
Abstract
Lung cancer is the number one cause of cancer-related deaths in humans worldwide. Environmental factors play an important role in the epidemiology of these cancers. Rodents are the most common experimental model to study human lung cancers and are frequently used in bioassays to identify environmental exposure hazards associated with lung cancer. Lung tumors in rodents are common, particularly in certain strains of mice. Rodent lung tumors are predominantly bronchioloalveolar carcinomas and usually follow a progressive continuum of hyperplasia to adenoma to carcinoma. Human lung cancers are phenotypically more diverse and broadly constitute 2 types: small cell lung cancers and nonsmall cell lung cancers (NSCLCs). Rodent lung tumors resulting from exposure to environmental agents are comparable with certain adenocarcinomas that are a subset of human NSCLCs. Human pulmonary carcinomas differ from rodent lung tumors by exhibiting greater morphologic heterogeneity (encompassing squamous cell, neuroendocrine, mucinous, sarcomatoid, and multiple cell combinations), higher metastatic rate, higher stromal response, aggressive clinical behavior, and lack of a clear continuum of proliferative lesions. In spite of these differences, rodent lung tumors recapitulate several fundamental aspects of human lung tumor biology at the morphologic and molecular level, especially in lung cancers resulting from exposure to environmental carcinogens.
Introduction
“If an experiment yields a clear-cut negative result, there is little discussion about the meaning or the meaninglessness of animal studies. When a clear-cut and strong positive result occurs, there is also little discussion. When the result is a slightly positive experiment, interpretation becomes difficult and discussion becomes lengthy. Biology, unfortunately, does not come only in black and white, but in many shades of gray, and in these gray areas disagreement is particularly evident.” (Rall 1988)
The above quote from Dr. David Rall, a pioneer and leader in environmental health sciences, is very germane when comparing rodent models to human diseases including lung cancer from environmental exposures (Rall 1988; Huff 1999). The goal of this brief review was to present pros and cons on the suitability of rodent models to study environmentally induced human lung cancers. Of course, as Dr. Rall suggested, we are all well aware that the issue of suitability of the rodent models to study environmentally induced lung cancers in humans is not black or white but has a gray scale spectrum. This mainly depends on the nature of exposures and on the unique biology of rodent species that differs from humans in certain aspects but is also similar in many ways.
Epidemiological studies provide direct information about potential associations between an environmental challenge and the risk of human cancer. However, there are several limitations for these studies, such as availability of suitable cohorts, long latency of cancer, limitations in the strength of causal inferences, poorly defined exposures, and several other biases (Cogliano et al. 2004). Epidemiological studies are very important in making unexpected and often logic defying associations of potential environmental exposures and human disease but in order to determine causality between these exposures and disease, animal studies involving chronic exposure to the environmental challenge are indispensable. However, cancer bioassays also have some limitations associated with methods of exposure, complex mixtures, limited genetic variation, and species-specific metabolism differences. Nonetheless, animal studies are essential for hazard identification and risk assessment of environmental exposures (Huff 1999). In this article, I will include some of the key points discussed during the 33rd STP Annual Symposium talk “Rodent vs. Human Lung Cancer: The Good, the Bad, and the Ugly!”. The main focus will be on the similarities and differences between human and rodent lung tumors as well as some issues related to the value of rodent models (rats and mice) for studying environmentally induced lung cancers.
Brief Overview on the Comparative Pathobiology of Rodent and Human Lung Tumors
Lung cancer is the number 1 cause of cancer-related deaths in humans worldwide. In the United States, lung cancer incidence is comparable with the combined cancer-related deaths resulting from the cancers of breast, pancreas, prostate, and colon and rectum (U.S. Cancer Statistics Working Group 2013). The majority (˜85%) of the lung cancer–related deaths are due to tobacco smoking; however, a significant number of lung cancer–related deaths are due to occupational or environmental exposures, such as radon, asbestos, crystalline silica, mixtures of polycyclic aromatic hydrocarbons, heavy metals, and air pollution (Travis et al. 1999). In recent times, because of a significant decrease in tobacco smoking, there is a corresponding decrease in the incidence of lung cancer. However, there is no decrease in lung cancer–related deaths due to factors other than smoking.
Lung tumors in rodents and humans are classified based on different criteria. Rodent lung tumors are primarily classified based on their cellular origin and/or location within the airways, whereas human lung tumors are primarily classified based on more descriptive morphologic features with prognostic value (Nikitin et al. 2004). As a result, as discussed subsequently, the rodent and human histological terminology cannot be used interchangeably.
Lung cancers in humans are usually divided into 2 major groups: nonsmall cell lung cancer (NSCLC) and small cell lung cancer (SCLC; Travis et al. 2011). NSCLC includes all epithelial lung cancers other than SCLC, most of which exhibit neuroendocrine morphology and markers. NSCLC comprise 80% of all lung tumors and have a variety of morphologies that include adenocarcinomas, squamous cell carcinomas, adenosquamous carcinomas, large cell carcinomas, and sarcomatoid carcinomas. Adenocarcinomas account for 30% to 40% of lung cancers and often arise in peripheral regions of the lung and may have a lepidic (neoplastic cells lining the alveolar walls), acinar, papillary, micropapillary, solid, and several other variants including invasive mucinous, colloid, fetal, and enteric morphologies (Travis et al. 2011). Squamous cell carcinomas constitute 20% to 25% of all lung cancers and arise centrally within large bronchi and have been largely associated with tobacco smoking. Large cell carcinomas account for 15% to 20% and occur diffusely in the lung and are characterized by large cells with large nuclei and prominent nucleoli and lack of glandular or squamous differentiation (Tuveson and Jacks 1999). About 18% of lung tumors may be categorized as SCLC that has a neuroendocrine morphology and a very high metastatic potential. The remaining 2% are neuroendocrine tumors consisting of typical and atypical carcinoids (Meuwissen and Berns 2005).
According to the recent classification of the human lung adenocarcinomas by the International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society, the term bronchioloalveolar carcinoma is no longer used for resected lung specimens. In its place, new terms for small solitary adenocarcinomas have been proposed: adenocarcinoma in situ (AIS) with pure lepidic growth and no invasion, minimally invasive adenocarcinoma (MIA) with predominant lepidic growth and with <5-mm invasion, and lepidic predominant adenocarcinoma with >5-mm invasion (Travis et al. 2011). The term bronchioloalveolar carcinoma used in rodents is not equivalent to the human lesion (Nikitin et al. 2004). Human lung cancer has a high degree of histologic heterogeneity within each tumor mass and may contain glandular, squamous, and neuroendocrine components, and the commonly diagnosed “mixed” subtype accounts for more than 90% of all resected lung adenocarcinomas. The recent classification also recommended not using the term mixed subtype and instead recommended classifying these invasive adenocarcinomas based on the predominant histologic pattern such as lepidic, acinar, papillary, micropapillary, and solid (Travis et al. 2011). Other differences between rodent and human lung tumors include greater stromal response and higher metastatic rates in human lung tumors compared with mouse lung tumors. The rodent lung tumors, especially the chemically induced tumors, have higher tumor multiplicity than human lung cancers (Nikitin et al. 2004).
On the contrary, the rodent lung tumors are predominantly of a single histologic type (Dixon et al. 1999; Boorman and Eustis 1990). The most commonly occurring lung tumors in rodents are termed bronchioloalveolar adenomas or carcinomas (Boorman and Eustis 1990; Dixon et al. 1999; Renne et al. 2009). This terminology is used in rodent lung tumors to primarily account for the location (bronchiolar alveolar region) of the tumors and the cell of origin (type II pneumocytes or Clara [club] cells) and is not typically comparable with the human lesion bearing the same name as described previously (Nikitin et al. 2004). The histologic heterogeneity of the rodent bronchioloalveolar tumors is very limited comparable with the human adenocarcinomas. The bronchioloalveolar tumors in rodents may be seen in vehicle control or chemically treated groups. However, other types of lung tumors such as cystic keratinizing epitheliomas and squamous cell carcinomas in rodents are observed with certain chemical inhalation exposures such as dioxin (2,3,7,8-tetrachlorodibenzo-para-dioxin [TCDD]) and particulates (titanium dioxide, talc, and nickel oxide) but not in unexposed control rodents (Dixon et al. 2008). Pulmonary neuroendocrine tumors (similar to SCLC in humans) that account for a significant number (˜20%) of tumors in tobacco smokers have not been reported in rodents (Nikitin et al. 2004).
Lung tumors in rats and mice originate primarily in peripheral lung involving the distal bronchioles and alveolar acini, and bronchial or proximal bronchiolar tumors are very rare. There is a well-documented and accepted paradigm of progression of the rodent lung tumors in the form of a continuum: hyperplasia to adenoma to carcinoma. In spite of the use of diagnoses of preinvasive lesions such as AIS and MIA, the progression of human lung adenocarcinomas from the preinvasive stage to malignancy is poorly understood. However, the lesion that was designated as atypical adenomatous hyperplasia in humans bears histologic similarities to alveolar hyperplasia in rodents and is thought to be a precursor lesion for peripheral lung adenocarcinomas. The term adenoma is reserved for only a few select, uncommon human lung tumors.
The chronic rodent (rat and mouse) bioassay is the standard test to assess carcinogenicity of environmental agents suspected of causing cancers in humans (Cogliano 2006). There are species, strain, and sex differences in the incidence of lung tumors in rodents (Boorman and Eustis 1990; Dixon et al. 1999). The male rodent has a higher lung tumor incidence than the female. Mice have higher incidences of spontaneous and chemically induced proliferative lung lesions (hyperplasia, adenoma, and carcinoma) than rats (Boorman and Eustis 1990; Dixon et al. 1999). There are strain differences in both species. In mice, the incidence of spontaneous lung tumors is strain dependent and the incidence of chemically induced lung tumors also follows the same strain dependency (Malkinson 2001). The order of decreasing incidences of spontaneous lung tumors in mice is A/J (82%) > SWR/J (47%) > BALB/c (33%) > CBA (17%) > C3H (9%) > C57BL6 (3%; Manenti and Dragani 2005). The higher lung tumor susceptibility of mice is due to the pulmonary adenoma susceptibility 1 (Pas1) locus, and the differential lung tumor susceptibility of various mouse strains has been attributed to Pas1 locus polymorphisms that can be of either an A/J- or C57BL/6J-type Pas1 haplotypes. The A/J-type haplotype has a higher spontaneous lung tumor incidence than the C57BL/6J-type haplotype. The National Toxicology Program (NTP)’s mouse model (B6C3F1) has the C57BL/6J-type haplotype since both the parent C57BL6 and C3H strains have the C57BL/6J-type haplotype (Manenti and Dragani 2005). The incidence of spontaneous lung tumors in male and female B6C3F1 mice is 27.7% and 9.5% (n = 950/sex; NTP 2013). The strain differences in the incidence of spontaneous lung tumors in the rat are not as striking as in the mouse. The order of decreasing incidences of spontaneous lung tumors in various rat strains is F344 (1.9%), Lewis (1.8%) > Osborne Mendel (0.7%), Brown Norway (0.6%) > Sprague Dawley (0.5%), Wistar (0.5%), CD (0.4%) > ACI/N (0%; Manenti and Dragani 2005). The status of Pas1 locus in various rat strains needs to be determined. The incidence of spontaneous lung tumors in male and female F344 rats is 3.6% and 1.4%, respectively (n = 700/sex; NTP 2013).
Lung cancer of rodents and humans shares several important morphologic and molecular similarities (Nikitin et al. 2004; Malkinson 2001). The bronchioloalveolar tumors in rodents are very similar to the human nonsmall cell carcinomas, adenocarcinoma subtype. The majority of the human lung tumors are due to tobacco smoke, and the tobacco smoke–induced lung tumors are primarily bronchial or central in origin and are predominantly squamous cell carcinomas or small cell carcinomas (Travis et al. 1999). However, a significant decrease in tobacco smoking, or cessation of smoking, improved diagnosis, and use of filtered cigarettes appears to have resulted in alterations in the incidences and types of lung tumors in humans. As a result of the above-mentioned factors, there is a reduction in the incidences of bronchial squamous cell carcinomas and small cell carcinomas and an increase in the incidences of adenocarcinomas (Devesa et al. 2005). Interestingly, cessation of smoking in previous smokers results in a decrease in the incidences of bronchial squamous cell carcinomas and small cell carcinomas but not adenocarcinomas (Khuder and Mutgi 2001; Jedrychowski et al. 1992). Better imaging technologies and diagnostic tests have also contributed to the higher diagnoses of adenocarcinomas.
There is a significant increase in the incidence of peripheral pulmonary adenocarcinomas in nonsmokers, women, and Asians (Devesa et al. 2005; Scagliotti, Longo, and Novello 2009). Since the rodent lung tumors are predominantly adenocarcinomas with comparable morphology with human adenocarcinomas (papillary, acinar, and solid), they are more relevant for the study of environmentally induced lung cancers. This is particularly relevant in the majority of the population that does not smoke but still is exposed to environmental carcinogens.
The KRAS gene is commonly altered in human and rodent lung tumors (Wakamatsu et al. 2007). The KRAS mutations in humans are primarily targeted within codon 12 followed by codons 61 and 13, and the same trend is seen in mouse tumors. The predominant KRAS mutation in pulmonary adenocarcinomas in nonsmokers and spontaneously arising bronchioloalveolar carcinomas in mice is a G to A transition. Interestingly, the pulmonary adenocarcinomas in smokers and chemically induced bronchioloalveolar carcinomas in mice usually harbor G to T transversions (Husgafvel-Pursiainen and Kannio 1996; Hong et al. 2007, 2008; Riely et al. 2008; Sills et al. 1999). Meta-analysis of transcriptomic alterations in human and mouse lung tumors revealed significant similarities in lung cancer pathways in both species (Stearman et al. 2005; Bonner et al. 2004; Pandiri et al. 2012). These data indicate that mouse lung tumors are similar to human adenocarcinomas at the morphologic and molecular levels and that mouse lung tumors are relevant in evaluating carcinogenic hazards associated with environmental exposures.
It is pertinent to note that rodents played a very important role in detecting environmental carcinogens even before epidemiologic studies suspected any association of these agents with human cancer. Examples include asbestos, beryllium, cadmium, 1,3-butadiene, bis(chloromethyl) ether, ethylene oxide, glass wool, sulfur mustard, radon gas, crystalline silica, vinyl chloride, and TCDD. In a recent workshop organized by the U.S. Environmental Protection Agency on mouse lung tumors, Dr. Dan Krewski from the University of Ottawa presented information on the human and rodent cancer site concordance of International Agency for Research on Cancer (IARC) group I agents (109; Krewski 2014). Tumors in the lung had greater site concordance than any other organ in the body, indicating that rodents are indeed most suitable to study environmental pulmonary carcinogens. Not surprisingly, the majority (~73%) of these 109 IARC group I agents are genotoxic carcinogens. However, this list also includes a significant number (~27%) of nongenotoxic carcinogens (Hernandez et al. 2009).
In subsequent sections, I will discuss some limitations of using rodent models, especially the mouse, in evaluating suspected pulmonary carcinogens.
Examples of Clear Human Carcinogens That Were Difficult to Model in Rodents
There are overwhelming epidemiologic data demonstrating the lung cancer hazard associated with exposures to arsenic and tobacco smoke. In fact, arsenic and tobacco smoke are IARC group I carcinogens that cause human lung cancer. However, unequivocal demonstration of lung cancer hazard using rodent models has proved elusive for a long time.
Arsenic and inorganic arsenic compounds have long been considered a conundrum in the field of chemically induced carcinogenesis. The first International Agency for Research on Cancer (IARC 1973) monograph indicated that inorganic arsenic exposure was consistently linked to human skin cancer but supporting animal carcinogenicity data were lacking (IARC 1973). The IARC (1987) document indicated that the evidence of carcinogenicity of arsenic in rodents was inadequate or limited. Since the 1990s, there is an increasing accumulation of data demonstrating the carcinogenicity of arsenic in rodents but even in the recent past, arsenic was considered a paradoxical carcinogen due to lack of unequivocal carcinogenicity in common laboratory animal models (National Science Foundation 2005). In a recent review on the topic, Tokar et al. (2010) noted that arsenic carcinogenicity has been demonstrated in rodents exposed to arsenic transplacentally and whole life, or in rodent models that were predisposed to cancer, which are not common protocols in the chronic rodent bioassays (Tokar et al. 2010; Waalkes, Ward, and Diwan 2004; Hayashi et al. 1998). In addition, recently, Waalkes et al. (2014) at NIEHS have demonstrated lung tumor induction in the CD1 mouse by whole-life exposure to sodium arsenite (50 ppb) at very low doses (in ppb) that are relevant at environmental exposure levels. In light of all these data, considering arsenic as a paradoxical carcinogen is no longer tenable (IARC 2012a).
Tobacco smoke is a known human carcinogen, but it was difficult to model pulmonary carcinogenesis in rodent models using tobacco smoke. Tobacco smoke is a complex mixture with more than 5,300 compounds, of which more than 70 constituents have been shown to have sufficient evidence of carcinogenicity in either laboratory animals or humans (IARC 2004). Tobacco smoke has been recognized as a cause of human lung cancer since the 1940s, but the unequivocal demonstration of lung tumors in rodents has not been very successful (Lorenz et al. 1943; Otto and Elmenhorst 1967; Harris and Negroni 1967). Some of the early studies may have been limited by using mouse strains with high background spontaneous lung tumor incidence and lack of sufficient challenge exposures. However, protocols involving lifetime exposures in C57BL6 mice (Harris et al. 1974), Snell’s mice (Leuchtenberger and Leuchtenberger 1970), and B6C3F1 mice (Hutt et al. 2005) have resulted in a significant increase in lung tumors. Whole-body exposure of Swiss albino mice to tobacco smoke within 12 hr of birth for 120 days have resulted in lung tumors (Balansky et al. 2007). Witschi et al. (2002) used an unconventional protocol of 5-month tobacco smoke exposure followed by a 4-month recovery period in air and found increases in lung tumor multiplicities compared with controls using Balb/c, SWR, and AJ mice. Tobacco smoke exposure studies in rats were also not uniformly positive. Lifetime exposure of tobacco smoke to Wistar rats did not increase lung tumor incidence (Davis et al. 1975). However, lifetime tobacco smoke exposure of F344 rats (Dalbey et al. 1980; Mauderly et al. 2004) and CDF rats (Finch et al. 1995) resulted in an increase in lung tumor incidence. Thus, life time, perinatal exposure or certain exposure protocols of rodents to tobacco smoke results in pulmonary adenomas and carcinomas, and the idea that tobacco smoke carcinogenesis cannot be replicated in rodents is not warranted (IARC 2012c).
Examples of Chemicals That Are Pulmonary Carcinogens in Only 1 Species
In a survey of 580 NTP chronic bioassays, there were only 67 bioassays where the same chemical was tested in both mice and rats and at least 1 species had a lung tumor response. Of these 67 bioassays, only 14 (21%) had lung tumor site concordance for both rats and mice. It shows that there is no absolute site concordance for lung (or for any other organ) in both rat and mouse chronic bioassays. Chemically induced lung tumor incidence in the mouse is slightly more than twice that observed in the rat (Table 1). Chemicals such as styrene, naphthalene, coumarin, ethylbenzene, cumene, and benzofuran are pulmonary carcinogens only in the mouse but not in the rat (NTP 1979, 1992, 1993, 1999, 2009, 1989; Chan 1992). With the exception of styrene, all these chemicals are carcinogenic in both rats and mice, but interestingly the lung tumors were observed only in mice but not in rats. The unique species specificity of the mouse at least to styrene is mainly due to the unique CYP isoform CYP2F2 in the mouse Clara (club) cells compared with CYP2F4 in rats and CYP2F1 in humans. CYP2F2 is highly efficient in metabolizing styrene to styrene 7,8-oxide (SO) and other metabolites. These metabolites are cytotoxic to Clara cells and cause regenerative hyperplasia and subsequently result in proliferative lung lesions including neoplasia (Cruzan et al. 2009). On the other hand, styrene is also metabolized by CYP2E1 in the liver in rats, mice, and humans resulting in the formation of SO and other metabolites that need further characterization. In addition, occupational exposure to styrene leads to the formation of O6-deoxyguanosine, O6-(2-hydroxy-1-phenylethyl)-2′-deoxyguanosine-3′-monophosphate, and N7-deoxyguanosine adducts in DNA. Low levels of these 2 adducts were also detected in liver of mice and rats exposed to styrene. Thus although there is no site concordance of styrene-induced mouse lung tumors with rats and humans, the presence of the DNA adducts, chromosomal damage, and some common metabolites that are genotoxic is of concern. As a result, IARC considers styrene as probably carcinogenic (group 2B) to humans, and the NTP Report on Carcinogens considers styrene to be reasonably anticipated to be a human carcinogen (NTP 2011; IARC 2002; National Research Council [NRC] 2014).
Species differences in chemically induced lung tumor response (with clear or some evidence of carcinogenicity) in NTP chronic bioassays.a
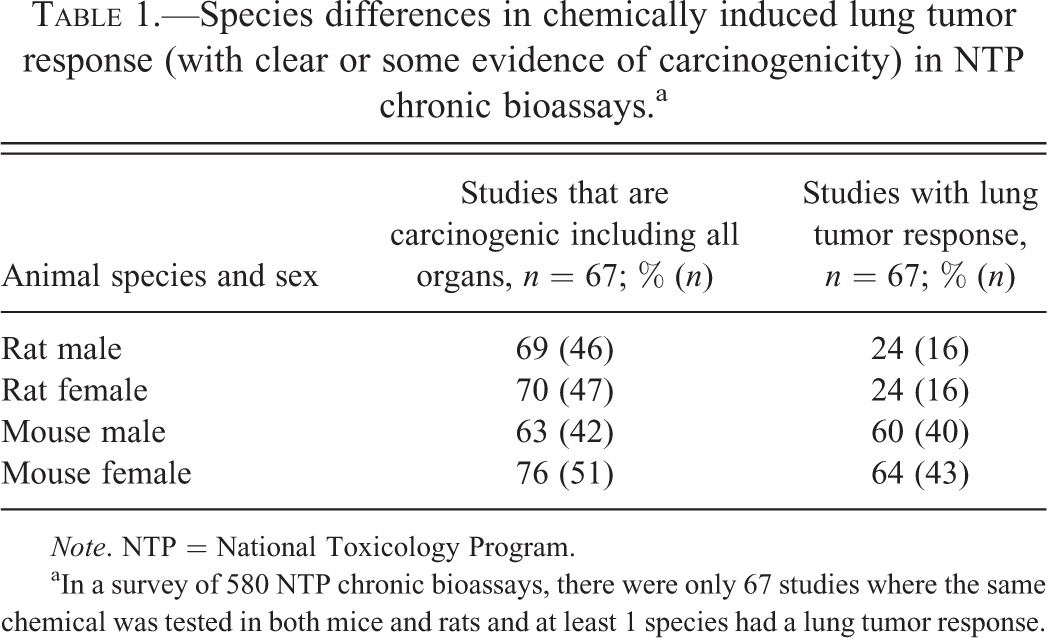
Note. NTP = National Toxicology Program.
aIn a survey of 580 NTP chronic bioassays, there were only 67 studies where the same chemical was tested in both mice and rats and at least 1 species had a lung tumor response.
Relevance of Cancer Site Concordance in Risk Assessment
The rodent bioassays are typically used as screens to identify hazards associated with chemical carcinogenesis. As discussed earlier, there is carcinogenicity site concordance for some organs more than others. However, an important issue that is repeatedly raised is the question if site concordance is really necessary to identify a carcinogenic hazard. According to the Environmental Protection Agency (2005) guidelines for carcinogen risk assessment, cancer site concordance is not always assumed between animals and humans since at the cellular level, growth control mechanisms are homologous among mammals, and cancer site concordance is not always observed with chemicals that are carcinogenic in both humans and animals. For example, vinyl chloride and benzene are carcinogenic to both humans and animals, but cancer site concordance is seen only with vinyl chloride but not with benzene (NRC 1994). In addition, the recent decision by the IARC to consider the use of mechanistic data in the absence of sufficient data on human carcinogenicity implies that site concordance is not essential to identify a carcinogenic hazard (Cogliano et al. 2008). The underlying premise for this decision is that certain mechanisms of action of chemical carcinogenesis are conserved across species and that cancer site concordance is not really necessary as long as the common plausible mechanism of action is demonstrated in both species. A classic example for this is the aryl hydrocarbon receptor (AhR) signaling by dioxin and dioxin-like chemicals, which results in changes in gene expression resulting in alterations in cell replication and apoptosis, causing tumors in a variety of organs in both species (NTP 2006). TCDD is one such chemical that was classified as group I carcinogen based on sufficient evidence of carcinogenicity in experimental animals and strong mechanistic data based on AhR activation. Similarly, 2,3,4,7,8-pentachlorodibenzofuran and 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) are complete carcinogens (capable of initiation and promotion of carcinogenesis) in experimental animals, and there is ample evidence that they act through the same AhR-mediated mechanism and hence were classified as group I carcinogens by IARC (Cogliano et al. 2008).
On some occasions, there may be multiple modes of action influencing the apical outcomes and/or conflicting data published on human relevance of a particular rodent carcinogen (Bond, Recio, and Andjelkovich 1995; Melnick and Kohn 1995). A good example of conflicting data on the human relevance of 1,3-butadiene-induced carcinogenicity are publications by Bond, Recio, and Andjelkovich (1995) and Melnick and Kohn (1995), where the former concluded that 1,3-butadiene will not be carcinogenic to humans at occupational exposure levels, while the latter concluded that it is carcinogenic (Cogliano et al. 2004). 1,3-Butadiene is considered a group I carcinogen and causes cancer of the hemolymphatic organs in humans and is also a multisite carcinogen in experimental animals. There is clear evidence of carcinogenicity in both rats and mice, but there was little tumor site concordance between the species. In mice, there was evidence of carcinogenicity within the hematopoietic system, circulatory system, lung, liver, forestomach, Harderian gland, preputial gland, and mammary gland, while in rats there was evidence of carcinogenicity in mammary gland and possibly the brain, thyroid, testis, uterus, Zymbal’s gland, and kidney (Melnick and Sills 2001; Melnick et al. 1999).
Another point to keep in mind regarding the cancer site concordance between humans and rodents is the limitations of the existing epidemiological studies. 1,3-Butadiene-induced mammary tumors were noted in both rats and mice, but there was no epidemiologic evidence supporting breast cancer risk in humans since all the people included in the epidemiologic studies were men in the industrial setting (IARC 2012b). Some epidemiologic studies have just started exploring the link between 1,3-butadiene exposure and breast cancer (IARC).
The above-mentioned examples demonstrate that a thorough examination of the epidemiologic data and the multiple modes of action of a chemical should be explored before making conclusive decisions on human relevance of chemical-induced tumors in animals.
Concluding Remarks
In general, there is good agreement between lung tumors in rodent bioassays and human lung cancer hazard due to environmental pulmonary carcinogens. The exceptions and controversies of disagreement between mouse lung tumors and human pulmonary carcinogenic hazard are usually centered on the mouse Clara (club) cell biology. Unlike the rat, the mouse is especially sensitive to a large number of metabolically activated pulmonary toxicants. Mice have higher numbers of Clara cells than humans, throughout the large and small airways. Species-specific qualitative and quantitative differences in CYP isoforms and their respective metabolizing abilities result in species-specific pulmonary toxicity and possibly neoplasia. Clara cells of mice have the CYP2F2 enzyme that plays a major role in producing toxic metabolites more efficiently than other related CYP isoforms such as CYP2F1 in humans and CYP2F4 in rats. Chemicals that mediate pulmonary toxicity and subsequent neoplasia in mice include styrene, naphthalene, coumarin, 3-methylindole (via CYP2F2 mechanism; Carlson 1997; Buckpitt et al. 1995; Born et al. 2002), 4-ipomeanol (via CYP4B1; Czerwinski et al. 1991), 1,1-dichloroethylene (via CYP2E1 and CYP2F2), and trichloroethylene (via CYP2E1; Odum, Foster, and Green 1992). There is a great diversity of oxidative enzymes expressed in lungs of various species including humans. In human lungs, various CYPs that are expressed include 1A1 (only in smokers), 1B1, 2A13, 2B6, 2C9, 2D6, 2E1, 2F1, 2J2, 2S1, 3A4, 3A5, and 4B1 (Bernauer et al. 2006; Hukkanen et al. 2002; Zhang, Wang, and Prakash 2006). Similarly, rodent lungs also express as many different CYPs with several qualitative and quantitative differences. Thus, there is no unequivocal standard for the risk assessment of pulmonary carcinogenicity in rodents and its relevance to human disease.
The immunohistochemical staining of chemically induced lung tumors in rodents is typically positive for type II pneumocyte markers (surfactant protein C) and to a lesser extent positive for Clara (club) cell markers (CC10). Some recent publications suggest that the cell of origin in KRAS-driven mouse lung tumors is a type II pneumocyte (Mainardi et al. 2014; Sutherland et al. 2014; Xu et al. 2012). However, the question of cell of origin in chemically induced pulmonary carcinogenesis needs to be resolved. This information is particularly relevant since Clara cell cytotoxicity and subsequent hyperplasia and neoplasia is the proposed mode of action of several pulmonary carcinogens in the mouse (Cruzan et al. 2009). On the other hand, it has been hypothesized that regenerating Clara cells may lose their CC10 expression and transdifferentiate into other cell types (Rawlins and Hogan 2006). Thus, immunohistochemical staining of tumors collected at terminal sacrifice may not provide an answer to the cell of origin of chemically induced lung tumors. Approaches such as cell lineage tracing experiments, as well as other investigative studies, are needed to understand the cell of origin in chemically induced pulmonary carcinogenesis that might give an insight into the mode of action.
Several issues that need to be resolved when evaluating the relevance of rodent lung tumors for human risk assessment include issues related to epidemiologic evidence in susceptible populations and life stages, exposure paradigms, species differences in tissue dosimetry, toxicokinetic data, mechanistic data, definitive characterization of species-specific phase I and II metabolizing enzymes, and the resulting metabolites, threshold issues, and so on. In spite of these unresolved issues, it is likely that the use of laboratory rodents in bioassays will continue to provide important information for hazard identification, as well as an insight into the mechanisms of chemically induced pulmonary carcinogenesis.
Footnotes
Acknowledgments
I would like to acknowledge the reviewers for their comments on this article. In addition, I would also like to thank Drs. Gordon Flake, Samuel Cohen, Janardhan Kyathanahalli, Charles Wood, David Malarkey, Rodney Miller, Jerry Hardisty, and Robert Sills for the discussions that helped me to prepare for the Society of Toxicologic Pathology presentation and this article.
Author Contribution
Arun Pandiri contributed to conception or design, data acquisition, analysis, or interpretation, drafting the article, critically revising the article; gave final approval; and agrees to be accountable for all aspects of work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.