RNA silencing drugs. Drisapersen is a rather proinflammatory compound as compared to other ASOs, and the 2′-oMe structure tends to have more immunomodulatory activity than other oligonucleotide therapies such as locked nucleic acid, bicyclic nucleic acid, silencing RNA modalities, or even other 2′-MOE PS platforms. Several other early generation ASOs also were associated with minimal mesangial expansion in monkeys in chronic studies, but generally lacked the notable hyaline glomerulopathy changes associated with drisapersen in mice. While immunomodulatory activity was less than drisapersen for most, these older legacy compounds still tended to have significant complement activation noted in monkeys. The number of ASO toxicity studies where preclinical glomerular changes have been noted in the intervening years has represented a relatively small percentage of the total toxicity studies performed with oligonucleotide therapies. Interestingly, before the publication with drisapersen was available, many biotechnology companies in the ASO field were already screening for proinflammatory activity and most of the candidates in this class in the last four years now have comparatively limited inflammatory potential in mice or monkeys. Despite the drive to limit proinflammatory activity, however, glomerular lesions continue to occasionally arise in chronic ASO preclinical toxicity studies (Engelhardt 2016), so their pathogenesis needs to be explained to help inform clinical risk.
While proinflammatory effects appear to drive the pathogenesis in both monkeys and rodents, the morphology of changes, initiating stimuli, and defining mechanisms are different between the two species. Glomerular alterations in mice are distinctly fibrillary with immune fragment deposition and amyloidosis. In contrast, membranoproliferative glomerulonephritis with immune complexes centered on basement membranes is the predominant morphologic pattern in monkeys. The mechanism in mice centers on species specific hypersensitivity to effects of oligonucleotides on pattern recognition receptors of innate immunity including TLR and RIG-like receptors (RLR) and subsequent local cytokine activation (Bauer et al. 2008; Burel et al. 2012; Henry et al. 2008; Frazier 2015; Richardt-Pargmann and Vollmer 2009; Senn, Burel, and Henry 2005). Rather than TLR or RLR stimulation, monkeys appear to be highly sensitive to complement activation of oligonucleotides as compared to humans or other species (Chi, Gatti, and Papoian 2017; Henry et al. 1997, 2008; Engelhardt et al. 2015). Complement activation in monkeys is dependent on peak plasma concentration of ASOs. The specific pathophysiologic mechanism is related to inhibition of negative regulators of the alternative pathway of the complement cascade (factor H) by many ASOs, but this appears to have very limited clinical relevance as human complement activation occurs at much higher ASO plasma concentrations (Engelhardt et al. 2015; Henry et al. 2002, 2008; Shen et al. 2014). CpG and other proinflammatory sequence motifs are avoided when selecting current oligonucleotide drug candidates, and most companies screen compounds using in vitro assays and longer-term animal studies to limit complement binding and TLR activation prior to selection for development (Choi, Chung, and Jung 2010; Farman and Korburst 2003; Henry et al. 2008). Even if glomerular changes are observed in monkey studies, it is unlikely that they will correspond to any clinical kidney lesions with newer ASO therapies. Mesangial hyperplasia and thickened glomerular basement membranes have been noted in preclinical toxicity studies with multiple 2′-MOE PS ASOs in monkeys, but clinical correlates have not been demonstrated (Engelhardt 2016). Therefore, in most cases, drug development has been allowed to progress and clinical trials initiated without clinical adverse outcomes.
Very rare clinical cases of glomerulonephritis during clinical trials with ASOs have been reported in the literature, and a few of these have been suspected of relationship to drug treatment, but clinical details of these cases have largely been unavailable to firmly establish a connection (Crooke et al. 2018; Frazier 2015; IONIS 2017). In a review paper by authors from the Food and Drug Administration (FDA), it was acknowledged that the few clinical cases of ASO-related glomerulonephritis have occurred in association with other renal comorbidities (Chi, Gatti, and Papoian 2017). Authors also stated that clinical dose regimens designed to keep plasma concentrations below the threshold for complement activation may limit clinical risk (Chi, Gatti, and Papoian 2017; Shen et al. 2014). This perspective is largely based on the experience with drisapersen, which is a 2′-oMe PS ASO that resulted in the previously described glomerular lesions in mice and monkeys. At a 2013 DIA/FDA Oligonucleotide Therapeutics meeting, Dr. K. E. Myers described a case of membranous glomerulonephritis in an Anti-neutrophil cytoplasmic antibody (ANCA)-negative patient on a clinical trial given by drisapersen (Frazier 2015). Complement and Ig deposition were noted in and around basement membranes. The case was considered possibly drug-related, but the patient had some nonspecified chronic underlying or preexisting glomerular pathology. Complement fixation was considered a potential causal mechanism based on the fact that glomerulonephritis was noted in both monkey and human with this particular compound (Chi, Gatti, and Papoian 2017).
Other clinical cases of glomerulonephritis have been reported in clinical trials without substantiated drug relationship. In the summary basis of approval document for mipomersen, a 2′-MOE PS ASO, a serious adverse event of glomerulonephritis was described. The patient was ANCA positive, with membranous glomerulonephritis characterized by small and rare subepithelial deposits positive for IgG, C1q, and κ and λ light chains by immunofluorescence. The patient had a history of Reynaud’s phenomenon and was on several concurrent medications. According to the summary basis of approval, “antibodies to proteinase 3 found in this patient’s serum is virtually diagnostic of Wegener’s granulomatosis, microscopic polyarteritis nodosa, related forms of vasculitis or idiopathic necrotizing and crescentic glomerulonephritis,” and CICs were consistently negative in patients on this drug. In review of postmarketing literature, it appears no further clinical glomerular cases have been attributed to mipomersen administration and there was no incidence of glomerulonephritis in either mice or monkeys preclinically, further suggesting that this case may have been unrelated to treatment. Therefore, all evidence in this case would argue against any relationship to drug. Although the sponsor felt this patient was spontaneous in origin, this case highlights the potential risk of confusing background glomerular disease in patients in ASO clinical trials with drug-related kidney effects (FDA 2012).
Most recently, clinical adverse events related to renal glomeruli were reported in a news release regarding patients given inotersen in a clinical trial, which is another 2′-MOE PS ASO (IONIS 2017; Crooke et al. 2018). Like mipomersen, there were apparently no glomerular lesions noted in monkeys or mice in preclinical studies with inotersen (S. Henry and J. Engelhardt, pers. comm., 2017). It should be noted that the intended patient population for inotersen are transthyretin amyloidosis patients, who have not only substantial preexisting renal pathology but also likely have disrupted immunoregulation (Crooke et al. 2017, 2018; Ferriera et al. 2016; Goncalves, Texera-Coehlo, and Sarava 2014; Hugli and Muller-Eberhard 1978; Sousa et al. 2001). Complete data sets, background information, and regulatory perspective on these patients await publication of regulatory review documents from the New Drug Application, as inotersen is currently under review by the FDA. Other than the sole and likely spontaneous mipomersen case previously discussed, ASO-related glomerulonephritis or other serious renal clinical events have not been previously associated with the 2′-MOE platform as a group (Crooke et al. 2016). Ionis recently published a comprehensive review of renal safety with their catalog of 2′-MOE PS ASOs that demonstrated no statistically significant increases in the incidence of renal events in their extensive experience with ASO clinical administration (Crooke et al. 2018). The cases related to inotersen were excluded, but comparing a total database of 2,435 patients derived from phases 2 and 3 trials, only minimal and clinically insignificant changes in measures of renal function were noted in the overall population or even in subpopulations of diabetes patients or those with baseline renal insufficiency (Crooke et al. 2018). In that recent review, the authors acknowledged that two patients given inotersen experienced serious renal adverse events (one having developed chronic renal insufficiency), but also recognized that many Transthyretin (TTR) amyloidosis patients have progressive renal dysfunction caused by amyloid deposits in the kidney unrelated to treatment. Significantly, while two other inotersen-treated patients discontinued treatment due to a predefined renal stopping rule, one placebo-treated patient also discontinued due to a predefined renal stopping rule (Crooke et al. 2018). It is unclear from the publication or press releases how many of the four patients taken off drug therapy were ascribed to rapidly RPGN (and hence considered potentially drug-related adverse events) or were instead related to underlying glomerular amyloidosis. The authors did conclude that other cofactors or comorbidities may be necessary for nephrotoxicity to be expressed or for clinical deterioration in renal function to become apparent while undergoing ASO therapy, especially the presence of underlying renal compromise. This is not surprising, given what is known about immune complex organ deposition. Tissues with tall fenestrated epithelium lining vessels such as ciliary body, choroid plexus, and pancreas are preferential sites for immune complex vasculitis, as the fenestrae allow complexes to be deposited and “be protected” from clearance factors (Frazier et al. 2015). Similarly, preexisting damage to the glomerular matrix may allow focal areas where further immune complex deposition could occur. Glomerular capillary endothelium is already fenestrated, and any pits or pockets which result from glomerular tuft pathology might enhance immune complex survival.
Taken together, knowledge accumulated to date provides some helpful conclusions regarding glomerulonephritis with ASOs. Most importantly, it appears that the pathogenesis of ASO-related immune complex deposition in glomeruli is completely different in animals and humans, and certainly that preclinical glomerulonephritis does not necessarily predict clinical glomerulonephritis. Compounds that do cause flagrant or robust glomerular injury in mice are those that tend to be most immunomodulatory and those that induce lesions in monkeys are likely to affect complement in that species. Preclinical cases of glomerulonephritis likely indicate the potential for increased proinflammatory activity but do not highlight any significant increased clinical risk in the kidney. A more likely manifestation would be greater potential for injection site reactions. Conversely, clinical glomerulonephritis is an exceedingly rare event with ASO therapy and in the rare instances where this has unfortunately transpired, the few published cases suggest patients likely had underlying glomerular pathology.
The question arises of how might preclinical activity relate to the few known clinical cases of glomerulonephritis and help inform potential clinical mechanism? The precise pathophysiologic mechanism for the human cases remains elusive due to their extreme rarity and incomplete availability of clinical data. Given recent experience, mechanisms in the two species appear to be separate, and in monkeys, it is focused on complement activity. The exquisite sensitivity for complement inhibitory effects in monkeys is related to 3-fold greater complement inhibition of monkey factor H as compared to the human protein based on a sequence variant in the monkey complement factor H gene (Shen et al. 2014). Further, human complement dyscrasias typically result in stereotypic syndromes such as C3 glomerulonephritis and CFHR5 nephropathy (Barbour, Pickering, and Cook 2013; Zhou and Silva 2007). Complement-based glomerulopathies are characterized by strong immunofluorescence staining for complement in glomeruli and are generally inherited or autoimmune diseases affecting the alternative pathway, with little or no Ig deposits. The characteristic morphology of these diseases does not appear to be consistent with descriptions noted in the drisapersen or inotersen clinical cases.
Critically, while antibodies to ASO’s may be formed in humans and animals, they are generally nonneutralizing and are not considered to result in immune complex–related glomerulonephritis as is the case with biologics (Crooke et al. 2017; Henry et al. 2008). There are thus profound differences between ASO and biotherapeutics in regard to immune activation. However, lack of ADA involvement does not rule out the possibility of preformed autoantibodies to some other antigen that might somehow be upregulated with ASO administration. Alternatively, CICs in a predisposed patient with underlying glomerular pathology might develop or persist if the tightly regulated balance of formation and clearance is somehow affected with the addition of an ASO therapy. Even if either explanation is plausible, the translation of this hypothetical risk into a clinical phenomenon of glomerular immune complex formation and activation appears to be exceedingly rare. Predicting such clinical events is not possible with our current lack of understanding of these processes. The risk:benefit ratio for a given disease indication will determine how or if such extremely rare cases arising in clinical trials will affect a drug development program, but scientifically based and optimally timed clinical monitoring for urine or plasma biomarkers (as noted for biologic therapies) and careful patient selection are likely to help minimize risk to patients.
Conclusion
There is a common factor in the pathogenesis of biotherapeutic- and ASO-mediated glomerulonephritis in all laboratory animal species. Normal clearance processes of immune complexes are saturable and with increased formation or impaired clearance mechanisms, the complexes deposit within glomeruli and drive local inflammation and complement damage. These processes and the underlying predisposing factors (ADA and/or sensitivity to immunomodulatory factors) may differ slightly between the two therapeutic modalities, but understanding their pathogenesis and the biology that drives species specificity are necessary to fully understand and appreciate clinical risk.
“Spectre” is a synonym defined as an apparition, a mental representation of a haunting experience. In that sense, the use of the term spectre in the title of this review is an apt metaphor for the dilemma faced by drug development project teams when glomerular toxicity is observed in a subchronic or chronic toxicity animal study. The preclinical finding is certainly observable, but the clinical translatability of the toxicity lacks corporeal substance. With appropriate context applied via informed clinical risk assessment, and potentially with supporting data derived from additional investigative studies, the preclinical toxicity can be explained mechanistically and managed clinically. Drug development need not be terminated and patients may be afforded the opportunity to receive candidate medications with potential medical benefit. Unexpected or unexplained immune activation by a drug candidate remains a regulatory concern, but in many cases, immune activation in preclinical species can be predicted and has limited human relevance. Specific properties of an antibody therapy will greatly enhance the chances of ADA formation or direct immunomodulation in nonhuman primates. In the case of ASOs, highly proinflammatory drugs or those that most affect complement pathways may also increase the risk for preclinical glomerular injury, but in either case the ghost of preclinical glomerulonephritis need not haunt the clinical program.
Footnotes
Acknowledgment
The authors would like to thank Scott Henry and Jeff Engelhardt for prepublication review of this article.
Author Contributions
All authors (KS, LO) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.