
Abstract
In nonclinical safety studies for new drug development, healthy animals have been commonly used. However, in some cases, the use of animal models of human disease is considered to be more favorable in evaluating risks in patients. To elucidate the current status of the use of animal models for nonclinical safety assessment, an internal questionnaire from the Japan Pharmaceutical Manufacturers Association and surveys (questionnaire period: August 27 to September 30, 2015) of both common technical documents and review reports of approved drugs (approval period: May 1999 to May 2017) disclosed by the Pharmaceutical and Medical Devices Agency were conducted. Although there were some concerns and limitations raised, the survey results revealed that animal models have been used in nonclinical safety assessment on a case-by-case basis and that nonclinical safety studies using animal models were included in the data packages of several approved drugs in Japan. The survey results also revealed that nonclinical safety studies using animal models have become more frequent in the past few years. In almost all cases, useful information, such as signs of toxicity under disease conditions and mechanisms of toxic change, was obtained from the results of nonclinical studies using animal models.
Note: This is an opinion article submitted to the Toxicologic Pathology Forum. It represents the views of the author(s). It does not constitute an official position of the Society of Toxicologic Pathology, British Society of Toxicological Pathology, or European Society of Toxicologic Pathology, and the views expressed might not reflect the best practices recommended by these Societies. This article should not be construed to represent the policies, positions, or opinions of their respective organizations, employers, or regulatory agencies.
Keywords
In the pharmaceutical industry, nonclinical safety studies are conducted during new drug development according to regulations, such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH 2009) M3(R2) guidance. These studies aim to provide the safety/toxicity information (e.g., target organs, dose-dependence relationships to exposure and reversibility) needed to support the testing of new drugs in human clinical trials as well as for postmarketing commitments. Healthy animals are commonly used in these nonclinical studies because of historical precedent in addition to the abundant historical background data available in these animals. However, sometimes animal models can provide additional information that cannot be obtained from healthy animals. For example, animal models might provide more relevant human risk assessment information regarding how the disease condition itself, genetic background, or other exposures might influence the toxicity profile of a drug (U.S. Food and Drug Administration [FDA], 2011).
Recently, several review articles regarding the use of animal models of human disease in nonclinical safety assessments were published, and the advantages and concerns are discussed (Boelsterli, 2003; Dixit and Boelsterli, 2007; Morgan et al., 2013; McGonigle and Ruggeri, 2014; Cavagnaro and Silva Lima, 2015; Morgan et al., 2017). Animal models used as tools to model certain aspects of human diseases may provide useful information with regard to pharmacological action, pharmacokinetics, disease-related expression of the target molecule, dosage, method of administration in a clinical setting, and human safety (Ministry of Health, Labour and Welfare [MHLW], Japan, 2012). However, there are limited numbers of publications regarding the detailed analysis of actual cases of the use of animal models in nonclinical safety assessments.
An innovation and quality (IQ) consortium focus group conducted a cross-company survey to evaluate the current practices and perceptions surrounding the use of animal models in nonclinical safety assessments (Morgan et al., 2017). Their survey determined that the majority of companies used animal models during drug discovery primarily as a tool for proactively assessing potential nonclinical safety issues prior to conducting traditional toxicology studies or to resolve issues when the target is only expressed in the disease state. Around the same time, our group (the Non-Clinical Evaluation Expert Committee [NEEC], Drug Evaluation Committee, Japan Pharmaceutical Manufacturers Association [JPMA]) conducted an internal questionnaire on the use of animal models in nonclinical safety assessments. Survey results of the internal questionnaire demonstrated that several companies have experience in the use of animal models in nonclinical safety assessments.
The Pharmaceuticals and Medical Devices Agency (PMDA) discloses common technical documents (CTDs) and review reports of approved drugs on their website. While they are in Japanese only, they provide us with valuable information and very important insights for toxicity assessment. In addition to the aforementioned JPMA internal survey, we conducted a survey of CTDs and review reports to elucidate actual cases of approved drugs where animal models were used for nonclinical safety assessments. This article includes the results of the JPMA internal questionnaire, opinions on the advantages and concerns from the questionnaire, the results of the CTD/review report survey, a brief assessment of representative examples of approved drugs that utilized animal models in development, and an overall general discussion on the use of animal models in nonclinical safety assessments. In this article, the term “animal model” is defined as animals with inherited spontaneous disease, genetic modification, drug- or food-induced disease, surgical operations, or other modifications for the specific purpose of evaluating the nonclinical safety of new drugs, excluding the healthy animals typically used in standard toxicity studies.
Materials and Methods
JPMA Internal Questionnaire
To clarify the current practice of usage of animal models in nonclinical toxicology studies of pharmaceutical products, a questionnaire letter was sent to 62 companies affiliated with the NEEC of the JPMA (questionnaire period: August 27 to September 30, 2015). To protect the profits of their affiliated companies, the JPMA internal questionnaire was conducted under the condition of anonymity, according to a general JPMA rule.
The NEEC asked each company whether they had experience with animal model studies. If they had experience, we asked the following details: therapeutic area, animal model type (species and manufacturing/procurement procedures), trigger for conducting the study, purpose of the study, timing of study conduct, and study design (route, dosing frequency and/or period, dose levels, and test parameters). Timing of study conduct was asked in order to group each study into one of the following categories: screening toxicity study stage for discovery and optimization of candidates, early glucagon-like peptide (good laboratory practice) study stage for phase 1 (first in human [FIH]) clinical study, before phase 2 clinical study, before initiation of phase 3 clinical study, late development after initiation of phase 3 clinical study, and postmarketing after drug approval. Route of administration was asked in order to select from the intended clinical route or another administration route. Dosing frequency and/or period included single dose, 4 weeks or less, or exceeding 4 week in duration. Dose levels were asked in order to determine toxic versus nontoxic doses or if some other classification were used. Examination parameters conducted in each individual study included the following categories: clinical signs, body weight, food consumption, water intake, hematology, blood chemistry, urine examination, ophthalmology, electrocardiogram, gross necropsy, organ weight, histopathology, toxicokinetics, or others. Cases were then divided by examination parameter count: studies including more than 5 examination parameters and studies including 5 examination parameters or less. The NEEC also asked whether the goal of study was achieved. In addition, we asked free opinions on animal model studies, including advantages, concerns, and relationships with the 3Rs (replacement, reduction, and refinement) principles of animal study.
The NEEC obtained 19 nonclinical safety studies using animal models which could be further subcategorized based on differences in animal model type, study trigger, study purpose, timings of study conduct, dose frequencies/periods, and dose level. However, in some instances, answers were left blank, and as a result, the sum of each category is not necessarily consistent with the total number reported in several figures and tables and this also resulted in the total number of studies being less than 19 in some instances. These data gaps were not considered to impact the quality of this survey. Additionally, one animal model study was still ongoing at the time of the response.
CTD/Review Report Survey
The PMDA discloses CTDs and their review reports for all the approved drugs in Japan on their website, in Japanese only, from 1999 to the present (http://www.pmda.go.jp/PmdaSearch/iyakuSearch/). To elucidate actual cases of the use of animal models for nonclinical safety assessments in approved drugs from May 1999 to May 2017, the NEEC conducted a survey for CTDs and review reports.
The total number of approved drugs with CTD/review reports was counted on the PMDA website, and they were classified into active pharmaceutical ingredients (APIs), new route of administration, new indication, and new prescription combination preparation with or without animal model studies. In addition, the number of drugs per year with animal model studies was counted.
The therapeutic area for the drug, information on the animal model (type of model, species, and manufacturing/procurement procedures), trigger for conducting study, purpose of study, and the study designs were surveyed from the CTD/review report for each case. However, detailed analysis of the timing of study conduct and study designs (other than dosing route) could not be obtained because of the limited information described in the CTDs. Based on the study objectives and conclusions described in the CTDs, we attempted to analyze whether the purpose of study was achieved in each case.
In addition, the NEEC tried to analyze the actual animal model studies for representative cases of CTDs/review reports from the following trigger categories: toxic signs in disease status anticipated based on mode of actions (MOAs) and class effects, insufficient extrapolation of toxic changes from healthy animals to human, insufficient sensitivity to toxic effects in healthy animals, toxic changes with concern observed in nonclinical safety studies, adverse events with concern observed in clinical studies, and requested from agency during development or review process.
Results
JPMA Internal Questionnaire
Results of the JPMA internal questionnaire
The NEEC received responses from 43 companies (35 Japan-based and 8 non-Japan-based companies), 12 of which reported using animal models for nonclinical safety assessment with a total of 19 studies overall. Table 1 shows the therapeutic areas for the drugs tested in these 19 studies. The number of animal model types, namely, “drug- or food-induced models,” “surgically operated models,” “spontaneous models,” and “genetic modification models,” is shown in Figure 1. The triggers for conducting the tests were as follows: “insufficient extrapolation of toxicological concerns from healthy animals to humans,” “toxicological changes of concerns observed in the nonclinical studies,” “MOAs of drugs or information of class effects,” and “insufficient sensitivity to toxic effects in healthy animals” (Figure 2). In addition, the categories of “adverse events of concerns observed in the clinical trials” and “request from regulatory authority” were noted in a few cases. For the purpose of conducting animal model studies, the count of “toxic signs identification in animal models (including comparison of response with healthy animals)” greatly exceeded that of “investigation of mechanisms of toxic changes” (Figure 3). Regarding the timing of animal model nonclinical safety studies, it was revealed that many cases were carried out prior to the FIH studies, most commonly during the drug discovery stage or during early development (Figure 4). Only, one study was implemented during the postmarketing period.
JPMA Questionnaire: Therapeutic Areas for the Drugs with Nonclinical Safety Studies with Animal Models.
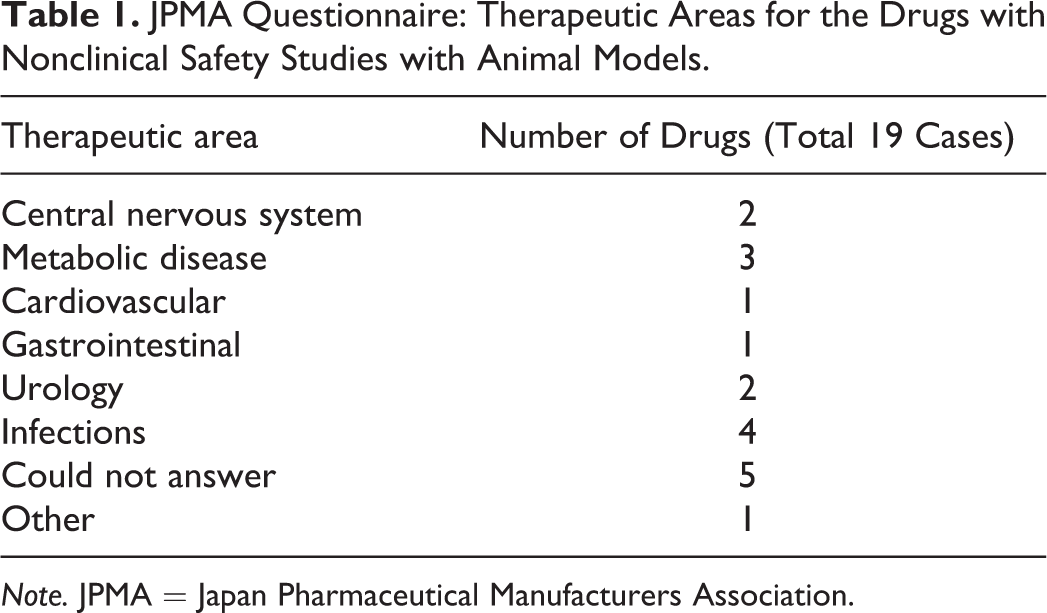
Note. JPMA = Japan Pharmaceutical Manufacturers Association.
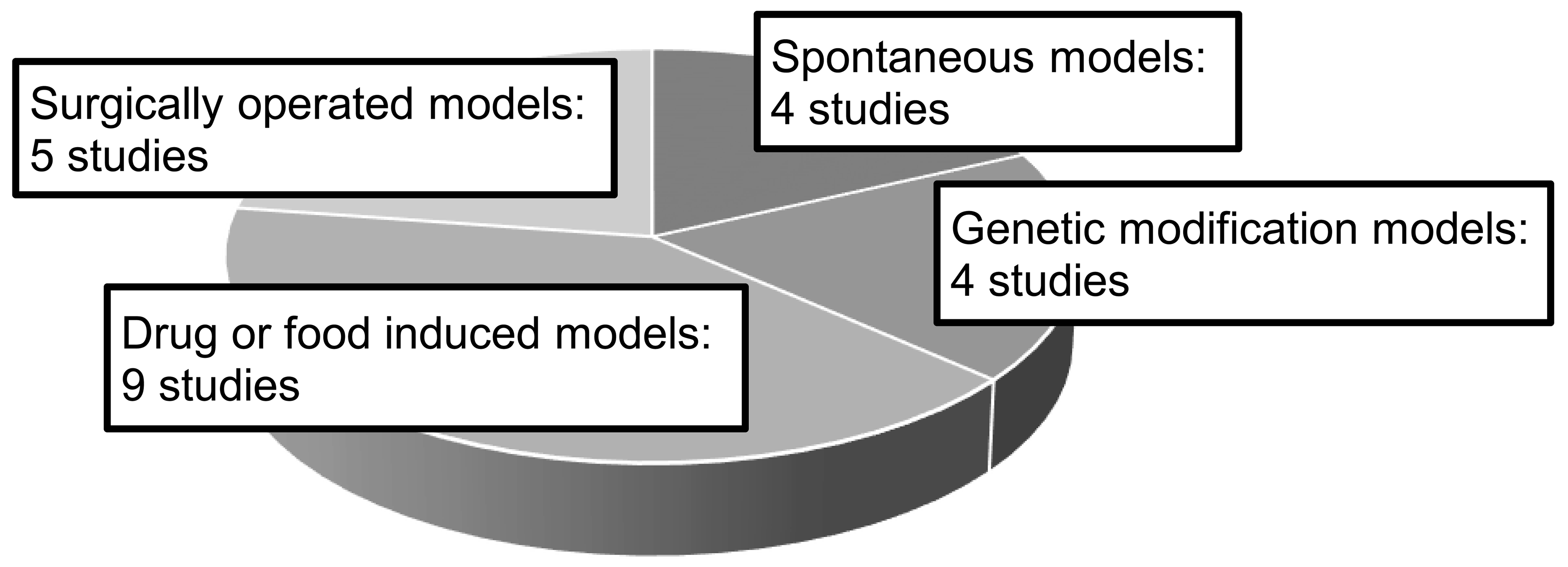
Japan Pharmaceutical Manufacturers Association questionnaire: Animal model types for safety assessment. Total 19 nonclinical safety studies with animal models are shown (including counting of duplicated answers).
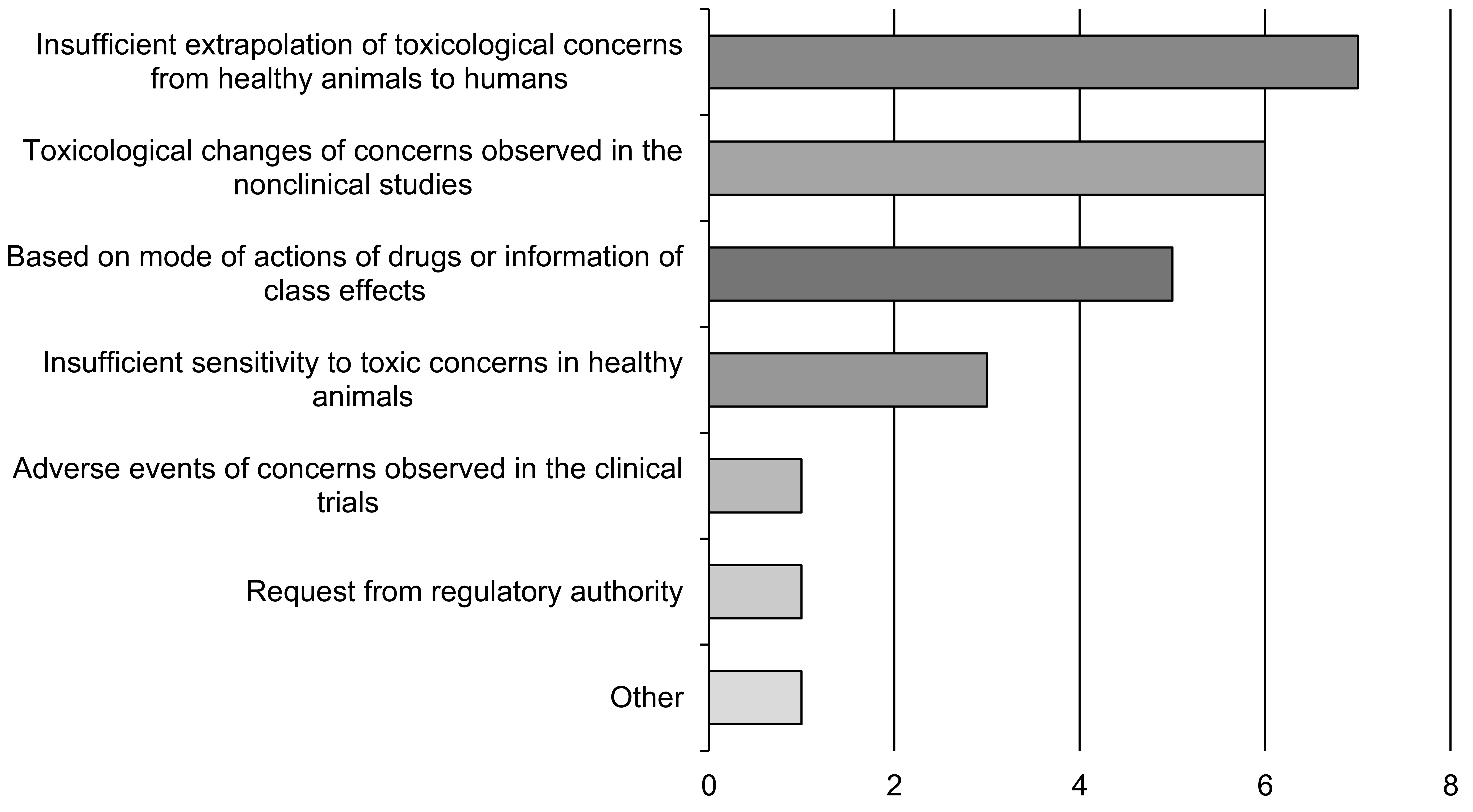
Japan Pharmaceutical Manufacturers Association questionnaire: Triggers of animal model studies. Total 18 nonclinical safety studies with animal models are shown (including counting of duplicated answers).
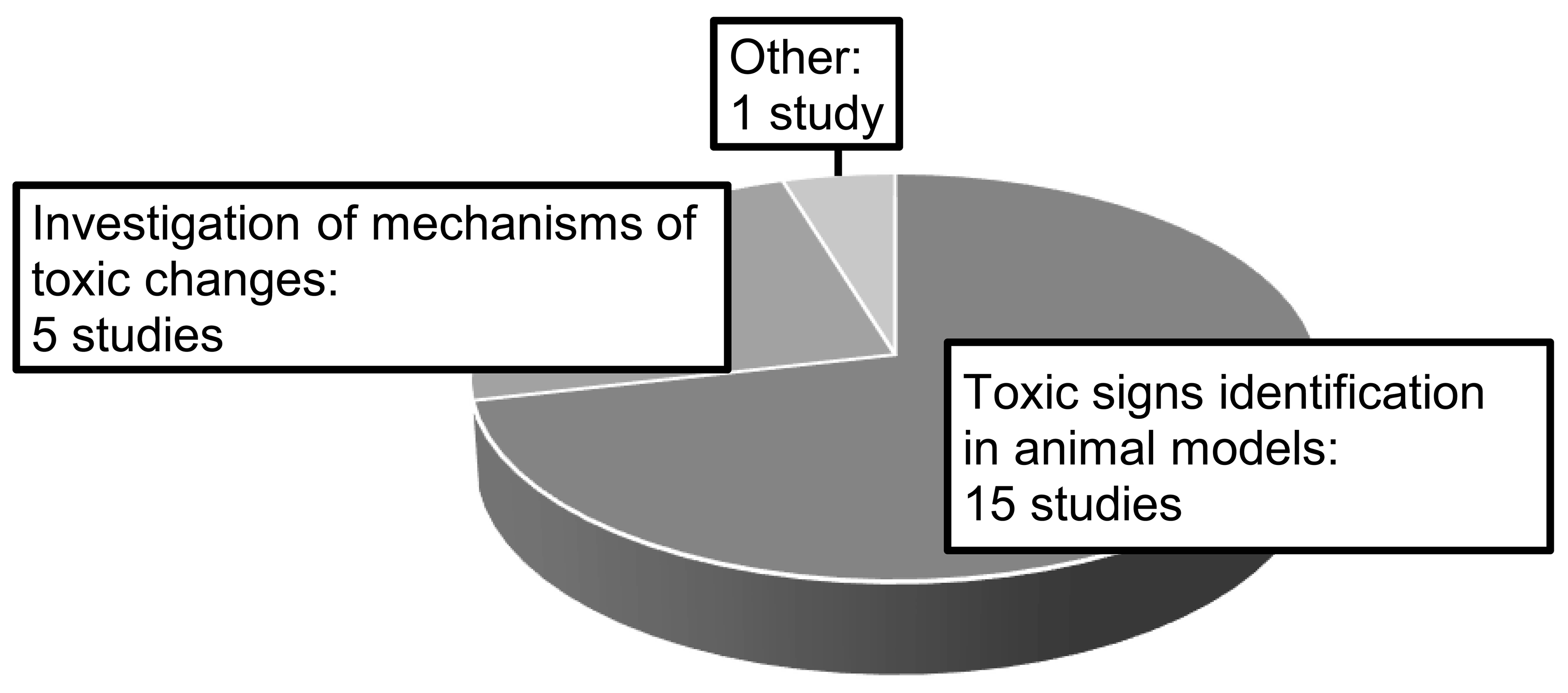
Japan Pharmaceutical Manufacturers Association questionnaire: Purposes of animal model studies. Total 18 nonclinical safety studies with animal models are shown (including counting of duplicated answers).
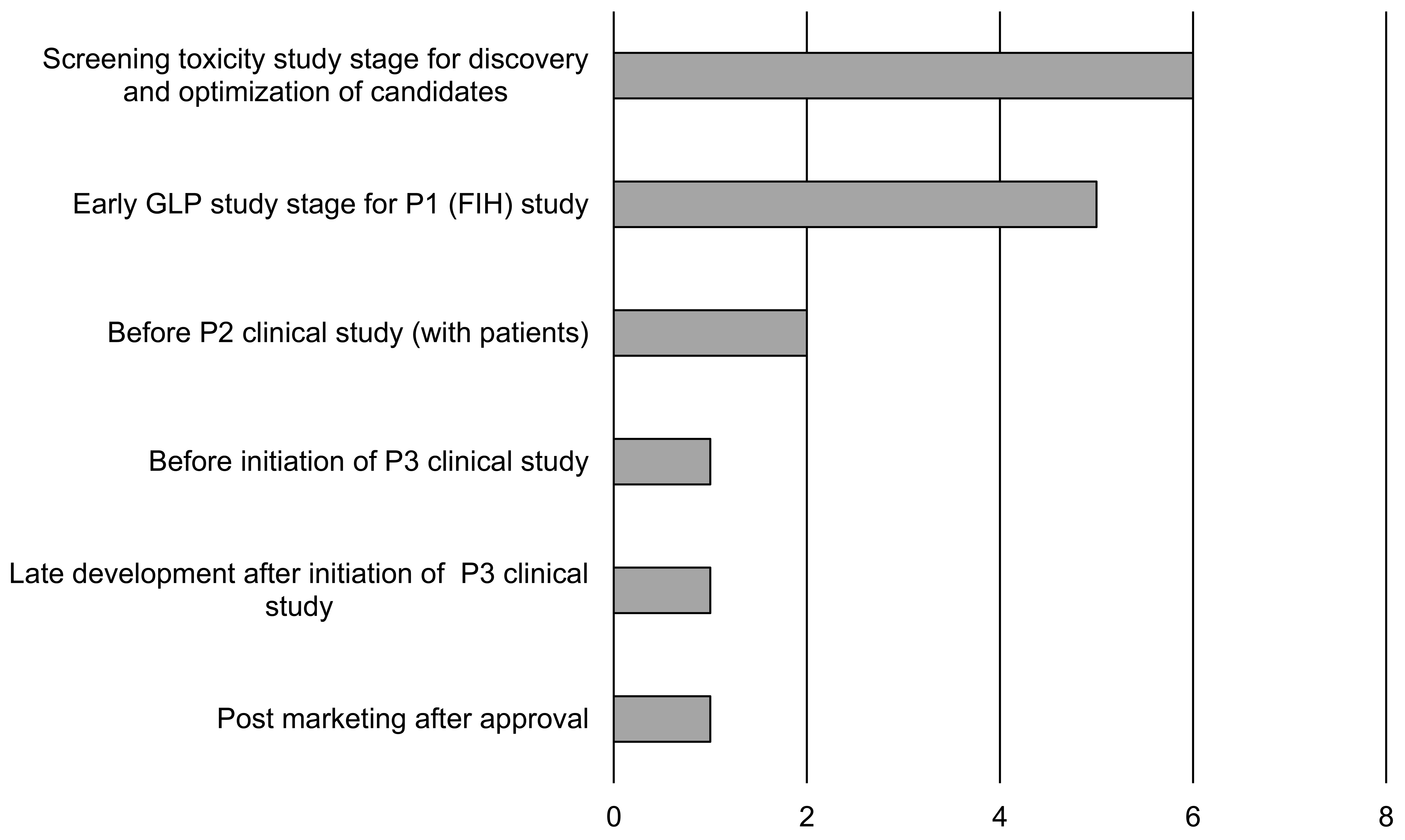
Japan Pharmaceutical Manufacturers Association questionnaire: Timing of animal model studies. Total 14 nonclinical safety studies with animal models are shown (including counting of duplicated answers).
A total of 18 studies were carried out by using the intended clinical route (Table 2), nine of which were conducted with more than five examination parameters (open circle in Table 3) with the remaining studies having less than five examination parameters (closed circle in Table 3). Studies with more than five examination parameters were similar in design to what’s commonly done in standard GLP repeat dose toxicity studies. All single dose studies had less than five examination parameters. Responses revealed that 17 of the 18 studies were completed and found the goals of these studies were achieved. One study was ongoing at the timing of the response and unknown if the goal was achieved.
JPMA Questionnaire: Study Designs of Nonclinical Safety Studies with Animal Models.
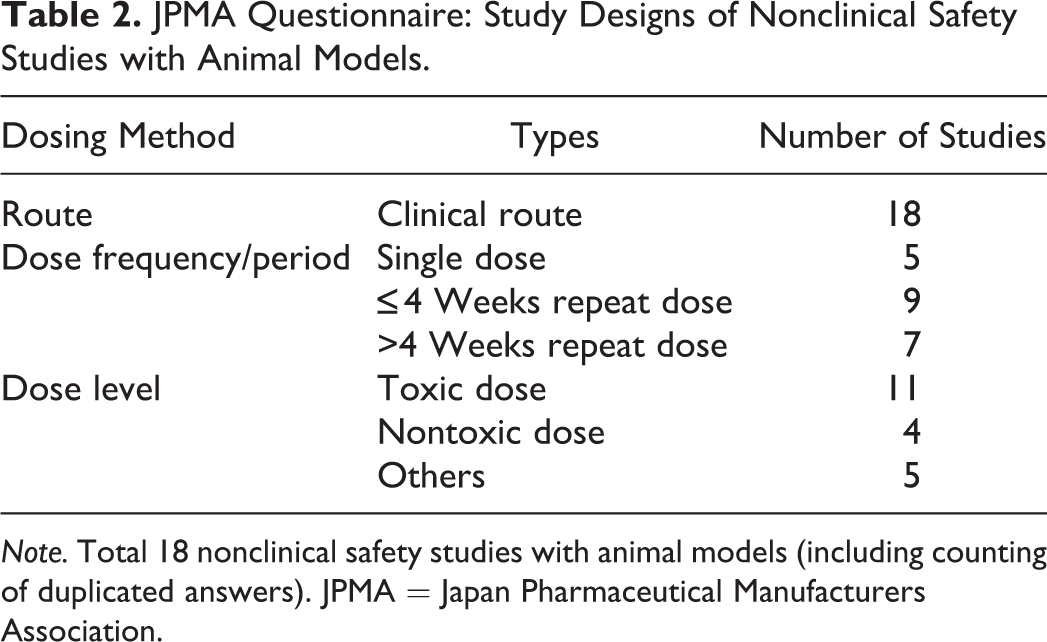
Note. Total 18 nonclinical safety studies with animal models (including counting of duplicated answers). JPMA = Japan Pharmaceutical Manufacturers Association.
JPMA Questionnaire: Examination Parameters Conducted in Nonclinical Safety Studies Using Animal Models.

Note. Study cases (number from 1 to 18) were collected through the JPMA internal questionnaire. Open circle = studies including more than five examination parameters (almost similar design to standard GLP repeated dose study); closed circle = studies including five examination parameters or less; JPMA = Japan Pharmaceutical Manufacturers Association; GLP = good laboratory practice.
Opinions for advantages and concerns from the JPMA internal questionnaire
Forty-three responses from JPMA’s member companies revealed a number of different advantages and concerns in using animal models for nonclinical safety assessments. The advantages included the opinion that using animal models could enable the detection of toxicological findings which could not be detected in healthy animals and that animal models were useful in simulating how the drug behave under conditions of disease state, especially with regard to pharmacokinetics. In addition, there were other advantageous aspects where models could be useful to elucidate mechanisms of toxicity as well as validate on- and off-target-based toxicologic findings or in instances where in silico or in vitro models were not available for safety evaluations.
Concerns extracted from the responses were: (1) animal model studies cannot accurately or precisely reflect the disease states in patients since the severity or stage of disease development can be quite variable in patients; (2) animal model study results must be carefully handled, since mechanisms of pathogenesis are not always consistent between human patients and animal models, even when they seem to be similar regarding disease manifestations; (3) execution of the studies is sometimes difficult, since the disease states of some animal models are more severe than those of human patients; (4) stable procurement of animal models is not easy; and (5) there are some cases where validation of the animal model was necessary before use.
Another opinion related to these concerns was that studies with animal models should not be readily conducted when adverse events in humans can be anticipated without the use of animal models or when the study is only being conducted for scientific interest. This viewpoint might be derived from the 3Rs principle of animal studies.
CTD/Review Report Survey
Results of the CTD/review report survey
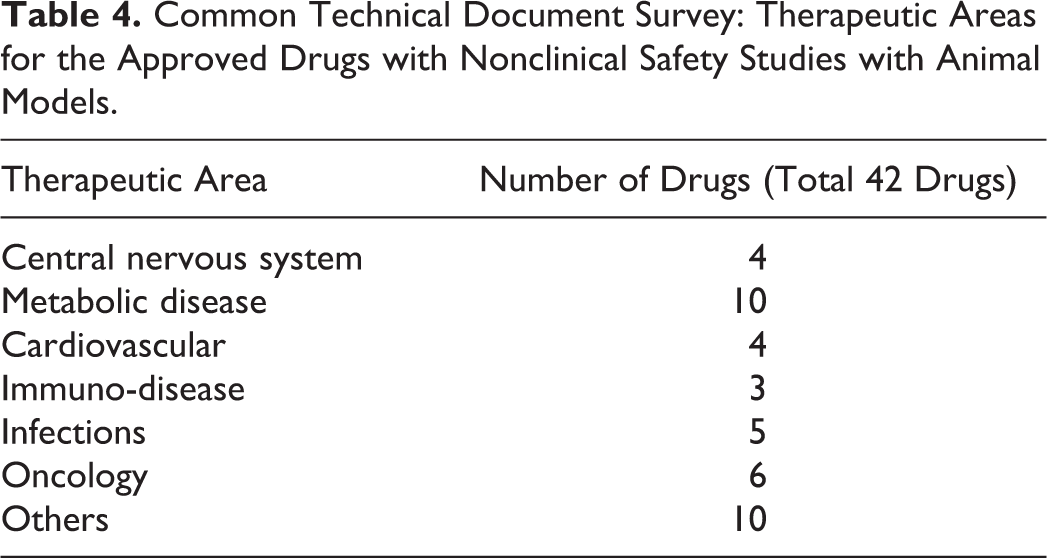
Animal model studies for safety assessment were found in 38 of the 577 CTDs, which accounted for 6.6% of new APIs. In addition to these 38 CTDs, animal model studies were found in 4 CTDs involving new routes of administration, new indications, new dosages, or new prescription combinations for already approved drugs. Annual trends of CTDs of approved drugs with animal model studies in nonclinical safety assessments are shown in Figure 5. There were no approved drugs with CTDs that included animal model studies from 1999 to 2002. Between 2003 and 2012, up to four approved drugs per year utilized animal model studies and four to ten approved drugs per year using them after 2013. Table 4 shows the therapeutic areas of the 42 drugs whose CTDs included animal model studies in nonclinical safety assessments, which included a total of fifty-nine studies. The most common types of animal models used were “drug- or food-induced models” and “surgically induced models,” followed by “spontaneous models” and “genetic modification models” (Figure 6). The major triggers for conducting animal model studies were “adverse events of concern observed in the clinical trials,” “MOA of drugs or information of class effects,” “insufficient sensitivity to toxic effects in healthy animals,” “insufficient extrapolation of toxicological concerns from healthy animals to humans,” and “toxicological changes of concern observed in the nonclinical studies” (Figure 7). A few triggers were “a request from regulatory authority.” For the purpose of conducting animal model studies, the number of cases of “toxic signs identified in animal models (including comparison of response with those in healthy animals)” greatly exceeded “investigation of mechanisms of toxic changes” (Figure 8). The majority of these studies (fifty-one) were carried out by using the intended clinical route with only 8 studies using other administration routes. Based on the study objectives and conclusions described in the CTDs, almost all studies were considered to have achieved the purpose of study, although a few of these studies were judged as having only slightly achieved the purpose of study.
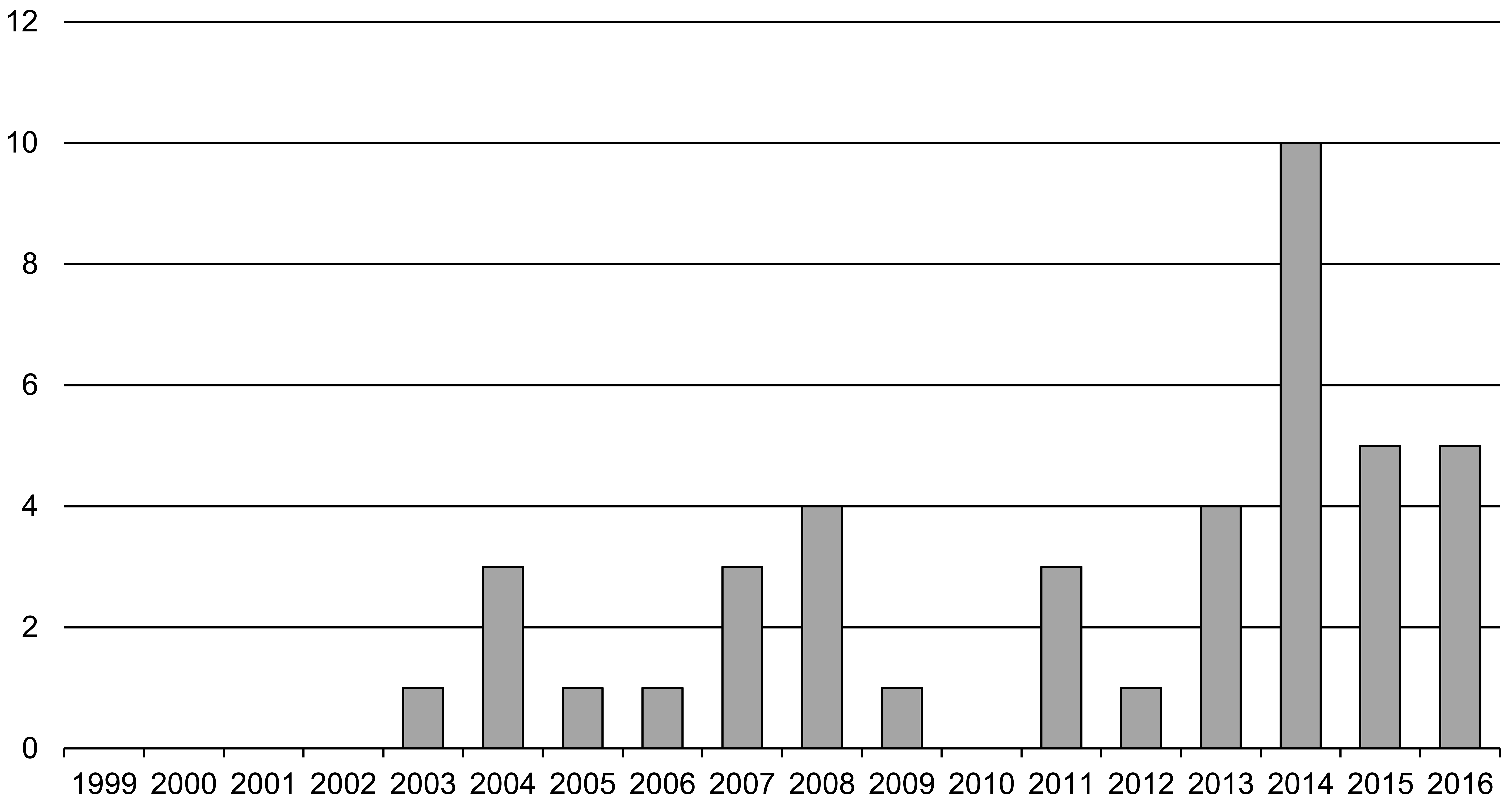
Common technical document survey: Chronological changes of approved drugs with toxicology studies using animal models.
Common Technical Document Survey: Therapeutic Areas for the Approved Drugs with Nonclinical Safety Studies with Animal Models.
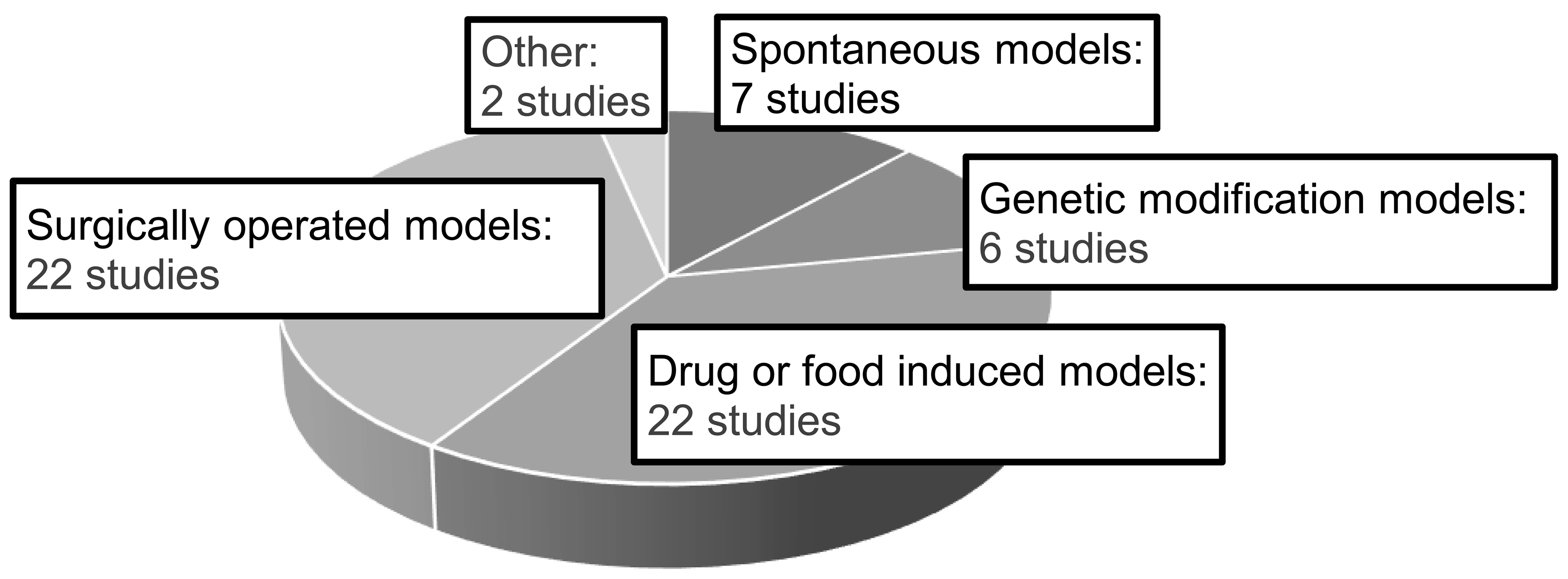
Common technical document survey: Animal model types for safety assessment. Study numbers shown in total 59 nonclinical safety studies with animal models (in 42 approved drugs).
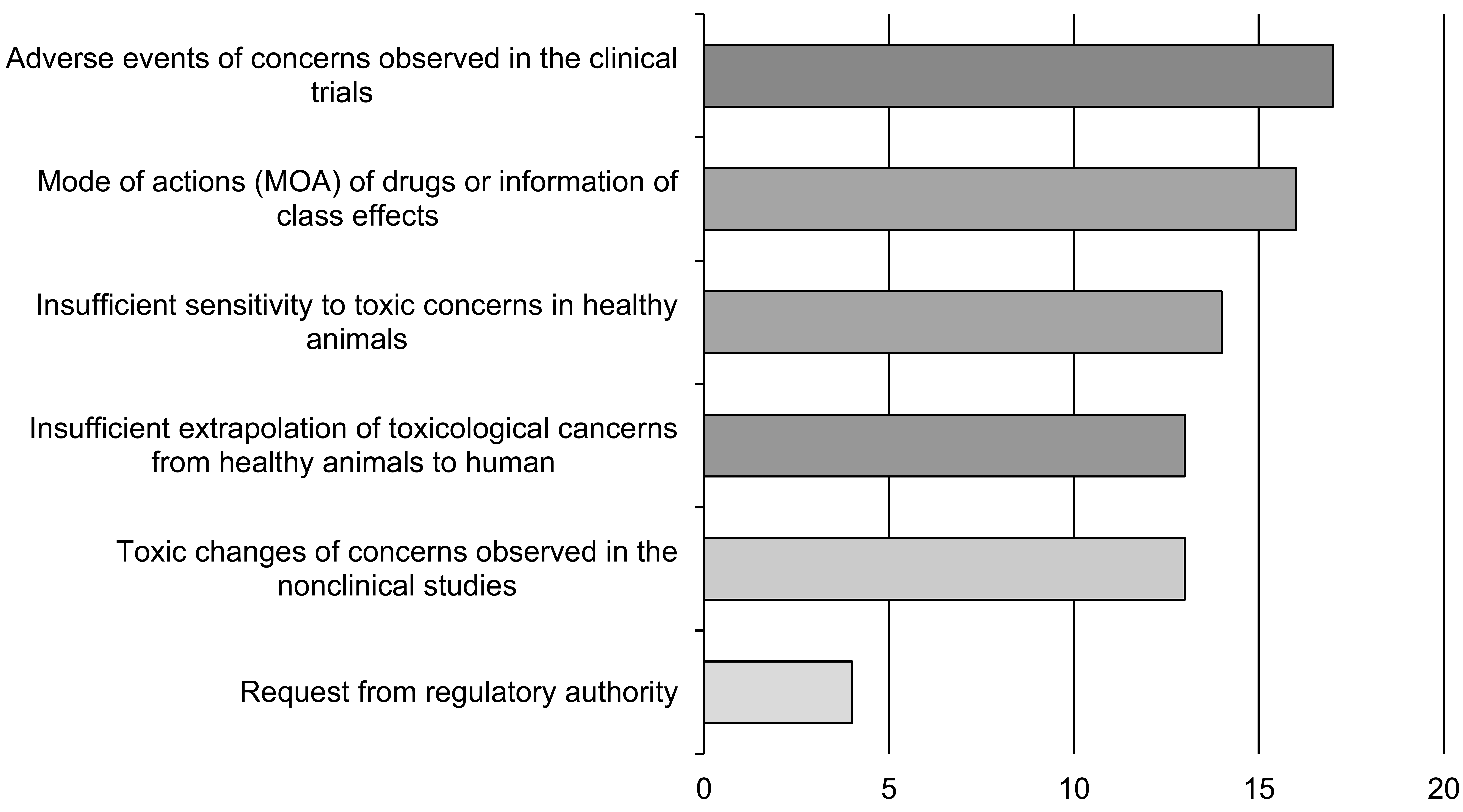
Common technical document survey: Triggers of animal model studies. Study numbers shown in total 59 nonclinical safety studies with animal models (including counting of duplicated studies).
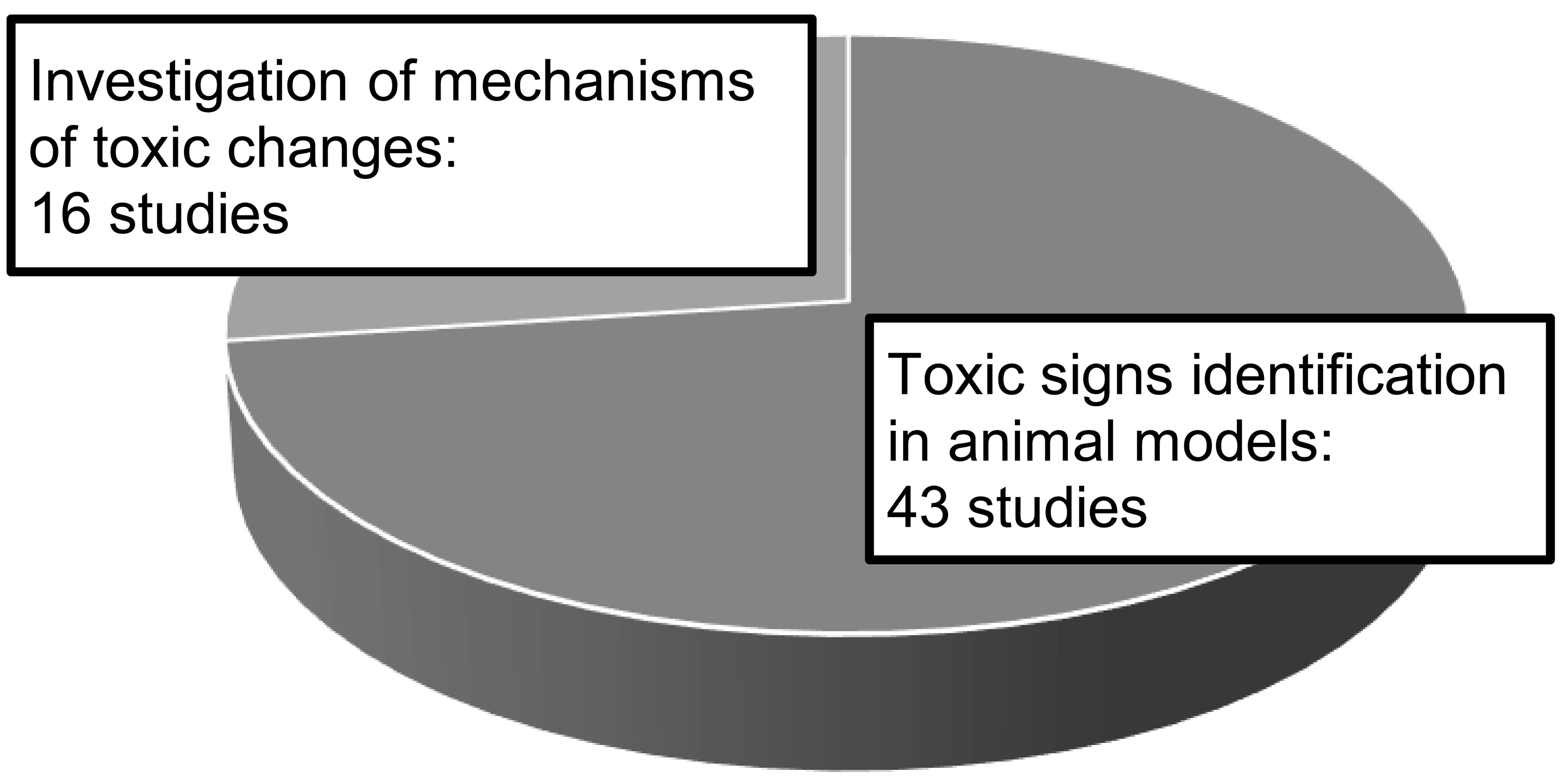
Common technical document survey: Purposes of animal model studies. Total 59 nonclinical safety studies with animal models are shown.
Brief assessment of representative study cases using animal models in approved drugs from CTDs/review reports
The NEEC tried to analyze actual study cases using animal models from CTDs/review reports. Table 5 shows the study rationales in these representative cases. This assessment included 6 examples as described below.
CTD Survey: Study Rationales for Representative Cases of Toxicity Studies Using Animal Models.

Note. CTD = common technical document; VEGF = vascular endothelial growth factor; SGLT = sodium glucose cotransporter.
Case 1: Toxic signs in disease status anticipated based on MOAs and class effects
Compound (Cpd) A is a human IgG1 monoclonal antibody against the extracellular domain of the human vascular endothelial growth factor receptor-2 (VEGFR-2), which was developed for unresectable advanced/recurrent gastric cancer. Since Cpd A inhibits angiogenesis mediated by the VEGFR-2 signal pathway, it was considered that Cpd A may delay wound healing in patients undergoing surgery. Therefore, the effect on wound healing was evaluated using cynomolgus monkey linear incision models. The linear incision animal model has been reported to reflect incisional wound as a clinically relevant model (Wilgus et al., 2008; Wilgus and DiPietro, 2012). The results showed that Cpd A had no effect on wound healing.
On the other hand, a conflicting result was reported from Cpd B, a humanized IgG1 monoclonal antibody targeting VEGF for the treatment of colorectal cancer. Because VEGF is involved in the wound healing process, the risk of delayed wound healing in patients treated with Cpd B after undergoing surgery was identified based on MOAs of Cpd B. In actuality, the result of a rabbit linear incision model showed that Cpd B had an effect on wound healing, but the result of the same linear incision model in monkeys indicated that Cpd B had no effect on wound healing. The applicant considered the reasons for the conflicted study results between species as being related to varied ages and body weights in monkeys while these remained uniform in rabbits. There are no descriptions regarding species differences of bioactivities in the toxicology part of CTD; however, the NEEC considered that bioactivities against model animals needed attention for the purposes of species selection.
Case 2: Insufficient extrapolation of toxic changes from healthy animals to human
Cpd C has been developed to treat postmenopausal osteoporosis and is a selective estrogen receptor modulator that produces estrogen-like effects on bone, reducing resorption of bone and increasing bone mineral density in postmenopausal women, thus slowing the rate of bone loss. Based on the biological activities of Cpd C, cynomolgus monkey was the selected species for toxicity studies.
Ovariectomized (OVX) animals are a typical model for investigation of postmenopausal osteoporosis due to estrogen deficiency, and it has been widely used for the evaluation and development of new drugs for postmenopausal osteoporosis treatments (Gao et al., 2014; Kobayashi et al., 2012). The U.S. FDA guidelines (1994) and World Health Organization (1998) guidelines noted that the OVX animal model is appropriate for clarification of effects on postmenopausal osteoporosis.
In a one-year oral administration toxicity study using healthy and OVX cynomolgus monkeys, changes such as reduction of body weights and uterus atrophy were observed only in healthy animals. The applicants considered these effects to be due to the estrogenic action of Cpd C since they were not observed in OVX animals, which have lower endogenous estrogen concentrations as compared to healthy animals. The applicant did consider these effects to have human relevance, given that endogenous estrogen concentrations were anticipated to also be low in patients.
Case 3: Insufficient sensitivity of toxic effects in healthy animals
Cpd D is an anti-obesity drug that acts by inhibiting pancreatic lipase, an enzyme that breaks down triglycerides in the intestine. Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and as a result they are excreted undigested.
In phase 1 repeated dose clinical study, Cpd D increased the alanine transaminase (ALT) and aspartate transaminase (AST) levels in healthy volunteers. In preclinical assessments, an increase in ALT was only observed at the highest dose (2,000 mg/kg/day) in the 26-week repeat dose study in rats. No additional ALT- and AST-related findings were shown in other toxicity studies. The increase in ALT/AST was thought to potentially be related to liver injury; however, it was speculated that this could be related to inhibition of fat absorption by Cpd D (the MOA of the compound), inducing an alteration of energy supply and, finally, excessive gluconeogenesis. Therefore, an additional 26-week study using gluconeogenesis-prone rats induced by a high-fat diet was conducted to evaluate the effects of Cpd D. The high-fat diet tended to enhance an increase in ALT/AST which was induced by treatment with Cpd D. In addition, the activities of enzymes for gluconeogenesis in the liver were elevated in high-fat diet rats and were even higher with compound treatment. No hepatic injury was observed. As mentioned above, the applicant considered that inhibition of lipid absorption by Cpd D enhanced gluconeogenesis, contributing to the increase of ALT/AST. In this case, the toxicity studies using healthy animals were not very predictive of the ALT and AST elevations noted in humans, although the NEEC had data indicating that the gluconeogenesis-prone animal model may be a better predictor of the changes seen in human clinical trials. As a result, the NEEC utilized these animal model observations in support phase 1 studies with Cpd D.
Case 4: Toxic changes with concern observed in nonclinical safety studies
Cpd E for hereditary tyrosinemia type 1 inhibits the enzyme 4-hydroxyphenylpyruvate dioxygenase at the second step of the tyrosine catabolism pathway. Corneal lesions including mainly keratitis were found in toxicity studies with Cpd E in healthy Sprague-Dawley rats and dogs. However, the findings were not observed in other animal species including mice, rabbits, or rhesus monkeys.
Wistar male rats were fed with a low-protein diet (the control group) or a low-protein diet containing 5% L-tyrosine (the test group) orally for 7 days. The plasma concentration of tyrosine was elevated significantly in the test group compared to the control group. In the test group, several ocular lesions were found from day 1 to day 7 of treatment, and keratitis and anterior uveitis were seen in histopathology examinations.
These ocular findings were similar to those observed in Cpd E-treated healthy animals. The applicant considered that these ocular lesions were due to tyrosine crystallization in lacrimal fluid as the result of elevated tyrosine concentration in the plasma by the pharmacological action of Cpd E. The applicant considered the data from these animal model studies to be more relevant for risk assessment in patients as compared with data generated in traditional toxicity studies using healthy animals. The NEEC considered that the risk of human use could be reduced based on the results of the animal model study and clinical pharmacokinetic information.
Case 5: Adverse events with concern observed in clinical studies
Cpd F was developed for type 2 diabetes with the mechanism being inhibition of the sodium-glucose cotransporter 2 (SGLT2). In an analysis of the phase 2b/3 clinical study, bladder cancers were reported as adverse events in 0.17% of patients treated with Cpd F versus 0.03% of those receiving placebo (Anderson, 2014; Ptaszynska et al., 2014). No concern was observed in genotoxicity tests nor in traditional carcinogenicity tests in healthy mice and rats (Reilly et al., 2014). An additional long-term study was conducted with SGLT2−/− (SGLT knockout [KO]) mice to evaluate the potential carcinogenic risk of inhibiting SGLT2, particularly in the urogenital tract.
In the 15-month study with SGLT2−/− mice, which were generated and bred as previously published (Vallon et al., 2011), pups were weaned at approximately 3 weeks of age, housed with free access to water and feed, and were not subjected to any therapeutic or pharmacologic interventions. There were no descriptions regarding study period in the CTD, but at 15 months, microscopic evaluations were conducted and compared between KO mice and wild-type mice.
No adverse effect attributable to SGLT2 gene deletion (SGLT2−/− mice) were observed upon microscopic evaluation, including in the urinary bladder and the kidney (Reilly et al., 2014). The applicant considered this to suggest that selective SGLT2 inhibition was not predicted to be associated with an increased cancer risk.
As mentioned above, the NEEC considered these animal model results using target-gene-deleted KO mice as an indication that the drug was not related to the adverse events observed in clinical trials.
Case 6: Requested from agency during development or review process
Cpd G was developed for type 2 diabetes with the mechanism being inhibition of dipeptidyl peptidase-4 (DPP-4). The same as other drugs that target incretin such as DPP-4 inhibitors and glucagon-like peptide-1 (GLP-1) agonists, the FDA requested a study of pancreatic toxicity in an animal model of diabetes as one of the conditions for registration of Cpd G (Morgan et al., 2017; Chadwick et al., 2014). To investigate the potential for drug-induced pancreatic injury, an exploratory 4-day repeat dose study and a 3-month repeat dose study were conducted in Zucker Diabetic Fatty (ZDF) rats as a model for type 2 diabetes. In the results of these studies, Cpd G did not show pancreatic injury in the ZDF rats, much the same as in study results with other DPP-4 inhibitors. Cpd G was not submitted to the FDA based on business reasons, but it was submitted and approved in Japan.
Discussion
The results of the JPMA internal questionnaire and the published CTD/review report survey revealed that animal models have been used on a case-by-case basis and if scientifically justified in nonclinical safety assessment of new drug candidates. Especially, the results of the published CTD/review report survey demonstrated that nonclinical safety studies using animal models were included in data packages of a number of approved drugs in Japan. This means there is an acceptability for using animal models by the PMDA in appropriate cases, and it is consistent with the guidance of the Japanese MHLW, which recommends the use of animal models to provide beneficial information for the pharmacological action and pharmacokinetics of drugs, disease-related expression of the target molecule, dosage and method of administration in a clinical setting, and safety (MHLW, Japan, 2012).
There were no approved drugs with animal model studies from 1999 to 2002, but several drugs with animal model studies were approved after 2003, and there is a trend of increasing the number of approved drugs in the past few years. Interestingly, this increasing trend of animal model use is consistent with the fact that a number of articles regarding animal models in nonclinical safety assessments have recently been published (Boelsterli, 2003; Dixit and Boelsterli, 2007; Morgan et al., 2013; McGonigle and Ruggeri, 2014; Cavagnaro and Silva Lima, 2015; Morgan et al., 2017).
Table 6 summarizes the list of animal models from the results of the JPMA internal questionnaire and the published CTD/review report survey. Because we have not yet tried to investigate the validity of these models, it is difficult to judge their appropriateness. However, these are expected to be helpful for an animal model selection.
Animal Models Used in Nonclinical Safety Studies.
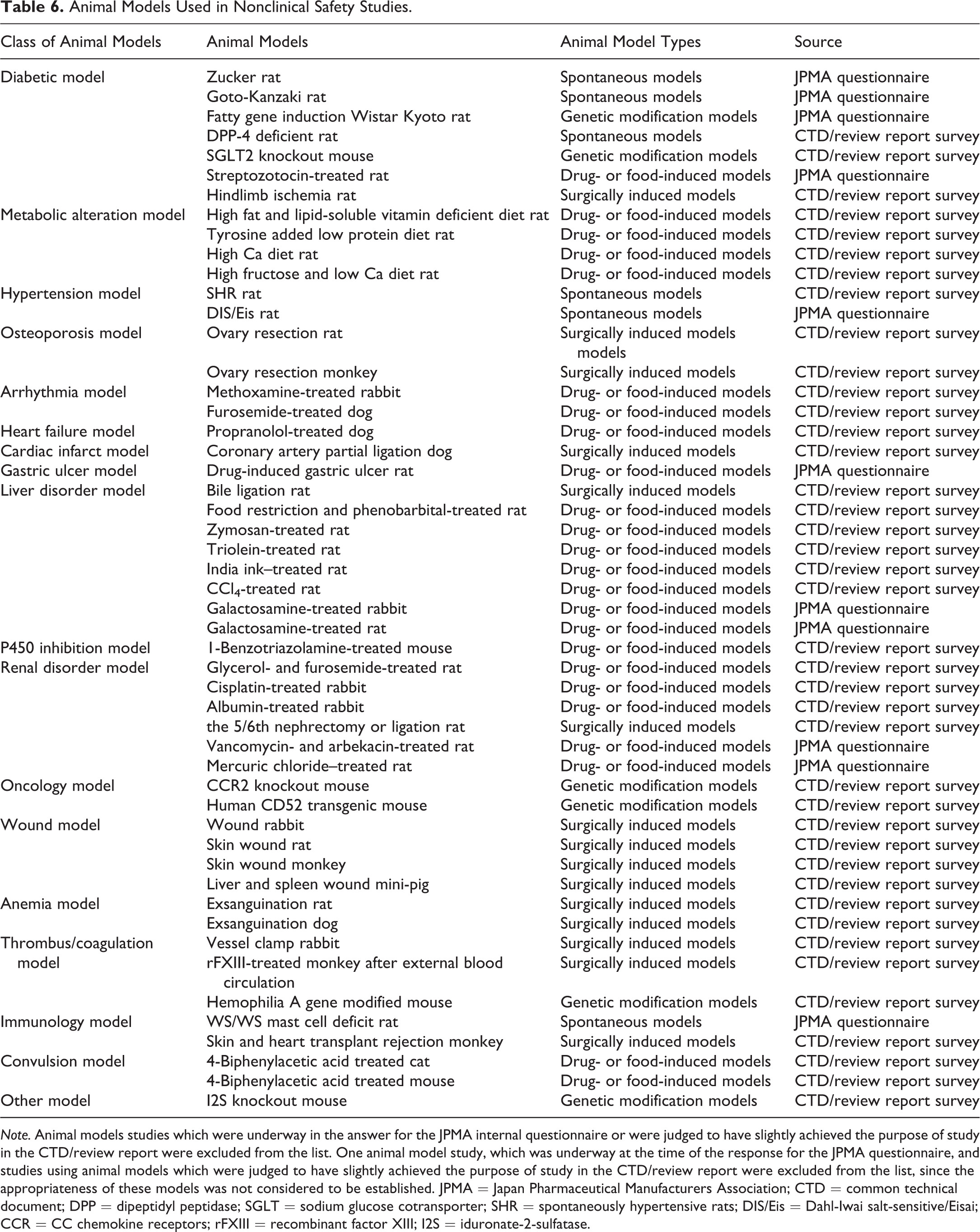
Note. Animal models studies which were underway in the answer for the JPMA internal questionnaire or were judged to have slightly achieved the purpose of study in the CTD/review report were excluded from the list. One animal model study, which was underway at the time of the response for the JPMA questionnaire, and studies using animal models which were judged to have slightly achieved the purpose of study in the CTD/review report were excluded from the list, since the appropriateness of these models was not considered to be established. JPMA = Japan Pharmaceutical Manufacturers Association; CTD = common technical document; DPP = dipeptidyl peptidase; SGLT = sodium glucose cotransporter; SHR = spontaneously hypertensive rats; DIS/Eis = Dahl-Iwai salt-sensitive/Eisai; CCR = CC chemokine receptors; rFXIII = recombinant factor XIII; I2S = iduronate-2-sulfatase.
The IQ consortium survey was conducted differently but used the same type of questionnaire (Morgan et al., 2017). While there is a limitation when directly comparing their results and ours, it is considered that such comparison will be helpful for bringing insight into the situations of use of animal models. Comparisons of the results of the two surveys are as follows. In the JPMA internal questionnaire, the use of drug-induced models was most common. The IQ consortium survey revealed that the use of laboratory-generated and transgenic models was most common (Morgan et al., 2017). Laboratory-generated models in the IQ consortium survey are considered to include both drug-induced models and surgically induced models. If we had collected data in the same way as the IQ consortium survey, the most common type of models would have been laboratory-generated models, which was consistent with our survey results. There is a difference in the frequency of use of genetic modification models between our survey results and IQ consortium survey results, but the reason for this is unknown.
The main triggers of animal model studies in the JPMA internal questionnaire were (1) insufficient sensitivity in healthy animals, (2) to investigate toxic changes observed in traditional nonclinical studies, and (3) to address the MOA/class effect-oriented changes. On the other hand, the main triggers of animal model studies in the IQ consortium survey were (1) requests from a global regulatory authority, (2) to derisk or understand safety issues that may be masked by excessive pharmacology in traditional animal models or when a target is only expressed in the disease state, and (3) to address target or program-specific concerns that important safety issues may be missed if data were collected using only healthy animals (Morgan et al., 2017). Almost all triggers of animal model studies were considered to be consistent between our survey and the IQ consortium survey results. On the other hand, there is a difference in rank order of triggers of animal model studies between the JPMA internal questionnaire and the published CTD/review report survey results. The published CTD/review report survey included studies for only approved drugs but did not include studies for terminated products during discovery and development phases or studies for actually developing products. This means that studies for the JPMA internal questionnaire include products in earlier discovery and development phases than those in the published CTD/review report survey. A reason for the difference in rank order for triggers of animal model studies might be related to those differences of development stages for each study. Requests from regulatory authorities were not a main trigger in the JPMA internal questionnaire. However, the published CTD/review report survey results and the IQ consortium survey results revealed that the FDA requested studies using diabetic animal models to address carcinogenic concerns in pancreas.
The IQ consortium survey revealed that animal model studies were generally conducted with a greater frequency during the drug discovery phase compared to the development phase (Morgan et al., 2017). The JPMA internal questionnaire survey also revealed that animal model studies were mainly conducted during the drug discovery and early development phases (before FIH study), and a tendency of timing for animal model studies was similar. In addition, one study conducted after drug approval was reported in the JPMA internal questionnaire survey. Nonclinical researchers should consider animal model studies, even if adverse events which cannot be detected from traditional nonclinical studies using healthy animals may occur during the postmarketing phase after approval. On the other hand, since almost all CTDs/review reports did not mention the timing of studies, it was difficult to analyze when the animal model studies were conducted.
From the results of our survey and the IQ consortium survey, it is suggested that each pharmaceutical company decides to use animal models in nonclinical safety assessment based on the abovementioned triggers. If there is guidance for the use of animal models in nonclinical safety assessment, decision-making might become much easier and unnecessary studies using animal models might be avoided. An issue of the voluntary guidance or reflection should be our next target, although Morgan et al. (2013) provided a decision tree for conducting nonclinical studies using animal models. They suggested that animal models are useful when there are questions that cannot be adequately answered with traditional studies, when there are appropriate animal models, and when sufficient consideration is made for animal model study design.
The results of the JPMA internal questionnaire and CTD/review report survey, especially in brief assessments of representative animal model studies, revealed many advantages for using animal models in nonclinical assessment. In the JPMA internal questionnaire, there were several opinions regarding advantages for using animal models in nonclinical safety assessments (e.g., higher sensitivity of animal models, pharmacokinetic simulation of disease states, investigation of mechanisms of toxicity, or others). On the other hand, the results of the JPMA internal questionnaire also provided concerns for using animal models (e.g., potential differences in disease status and in mechanisms of pathogenesis between animal models and patients, more severe conditions than in patients, or others). Boelsterli (2003) and Morgan et al. (2013) discussed the advantages and disadvantages for nonclinical studies using animal models. Advantages included improvements to sensitivity, closer to human patient situations, a better understanding of the mechanisms of toxic changes, they might explain clinical findings, an improvement in candidate selection, and so on. These advantages are considered helpful in understanding safety risks of compounds in nonclinical and clinical development. Disadvantages included false-positive data, additional costs/time, heterogeneity in disease expression, a limited life span of animals, and so on. In several cases, animal models are more sensitive to toxic responses than healthy animals, and the use of animal models might be able to detect toxic changes which cannot be observed in healthy animals (Dixit and Boelsterli, 2007; Boelsteril, 2003). McGonigle and Ruggeri (2014) mentioned that animal models have been used to estimate clinical dosing regimens and determine relevant safety margins but also pointed out a potential pitfall in predicting clinical efficacy and safety margins based on the nonclinical study results with the use of single inbred strains of animals.
Selection of animal models is an important decision in conducting nonclinical studies. Sometimes, manufacture or procurement of animal models with appropriate quality is difficult. In these cases, appropriate validation of animal models might be required. Historical data for animal models are generally less abundant than for traditional healthy animals (McGonigle and Ruggeri, 2014; Boelsterli, 2003). Further detailed analyses (e.g., manufacturing/procurement procedures, background information, validity and others) for each animal model are considered to be necessary to utilize broadly in nonclinical safety assessments.
Nonclinical studies using animal models are conducted as supplements to traditional studies using healthy animals in many cases; unfortunately, this means increases in the number of studies and animals used. From the 3Rs principle of animal study, this situation is considered unfavorable, and we need to avoid conducting unnecessary or less-needed studies. We must consider carefully the expectations of studies from the viewpoints of their pros and cons. When nonclinical studies using animal models are conducted, in-depth analysis of the animal model selection and study design is necessary. Several cases of nonclinical studies using animal models during the discovery and early development phases might be combined with in vivo pharmacology assessment studies, which might be effective for the selection of candidates.
There are two nonclinical assessment cases without healthy animal studies or with only animal model studies: one case in the JPMA internal questionnaire survey results and the other case of the CTD/review report survey results. Those cases could avoid an increase in the number of study animals and seem to be among the favorable options, if appropriate for the safety assessment of new drugs.
Conclusions
It is the opinion of the authors that results of the JPMA internal questionnaire and the published CTD/review report survey revealed that animal models have been used on a case-by-case basis in the nonclinical safety assessment of drugs, and data of the studies conducted in animal models were included in the data packages of several approved drugs in Japan. In the past few years, the number of nonclinical studies using animal models has increased, and a further increase in the future is anticipated. In almost all cases, useful information, such as signs of toxicity under disease conditions and mechanisms of toxic changes, can be obtained from the results of nonclinical studies using animal models. Even with the abovementioned positive implications, the results from these surveys indicate that we should consider carefully the expectations of such studies, including pros and cons from the 3Rs principle of animal study, and that in-depth thinking concerning the selection of animal models and study design is necessary.
Footnotes
Acknowledgment
The authors would like to thank Akio Kobayashi (Japan Tobacco Inc.), Yoshihiko Esaki (Santen Pharmaceutical Co., Ltd.), Yamato Ogino (Toa Eiyo Ltd.), and Tetsuya Yamamoto and Yusuke Nozaki (Toyama Chemical Co., Ltd.) for their cooperation regarding the survey-related work regarding animal model studies.
Author Contributions
Authors (MT, TO, YK, KK, MM, YM, KM, RY, SS, MF, KW) contributed to conception or design; data acquisition, analysis, or interpretation (MT, TO, YK, KK, MM, YM, KM, SS, MF, KW); drafting the manuscript (MT, TO, YK, KK, MM, YM, KM, RY, SS, MF, KW); and critically revising the manuscript (MT, TO, YK, KK, MM, YM, KM, RY, SS, MF, KW). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declare no potential, real or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.