
Abstract
Drug-induced vascular injury (DIVI) is a recurrent challenge in the development of novel pharmaceutical agents. Although DIVI in laboratory animal species has been well characterized for vasoactive small molecules, there is little available information regarding DIVI associated with biotherapeutics such as peptides/proteins or antibodies. Because of the uncertainty about whether DIVI in preclinical studies is predictive of effects in humans and the lack of robust biomarkers of DIVI, preclinical DIVI findings can cause considerable delays in or even halt development of promising new drugs. This review discusses standard terminology, characteristics, and mechanisms of DIVI associated with biotherapeutics. Guidance and points to consider for the toxicologist and pathologist facing preclinical cases of biotherapeutic-related DIVI are outlined, and examples of regulatory feedback for each of the mechanistic types of DIVI are included to provide insight into risk assessment.
Keywords
Introduction
Drug-induced vascular injury (DIVI) is a recurrent challenge in the development of novel pharmaceutical agents. Many small molecule drugs are known to cause DIVI in preclinical species (Clemo et al. 2003; Joseph, Rees, and Dayan 1996; W. Kerns et al. 2005; Zhang et al. 2008). Despite extensive efforts to define the relevance of DIVI in preclinical studies to humans, and to identify biomarkers for vascular injury across species, the translatability of DIVI from animals to humans remains unclear (Mesfin et al. 1996). Because of the uncertainty about whether DIVI in preclinical studies is predictive of effects in humans and the lack of robust biomarkers of DIVI, preclinical DIVI findings can cause considerable delays in or even halt development of promising new drugs.
Although DIVI in laboratory animal species has been well characterized for vasoactive small molecules, there is little available information regarding the vascular lesions associated with biotherapeutics such as recombinant peptides/proteins or monoclonal antibodies (mAbs). The Society of Toxicologic Pathology (STP) tasked a working group comprised of experts in the fields of vascular toxicology and pathology, and biotherapeutic-induced vascular injury to address this issue and provide points to consider in a comprehensive review. The objective of this review is to describe mechanisms of DIVI for the biotherapeutic class of drugs and provide guidance on risk assessment strategies for toxicologic pathologists, toxicologists, and pharmaceutical companies. Although a brief background on DIVI associated with small molecule drug therapy is included for comparison, the major scope is focused on vascular injury associated with the use of biotherapeutics. Vascular injury associated with antisense oligonucleotides is discussed in the companion to this article (Engelhardt et al. 2015). This review includes a description of the characteristic microscopic findings and mechanisms with actual examples from drug development. This review also includes information on standard DIVI terminology, comparisons to spontaneous vascular diseases in animals, and a brief synopsis of drug-induced vasculitis in humans as it relates to preclinical vascular changes. The difference in pathophysiologic mechanisms between small molecule DIVI, which are often considered direct injuries to the vasculature, and those related to biotherapeutics, which are mainly immune-mediated indirect mechanisms, are discussed. Since there have been several recent reviews of potential biomarkers for DIVI (Bendjama et al. 2014; C. Louden et al. 2006; Mikaelian et al. 2014; Zhang et al. 2010), this article will only briefly summarize work in vascular biomarkers related to small molecule development and will instead focus on recent biomarkers not previously reviewed and those especially relevant to biotherapeutics. Examples of regulatory feedback are included to provide insight into risk assessment. Finally, a series of bulleted “points to consider” are presented to aid the toxicologist and pathologist in dealing with preclinical findings of DIVI related to biotherapeutic administration.
Anatomic Considerations and DIVI Standard Terminology
Endothelial cells (ECs) lining vessels help to maintain the integrity of the vascular wall, participate in immune reactions via expression of adhesion molecules, and regulate local vascular tone by release of nitrous oxide (NO; Arnal et al. 1999). They also synthesize anticoagulants, cytokines, and growth factors in response to vascular dynamics and shear stress (Annas and Brittebo 1998; Shyu 2009). ECs do not form a continuous layer but have fenestrations that vary depending on anatomic site. These properties influence the susceptibility of vessels to injury depending on their anatomic location. Smooth muscle cells (SMCs) regulate vascular tone and have receptors for endogenous vasoconstrictors such as angiotensin, endothelin, and epinephrine, and are also a target for NO and ion channel openers (Nguyen Dinh Cat and Touyz 2011; Galley and Webster 2004; Mehta and Griendling 2007). The adventitia participates in inflammatory and proliferative responses and may also influence vascular damage through paracrine effects (Dalmas et al. 2011; Gollasch 2012). The size of interendothelial gaps as well as electric charge and composition of basement membrane components (e.g., as in the glomerulus) can influence the deposition of large immune complexes and can affect susceptibility to vascular damage (Mannik 1987; Rojko et al. 2014).
Lesions in the vascular system can be categorized as degenerative/necrotic, inflammatory, and proliferative (Mikaelian et al. 2014); these are not specific for drug-induced injury and can occur in spontaneous vascular disease. Similar vascular lesions are found in laboratory animals and humans (Bendjama et al. 2014). Changes specifically in the endothelium include degeneration, apoptosis/necrosis, hypertrophy, and hyperplasia. EC degeneration may appear as vacuolation or alterations of cytoplasmic tinctorial characteristics. EC necrosis/apoptosis may be characterized by nuclear karyorrhexis/loss and desquamation or may be mainly apparent through secondary findings such as thrombosis and vascular leakage. Often, verification of EC injury or loss requires additional methods beyond routine histology (Lim, DeLano, and Schmid-Schonbein 2001; Mikaelian et al. 2010; Sharma and Kiyatkin 2009). Hypertrophy and hyperplasia of ECs may be a primary change or occur later in the time course of vascular injury following EC loss (Morton et al. 1997). Both necrosis and hypertrophy can occur simultaneously and may accompany proliferation of fibroblasts and SMCs within the tunica media (Mikaelian et al. 2014). Necrosis and/or apoptosis of SMCs of the tunica media is often seen as the hallmark of small molecule induced DIVI, whereas it is a less frequent morphologic observation with biotherapeutic-induced DIVI (Zhang et al. 2008). Medial arterial necrosis is the preferred term for this lesion in the current literature and the pending International Nomenclature and Harmonization of Nomenclature and Diagnostic Criteria (INHAND) terminology (i.e., artery, necrosis, and medial). Medial necrosis of veins is less common and less conspicuous. Medial arterial necrosis can appear as individual necrotic cells with pyknotic or karyorrhectic nuclei, or as fibrinoid necrosis of a portion of the media that is accompanied by local accumulation of plasma proteins, giving the media a hypereosinophilic, homogenous to fibrillar appearance. Medial necrosis is often accompanied by mural and perivascular hemorrhage. Hypertrophy and hyperplasia of SMCs occurs as a late stage vascular change and is often accompanied by intimal and adventitial fibrosis (Greaves 1998). Both hypertrophy and hyperplasia usually occur together and are characterized by increased number, size, and basophilia of SMCs causing medial thickening. Inflammation associated with DIVI is characterized by perivascular edema, hemorrhage, and inflammatory cell infiltration either transmural or localized to the intima, media, or adventitia. In most types of vascular injury in laboratory animals, mixed inflammatory cell infiltrates are most common and may include macrophages, neutrophils (Albassam et al. 1999; C. S. Louden et al. 2000; Joseph, Rees, and Dayan 1996), eosinophils (Anderson and Hayes 1989; Bregman et al. 1987), and lymphocytes (Figure 1). Adventitial fibrosis and neovascularization can occur with chronic lesions, especially in large vessels, with differentiation of fibroblasts to myofibroblasts (Siow, Mallawaarachchi, and Weissberg 2003; Zalewski and Shi 1997). Rarely, plexiform vasculopathy, that is, the increased size and number of vasa vasorum, can be observed after chronic administration of vasoactive substances (Collins et al. 1988; Nyska et al. 1998; Westwood, Iswaran, and Greaves 1990; Zhang et al. 2008).
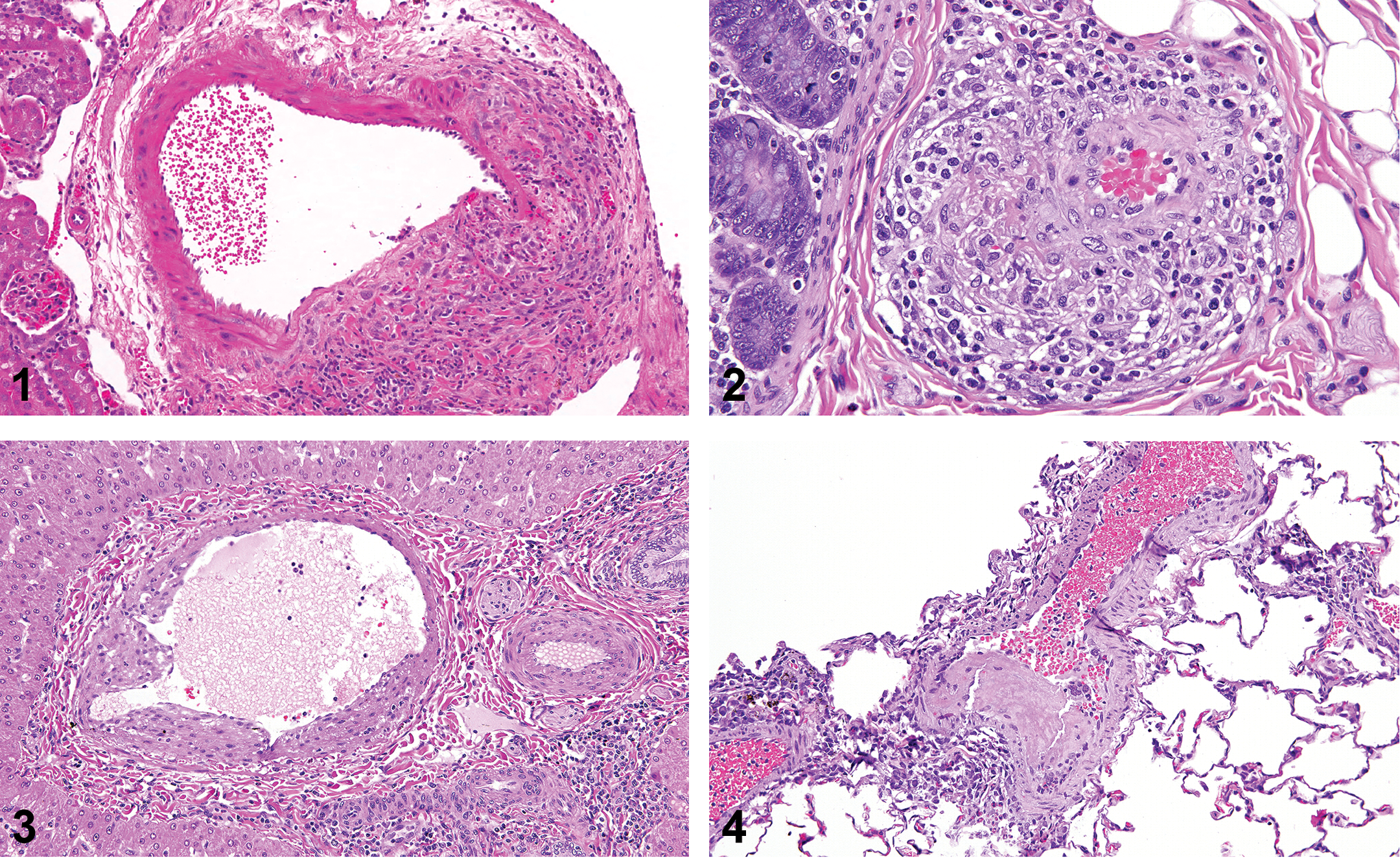
Kidney. H&E stain. Spontaneous arteriolar lesion in a mouse kidney (polyarteritis nodosa). Transmural inflammation, loss of endothelium, and medial degeneration. Figure 2.—Colon. H&E stain. Arteriolar lesion in a monkey after administration of a monoclonal antibody in an 8-week good laboratory practices study. Perivascular and adventitial infiltrate of predominantly mononuclear leukocytes and rare neutrophils as well as reactive endothelial cells in the tunica intima. Figure 3.—Liver. H&E stain. Portal vein lesion in a monkey after administration of a monoclonal antibody in an 8-week good laboratory practices study. Segmental injury to the tunica intima and media with neointimal proliferation, overlying reactive endothelial cells, stromal vacuolation, and minimal mononuclear leukocyte infiltrate and karyorrhectic debris. Figure 4.—Lung. H&E stain. Arteriolar lesion in a monkey after administration of a monoclonal antibody in an 8-week good laboratory practices study. Segmental necrosis of the tunica intima with thrombus formation and minimal subendothelial neutrophilic infiltrate as well as associated medial vacuolation and minimal perivascular neutrophilic infiltrate.
The INHAND terminology for the vascular system reflects the nomenclature of changes noted in other organs (Mann et al. 2012). When using morphologic diagnoses in toxicity studies, lesions in the vessels are generally lumped together, with the overriding morphologic term representing the major histologic lesion. However, there are many instances where multiple diagnoses or descriptors are necessary in delineating the toxicologic process. The authors suggest the following current terminology guidelines set out in INHAND and Standard Exchange of Nonclinical Data (SEND) initiatives (Mann et al. 2012), using one or more of the preferred terms: artery, necrosis, medial; artery, inflammation, mural; artery, inflammation, perivascular; and artery, intimal hypertrophy/hyperplasia. “Inflammation” and “inflammatory infiltrate” are considered equivalent and interchangeable. This terminology is applicable to DIVI associated with biotherapeutics, but since the media is less commonly affected than with small molecule therapies, the term “artery, necrosis, medial” is usually not as commonly applied with biotherapeutic-related DIVI as it is with vasoactive archetypal DIVI where it represents the hallmark or major category of change.
Role of EC Activation in Vascular Injury
EC activation refers to the profound and diverse changes in EC phenotype (i.e., hypertrophy, protrusion into the vascular lumen, and immunohistochemical marker expression) in response to different cytokines, as well as the response to external mechanical forces (Davies 2007; Pober et al. 1990a). Although EC activation is not equivalent to—nor does it always lead to—vascular injury, the two processes are thought to overlap, with activation being a common prerequisite to vascular injury (Pober et al. 1990b). EC activation is considered critical to the mechanism of vascular injury with many agents, including small molecules, biotherapeutics, and antisense oligonucleotide therapeutics (Cines et al. 1998; Frazier 2014; Joseph 2000; Dai et al. 2004; Gimbrone, Nagel, and Topper 1997; Topper et al. 1996). Different stimuli can influence EC activation and can affect EC properties such as cell adhesion, cell shape, and immune function (Bach, Hancock, and Ferran 1997; Pober et al. 1990a; 1990b; Rondaij et al. 2006). Inflammatory cytokines, such as interleukin (IL) 1 and tumor necrosis factor (TNF) α, can activate ECs and modulate their phenotype by inducing the expression of cell surface markers (i.e., E-Selectin, P-Selectin, vascular cell adhesion molecule [VCAM] 1, and intercellular adhesion molecule [ICAM] 1) promoting leukocyte interactions (Cybulsky 2007). The activation of ECs causes an array of responses to biochemical and physical stimuli contributing to propagation of the immunological and inflammatory response (Zhang et al. 2010). Initial activation/injury of the EC and persistent local inflammation is considered to contribute to vascular injury by a variety of means including those related to damage from cytokines, immune complexes, complement dysregulation, or T-cell activation (Blann et al. 2005; Constans and Conri 2006; Day, Hewins, and Savage 2003; Filer et al. 2003; Kollmeier et al. 1998; Langford 2003; Sibelius et al. 1998) as well as additional effects on leukocyte trafficking, vascular permeability, and changes in vasomotor tone (Stewart and Marsden 1994).
ECs have been previously shown to be activated by pharmacologic agents such as immunosuppressive drugs and vasoactive drugs (Joseph 2000; Trapp and Weiss 2005; Zhang, Herman, Knapton, et al. 2002, Zhang et al. 2006, 2008). After drug administration, EC morphology becomes characteristic for activation, as demonstrated by plump appearance, cuboidal shape, protrusion into the lumen, and prominent organelles, followed by apoptosis and eventually EC loss (Zhang, Herman, Knapton, et al. 2002; Zhang, Herman, Holt, et al. 2002; Zhang et al. 2008, 2010).
Role of the Complement System in Vascular Injury
Drug-related vascular changes may often occur via an immune response that is mediated through complement activation rather than by cytokine activation or directly via antibody binding. Complement-related vascular effects are an important component in the development of vasculitis accompanying various antibody and peptide biotherapeutics. General details on specific complement biology will not be reviewed in this article but can be found in relevant review articles (Harboe et al. 2004, 2009; Noris and Remuzzi 2013; Ricklin et al. 2010; Ricklin and Lambris 2013; Volanakis 2014).
The precise mechanism by which biotherapeutics induce complement activation varies with the particular molecule. In some cases, complement activation may be associated with large immune complexes following formation of antidrug antibodies (ADAs) that may be deposited in vascular beds. In other cases, complement activation may occur without circulating immune complexes and can be directed locally at the vascular interface as a result of peptide or antibody interactions with ECs. Regardless of the initiating pathway for complement activation, biologically active soluble complement factors interact with membrane-bound receptors on leukocytes, ECs, and vascular SMCs. The major vascular complement effectors are the following: (1) anaphylatoxins (C3a and C5a), which act as proinflammatory molecules that cause leukocyte chemotactic migration and activation, and degranulation of mast cells and ECs; (2) opsonins (C3b, iC3b, and C3d), which facilitate the removal of target cells and immune complexes; and (3) the terminal membrane attack complex (MAC, C5b-9) that directly causes the lysis of damaged cells. As a consequence of complement activation, several events occur including EC activation, adhesion, and chemotaxis of leukocytes, but importantly in this context, vascular changes such as wall disruption and increased vascular permeability ensue. The location and activity of complement is tightly regulated by glycoproteins, such as C1 inhibitor, decay-accelerating factor (DAF), factors H and I, and several other molecules acting as negative regulators. Properdin, a plasma glycoprotein, is the only currently known positive regulator of complement cascade and acts through stabilizing C3 convertase of the alternative pathway (Lesher, Nilsson, and Song 2013). Therefore, any functional disruption of the regulatory mechanisms can cause a dysregulated cascade of complement activation, which has the potential for deleterious downstream effects in the vascular wall, including severe inflammatory changes in and around vessels.
Structural and functional changes in ECs due to complement activation are mainly mediated by C5a and MAC, causing activation and vascular injury. They confer a proinflammatory and procoagulant phenotype to ECs and, therefore, may drive inflammatory, proliferative, and thrombotic reactions of the vascular wall. Following the interaction with its receptor on the endothelial surface, C5a can directly cause EC retraction, increased permeability and transient expression of P-selectin, von Willebrand factor, and coagulation factor V. P-selectin exposed on the luminal surface of ECs initiates their interactions with circulating leukocytes that ends in the transmigration of leukocytes through the endothelial layer and their accumulation in the vascular wall and/or the surrounding tissue. Complement-mediated activation of leukocytes (such as through C5a on neutrophils) can lead to generation of reactive oxygen species and proteases which cause the release of heparin from the endothelial surface and disrupt the interendothelial junctions, therefore, respectively, compromising the anti-thrombotic and barrier functions of the endothelium. The interaction between MAC and ECs also causes necrosis of the latter, and stimulates the secretion of von Willebrand factor and the synthesis/secretion of IL-1. IL-1, via an autocrine mechanism, stimulates the expression of E-selectin, plasminogen activator inhibitor-1, tissue factor, and other procoagulant and proinflammatory factors. Additionally, the kinin system can interact with ECs in induction of vascular leakage and, although the 2 systems may act independently, there is cross-talk, which involves different components of the complement and kinin systems (Bossi, Bulla and Tedesco 2008, Bossi et al. 2011; Volanakis 2014). Consequently, infiltrating inflammatory cells and structural changes of the vascular wall become apparent at the light microscopic level, and the ensuing morphologic response described with several biotherapeutics is generally indistinguishable from other immune-mediated vascular changes.
Preclinical DIVI Associated with Small Molecules
To put biotherapeutic-related DIVI in context, a brief review of small molecule induced DIVI is necessary. Small molecule–related DIVI in preclinical species can be separated broadly by three causal mechanisms. The first group includes vascular injuries due to a direct mechanism of toxicity to the endothelium. An example is the multiorgan endothelial injury due to anticancer drugs (Bregman et al. 1987; Mikaelian et al. 2010), and features will be described later in this article as they apply to relevant biotherapeutics. The second archetypal lesion of DIVI occurs across a number of classes of vasoactive drugs and is considered related to hemodynamic properties of the molecules, including local or systemic vasoconstriction or vasodilation. The third group is immune mediated, and rare as a cause for small molecule DIVI.
Although the most studied of the three types, there is no generally accepted mechanism for preclinical toxicity with archetypal DIVI related to vasoactive small molecules. Several concepts have been proposed including physical injury due to excessive relaxation or excessive shear stress (Joseph, Rees, and Dayan 1996), and these mechanisms have been recently reviewed (Bendjama et al. 2014; Mikaelian et al. 2014). A unifying feature in common with these proposed mechanisms is the observation of high production of NO within tissues after exposure to many compounds producing vascular effects. Many drugs also have signaling pathways that can pass through endothelial nitric oxide synthase (eNOS), and modulation of NO production can affect lesion severity (Tobin et al. 2014).
The anatomical distribution for DIVI lesions related to vasoactive compounds is species related, and often noted as only focal lesion within organ-specific vascular beds. In the rat, effects are primarily observed in the smaller arterioles of the mesentery and less commonly in pancreatic or testicular arterioles. In dogs and nonhuman primates (NHPs), the lesions are generally noted in the cardiac arterioles and arteries. Chronic administration of these vasoactive agents may result in extensive acute lesions (Westwood, Iswaran, and Greaves 1990) or lesions similar to those observed in spontaneous lesions of polyarteritis nodosa (PAN). DIVI lesions associated with vasoactive compounds are very rare in the mouse but have been observed commonly in rats following treatment with phosphodiesterase (PDE) 3 and 4 inhibitors, adenosine receptor agonists, dopaminergic agonists, endothelin receptor antagonists, angiotensin, bradykinin, α-adrenergic agonists, and Kir channel antagonists (Herman et al. 1989; Johansson 1981; W. Kerns et al. 2005; Losco et al. 2004; Mikaelian et al. 2014).
The histologic lesions of small molecule–related DIVI have been well described (Dietsch et al. 2006; Hanton et al. 2008; Joseph, Rees, and Dayan 1996; Joseph 2000; W. D. Kerns, Arena, and Morgan 1989; C. Louden and Morgan 2001; Weaver et al. 2008; Yuhas et al. 1985), and include perivascular edema, inflammatory cell infiltrates (frequently including macrophages and neutrophils), and mural or perivascular hemorrhage together with degenerative or necrotic changes of the tunica media. Loss of SMCs results in fibrinoid necrosis and/or hemorrhage. Fibrinoid necrosis with vasoactive small molecule–related DIVI is usually segmental and of limited extent and may not be readily observed in tissue sections. Some compounds, like the dopaminergic agonist fenoldopam, cause single cell necrosis and apoptosis of the media rather than classical fibrinoid necrosis. Thrombosis is generally not observed with this type of DIVI. Depending on the drug and duration of exposure, a mixed perivascular inflammatory cellular response can be observed. Perivascular fibrosis with intimal and medial hypertrophy is noted chronically (Losco et al. 2004). Fulminant lesions can be observed as soon as 12 hr (Zhang et al., 2002a), but more typically several days of dosing are required to induce the full spectrum of morphologic changes (Zhang et al. 2008).
The third form of preclinical DIVI corresponds to immune-mediated vasculitis. This form is rare with small molecule drug administration, and damage to the vasculature is secondary to inflammation and/or immune complex formation. With small molecules, this may occur via haptenic binding, such as has been demonstrated with penicillin, allopurinol, or propylthiouracil. The mechanism for this type of secondary DIVI can result from recognition by a therapeutic antibody or induced endogenous antibody of an epitope expressed by ECs (type II hypersensitivity); by binding of a soluble target followed by immune complex formation and deposition (type III hypersensitivity); by binding of drug aggregates to complement; or by the direct activation/dysregulation of complement by the drug (Rojko et al. 2014). The changes are frequently not dose or time dependent. These mechanisms of secondary DIVI are relevant to biotherapeutic-related preclinical DIVI and will be discussed as they relate to vascular injury.
Although detailing the risk assessment strategies for small molecule DIVI is outside the scope of this article, regulatory agency experiences with small molecules causing vascular injury are relevant in DIVI cases related to biotherapeutic administration based on similar principles. It is well recognized that with few exceptions, like dopamine, small molecule drugs that result in preclinical DIVI have not been shown to cause vascular injury in humans and have long records of safe clinical administration (Bendjama et al. 2014; Joseph 2000). Sporadic clinical cases of drug-induced vasculitis cannot always be differentiated from spontaneous idiopathic vasculitides (Radic, Martinovic Kaliterna, and Radic 2012). Hence, regulatory agency responses to sponsors have been tempered with the understanding that clinical relevance of these preclinical findings was uncertain or absent. In some cases of preclinical DIVI with vasoactive agents, drug development has been allowed to progress in clinical trials by regulatory authorities, especially where only a single preclinical species has been involved, safety margins were deemed sufficient, and there has been no clinical evidence of human vascular changes. Without standardized end points, however, clinical monitoring for vascular changes has been problematic. As there are currently no accepted biomarkers for vascular injury, the problem of monitoring has inhibited development of many compounds and posed a major regulatory hurdle. A description of ongoing biomarker work related to DIVI and their specific relevance to biotherapeutics is included later in this article.
An example of regulatory feedback of a small molecule that induces preclinical DIVI is provided by the recent approval of apremilast, a PDE4 inhibitor indicated for the treatment of severe psoriatic arthritis (Food and Drug Administration [FDA] 2014). There was no evidence of vasculitis or related vascular syndromes in human patients during clinical trials. Arteritis was noted in mouse toxicity studies, particularly prominent in the aortic root and heart, as well as to a lesser degree in other organs including the thymus. Pulmonary perivascular inflammation of the lung was also observed. Although the No Observed Adverse Effect Level (NOAEL) in the 6-month mouse study was considered 10 mg/kg/day, based on arteritis at higher doses, the area under the curve (AUC) exposure margin for the maximum recommended human dose of 30 mg twice daily at the NOAEL was subclinical. Several occurrences of vasculitis were also noted in 4-week monkey studies but not in longer studies of 13 weeks or 52 weeks with doses of up to 300 or 600 mg/kg/day, respectively. Based on a 600-mg/kg/day NOAEL in the 52-week monkey study, the AUC exposure margin for the maximum recommended human dose was 4.7-fold over the NOAEL, but based on an NOAEL of 50 mg/kg/day in the 4-week study, the clinical margin was much lower. Although the animal to human exposure margin in both chronic mouse and monkey studies were near parity, the FDA pharmacology reviewer concluded that “the NOAELs provided adequate systemic safety margins for the maximum dose of apremilast” and the new drug application (NDA) was approved despite these preclinical findings (FDA 2014).
DIVI Associated with Biotherapeutics
Histopathology of biotherapeutic-related DIVI
Although there are several different causes or subtypes of vasculitis associated with the administration of biotherapeutics, the microscopic changes and distribution tend to be remarkably similar as a group. Biotherapeutic-induced DIVI is most often attributable to immunogenicity of the drug and results in vasculitis through immune-mediated effects of cytokine release, complement fixation, and/or secondary thrombosis (Rojko et al. 2014). Due to the ubiquitous use of the NHP in preclinical toxicity studies for this class of agents, most cases of preclinical DIVI attributable to biotherapeutic administration are noted in monkeys. Site predilection of preclinical DIVI with biotherapeutics is surprisingly consistent, with vessels prone to immune complex deposition more often affected, including those with high flow rates and especially vessels lined by tall fenestrated endothelium. Most frequent organ sites in monkeys are small to medium caliber arteries in the gastrointestinal tract, gall bladder, pancreas, kidney, and heart (Rojko et al. 2014), but skin, eye, synovium, lung, choroid plexus, and liver, as well as reproductive tract, are also encountered, although less frequently. In the kidney, the renal glomerular capillaries are primarily affected, while in the eye, immune complexes are generally localized to the ciliary body and uvea. In addition, vascular lesions can frequently be noted in areas where there is pronounced vascular turbulence such as aortic bifurcation and other branch points.
Although small molecule DIVI tends to involve specific vessels (especially small- to medium-sized arterioles) in particular vascular beds (W. Kerns et al. 2005; C. Louden et al. 2006), biotherapeutic-mediated DIVI lesion distribution is often more widespread and vascular lesions can occur in capillaries, venules, and occasionally larger arteries. Thickening of the vascular intima is often noted, with loss of endothelium accompanying EC activation, and disruption of the underlying elastic lamina. There is usually a prominent mural or adventitial/perivascular inflammatory infiltrate, which is predominated by lymphocytes and may include neutrophils and often hemorrhage (Figures 2–4). Macrophage infiltration occurs with chronicity. Unlike small molecule–related DIVI, medial changes such as degeneration or fibrinoid necrosis are uncommon until lesions are very advanced, but mural hypertrophy is frequently noted and mural fibrosis and neointima formation may develop over time. Thrombosis may be present, especially in smaller vessels, and when occlusive, this may result in secondary necrosis of surrounding tissue. Lesions may resolve after drug withdrawal, but due to long half-lives of many of biological agents and the length of time necessary for vascular healing, complete recovery may take several weeks despite rather rapid clearing of immune complexes. Simultaneous occurrence of glomerulonephritis is a very common feature with many forms of DIVI related to biotherapeutic administration due to similar pathogenesis (immune complex formation) in glomerular capillary tufts and resolution of these lesions follows a similar course.
Although many cases of biotherapeutic-related DIVI manifest primarily as intimal or mural changes, others tend to present with inflammatory infiltrates limited to the adventitia and/or perivascular stroma. The reason for this difference may be at least partially related to the composition of the immune complexes responsible for the vascular damage. Immune complexes differ in size and charge, and both may play a role in whether the immune complexes deposit in a tissue (Nangaku and Couser 2005). Furthermore, if they do deposit, the localization and ability to activate complement are also impacted by size and charge. Immune complexes of larger sizes are more likely to be deposited in or around the intima with smaller immune fragments able to move more freely through the vascular wall, especially with injury-induced increased endothelial permeability. Cationic antigens are more likely to activate complement. In situ formation of immune complexes also may occur. When a circulating antigen diffuses slowly into the wall of a vessel of an animal with preexisting circulating antibodies against it, there is also a possibility for in situ immune complex formation within the vascular wall at various levels. Regardless of the mechanism of immune complex formation, the subsequent activation of complement and induction of inflammation can result in tissue damage, including subendothelial or mural inflammatory cell infiltrates, fibrinoid necrosis of the media, and/or marked adventitial inflammation, depending on localization of the immune complexes. Different classes of antibodies (immunoglobulin [Ig] G1, IgG3, IgM, IgA, etc.) have varying capacities for activating either classical or alternative pathways of complement, and this may further affect amount of immune complex deposition within an organ or even within a vessel. Immune complexes also interact differently with neutrophils and macrophages, depending on their formation, and this can have a profound effect on the morphologic location of injury, due to paracrine cytokine release. It should be noted that immune complexes within individuals are a divergent mixture of structures—that is, they are not the same. In addition, immune responses can vary over time. Finally, different individuals have different immune responses. All of these differences in immune complex makeup and varying potential for sequestration/activation in leukocytes likely explain the diverse localization of tissue injury (i.e., artery vs. vein, and different substructures within the vessels) and different individual animal responses.
ADAs and Immune Complex Formation
Mechanisms of immune complex–mediated disease associated with biotherapeutics have recently been reviewed in detail (Rojko et al. 2014). ADAs have been a recurrent problem with protein-based biotherapeutics and typically occur within 10 to 14 days after initiation of dosing (Clarke 2010; Leach 2013). ADAs form relatively often in monkeys because recombinant human proteins are sufficiently different from homologous monkey proteins to result in immunogenicity (Brinks, Jiskoot, and Schellekens 2011; Leach et al. 2014; Rojko et al. 2014). Rarely, preexisting, circulating antibodies to the test article may be present prior to dosing, such that immune effects can occur rapidly after administration of drug for the first time (Leach et al. 2014). In addition to varied effects on the pharmacologic activity of a given biotherapeutic, the immune response resulting from ADAs can result in a variety of hypersensitivity reactions that may manifest in vascular injury (Kumar et al. 2010). These may include type II (antibody-dependent) hypersensitivities, but more typically are type III (immune complex–mediated) hypersensitivities. Type I (IgE-mediated) hypersensitivities may also occur after biotherapeutic administration in monkeys (Van Scott et al. 2008), but these generally do not manifest as vascular injury. Although other isotypes may be involved, IgG2 generally is considered the more common antibody isotype for ADA-related immune reactions. Irrespective of isotype, humanized antibodies seem to be associated with the greatest incidence of ADA-related vasculopathies in monkeys. Importantly, ADA in the NHP does not consistently correlate with the occurrence of ADA to a biotherapeutic in humans (Van Meer et al. 2013). According to regulatory guidance and per International Conference on Harmonization—ICH S6R1, “the demonstration of ADAs in any nonclinical species is not considered predictive for ADA response in humans.” Rather, in humans, immune-mediated reactions to biologic agents tend to depend on the foreign character of the molecule, with murine antibodies and chimeric antibodies having much greater potential than humanized antibodies for clinical adverse reactions.
Histopathologic and immunohistochemical evaluation may be very helpful in the identification of these types of drug-induced immune-mediated vascular injuries, based on the demonstration of immune complexes, sometimes containing the implicated biotherapeutic protein, within affected tissues. For instance, coronary arterial injury was noted in bovine serum albumin (BSA)-immunized monkeys that developed anti-BSA antibodies (Stills, Bullock, and Clarkson 1983). Although immune complexes with IgM were identified in lesions of thickened intima within the aorta, glomerular capillaries also demonstrated immunofluorescent granular antigen deposits composed of IgG, IgM, C3, and C4. Several other examples of immune complex–related vasculitis with therapeutic mAbs have been documented using immunohistochemistry (Rojko and Price-Schiavi 2008; Rojko et al. 2014). Characteristically, discrete or aggregated, densely staining granular deposits are found within the intima and/or media in affected small arteries/arterioles (less often small veins/venules) or as globular intravascular microthrombi (Rojko et al. 2014). They are most prominent at vascular branch points and specifically at the level of the internal elastic lamina.
When biotherapeutics are injected, an antibody response may be generated in the monkey, and immune complexes of various sizes can form (Wener 2007). The rate of infusion has an effect, with rapid infusions resulting in greater immune complex formation and hence the potential for more severe immune reactions from biotherapeutic administration. Naturally occurring, circulating immune complexes that persist in the serum are more common in older monkeys as compared to juveniles, in those with underlying disease and in macaques of some origins as compared to others. These factors may increase the risk for drug-related immune complex persistence in serum (Alexander, Clarkson, and Fulgham 1985; Hebert et al. 1991, 1994). Higher concentrations of antigen and antibody generally increase the risk for immune complex formation and vascular injury, but the amount and size of the complex or lattice depend on antigen to antibody ratios (Ponce et al. 2009). If there is a large excess of either foreign antigen or antibodies, complexes may be small, in which they generally either remain in circulation or are rapidly cleared posing little risk for vascular injury. As ratios of antibodies increase in relationship to antigen, somewhat larger, but still soluble immune complexes form, and they may persist in the circulation and result in immune complex vascular wall deposition and/or complement activation, resulting in vascular injury in several tissues/organs. If there are relatively equal molar equivalence of antigen and antibody, large insoluble complexes form, which quickly precipitate and are cleared by phagocytosis. However, if clearance mechanisms are impeded or saturated, these large complexes may also deposit in vascular beds, activate complement, and result in tissue damage (Rojko et al. 2014). The immune complexes may activate the classical or alternative complement pathways, depending on isotype makeup, or may be cleared via the phagocytic system in the liver and spleen following immune complex binding to erythrocytic CR1 via C3b. Characteristics of the biotherapeutic or immune complex itself may result in a propensity for delayed clearance, and in these cases, fixed complement at the vascular interface from persistent complexes may result in vascular wall damage. Immune complexes may also bind and deposit via CD32 expressed on inflammatory cells in monkeys, resulting in sequestration of neutrophils and monocytes in tissue capillaries and local cytokine release that may result in vascular damage (Hart, Alexander, and Dransfield 2004). Platelet activation may also occur, resulting in fibrin microthrombi and possible disseminated intravascular coagulation (DIC).
An example of this type of DIVI in monkeys occurred with a humanized monoclonal antibody. During 2 separate 6-month duration toxicity studies, immune complex–mediated vasculitis was noted following administration of RN6G (Heyen et al. 2014). In one study, 3 monkeys in the mid-dose group died or were euthanized. RN6G, Ig, and complement components were localized by immunohistochemistry within glomeruli and vessels, consistent with immune complex disease. Affected monkeys had lower plasma drug levels than cohorts (decreased by up to 90%) and only these 3 monkeys demonstrated ADA. Perivascular neutrophilic infiltrates were noted in the synovium of 1 monkey and immune complex deposits were found in the glomerulus of all 3 monkeys. A subsequent investigational study was run to assess onset and reversibility, but only 1 female had findings consistent with immune complex disease, and ADA was noted in the same animal. All biomarkers signaling immune complex disease, with the exception of ADA, returned to near normal within a week of dose cessation. Upon submission of the data from the investigative study, the concerns from the regulatory agency appeared alleviated, a clinical hold was lifted, and development of the compound continued (Heyen et al. 2014). The authors considered the severe onset of clinical signs in monkeys related to systemic complement activation, and the clinical risk from this mechanism was considered quite low.
Some preclinical toxicity studies with biotherapeutics are performed in rodents, and marked immunogenicity can result in immune complex disease and hence vasculitis, similar to that in monkeys. For instance, a good laboratory practices (GLP)-1 agonistic peptide-human albumin fusion protein was tested in a 2-week subcutaneous injection toxicity study in mice, and arteritis was noted in 1 high-dose female. The mouse was selected as the preclinical toxicity species (in addition to monkey) based on pharmacodynamic responsiveness and target-related pharmacology data. Vascular mural hemorrhage, mixed inflammatory infiltrates, and medial degeneration were noted in small arteries in the thyroid, salivary gland, thymus, vagina, and skeletal muscle. Consistent with an immune response to an injected foreign protein, there were also injection site inflammatory infiltrates and lymphoid hyperplasia in the lymph nodes and spleen, and increased serum globulins. Concurrent glomerulonephritis was noted in the kidney. The drug exposure (AUC) in this mouse was very low as compared to other mice, and ADA was present in high titers. The sponsor concluded the response was a typical ADA-mediated systemic vascular immune response, and in regulatory documents noted the immunogenicity and rapid emergence of clearing ADAs enabled monitoring for DIVI in chronic rodent studies. The sponsor also was able to demonstrate convincingly that the agent had low immunogenic potential in humans. The arteritis was considered adverse and treatment related and was used to calculate the NOAEL, which was 18-fold higher than the highest expected human exposure. This approach was accepted by all regulatory agencies and the demonstration of drug-induced vasculitis in rodents with this agent has not hindered further drug development.
Nonendogenous Immune Drug Aggregates
Occasionally, large complexes are formed from the administered drug itself, rather than as a result of complexing with endogenous antibodies. Drug or protein aggregates within the dosing solution can further enhance the immune recognition of a substance as “foreign” even without the participation of ADA (Rosenberg 2006; Leach 2013). Nonendogenous IgG aggregates, including those derived from mAb, can simulate immune complexes within the bloodstream, stimulate a local immune response, and result in local hypersensitivity reactions (Vazquez-Rey and Lang 2011). These aggregates can form during production or formulation of a biotherapeutic (Leach et al. 2014; Liu et al. 2011). The tendency for aggregation is further enhanced by heat, shear stress, or agitation and may be reduced by filtration or inclusion of certain vehicles and excipients (Rojko et al. 2014; Vazquez-Rey and Lang 2011). Repackaging has also been associated with increased mAb aggregation due to exposure to silicone particulates, as in the case of bevacizumab (Kahook et al. 2010). Aggregated exogenous mAbs with intact functional capacity in serum can fix complement directly resulting in the potential for tissue deposition and damage along the capillary wall. Circulating large aggregates that lodge within small capillaries in organs and tissues may thus cause local immune-mediated vascular injury independent of ADAs.
In one of the uncommon instances where these nonendogenous immune drug complexes have resulted in vasculitis (from an unpublished study), a monoclonal humanized IgG therapy was associated with morbidity in a single monkey after 24 days during a 28-day toxicity study. Glomerulonephritis was noted in the kidney, and vascular and perivascular inflammation were noted in heart, heart valves, kidney, and skin. Immunohistochemical examination of kidney and dermal vessels in this monkey identified immune complex disease, with mAb drug substance and complement fragment deposition locally. Analysis of the drug material demonstrated that there were large complex aggregates in the administered drug substance. The formulation was reworked and additional subacute and subchronic toxicity studies in monkeys failed to replicate any further vascular lesions.
Vasculopathies Related to Direct or Indirect Pharmacology
Although secondary mechanisms of DIVI tend to be much more common in preclinical toxicity studies with biotherapeutics than primary effects on endothelium, adverse drug reactions after dosing with a biotherapeutic could theoretically occur as a result of direct dose-dependent toxicity or suprapharmacologic effects, especially if ECs or other components of the vessel wall are the target of the agent. These types of vascular injury do not fit into the two major categories of preclinical DIVI—archetypal vasoactive DIVI or secondary DIVI related to immune complexes, complement damage or ADA—and are considered to be rare preclinically. An example of primary pharmacologic effects on endothelium is demonstrated by the small molecule anticancer agents, vincristine and colchicine, which bind tubulin and cause disruption of the microtubule apparatus within ECs and at high doses can result in vascular lesions in preclinical studies (Mikaelian et al. 2010).
Pharmacologic activity of some peptides, particularly those with immunomodulatory activity, can result in secondary damage to the endothelium or vessel wall via immune activation independent of immune complexes or drug aggregates. Although this can result from direct effects of recombinant cytokines on ECs, more commonly vascular damage is a result of upregulation of downstream cytokines via other inflammatory pathways. As an example, recombinant TNF (rTNF) caused endothelial activation resulting in recruitment of leukocytes to the vascular surface, transmural migration, and rarely microthrombi formation (Gribble et al. 2007). Necrosis of the vascular wall and extension of the necrosis to surrounding organ parenchyma have also occurred with this type of injury. High doses of a separate rTNF induced vascular/perivascular inflammatory infiltrates in multiple organs and mortalities in monkeys, suggesting a pharmacologic basis of the effect. In addition to rTNF, recombinant human IL-4 (rhuIL-4), a potent immune stimulator targeting cancer, induced dose-dependent proliferative and inflammatory vascular changes centered on arteries in multiple organs when given to monkeys for 1 month by daily subcutaneous injection (Barbolt, Gossett, and Cornacoff 1991; Leach et al. 1997). Arterial and periarterial changes, including necrosis and inflammation, were noted, and after a 2-week recovery period, arterial obliterative fibrosis, intimal hypertrophy, and intimal/medial basophilia persisted in several organs. The authors concluded that rhuIL-4-related lesions resulted from activation of the immune system and secondary effects on ECs (Leach, Rybak, and Rosenblum 1997), and the rhuIL-4-related clinico-pathological changes were considered to be reversible (Gossett et al. 1993). Because of the lack of pharmacologic activity of many biologics or recombinant peptides in rodent species, the use of rodent surrogate molecules may be the only way of testing such effects preclinically when additional species other than monkey are required. Overall, direct cytokine-related vasculopathies appear to be uncommon in any preclinical species.
Clinical cases where marketed immunomodulatory biologics have induced vasculitis through cytokine-mediated pathways are also uncommon, but examples include reports with etanercept and rituximab (Dereure et al. 2001; Galaria, Werth, and Schumacher 2000). Etanercept is a soluble rTNF receptor: Fc-fusion protein for rheumatoid arthritis. Leukocytoclastic vasculitis has been noted in humans as a very rare potential complication of therapy. Dysregulation of cytokine cascades was suspected, because etanercept is considered immunosuppressive rather than proinflammatory. Rituximab is a murine anti-CD20 chimeric antibody targeting lymphoid malignancies. Leukocytoclastic vasculitis has been reported soon after administration in a few patients, and the lack of CD20 expression on ECs and other ex vivo data has led to the conclusion that the mechanism for these rare clinical effects are also through cytokine release even though in other clinical cases, immune complexes have been identified (Dereure et al. 2001; Rojko et al. 2014). No vascular effects were reported in preclinical studies with etanercept or rituximab, even though etanercept cross reacts with and neutralizes TNF from multiple species. This reaffirms the often held contention that clinical immune-mediated reactions are hard to predict from preclinical data (Bugelski and Tracy 2004; Martin and Bugelski 2012).
Pharmacologically related injury to the vasculature also may occur via a mechanical mechanism whereby there are physiologic changes in the vascular wall. A generalized vascular leakage syndrome (VLS) involving angiocentric inflammation has been described after administration of IL-2 both clinically and preclinically, which has limited its use as an antineoplastic agent (Anderson and Hayes 1989). In mouse models, recombinant IL-2 also induced VLS, evidenced by inflammatory cell infiltration and fluid extravasation into the lung and liver and increased levels of TNFα, interferon γ, IL-5, MCP1, and IL-6 in serum (Sivakumar et al. 2013). In animal models, the progression of vascular leakage can be assessed using the accumulation of Evans Blue dye in tissues such as lung. Less pronounced VLS also has been noted with administration of other ILs and cytokines such as rIL-21 and GM-CSF.
Probably, the most publicized case of clinical vascular damage due to proinflammatory peptide therapies involved a clinical trial in patients administered the anti-CD28 product TGN1412, and which resulted in effects that were not predicted preclinically (Suntharalingam et al. 2006). Although there were vascular lesions associated with this “cytokine release syndrome” (CRS), the changes were more generally associated with generalized organ failure, and these types of responses should be considered distinct from classical DIVI. Other mAb therapeutics known to induce CRS or first infusion reactions include OKT3, Campath-1H, and Herceptin, but like TGN1412, these agents do not necessarily result in vasculitis or in direct injury to endothelium, and hence such agents should not be classified with others that may induce DIVI (Dhir et al. 2012).
Proinflammatory monoclonal antibody and peptide therapies can also result in vasculitis or perivasculitis related to pharmacological activity on resident lymphocytes. An example includes ipilimumab, a marketed anti-melanoma agent that inhibits cytotoxic lymphoid antigen–4 (CTLA–4; Minor, Bunker, and Doyle 2013). The blockade of the CTLA-4 immune checkpoint elicits an effective anticancer immune response in a range of preclinical models and humans, and since it is believed that specific lymphocytes targeting tumor associated antigens are primed or released by the actions of this drug in order to attack malignant cells, the incidence of vascular/perivascular infiltrates corresponds favorably with efficacy. Some patients have shown autoimmune antibody titers and infiltrates of lymphocytes within and around vessels in various organs including gastrointestinal tract, pancreas, and reproductive tracts (Minor, Bunker, and Doyle 2013). Perivascular organ infiltrates of lymphocytes were also noted in preclinical studies with this agent.
Less severe inflammatory responses and inflammatory infiltrates have occurred with other proinflammatory anticancer agents, such as lambrolizumab and BMS-936558, both antibody therapies that specifically block programmed death antigen 1 (PD-1). Blockade of PD-1, a receptor expressed by T cells, modulates immune responses, and organ inflammation has been noted preclinically with this class of agents due to pharmacologic activity (Topalian et al. 2012). Although vascular/organ inflammation has also been noted in patients with these various biotherapeutics, there are effective clinical strategies in place to combat adverse events, and regulatory authorities have accepted the vascular toxicity profiles in preclinical and clinical packages as part of the risk:benefit for many of these proinflammatory antineoplastic antibodies. In the case of some of the anticancer (and potentially life-threatening) indications noted earlier, antineoplastic agents have received FDA approval to progress in trials with only a 2-fold margin over NOAELs. However, for other agents used for other less severe indications, the regulatory agency has requested a 10-fold or greater safety margin above the NOAEL. In a few cases, the FDA also has requested investigative studies to support sponsor’s mechanistic hypotheses and documentation of ADAs—or in this case, the lack thereof—both preclinically and in patients, to rule out other immune-mediated drug-induced vascular reactions. Due to the pharmacologic pathogenesis of this particular type of injury, the FDA requested inclusion of arms to some investigative studies using rodent homologues of peptides/antibodies with demonstrated pharmacologic activity in that species, to help provide evidence for the pharmacologic basis of injury.
Vasculitis Due to Off-target Pharmacology
Vascular injury due to off-target pharmacology involving toxicity to ECs or SMCs is a more recently identified mechanism related to administration of therapeutic antibodies. Due to the relative specificity and receptor targeting of peptide therapies, off-target interactions are considered rare when compared to small molecules (Gribble et al. 2007). Due to their catabolism to small peptides and amino acids, and in contrast to small molecules, biotherapeutic metabolites generally lack pharmacology (Rojko and Price-Schiavi 2008). There is always the potential for unintended toxicity if the targeted epitope of a biotherapeutic agent is shared with endothelial or other vascular epitopes. For example, alemtuzamab binds to CD52 on B and T cells, but is widely distributed across leukocyte lineages, and consequently, granulocytopenia is a common side effect (Gribble et al. 2007). As noted previously, immune-mediated vascular toxicity has been induced by a variety of compounds that possess proinflammatory potential, but these cytokine-mediated and superagonistic effects do not represent truly unexpected off-target pharmacology, but rather target-related toxicity associated with immune system activation.
One example of off-target localized DIVI involved a monoclonal antibody that targeted a circulating antigen and was developed as an anti-inflammatory agent. When administered intravenously (IV) as a single dose or subcutaneously for multiple doses in a monkey safety study, it caused unanticipated acute vascular injury (R. Pai, personal communication, January, 2014). The IV injection site had mural thickening and focal adventitial necrosis with inflammatory cell infiltration of the vein. These findings resolved, except for residual hemosiderin accumulation, in the recovery phase. Immune complexes were not noted with tissue immunohistochemistry at either time point. Both in vivo and in vitro mechanistic investigations demonstrated a lack of typical mechanisms of antibody-induced vascular injury (including cytokine, proinflammatory, or complement-mediated), but instead supported an unanticipated off-target effect of the monoclonal antibody on ECs and consequent downstream effects, including alterations in vascular SMCs. Ligand binding of the antibody activated NO, eventually resulting in increased endothelial permeability. This mechanistic understanding, in addition to the narrow safety margin, acute onset of the severe toxicity, and likely relevance to humans, precluded further development of the monoclonal antibody.
In another unpublished example, a monoclonal antibody that targeted a circulating antigen was being developed as an oncology therapy. Vascular injury was observed following repeated IV administration in monkey and rat GLP safety studies. In the monkey, a mixed inflammatory vasculitis and perivasculitis with various morphologic appearances of proliferative and degenerative changes affected multiple organs and vessel types. In the rat, multifocal splenic necrosis and inflammation and multifocal bone marrow hypocellularity were interpreted as vascular in origin. These findings were not observed in the recovery groups in either species. Vascular injury was not associated with a consistent circulating cytokine profile, elevated levels of circulating ADAs, or co-localization of the monoclonal antibody by IHC. Studies with small molecules sharing the same pharmacologic target did not share a similar toxicity profile. Together, these findings suggested the mechanism was not mediated by proinflammatory or cytokine signals, immune complex formation, or direct pharmacologic activity. Mechanistic investigations using an in vitro human EC system and tool molecules with in vitro and in vivo studies further excluded antibody-dependent cell cytotoxicity (ADCC)- or complement-dependent cytotoxicity (CDC)-related mechanisms. Rather, data pointed to an off-target mechanism involving binding of heparan sulfate proteoglycans on the plasma membrane of ECs with consequent signaling through extracellular signal-regulated kinases and EC proliferation. Collectively, the results indicated that vascular injury induced by this monoclonal antibody in two pharmacologically relevant preclinical species was an off-target toxicity that was not monitorable, had a narrow safety margin, had potentially life-threatening sequelae, and was likely relevant to humans; therefore, further development of this anticancer biotherapeutic was not pursued.
Differentiation from Spontaneous Vascular Disease in Laboratory Animals
Spontaneous vascular inflammation is an uncommon event but has been described in a variety of laboratory animal species. In preclinical species, lesions can occur either in isolated vessels or in multiple vessels and tissues where they are commonly lumped under the term “PAN.” The morphologic spectrum of spontaneous inflammatory vascular lesions of PAN described in common laboratory animal species (including mice, rats, dogs, minipigs, and cynomolgus macaques) is similar to those noted in humans, suggesting a similar etiology and pathogenesis.
In rats, spontaneous vascular lesions are most often seen as incidental findings in aging animals with localization primarily to the medium-caliber mesenteric arteries, but may also be found in pancreatic, hepatic, spermatic, uterine, ovarian, cerebral, and adrenal arteries (Carlton and Engelhardt 1991). Macroscopically affected vessels may be thickened or have nodular swellings that may contain thrombi. Histological features vary with the age of the lesion and are thought to begin with fibrinoid necrosis of the media and internal elastic lamina followed by infiltration of a mixed inflammatory cell population, consisting of lymphocytes, plasma cells, and macrophages, within the media and adventitia. In some arteries with well-established lesions, the media and/or adventitia may be thickened due to fibrosis and extension of the inflammatory cell infiltrates into periarterial tissues. Lesions are more common in certain strains and stocks of laboratory rat, indicating a potential role of genetic background in predisposition for the development of these vascular alterations. A host of rat models of disease may also demonstrate vascular lesions without drug treatment, including spontaneous hypertensive rats (SHR) and various models of autoimmune disease (Amano, Hozama, and Hamaskima 1979; Brinks, Jiskoot, Schellekens 2011; Lim, DeLano, and Schmid-Schonbein 2001; Usui et al. 2012). Although the vascular lesions in these rat models are not truly idiopathic, they should be considered in the background pathology of the various strains and can be confused with DIVI in efficacy studies when using these models.
In mice, vascular lesions are similar to the rat, with the exception that in some strains spontaneous vascular changes are more frequently localized to the renal blood vessels (Faccini, Abbott, and Paulus 1990; Figure 1), and somewhat less commonly in vessels of the pancreas and thymus. Spontaneous vascular lesions have also been seen in the epididymis and vas deferens in aging BALB/c mice and are of unknown pathogenesis (Itoh et al. 1999). In both rats and mice, lesions of PAN may be noted more commonly in animals with concurrent chronic progressive nephropathy. Similarly, vascular lesions are often noted in aged mice with amyloidosis more frequently than in those without renal pathology. Presumably, the mechanism involves alterations in renal–vasomotor interactions, but there are currently no data to support this theory. There are many mouse models of vasculitis involving strains such as MRL/lpr, NZB, PN, and SNF1 (Holmes, and Burnet 1963, Luzina, Knitzer, and Atamas 1999, Kalled et al. 1998; Moyer, Strandberg, and Reinisch 1987) with lesions consisting of medial necrosis, and neutrophilic and/or mononuclear mural and adventitial infiltrates. Vascular lesions may also spontaneously develop in some strains of mice prone to develop lupus or hypertension, as in similar rat models.
The so-called Beagle Pain Syndrome or canine juvenile polyarteritis syndrome (CJPS) occurs most often in young beagle dogs and shows similar inflammatory changes in vessels with consistent involvement of small- to medium-caliber cardiac, cranial mediastinal, and cervical meningeal vessels (Hayes, Roberts, and Halliwell 1989; Snyder et al. 1995). Vasculitis may also be observed in tracheobronchial lymph nodes, testes, small intestine, stomach, diaphragm, esophagus, and urinary bladder. The pattern of vascular lesions can vary from infiltration of lymphocytes and macrophages into the tunica adventitia, to transmural inflammation accompanied by fibrinoid necrosis and vascular thrombosis. Inflammatory cell infiltrates may be present segmentally or form complete cuffs around affected vessels.
Spontaneous arteritis has been described in minipigs in multiple organs including mesentery, heart, lungs, spleen, pancreas, kidney, salivary glands, and both male and female reproductive tracts (Jeppesen and Skydsgaard 2014). Vasculitis associated with an idiopathic thrombocytopenia syndrome has also been described, particularly in the Gottingen breed (Swindle et al. 2012). In either case, the lesion is similar morphologically to PAN in other species and is characterized by medial smooth muscle degeneration, intimal hyperplasia with EC hypertrophy, and perivascular or transmural mixed inflammatory infiltrates in scattered, small- to medium-sized arterioles and arteries. Necrosis or thrombosis may occur and laminar thickening of the adventitia has been described (Swindle et al. 2012).
Spontaneous vascular lesions in cynomolgus macaques are noted occasionally in preclinical studies, but systemic vascular syndromes involving multiple vessels are much less common (Chamanza et al. 2006; Sato et al. 2012). In a case report involving an untreated cynomolgus macaque (Porter, Frost, and Hubbard 2003), small- to medium-caliber arteries in the kidney, small intestine, colon, heart, spleen, mesentery, urinary bladder, and pancreas had varying degrees of inflammation. The mildest changes described were infiltration of the tunica adventitia and tunica media by small to moderate numbers of lymphocytes and macrophages admixed with a few neutrophils, eosinophils, and plasma cells. As severity increased, the inflammatory infiltrate was accompanied by intimal proliferation and fibrinoid necrosis and loss of internal elastic lamina in the most severely affected vessels. In another case report in an untreated cynomolgus macaque (Albassam, Lillie, and Smith 1993), lesions were also present in small- to medium-caliber arteries in the kidney, heart, gall bladder, stomach, pancreas, large intestine, and uterus with a similar spectrum of inflammatory cell infiltration but no fibrinoid necrosis.
Differentiating drug-induced changes from these spontaneous background lesions in preclinical studies is complicated when changes potentially caused by treatment are histologically indistinguishable from spontaneous lesions and are present in incidence and/or severity greater than expected for spontaneous lesions (Clemo et al. 2003). Histomorphology alone is unlikely to be definitive in making the case for either drug-induced or spontaneous origin, so careful consideration of historical background incidence in a given species/strain is often necessary for correct interpretation if multiple individuals are affected. Although dose response may be helpful in differentiating small molecule–related DIVI from background lesions, this is problematic with DIVI associated with either biotherapeutics or antisense oligonucleotides since neither generally follow a typical dose response pattern.
DIVI in Humans
Vasculitis in humans may occur without a recognized cause (idiopathic), may be drug-induced, or may accompany systemic disorders (neoplastic, infectious, autoimmune, and rheumatic diseases). In humans, DIVI accounts for about 3% of all vasculitides (Biasucci and Cardillo 2012), and the skin and kidneys are most frequently affected. Many marketed drugs have been implicated with clinical DIVI, including small molecules and biotherapeutics (Doyle and Cuellar 2003; Radic, Martinovic Kaliterna, and Radic 2012; Taborda, Amaral, and Isenberg 2013). The typical histologic appearance consists of inflammation of small- to medium-sized arteries, predominantly by lymphocytes or, more rarely, with giant cells (Dubost, Souteyrand, and Sauvezie 1991; Jennette and Falk 1997). Microscopic changes are often segmental and focus on vascular branching points. Human DIVI may mimic a spectrum of spontaneous vascular diseases like PAN, Wegner’s granulomatosis, or Churg–Strauss syndrome (Doyle and Cuellar 2003; Merkel 2001; Radic, Martinovic Kaliterna, and Radic 2012), and in these cases can involve any type of artery, vein, or capillary including the aorta.
The etiology and pathogenesis of spontaneous vascular lesions in humans are not well understood but are believed to be immune mediated (Barron et al. 1988; Conn et al. 1976; Felsburg et al. 1992). Lesions of PAN in humans resemble those in preclinical species (Schoen 2005). Additional qualification of this disease in man has been proposed in 2012 by the International Chapel Hill Consensus Conference on Nomenclature of Vasculitides (ICHCCNV), which defined PAN as “necrotizing arteritis of medium or small arteries without glomerulonephritis or vasculitis in arterioles, capillaries, or venules, and not associated with antineutrophil cytoplasmic antibodies.” Antineutrophil cytoplasmic antibodies (ANCA) are autoantibodies against myeloperoxidase and proteinase-3 contained in the azurophilic granules of neutrophils and peroxidase-positive lysosomes in monocytes, and can be expressed at the surface of TNF-primed neutrophils. The binding of ANCA to primed leucocytes induces the production and release of many cytokines such as IL-1, MCP-1, and IL-8 (Berger et al. 1996; Brooks et al. 1996; Casselman et al. 1995; Ralston et al. 1997). ANCAs are typically absent in patients with PAN whereas ANCA are frequently noted in the serum of human patients with small molecule DIVI. They are therefore considered a useful discriminator, because PAN- and ANCA-associated vasculitis in man can exhibit clinically and pathologically indistinguishable necrotizing arteritis of medium and small arteries (Amano, Hozama, and Hamaskima 1979; Fauci, Haynes, and Katz 1978; Jennette, Falk, et al. 2013). It further appears that preexisting systemic inflammation in sensitive individuals increases the severity of vascular lesions in commonly affected locations (Jennette, Falk, et al. 2013). This observation is important when considering the risk to humans of vascular inflammation seen with a variety of biotherapeutics in laboratory animal species.
A revised nomenclature for human vasculitides has recently been proposed by the ICHCCNV (Jennette, Falk, et al. 2013). There are 2 human categories of vasculitis related to drug administration, namely, “drug-associated ANCA-associated vasculitis (AAV)” and “drug-associated immune complex vasculitis” (ICV). Although ANCAs are often a hallmark of human AAV, their incidence and association with DIVI in laboratory animal species is largely unknown, as ANCA analysis has not generally been performed in preclinical studies due to the lack of validated assays. AAV has been defined as “necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels (i.e., capillaries, venules, arterioles, and small arteries), associated with ANCA” (Jennette, Falk, et al. 2013). The pathogenesis of AAV in man is still not fully understood and the involvement of different factors such as drugs (e.g., prophylthiouracil, hydralazine, levamisol adulterated cocaine,
The other type of DIVI in humans, ICV, is most often related to the administration of biotherapeutic agents and can involve antibodies to a wide variety of antigenic epitopes. It has been noted with several different antibody therapies, including rituximab and natalizumab. The pathogenesis often involves ADAs and has many parallels with many or most cases of drug-induced immune complex formation in preclinical species and is therefore most relevant to preclinical biotherapeutic-related DIVI. This type of vascular injury can also occasionally be associated with some non-Ig therapeutic products. For instance, immune complex reactions have resulted rarely from dextran infusion in clinical patients with high antidextran IgG levels (Bircher, Hedin, and Berglund 1995).
Regardless of type, treatment of DIVI in humans consists of drug withdrawal, which can often lead to spontaneous recovery, and immunosuppressive therapy, with clinical improvement generally observed within days or weeks. Although the prognosis is generally good, especially when the skin is the only affected organ, multiorgan involvement may have a worse prognosis, and death occurs in about 10% of these types of systemic cases (Taborda, Amaral, and Isenberg 2013).
Biomarkers of Biotherapeutic-associated DIVI
As previously noted, hemodynamic parameters such as heart rate and blood pressure have been successfully used as surrogate biomarkers of DIVI for many small molecule drugs (W. D. Kerns, Arena, and Morgan 1989; W. D. Kerns et al. 1989; W. Kerns et al. 2005; Mesfin, Shawaryn, and Higgins 1987, Mesfin et al. 1989; 1996; Nyska et al. 1998). However, not all small molecules that are associated with DIVI alter clinically measurable hemodynamic parameters (Albassam et al. 1999, 2001; Dalmas et al. 2011; Jones et al. 2003). Hence, there has been an ongoing search for circulating biomarkers that can detect the onset, progression, and reversibility of this type of drug-induced damage. There are currently no validated circulating biomarkers for DIVI, although there have been many potential or candidate biomarkers proposed for identifying preclinical small molecule DIVI. These have included circulating proteins, and even genomic markers, but their utility has been limited by their potential qualification with only a very limited number of compounds, most of which are vasoactive molecules (Brott et al. 2005; Daguès, Pawlowski, Sobry, et al. 2007, Daguès, Pawlowski, Guigon, G, et al. 2007; Dalmas et al. 2008, 2011; Enerson et al. 2006; Joseph, Rees, and Dayan 1996; W. Kerns et al. 2005; C. Louden et al. 2006; Newsholme et al. 2000; Stagliano et al. 2001; Thomas et al. 2009; Thomas et al. 2012; Turk 2010; Weaver et al. 2008; 2010; Zhang et al., 2002b; Zhang et al. 2006; 2008; 2012). The majority of candidate biomarkers have included factors involved in EC activation (Zhang et al. 2010), edema, inflammation, tissue remodeling, (Blake, and Ridker 2001; Dalmas et al. 2008; Daguès, Pawlowski, Guigon, et al. 2007; Daguès, Pawlowski, Sobry, et al. 2007) and maintenance of vascular tone (C. Louden et al. 2006). The search for DIVI markers has been formalized, with the formation of a collaborative consortium of industry, academic, and regulatory experts into The Predictive Safety Testing Consortium (PSTC) Vascular Injury Working Group (VIWG), and a panel of biomarkers for DIVI has been proposed (Mikaelian et al. 2014). The panel was based on vasoactive small molecules, but many of the proposed markers may be applicable to biotherapeutic therapies, especially those related to EC or SMC activation or inflammatory pathways. Ongoing work among the PSTC VIWG involves testing the panel of potential biomarkers in more definitive biological qualification studies with a wide range of nonvasoactive compounds known to induce vascular lesions in rats, regardless of pathophysiologic mechanism. Other groups such as the European Union based Safer and Faster Evidence-based Translation (SAFE-T) consortium are doing clinical validation work in parallel (Bendjama et al. 2014). Based on mechanistic studies of complement activation, T cell activation, and cell immune responses associated with EC injury (Blann et al. 2005, Constans and Conri 2006; Drouet et al. 2003; Kollmeier et al. 1998; Sibelius et al. 1998), several potential biomarkers for detecting DIVI have recently been proposed by these groups and lists of candidates are available (Bendjama et al. 2014; Mikaelian et al. 2014). Changes in other markers such as matrix metalloproteinases (MMP-1, MMP-3, and MMP-9), Vascular Endothelial Growth Factor, thrombomodulin, E-selectin, and P-selectin have been evaluated in human patients with vasculitis (Monach et al. 2011). In that study, circulating levels of MMP-3 outperformed CRP and were able to distinguish active disease from remission. MMP-3 and CRP were also used effectively in a separate clinical study of immune-mediated vasculitis patients (Matsuyama et al. 2003); therefore, these biomarkers are gaining interest in preclinical DIVI investigative studies where an immune component is suspected. However, no vascular biomarkers have risen to the level of preclinical or clinical validation. Thus, such exploratory analytes should be interpreted with caution and should not be the sole basis for clinical decision making (Frazier and Seely 2013).
Specific forms of human vascular injury are associated with ANCA antibodies, which involve direct activation of cytokine-primed neutrophils and monocytes and results in their degranulation and subsequent damage to the adjacent endothelium (Falk et al. 1990; Gao and Zhao 2009; Jennette, Falk, and Wilkman 1995; Radic, Martinovic Kaliterna, and Radic 2012). The contribution of ANCA in preclinical cases of DIVI in toxicity studies remains to be determined. Different animal models of vasculitis associated with ANCA antibodies have been developed where the production of autoantibodies against myeloperoxidase, proteinase-3, and other proteases was obtained in mice and rat (Coughlan, Freeley, and Robson 2012; Salama and Little 2012). If similar mechanisms are operative in cases of preclinical DIVI, they have not yet been conclusively demonstrated other than in these few animal models, and as in clinical cases, are likely to be associated more commonly with specific small molecule administration than with biotherapeutics.
There are a number of other inflammatory markers that may be of relevance to biotherapeutic-associated DIVI due to their pathogenesis involving immune deposition (Sams 1985). As previously noted, immune complex–related DIVI may involve recognition of endothelial epitopes, complement activation, and at least in the case of peptide therapies, the generation of ADA and potentially antibody or immune complexes within the vasculature. However, since ADA may be present in many animals without any evidence for vascular injury, it should not be regarded as a useful biomarker (Alpers 2009; Nangaku, and Couser 2005). Rather, a host of other circulating inflammatory mediators may be potential candidates in DIVI, based on the specific cell types and inflammatory cascades involved and their similarity to pathophysiology associated with biotherapeutic-related DIVI. The list of potential circulating cytokines that can be used as biomarkers in this regard is exhaustive, but there are excellent reviews available with descriptions of their benefits and pitfalls (Tarrant et al. 2010). In addition, since biotherapeutic-related DIVI is often accompanied by the simultaneous occurrence of immune-mediated glomerulonephritis (due to similar pathophysiologic mechanisms; Demeule, Gurny, and Arvinte 2006), proteinuria and/or albuminuria may be useful monitorable surrogate biomarkers for these effects (Frazier and Seely 2013).
In addition to more conventional analytes such as soluble proteins or cytokines noted in the previous references, other less routine sources of vascular biomarkers are being developed that hold promise in the large molecule arena, including endothelial cell microparticles (EMP; Enerson et al. 2010, Erdbruegger et al. 2008). EMP can serve as surrogate markers of endothelial function and elevated levels of EMP have been seen in patients with vascular diseases, and may correspond to vascular injury in patients with cardiovascular disease and/or vasculitis (Ardoin, Shanahan, and Pisetsky 2007; Brogan et al. 2004; Clarke et al. 2010; Dignat-George and Boulanger 2011; Erdbruegger et al. 2008, Erdbruegger, Dhaygude, and Woywodt 2011; Mesri and Altieri 1998; 1999). Circulating endothelial cells (CEC) have also been proposed as biomarkers of DIVI (Thomas et al. 2009) and are thought to be detached following injury and hence detectable in the peripheral circulation. Blood levels of CECs are increased in many conditions associated with endothelial dysfunction including hypertension, diabetes, preeclampsia, and chronic kidney failure (Boos et al. 2007; Canbakan et al. 2007; Koc, Bihorac, and Segal 2003; McClung et al. 2005; Woywodt et al. 2002). The degree of vascular injury has been shown to correlate with increases in the number of CECs in circulation (Gill et al. 2001; Wu, Chen, and Hu 2007). Recent work has also demonstrated a method for identifying and enumerating endothelial progenitor cells in DIVI in rat toxicology studies (Thomas et al. 2009).
Points to Consider for the Interpretation of Biotherapeutic-related DIVI
The following bulleted list incorporates several points to consider as they specifically relate to DIVI associated with biotherapeutics. The list is not meant to be comprehensive but instead provide some considerations for toxicologists and pathologists when vascular effects are noted in preclinical toxicity studies with biotherapeutics. Vascular toxicity in preclinical species is not always translated to human; therefore, the presence of preclinical DIVI should not necessarily halt development or progression of a potential drug, but rather signal the need for further investigative work. Cautious clinical dose selection/escalation with appropriate safety margins is necessary once clinical progression is instituted. Although the precise pathogenesis of DIVI associated with biotherapeutic administration varies with the agent, attempts to demonstrate causal mechanisms and clinical relevance are important in all cases. Consultation with regulatory authorities may be important to help select the most appropriate study designs and end points. For biotherapeutics where the pathogenesis is related to ADA, the incidence of lesions are often idiosyncratic in dose response (i.e., do not show a strict dose relationship in terms of incidence and severity). Due to a variety of factors including pathogenesis and species predisposition, DIVI noted in rodents with a biotherapeutic is generally of less concern or clinical relevance than biotherapeutic-related DIVI in monkeys. However, there are situations where rodent DIVI may have greater clinical relevance, for instance, with molecules intended to be immunostimulatory or those with pharmacologic effects on endothelium. When vascular injury is noted following biotherapeutic administration in preclinical toxicity studies, it is important to compare and correlate individual animal ADA with incidence of vascular injury. It may also be useful to attempt to demonstrate deposits of Ig and/or complement components (suggestive of immune complexes) via tissue immunohistochemistry in affected vessels. The use of a panel of preclinical biomarkers for DIVI, while not necessarily qualified nor validated, may be helpful in determining which potential analytes might also be included in clinical trials and provide a greater level of patient safety. With any mechanism of DIVI, a panel of prospective markers of endothelial activation (i.e., VCAM) or medial injury may also be of benefit. The authors recommend following current terminology guidelines set out in INHAND and SEND initiatives, which provide consistency in morphologic terms for biotherapeutic-associated DIVI:
artery, inflammation, mural; artery, inflammation, perivascular; and/or artery, hypertrophy/hyperplasia, intima.
Inflammation and inflammatory infiltrate are equally acceptable and considered interchangeable.
Lesions of DIVI associated with biotherapeutics generally occur randomly in vessels in many organs. Hence, routine GLP tissue lists are considered sufficient to identify DIVI with these agents and there is little to be gained from sampling additional nontraditional vascular beds (e.g., mesenteric plexi in rats).
Due to the idiosyncratic nondose responsive nature and random distribution of vasculitis with biotherapeutic administration, all routine organs (not just target organs) in low- and mid-dose group monkeys should be examined in GLP studies once vasculitis has been observed with a compound in a high-dose group. In non-GLP studies where tissue evaluations are typically limited, examination of liver, kidney, heart, lungs, eyes, synovium, and brain (specifically choroid plexus) are recommended as they are most likely to demonstrate vascular lesions associated with biotherapeutic agents.
Recovery groups with a minimum of 2 animals per sex and dose group should be included in subsequent subchronic or chronic studies when vascular lesions are noted in preclinical species with biotherapeutic administration that are not related to ADA. A variety of tissues may be required to be examined in these groups due to the variability in localization of DIVI lesions. If ADA is implicated in the pathogenesis, recovery groups may not be necessary or even applicable, depending on the circumstance.
Due to the protracted nature of immunomodulatory stimuli and resolution rate in vessels, an off-dose period of 6 to 12 weeks in recovery groups may be required for complete reversal.
Conclusions
Despite numerous years of study, DIVI remains a significant challenge for the development of new and novel pharmaceutical agents. Much has been learned regarding the vasoactive mechanisms of toxicity for small molecules and human risk. For biotherapeutics such as recombinant peptides or antibodies, a variety of immune and inflammatory mechanisms appear to be involved, so the risk assessment is less clear. This review has discussed the various mechanisms involved and how they may or may not be relevant to humans. The role of spontaneous vascular inflammation in common laboratory species and humans has been presented as a potential confounding factor. The collective experiences of the review team suggest that the toxicologist and pathologist can tease apart the various effects seen in general toxicity studies to make an informed risk assessment. Additional parameters to those routinely collected in toxicity studies may be needed in a weight-of-evidence approach and to support plausibility of mechanisms.
Footnotes
Acknowledgments
The authors thank Reina Fuji and Bev Maleef for help with images and/or providing some of the photomicrographs.
*
This review is a product of a Society of Toxicologic Pathology (STP) Working Group commissioned by the Scientific and Regulatory Policy Committee (SRPC) of the STP. It has been reviewed and approved by the SRPC and Executive Committee (EC) of the STP. The article does not represent a formal best practice recommendation of the society but provides expert guidance on key principles to consider in designing, conducting, and reviewing regulated toxicity studies. The views expressed in this article are those of the authors and do not necessarily represent the policies, positions, or opinions of their respective agencies and organizations. Readers of Toxicologic Pathology are encouraged to send their thoughts on these articles or ideas for new topics to the editor.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author Contribution
All authors (KF, JE, PF, SG, SH, ML, CL, MS, JW, TZ) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be considered to represent any agency determination or policy.