
Abstract
Mass spectrometry (MS) has become a key platform in the clinical pathology laboratory and is being used more frequently for clinical pathology assessments in preclinical species for drug development studies. MS assays are being utilized for some traditional clinical pathology end points as well as novel biomarker analyses. For effective deployment in drug development toxicology studies, assays must be validated for use, and these validations are not very different from other bioanalytical platforms commonly found in the clinical pathology laboratory. Validations for MS assays include accuracy and precision assessments, analyte stability evaluations, carryover determinations, and recovery measures. The MS platform does present some unique challenges that should be considered, including ion suppression and availability of reference standards with MS data. Understanding the caveats of the MS platform is important for thorough validations and effective deployment.
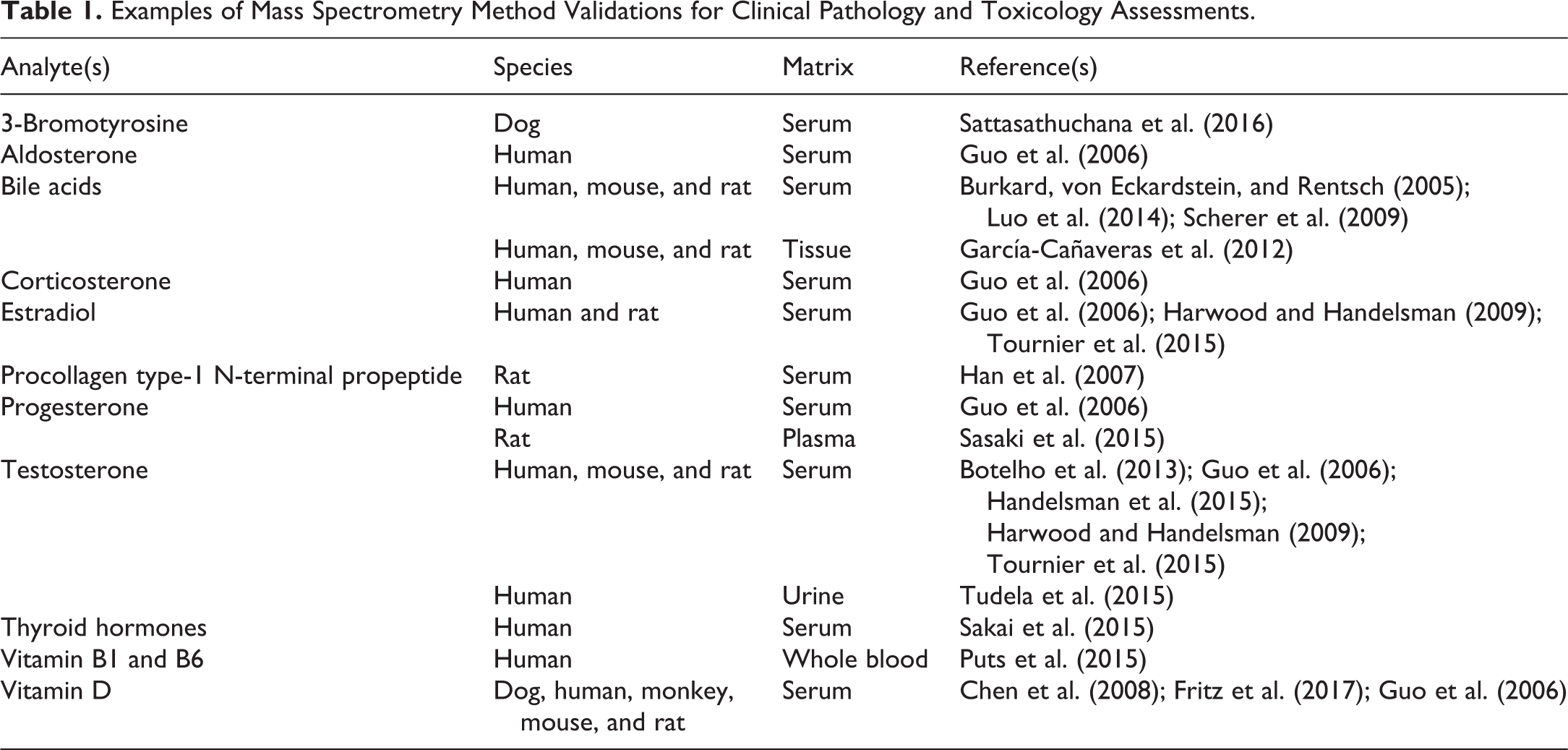
Mass spectrometry (MS) has become a platform that can be fully integrated into the clinical pathology laboratory over the past 5 years. This platform has become the gold standard for some clinical chemistry tests that directly measure the analyte of interest, such as vitamin D, triiodothyronine (T3), and thyroxine (T4), and is widely used for others, including newborn screening tests, drugs of abuse, testosterone, and other steroids (Wu and French 2013). As the translation of these tests is evaluated from clinical models to animal models, the application of liquid chromatography–mass spectrometry (LC-MS) assays in preclinical species for drug development studies is beginning to take hold. Table 1 provides a representative list of validated assays that have been deployed in the clinical pathology laboratory and drug development applications, and many other examples can be found in the literature(Jenkins 2015; Jones 2012; Moein 2017; Tournier 2015; Van Eeckhaut 2009). Assays developed for human samples can be modified for application in preclinical species. Typically, the major difference is the amount and/or type of sample available and the calibration range required. These data will enable drug-induced toxicity findings to be better understood in the preclinical models and how it may translate to humans for safer drug development.
Examples of Mass Spectrometry Method Validations for Clinical Pathology and Toxicology Assessments.
What makes the MS platform attractive for application in drug development toxicology studies is its ability to be applied to multiple species. Once an assay is developed for a particular analyte in one species, it can typically be applied to other species with little or no additional development. Often the difference is only the required range of detection, as the concentrations of endogenous molecules may differ between large and small animals. This capability often allows the laboratory to validate and maintain a single assay. Another benefit of MS is that typically no special reagents are required to monitor multiple species, as can be the case for many immuno-based assays that often require a different antibody for each species of interest. Although the equipment cost required for an LC-MS platform is relatively high in comparison to other quantitative systems, the overall reagent cost is low, making it an attractive alternative.
The LC-MS platform is also gaining wider use in toxicology with its ability to discover, validate, and deploy biomarkers. Laboratories are deploying metabolomics and proteomics assessments by LC-MS techniques to identify promising molecules. These techniques are then modified for further assessments of the potential biomarker, and if successful, into a validated assay that can be applied in drug development studies.
With the increased use in drug development toxicology programs, a wider variety of MS assays and applications are being deployed and integrated into toxicology studies (Table 1). Anatomic pathologists and clinical pathologists are using these data with histopathology assessments and traditional clinical chemistry end points for the interpretation of findings. This review provides general background information on MS assay validations with a focus on the LC-MS platform, yet many of the premises can be extrapolated to gas chromatography–mass spectrometry assays. A general overview of the LC-MS quantitative assay will be provided, followed by a description of typical validation procedures. Then challenges specific to the validation of LC-MS assays will be described.
LC-MS Quantitative Assays
Quantitative assays developed on LC-MS platforms have a similar overall experimental flow (Figure 1). An internal standard (IS) is added to the biological matrix in which the analyte is to be assessed. Typically, the IS is a stable-labeled version of the analyte to be measured. The samples are processed and injected onto the LC-MS platform. The liquid chromatography (LC) system separates the analytes from other endogenous molecules and then the MS monitors the effluent for the intact ion and a fragment ion unique to the analyte of interest. Assays have the capability to quantitate one analyte or multiple analytes.

Liquid chromatography–mass spectrometry (LC-MS) assay validation basics. The validation process for LC-MS assays is very similar to those for other bioanalytical platforms. First, an experimental procedure for the assay is developed. Second, appropriate samples for analysis are identified to conduct the validation. Finally, samples undergo the experimental procedure to conduct the validation and determine is the assay meets predetermined criteria. Additional validation parameters may be required depending on the nature of the molecule and the application of the assay.
The step for which most assays differ is the sample preparation. This preparation can be as simple as a dilution or a complex combination of extractions. Commonly used methods include protein precipitation, derivatization, and extraction. Different types of extractions include solid phase extraction, liquid/liquid extraction, supported liquid extraction, and immunoprecipitation. The method chosen depends on the complexity of the biological matrix, the characteristics of the analyte, and the concentration of the analyte in the matrix.
What makes LC-MS selective is that the analyte is separated by 3 analytical characteristics: its retention time on the LC column, the intact mass of the analyte, and the mass of a fragment ion. These characteristics are used to uniquely identify the analyte and confirm its identity with the use of known standards. Standards and biological samples are evaluated during method development to ensure that other endogenous molecules do not interfere with the signal. However, structural isomers can sometimes be difficult to separate. Instrument parameters are utilized to enable separation from other molecules and detect the molecule of interest.
The selection of the IS is another important aspect of assay development. A stable-labeled version of the analyte is the most preferred. For commonly analyzed small molecules, often stable-labeled versions are available that are high in purity. Finding commercial standards for peptides and proteins for preclinical species can sometimes be a challenge. Other alternatives for ISs include molecules with similar structures or those with similar column retention and physical–chemical properties.
Standard Assay Validation Parameters
The validation of an LC-MS assay is not that different from other bioanalytical assays (Table 2). Standards are used to construct a calibration curve for quantitation that covers the necessary range. Quality control (QC) standards are then used to evaluate accuracy, intra-assay precision (repeatability), and interassay precision (intermediate precision) at 4 concentrations over the calibration range. Often these standards are run at the high, middle, and low ends of the curve, as well as at the lower limit, in replicates of 6. Assessments are conducted in 3 to 6 runs over nonconsecutive days. If accepted reference standards are available, bias can be evaluated, too. Acceptable performance criteria should be defined prior to validation, and the application of the assay should be taken into consideration. To further evaluate assay precision, the validation should also include replicate analyses in a pooled sample that represents the samples for which the assay will be deployed.
Resources with Guidelines for the Validation of Quantitative LC-MS Assays.

Note: LC-MS = liquid chromatography–mass spectrometry; MS = mass spectrometry.
Many other aspects of the validation for quantitative LC-MS assays are similar to validations on other platforms. Stability of the analyte needs to be assessed, and typically includes long-term storage, freeze/thaw, in-process (or benchtop), and processed, in case a need for reanalysis occurs. Carryover can occur during sample processing as well as on multiple sites within the LC-MS platform. To evaluate whether or not carryover is present, the standard with the highest concentration is processed and injected on the system, followed by a blank injection. Carryover is calculated as a percentage of the signal from the blank to the highest standard. If carryover is present, the result is compared to the signal of the lowest standard to determine whether it will interfere. Recovery of the analyte from the sample preparation is another aspect that should be considered, especially if the process is complicated. This assessment is performed not only for the analyte but also for the IS. Often no guidelines are given for acceptable recovery other than the process be reproducible.
One aspect of the validation process that is often overlooked is biological verification of the assay, if applicable. This step is not the same as the qualification of the analyte as a biomarker yet provides valuable information for the deployment of the assay. Demonstration that the assay can detect differences that are biologically relevant is important. Samples that are expected to have different concentrations of the analyte (i.e., testosterone in male serum compared to female serum) should be identified.
Other factors may need to be considered for evaluation in the validation. Properties of the molecule, such as light sensitivity, may need to be built into the experimental plan. Sample collection can also be a variable to be considered. The site of collection, the method of collection, and the time from collection to processing have the potential to affect the concentration of the analyte in the sample.
Quantitation of multiple analytes in a sample is possible with LC-MS assays (Kruve et al. 2015a). Calibration curves are built with standards independently, or standards for different analytes can be combined. The QC standards are typically a mixture of the analytes at varying concentrations. The accuracy and precision of the different analytes can vary due to physical and chemical differences. Molecules with similar physical and chemical properties are easier to multiplex. Typically, favorable accuracy and precision values are more difficult to obtain, as the number of analytes in the assay increases. More analytes also means that the assay can be more difficult to develop and more challenging to pass validation criteria. Individual bile acids, steroids, and multiple forms of vitamin D are some examples of analytes that are multiplexed with LC-MS assays.
Validation Challenges Specific to LC-MS Assays
Many of the challenges that occur during the validation of LC-MS assays for animal models are also encountered with other bioanalytical platforms. Establishing the calibration curve for an endogenous molecule in the matrix of interest can be a challenge when the concentration of that molecule is relatively high. Surrogate matrices can be utilized, such as charcoal stripped or artificial serum (Jones et al. 2012). Obtaining enough volume from small animals for validation purposes can be difficult, especially in genetically modified mice. Also, for analytes at very low concentrations, reaching the necessary detection limits may not be possible.
Ion suppression or enhancement is one of the unique challenges encountered in the development and validation of LC-MS assays. These matrix effects occur when a substance in the matrix affects the ionization of the analyte in the mass spectrometer (Annesley 2003; Van Eeckhaut et al. 2009). This effect is often due to the affinity of the analyte and the substance to ions in the matrix. Both ion suppression and enhancement can be difficult to detect and are not common occurrences. In some instances, compensation of the ionization difference can be taken into account, but in others, quantitation becomes difficult, if not impossible.
Another challenge in the MS community is defining the lower limit of quantitation (LLOQ; Kruve et al. 2015a). Most of the guidelines for assay validation provide an acceptable signal to noise ratio to determine the LLOQ for the analyte. A minimal signal to noise requirement for the LLOQ is often 3:1 or 5:1 depending on the application of the assay. However, laboratory practices vary in the identification of the region of noise and the calculation of this signal, which can complicate comparing data between laboratories, especially when defining the assay’s sensitivity. Having a consistent, defined method would be beneficial.
As more LC-MS assays are being used in the clinical pathology laboratory, results between this platform and other platforms are being compared and evaluated (Sakai et al. 2015; Handelsman et al. 2015). Laboratories are determining which platforms are best suited for their applications based on sensitivity, scientific rationale, and operational efficiency. Every assay developed and platform utilized have different benefits depending on the capabilities and application. Whatever the reason for the comparison of an assay between platforms, different measurements can be observed, and scientists want to understand those differences. However, understanding the bias between assays is not straightforward, and these differences can exist due to many different reasons, such as differences in the detection method or the quality of the samples used for the assessment. Identifying the source of bias can be time-consuming and not always possible, which can make platform or assay selection challenging.
Calibrated reference standards are one tool that can aid in understanding bias between platforms, enabling platform comparisons and cross-laboratory comparisons (Bower et al. 2014). The number of certified standards available commercially is limited, and data from LC-MS platforms are even more limited. Efforts to standardize certain analytes, such as hormones and vitamin D, are aiding in the progression of more consistent data between laboratories for more accurate measures.
Summary and Conclusions
Properly validated LC-MS assays are necessary for the successful evaluation and integration of results from drug development toxicology studies. Considerations for the validation of LC-MS methods are very similar to those for other bioanalytical methods. The entire validation must demonstrate that the assay is suitable for its intended application to provide confidence in the data generated. As with any platform, understanding the caveats of MS methods is important to ensure the validation encompasses the proper evaluations. As LC-MS assays are deployed in toxicology studies, robust validations will be necessary to ensure data integrity and regulatory acceptance.
Footnotes
Acknowledgments
The author thanks the individuals in the Pfizer Drug Safety Research and Development Biomarkers group, especially Jiri Aubrecht, Carol Fritz, Lina Luo, and Kimberly Navetta, for their support of this article. The author also thanks Christopher Colangelo for his review and input.
Author Contribution
JC contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. The author gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.