
Abstract
Drug-induced vascular injury (DIVI) is a recurrent challenge in the development of novel pharmaceutical agents. In recent years, DIVI has been occasionally observed in nonhuman primates given RNA-targeting therapeutics such as antisense oligonucleotide therapies (ASOs) during chronic toxicity studies. While DIVI in laboratory animal species has been well characterized for vasoactive small molecules, and immune-mediated responses against large molecule biotherapeutics have been well described, there is little published information regarding DIVI induced by ASOs to date. Preclinical DIVI findings in monkeys have caused considerable delays in development of promising new ASO therapies, because of the uncertainty about whether DIVI in preclinical studies is predictive of effects in humans, and the lack of robust biomarkers of DIVI. This review of DIVI discusses clinical and microscopic features of vasculitis in monkeys, their pathogenic mechanisms, and points to consider for the toxicologist and pathologist when confronted with ASO-related DIVI. Relevant examples of regulatory feedback are included to provide insight into risk assessment of ASO therapies.
Keywords
The class of RNA-targeting therapeutic drugs is diverse and increasingly so with the recent development of new chemical variants and liposomal or nanoparticle-associated delivery systems for various types of antisense oligonucleotide therapies (ASOs). Vasculitis associated with administration of RNA-targeting therapeutics has been noted sporadically in preclinical toxicity studies with ASOs (Frazier 2014), but a comprehensive review of these lesions has not been previously published. In contrast, preclinical drug-induced vascular injury (DIVI) has been well characterized for vasoactive small molecules and immune-mediated responses against large molecule biotherapeutics resulting in vascular injury have been previously described. The Society of Toxicologic Pathology (STP) tasked a working group comprised of experts in the fields of vascular toxicology and pathology, ASO-related toxicology, and vascular injury to address this issue and fill current gaps in knowledge. The objective of this review is to describe mechanisms of DIVI for ASOs and provide guidance on risk assessment. Examples of regulatory feedback are included, and a series of bulleted points to consider are presented to aid the toxicologist and toxicologic pathologist in dealing with preclinical findings of DIVI related to ASO administration. In the first segment of this two-part series (Frazier et al. 2015), similar characteristics of biotherapeutic-associated DIVI and small-molecule DIVI were discussed, and the reader is referred to that article for more in-depth comparison of histomorphology and mechanism.
Background: ASO-related DIVI in Nonhuman Primates
Vascular changes have been noted with only some subclasses of RNA-targeting therapeutics, typically those related to pharmacologic inactivation of target messenger RNA through RNAse H, such as 2nd-generation ASOs including 2′-O-methoxyethyl (MOE) or 2′-O-methyl (OMe) phosphorothioate chemistries and locked nucleic acid (LNA) backbones. Not coincidentally, the same backbone chemistries may have individual compounds within their respective subclasses that have marked proinflammatory activity (Krieg 1999). Other types of therapeutic oligonucleotides, such as short interfering RNAs (siRNAs), have been associated with preclinical DIVI much less commonly, and in the case of some of these agents, have been limited to only those molecules with intended proinflammatory activity. Therefore, it is important to differentiate ASOs and other therapeutic oligonucleotides by backbone chemistry when considering their vasculotoxic potential. In addition, the propensity for inducing vasculitis in nonhuman primates (NHPs) may vary dramatically between molecules even among members of the same ASO subclass, so that the following information should not be considered to represent a true “class-wide toxicity” common to all RNA-targeting therapeutics. However, for the individual ASO molecules that exhibit DIVI, the morphologic, mechanistic, and pathophysiologic presentation tends to be similar. For the purposes of this article, ASO-related DIVI is the nomenclature utilized for all vascular injury induced by RNA-targeting therapeutics, regardless of backbone chemistry, even those that are technically of a different class, like siRNAs.
The interpretation of drug-induced vascular lesions in NHPs is made challenging by the occasional observation of spontaneous vascular lesions in cynomolgus macaques (Frazier et al. 2015). Such lesions may be observed in isolated vessels from individual animals in preclinical studies, but systemic vascular syndromes involving multiple vessels are much less common. In the authors’ experience, these isolated, spontaneous vascular lesions are noted most frequently in individual vessels of the liver, heart, lung, kidney, or female reproductive tract from cynomolgus macaques in preclinical studies. Published reports have also described multiple (systemic) vascular lesions in tissues from untreated cynomolgus macaques (Albassam, Lillie, and Smith 1993; Chamanza et al. 2006; Porter et al. 2003), and the distribution and character of the spontaneous vascular lesions described in these publications were quite similar to those seen in monkeys treated with ASOs.
The sites of predilection for ASO-associated vasculitis are small- and medium-sized arteries and arterioles. Occasionally, small veins can also be affected, whereas large vessels, such as elastic arteries (e.g., the aorta and the pulmonary artery) and large veins, are generally spared. The pattern of involved tissues and the morphologic appearance are often very similar to the spontaneous lesions reported in monkeys, albeit in increased incidence or severity in ASO-treated animals. ASO-related vasculitis generally presents as an endarteritis that may progress to include mural and perivascular changes, or as only perivascular inflammation (Figure 1). Chronic, repeated ASO administration (
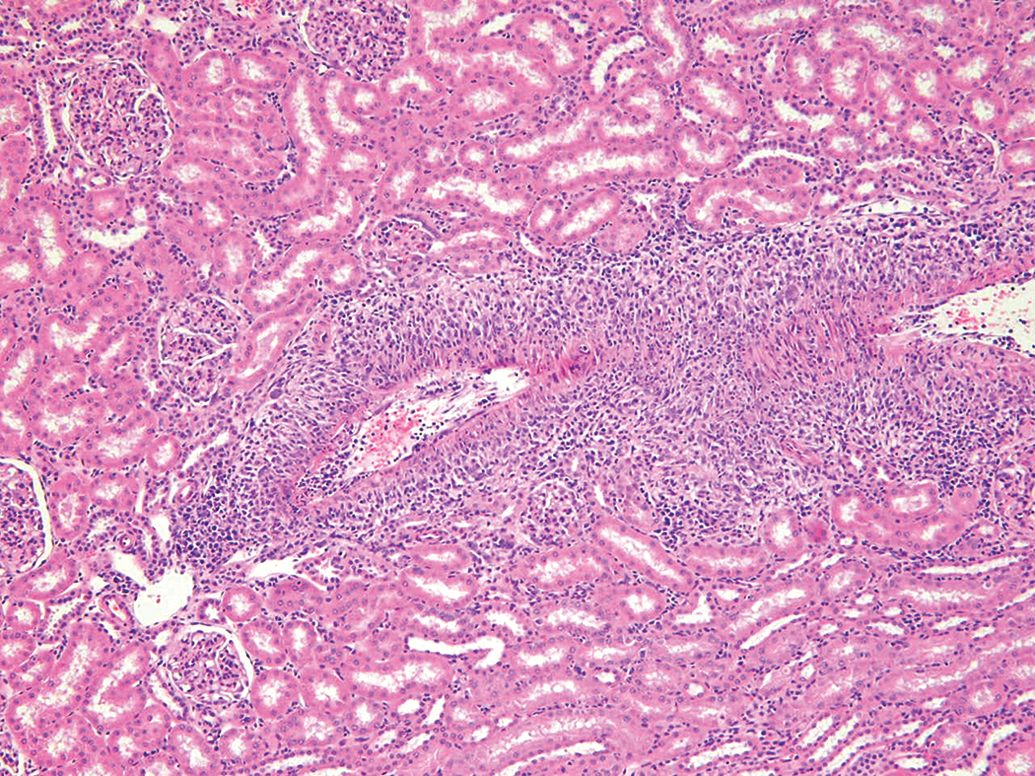
Kidney. H&E. Drug-induced vascular lesion in a monkey given an antisense oligonucleotide therapeutic for 9 months (30 mg/kg/week SC).
Despite the presence of well-established intimal, adventitial, and/or perivascular inflammatory changes, the media of the arterial wall is initially, typically unaltered. The tunica media seems to be only secondarily affected since this generally occurs only when the intimal and/or adventitial cell infiltration becomes prominent. Likewise, the internal elastic membrane generally remains intact and other changes, such as medial hemorrhages and fibrinoid necrosis, are not common features of early ASO-related arteritis, although both may occur in severely affected vessels when prominent full-thickness mural inflammation develops. At that stage, the medial layer is segmentally or circumferentially disrupted, hyperplastic, and/or edematous and the smooth muscle cells are disarranged. Medial necrosis can be observed at this stage but is uncommon (Figure 2). Severe intimal changes may be accompanied by fibrinous thrombi that can partially or completely occlude the vascular lumen.
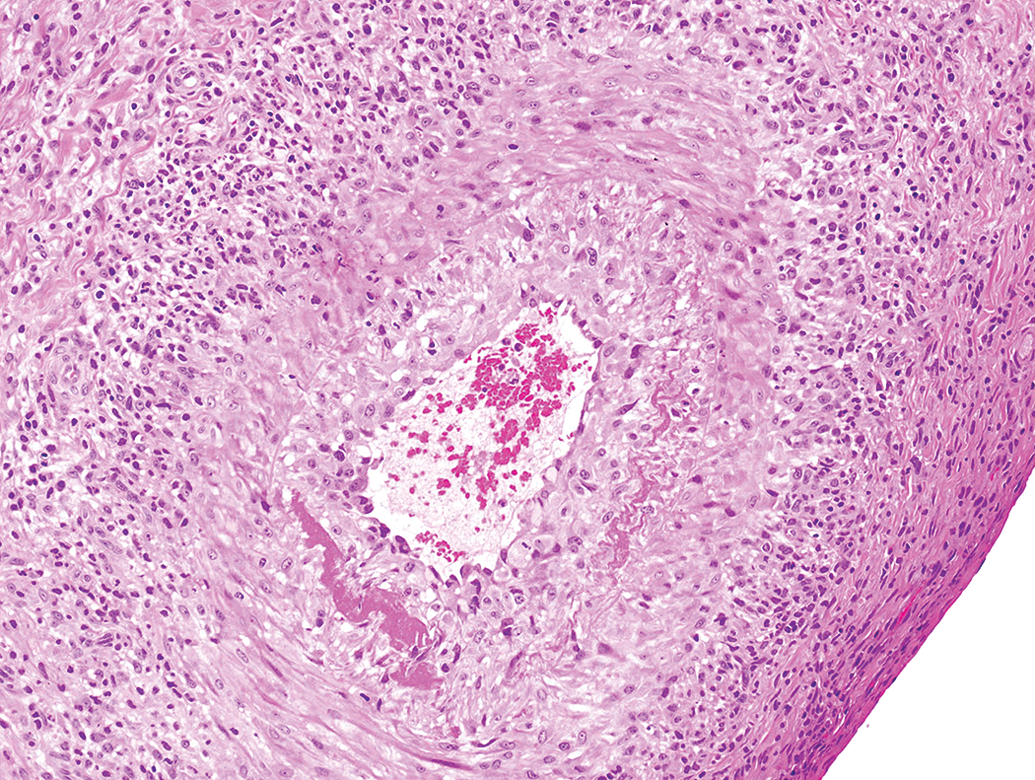
Heart. H&E. Drug-induced vascular lesion in a monkey given an antisense oligonucleotide therapeutic for 9 months. Note both intimal and medial changes, including medial fibrinoid necrosis, as well as mural and adventitial mixed inflammation (30 mg/kg/week SC).
Recovery from ASO-related vascular injury can be prolonged. Complete recovery of these vascular changes can be observed after several weeks off-dose with many of these compounds; however, in some cases, monkeys treated with ASOs chronically and then allowed a treatment-free period have still exhibited some vascular inflammatory changes weeks or months later. Generally, these vascular effects were of lower severity and/or incidence than those seen at the end of the treatment period. When present after an off-dose period, their identification and interpretation may be complicated by the concurrent presence of background vascular alterations in monkeys. The persistence of vascular changes in some of these cases may reflect the physiologically slow resolution rate of vascular wall inflammation and, at least in part, the long half-life and relative accumulation of these therapeutics within some tissue compartments in the body. Thus, the lack of complete recovery may be partially related to continued drug exposure despite being “off-dose.”
The above-mentioned arterial mural and periarterial inflammation can be observed in virtually all organs; however, the kidneys, liver, and heart (as well as the skin when ASOs are administered by subcutaneous injection) are most often affected. In the kidneys, the arteritis is generally seen at the cortico-medullary junction (e.g., arcuate and interlobular arteries), but occasional foci of capsular/subcapsular arteritis may also be seen. Diffuse inflammatory cell infiltration and edema often occur in the renal pelvic adipose-connective tissue accompanying the inflammation in segmental arteries, whereas the renal papillary vessels are generally unaffected. In the liver, the arteritis generally occurs in large portal tracts and at the junction between the liver and gallbladder. Dermal and subcutaneous arteritis is observed at injection sites when NHPs are treated by repeated subcutaneous administration with ASOs and may be accompanied by perivascular lymphoid aggregates and/or secondary lymphoid follicles. Prominent vascular inflammatory changes may also be observed in the heart, especially in extramural coronary arteries and small arteries in the coronary grooves and ventricular epicardium. These vascular changes may be associated with extension of inflammation into the myocardium and/or into the valves. When prominent multiorgan arteritis occurs, NHPs may present with clinical signs of discomfort or be moribund. Inflammatory vascular lesions in monkeys are known to occur spontaneously in the heart and kidneys, as well as several other organs, whereas the liver does not appear to be a typical anatomical site of spontaneous vasculitis (Chamanza et al. 2006; Foster 2005; Porter et al. 2003; Sato et al. 2012).
While the ASO-induced lesion of DIVI is most correctly described as a vasculitis and in early stages often specifically as an endarteritis, the morphologic terms that a toxicologic pathologist most commonly utilizes in daily practice tend to be descriptive rather than mechanistic. Consistency in nomenclature among pathologists is considered critical. Hence, the authors suggest following current terminology guidelines set out in International Harmonization of Nomenclature and Diagnostic (INHAND) and Standard Exchange of Nonclinical Data (SEND) initiatives (Mann et al. 2012), using one or more of the following preferred terms to best describe this pattern of injury in preclinical toxicity studies: artery, inflammation (or inflammatory infiltrate), mural; artery, inflammation (or inflammatory infiltrate), perivascular; and/or artery, hypertrophy/hyperplasia, intima. Similar or identical terms are also applicable to DIVI associated with biotherapeutics. Since the tunica media is less commonly affected with ASO-related vasculitis in monkeys, terms such as “artery, necrosis, medial” are usually not applicable as it is with vasoactive archetypal DIVI except in very advanced lesions.
Incidence of ASO-related DIVI
The incidence of ASO-associated vasculitis in monkeys does not follow a typical dose response and typically involves a small number of affected animals at doses above a certain threshold for a given ASO compound (Frazier et al. 2014). Above this threshold, the incidence and severity is often highly variable between animals and can be inconsistent between studies even with the same ASO. This would suggest that individual animal predilection or susceptibility may play a central role in development of vascular injury related to ASO treatment. In contrast, peptide- or antibody-associated vascular changes often tend to occur at an “optimum dose” where there is an optimal antidrug antibody (ADA) to antigen ratio (Clarke 2010; Ponce et al. 2009). Interestingly, and at least anecdotally, there does seem to be a difference in the susceptibility of cynomolgus macaques of certain origins for vasculitis as compared to others. This should not be surprising since the genetic backgrounds of NHP populations are different (Vidal et al. 2010), and hence immunocompetence, complement activation, and overall immune response are likely to vary. Marked differences have previously been described between cynomolgus macaques of various origins in regard to immune responses to diseases and drug administration (Menninger et al. 2002; Migot-Nabias et al. 1999). Therefore, it appears that innate genetic factors are an important component for the predisposition, expression, or at least magnitude of the vascular immune response to ASOs.
Comparison to Small Molecule and Biotherapeutic-related DIVI
There are a number of features of ASO-related vasculitis that differ substantially from vasculopathies associated with administration of small molecules or biopharmaceutical products. ASO-related morphologic changes in vessels tend to be more endothelial-centric whereas small molecule vascular lesions are generally centered on the tunica media (Dalmas et al. 2008; Kerns et al. 2005; Weaver et al. 2008). Further, while ASO-related vascular lesions often have pronounced adventitial inflammatory infiltrates, they lack the medial/transmural inflammation or hemorrhage noted with many small molecule vasculotoxicants. Mechanistically, ASOs lack vasopressor or vasodilatory activity that is a common feature of most small molecules that induce vasculopathy (Dalmas et al. 2011; Kerns et al. 2005; Louden et al. 2006), and with few exceptions, lack the immunogenicity and ADA response noted so commonly in cases of vasculitis associated with biotherapeutics. In fact, where ADAs have been noted with ASO therapies, the antibodies have been shown to be nonneutralizing and transient in nature, and therefore considered unrelated to any concurrent toxicity. There is also no known association with antibody aggregates or complexes with ASOs, unlike those that have been implicated in the pathogenesis of some cases of biotherapeutic vasculitis (Clarke 2010; Leach 2013). While small molecule-related vasculitis is generally a consistent and dose-responsive toxicity, the pattern of ASO-associated DIVI is very heterogeneous across animals within a dose group. A typical dose relationship not present, but there is a general threshold dose required to induce vascular changes. The incidence above the threshold is highly variable, suggesting other contributing factors lead to individual animal sensitivity. This is similar to the variability in immune-mediated DIVI seen for biotherapeutic compounds. The predilection sites for injury also differ among the various types of vasculotoxicants. With small molecules, this can vary with the particular agent, but coronary vessel in dogs or the mesenteric bed in rats are often implicated (Dalmas et al. 2011; Kerns et al. 2005). ASO-related vascular lesions tend to show random distribution of organ localization. With biotherapeutic-related DIVI, the distribution is relatively consistent, with vessels prone to immune complex deposition more often affected, including those with high flow rates and especially vessels lined by tall fenestrated endothelium. Most frequent organ sites in monkeys with biotherapeutic-related DIVI are small to medium caliber arteries in the gastrointestinal tract, gall bladder, pancreas, kidney, and heart (Rojko et al. 2014), but skin, eye, synovium, lung, choroid plexus, and liver, as well as reproductive tract are also encountered, although less frequently. Of these, only the renal interstitium and myocardium are common locations for endothelial or vascular damage noted with ASO therapy, but within the kidney, the glomeruli are also often affected with either biotherapeutic- or ASO-related DIVI. Finally, the type of systemic inflammatory response is different among the various compound classes, as suggested by their pattern of cytokine induction. Taken together, these attributes provide clues to a distinct mechanism of ASO-induced vascular injury as noted subsequently.
Potential Mechanism for ASO-related Vascular Inflammation
The pattern of vascular pathology described in monkeys treated with ASOs is clearly inflammatory in nature, and there are well-recognized proinflammatory properties of ASOs (Henry et al. 1999; Monteith and Levin 1999; Ravindran, Jeng, and Liang 2010), with known quantitative differences based on backbone structure and/or base sequence (Krieg 2002). The primary source of inflammation when high doses of an ASO are administered to monkeys involves activation of the alternative complement cascade (described subsequently). Effects on complement are a common class effect for phosphorothioate-modified ASOs, and there is also a potential of some sequences of phosphorothioates to induce innate immune cell activation. A mechanism related to complement and immune activation has recently been proposed for effects of 2′-OMe or 2′-MOE ASOs on glomerular endothelium in monkeys (Frazier et al. 2014; Shen et al. submitted to Toxicol Pathol). Immune-related pathogenesis for ASO-related systemic vasculitis is supported by the common demonstration of increased leukocyte counts and histologic effects in lymphoid organs such as lymphoid hyperplasia in lymph nodes and spleen in monkeys. Increases in systemic inflammatory proteins have also been reported with chronic ASO therapy in monkeys, including MCP-1, C-reactive protein (CRP), fibrinogen, and haptoglobin (Burel et al. 2013; Frazier et al. 2014). Inflammatory cell infiltrates also tend to be increased in many tissues of monkeys that develop ASO-associated vasculopathy.
The key factor in the immune-mediated pathogenesis of this type of vascular injury in monkeys is believed to be complement activation. Complement activation is a widely recognized complication of ASO therapy in monkeys and is related to direct inhibition of negative regulators (specifically Factor H) of the alternative pathway of complement (Henry, Giclas, et al. 1997; Henry et al. 2008). Factor H is a plasma glycoprotein synthesized by the liver and other tissues, including endothelial and glomerular mesangial cells. Additionally, activated platelets can release Factor H from their α-granules. Although Factor H is considered to act in the blood fluid phase, it binds to the endothelial cell surface through interaction with integrins (CD11b/CD18) and polyanionic glycopeptides that are present on cell surfaces. The bound Factor H maintains its complement regulatory activity and therefore plays a central role in the protection of cell/tissue surfaces against complement activation (Józsi et al. 2004). The interactions between ASOs and Factor H are peak plasma concentration-dependent and readily reversible (Henry, Novotny, et al. 1997; Henry et al. 2008). Factor H acts to inhibit the constitutive activity of the alternative pathway. Therefore, Factor H inhibition by an ASO can result in activation of C3 convertase and uncontrolled turnover of the C3 fraction of complement (Barbour, Pickering, and Cook 2013). Activation of complement results in the production of anaphylactic split products such as C3a and C5a, and dysregulated complement cascade triggers both inflammatory and hemodynamic responses within the vascular bed. Shortly after administration of ASOs, monkeys may exhibit a transient decrease in systemic blood pressure. This generally reflects endothelial changes and vascular leakage induced by complement factors (e.g., C3a and C5a) rather than an effect of ASOs on tunica media, as is observed after administration of vasoactive small molecules.
When unrestrained activation of the complement pathways is persistent, such as following chronic, repeated ASO administration, consumption overwhelms synthesis and essential plasma complement factors such as C3 are depleted. After repeated ASO administration, regular activation of the alternative pathway can result in sustained decreases in plasma C3 concentrations (Shen et al. Submited to Toxicol Pathol). This results in markedly reduced clearance of immune complexes from the bloodstream and is widely believed to be the ultimate mechanism for complement-mediated vascular damage with ASOs (Shen et al. 2014). Increasing concentration of a complement blocker (CAB-2) effectively prevented oligonucleotide induced complement activation in monkeys (Henry et al. 2002). Cytokine release has been repeatedly demonstrated in monkeys treated with oligonucleotides (Frazier et al. 2014; Henry et al. 2002). In one study, increases in interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and interleukin-12 (IL-12) occurred maximally at 120 min after the start of infusion, but were transient and returned to baseline after approximately 4 hr. This emphasizes the importance of sampling in this critical time window after dosing to get solid evidence of complement activation. Interestingly, pretreatment with CAB-2 in monkeys given ASOs generally prevented the increases in serum concentration of IL-6, MCP-1, and IL-12 and this further suggests that the increase in circulating cytokines was secondary to complement activation (Henry et al. 2002). While macaques (both cynomolgus and rhesus) are particularly sensitive to this complement-related effect (Farman and Kornbrust 2003; Henry et al. 2002; Henry et al. 2008), there are no published reports of a similar direct ASO-related complement activation in humans or other species (Shen et al. 2014). Not coincidentally, ASO-related vascular changes have not been observed in laboratory animal species other than monkeys, to the best of the authors’ knowledge.
An example of the relationship between complement activation and the vascular pathology is illustrated in a study where cynomolgus macaques were given ISIS 325686 (a 2′-MOE ASO) at 30 mg/kg weekly by subcutaneous injection for 9 mo. At this dose level, transient complement activation occurred after each weekly dose. Over time, there tended to be a cumulative effect on the inflammatory cytokine profile and complement fragment profile occurring secondary to enhanced complement activation, along with a tendency for decreased circulating levels of C3. Additionally, there was a correlation on an individual animal basis between the occurrence of vascular pathology and sustained decreases in C3 serum concentration as well as increases in inflammatory markers (Table 1).
Correlations of vascular pathology with complement profiles and other inflammatory markers in monkeys after chronic ASO treatment (fold increase or decrease compared to baseline).
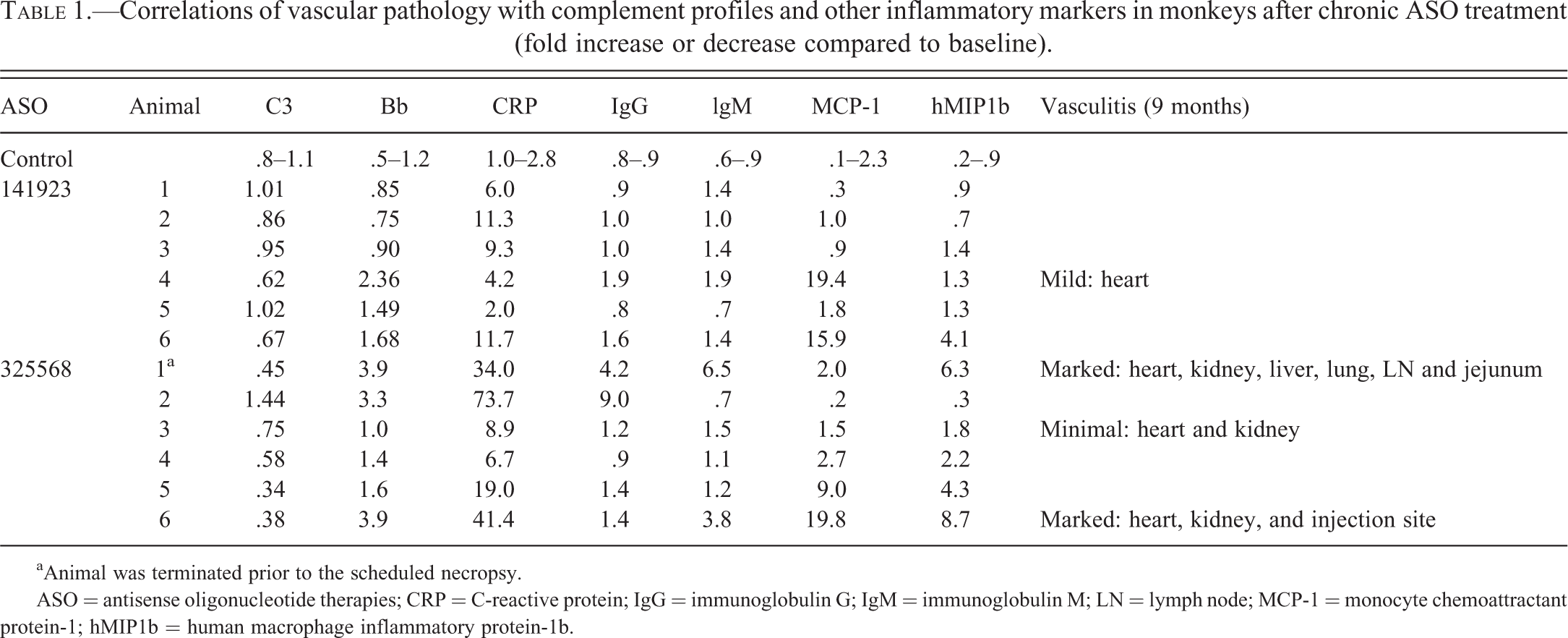
aAnimal was terminated prior to the scheduled necropsy.
ASO = antisense oligonucleotide therapies; CRP = C-reactive protein; IgG = immunoglobulin G; IgM = immunoglobulin M; LN = lymph node; MCP-1 = monocyte chemoattractant protein-1; hMIP1b = human macrophage inflammatory protein-1b.
As discussed earlier, direct activation of the alternative pathway for complement via Factor H inhibition is at present the most likely explainable mechanism for these types of effects, but there are other proinflammatory pathways initiated by ASOs that may also interact with and/or augment complement activation. Immunomodulatory effects of ASOs involve stimulation of multiple receptors of the innate immune system, such as toll-like receptors (TLRs), and especially TLR-9. Cross talk between complement pathways and TLRs has been demonstrated, which reinforces innate immunity and inflammation through synergistic interactions (Hajishengallis and Lambris 2010). This innate immune stimulation is highly dependent on nucleotide sequence and can vary widely between sequences from little immunomodulatory activity to quite strong. Certain backbone modifications can reduce proinflammatory effects, and alteration of base sequence can alter the degree of complement activation. ASOs containing cytosine-guanine dinucleotides (CpG motifs) are well known for their prominent proinflammatory effects through activation of TLR-9 receptors (Heikenwalder et al. 2004; Senn, Burel, and Henry 2005). When the CpG motif is flanked by 2 purines on the 5′ end and 2 pyrimidines on the 3′ end, especially potent proinflammatory activity and B cell as well as macrophage activation have been observed (Henry et al. 2008; Krieg 1998). The CpG containing ASOs in this regard can work as an activating ligand for TLR-9 simulating a bacterial pathogen (Richardt-Pargmann and Vollmer 2009) and can induce specific TLR-9 dependent immune stimulatory effects in a variety of species. This approach has been utilized in industry to deliberately design ASOs which target immunodeficient states or as adjuvants for vaccine formulations. After injection of one such ASO in C57BL/6 mice, substantial increases in IL-6, TNFα, and interferon-λ have been observed in the serum (Heikenwalder et al. 2004). Most ASOs that progress into clinical trials have avoided CpG dimers, and are selected for minimal inflammatory effects, as much as possible. Nonetheless, there is typically some local inflammation at SC injection sites and, possibly, draining lymph nodes. A contributory effect of this inflammation toward exacerbating the background vascular inflammation in monkeys cannot be excluded. Proinflammatory activity by ASOs mediated through pathways involving innate immune system receptors other than through CpG motifs/TLR-9 have been noted in mice (Burel et al. 2012; Choi, Chung, and Jung 2010; Ravindran, Jeng, and Liang 2010; Senn, Burel, and Henry 2005), but it is unknown whether such pathways are operative in the monkey. Importantly, no vascular inflammation has been observed in chronic mouse studies with ASOs despite the presence of this direct innate immune cell stimulation, and no complement activation has been demonstrated.
Risk Assessment Associated with ASO-related DIVI
Numerous ASOs are currently being evaluated in clinical trials for the treatment of cancer, inflammation, neuromuscular disorders, metabolic, or viral diseases. One systemically administered ASO, Kynamro™ (mipomersen, a 2nd-generation MOE-phosphorothioate oligonucleotide indicated for homozygous familial hypercholesterolemia), has received Food and Drug Administration (FDA) regulatory approval for marketing. Additionally, 2 locally administered therapeutic oligonucleotides are currently marketed—Vitravene™ (fomivirsen, a 1st-generation phosphorothioate ASO) for treatment of cytomegalovirus retinitis in AIDS patients, and Macugen™ (pegaptanib, an oligonucleotide aptamer) for wet age-related macular degeneration. In preclinical studies with mipomersen, 2 cynomolgus monkeys given the highest dose (30 mg/kg/week) were noted with vasculitis and/or perivasculitis in multiple tissues and 1 of these died due to comorbidities (Food and Drug Administration 2012). Proinflammatory effects and complement activation were noted in monkeys given
Unpublished regulatory experience with other ASOs currently in clinical trials, which have undergone review of investigatory brochures, has generally mirrored the situation with mipomersen pertaining to cases of monkey vasculitis, complement activation, and human risk. Although complement activation has been noted in individual monkeys in preclinical toxicity studies with multiple different ASOs presently in development, this has led to a clinical “hold” or halting of dosing in only early, initial cases. As the regulatory reviewers have become more familiar with ASOs and their effects, and cognizant of apparent sensitivities of monkeys to develop complement-mediated vascular changes, cautious dose escalation and careful clinical monitoring for signs of inflammation have generally allowed clinical trials to proceed. While the FDA and European Medicines Agency have allowed dosing of patients with ASOs that have been associated with vasculitis in monkeys, several of these agents have been for orphan or life-threatening indications. Hence, risk/benefit must still be carefully considered. Well-rationalized clinical monitoring of inflammatory responses and/or complement activation with other proposed vascular biomarkers in trials may provide the ability to detect inflammatory cascades that may predispose individuals to ASO-related DIVI. Until there is more robust preclinical and clinical qualification and/or validation of direct or surrogate biomarkers of vascular injury, their use in clinical trials will likely be on an exploratory basis only, and not as a tool for clinical decision making or dose escalation. As a greater number of ASOs reach phase II or III clinical trials, more data may be available to determine whether the clinical risk for DIVI is apparent, or if there is greater confidence that the vascular changes are peculiar to the NHP based on its sensitivity to systemic complement activation.
Biomarkers of ASO-related DIVI
Based on the proposed immune pathogenesis of these vascular lesions, biomarkers of chronic immune stimulation and especially complement activation are likely to be valuable as surrogates for monitoring for this type of injury. Excellent reviews of these types of markers are available (Tarrant 2010). As noted elsewhere in this article, assessment of plasma complement activation, particularly regarding the alternative pathway, are critical in chronic monkey studies with proinflammatory molecules where prominent complement activation is suspected. Multiple assays for complement activation have been utilized and/or proposed in monkeys on ASO treatment, including total complement (CH50), and complement split products Bb, C3a, and C5a. Plasma MCP-1 and CRP have been utilized successfully in monkeys to assess systemic immune stimulation, and other markers such as fibrinogen, haptoglobin, and globulin have also been increased in monkeys with vascular injury when administered proinflammatory ASOs. In addition, since vasculitis associated with ASO administration is often accompanied by the simultaneous occurrence of complement-mediated glomerulonephritis, proteinuria, and/or albuminuria may be useful monitorable surrogate biomarkers for these effects (Demeule, Gurny, and Arvinte 2006; Frazier 2014).
Other potential vascular biomarkers, currently under investigation, are also likely to be of benefit in clinical monitoring of patients given ASOs, including those proposed by The Predictive Safety Testing Consortium (PSTC) Vascular Injury Working Group (VIWG) including Angiopoietin-2, Endothelin-1, E-selectin, Thrombospondin-1, Vascular endothelial growth factor alpha, Calponin-1, Tissue inhibitor of metalloproteinase 1, Lipocalin 2, Cytokine-induced neutrophil chemoattractant 1, Alpha-1 acid glycoprotein 1, and total nitric oxide (Mikaelian et al. 2014). It is important to note that this proposed panel is based on vasoactive compounds (i.e., small molecules), but many of the proposed markers may be applicable to ASO therapies. Other groups such as the EU-based Safer And Faster Evidence-based Translation (SAFE-T) consortium are doing clinical validation work in parallel on these and other potential vascular biomarkers (Bendjama et al. 2014). This clinical candidate list for biomarker validation is more comprehensive and includes Von Willebrands factor, intercellular adhesion molecule (ICAM), vascular cellular adhesion molecule-1 (VCAM-1), interleukins, monocyte chemotactic protein-1, serum amyloid A, CRP, fibrinogen, plasminogen activator inhibitor-1, and caveolin, among others (Bendjama et al. 2014). However, none of these potential biomarkers on either preclinical or clinical list have risen to the level of validation or qualification for vascular injury and their ubiquity in systemic inflammatory processes potentially limits their selectivity. Changes in markers such as matrix metalloproteinases (MMP-1, MMP-3, and MMP-9), vascular endothelial growth factor, thrombomodulin, E-selectin, and P-selectin have been previously evaluated in human patients with vasculitis (Monach et al. 2011), and these types of markers may also be applicable in ASO-mediated vasculitis both preclinically and clinically as exploratory surrogate markers.
Points to Consider for the Interpretation of ASO-related DIVI
The following bulleted list incorporates the accumulated learnings from experts in the field of vascular injury as they specifically relate to ASO therapeutic administration. The list is not meant to be comprehensive, but instead provide some points to consider for toxicologists and pathologists when vascular effects are noted in preclinical toxicity studies with ASOs. Since vascular toxicity in preclinical species is not always translated across the range of species to human, the presence of preclinical DIVI should not necessarily halt development or progression of a potential drug, but rather signal the need for further investigative work. Cautious clinical dose selection/escalation with appropriate safety margins is necessary once clinical progression is instituted. To date, ASO-mediated DIVI has been limited to monkeys, due to a variety of factors including pathogenesis and species predisposition for complement activation. DIVI associated with ASO administration shows parallels to biotherapeutic-related DIVI, in that the lesions have an immune/inflammatory pathogenesis, often show random distribution of organ localization, and may lack a typical dose relationship. Attempts to demonstrate causal mechanisms and clinical relevance are important. For ASOs, the incidence of DIVI is typically threshold-dependent, with a dose below which lesions are not noted; however, individual animals across dose groups may be affected above this threshold dose, so a typical dose response relationship may not be evident. Lesions of ASO-related DIVI are most often observed after chronic administration (≥6 months of treatment). Complement activation analysis is critical for monitoring DIVI in monkeys given ASO therapeutics. When calculating doses for chronic preclinical studies, toxicologists should aim for a mild degree of complement activation at the high dose. Complement activation should be monitored at multiple times during the course of a study. In the context of observed DIVI, the assessment should include both alternative pathway split products (i.e., Bb) and the plasma complement fragment levels over time. The use of a panel of preclinical biomarkers for DIVI, while not necessarily qualified nor validated, may be helpful in determining which potential analytes might also be included in clinical trials for a specific agent. Such markers could be used to detect or exclude DIVI in the clinical setting providing a greater level of patient safety. In the case of ASO-related DIVI, complement pathway analysis and other inflammatory markers (i.e., CRP, MCP-1, MMP-3, and/or other cytokines or chemokines) may be helpful in subchronic preclinical studies. With any mechanism of DIVI, a panel of prospective markers of endothelial activation (e.g., VCAM) or medial injury may also be of benefit. The authors recommend following current terminology guidelines set out in INHAND and SEND initiatives for describing the lesion of ASO-related vascular injury, using any of the following terms: artery, inflammation, mural; artery, inflammation, perivascular; artery, intima, hypertrophy/hyperplasia; “Inflammation” and “inflammatory infiltrate” are equally acceptable and considered interchangeable. Lesions of DIVI associated with ASOs occur randomly in vessels in many organs. Hence, routine good laboratory practices (GLP) tissue lists are considered sufficient to identify DIVI with these agents and there is little to be gained from sampling additional vascular beds. Due to the idiosyncratic nondose-responsive nature and random distribution of vasculitis with ASO administration, all routine organs (not just target organs) in low- and mid-dose group monkeys should be examined in GLP studies once a vascular lesion has been observed with a compound in a high dose group. In non-GLP studies, where the list of tissues to be examined can be limited, examination of kidney and heart are most likely to demonstrate vascular lesions caused by ASOs. Recovery groups with a minimum of 2 monkeys per sex, per dose group should be included in subsequent subchronic or chronic studies when vascular lesions are noted in monkeys given ASOs in preclinical toxicity studies. A variety of tissues may be required to be examined in these groups due to the variability in localization of DIVI lesions. Due to the protracted nature of immunomodulatory stimuli and long tissue half-life of ASOs, an off-dose period of several weeks to months may be required to demonstrate complete reversal. The lack of complete recovery may be partially related to long-lasting drug exposure of previously treated animals or reflect the physiologically slow resolution rate of vascular wall inflammation.
Conclusions
Despite numerous years of study, drug-induced vascular injury remains a significant challenge for the development of new and novel pharmaceutical agents. With RNA-targeting therapeutics, particularly ASOs, immune-mediated and inflammatory mechanisms appear to be involved so the risk assessment remains a challenge. As vascular effects appear to occur through secondary mechanisms involving complement activation, this type of DIVI needs to be differentiated from “classical” or “archetypal” DIVI associated with vasoactive small molecules. The current review has discussed the pathophysiology involved in this type of vascular injury and how these effects may be relevant to humans. The role of spontaneous vascular inflammation, particularly in NHPs, has been presented as a potential confounding factor with ASOs. As discussed, the collective experiences of the review team suggest that the toxicologist and pathologist can tease apart the various effects seen in general toxicity studies to make an informed risk assessment. Additional parameters to those routinely collected in toxicity studies may be needed in a weight-of-evidence approach and to support plausibility of mechanisms.
Footnotes
*
This review is a product of a Society of Toxicologic Pathology (STP) Working Group commissioned by the Scientific and Regulatory Policy Committee (SRPC) of the STP. It has been reviewed and approved by the SRPC and Executive Committee of the STP. The article does not represent a formal best practice recommendation of the Society but provides expert guidance on key principles to consider in designing, conducting, and reviewing regulated toxicity studies. The views expressed in this article are those of the authors and do not necessarily represent the policies, positions, or opinions of their respective agencies and organizations. Readers of Toxicologic Pathology are encouraged to send their thoughts on these articles or ideas for new topics to the editor.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author Contribution
All authors (KF, JE, PF, SG, SH, ML, CL, MS, JW, TZ) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the article; and critically revising the article. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be considered to represent any agency determination or policy.