
Abstract
The International Consortium for Innovation and Quality (IQ) in Pharmaceutical Development is a science-focused organization of pharmaceutical and biotechnology companies. The mission of the Preclinical Safety Leadership Group (DruSafe) of the IQ is to advance science-based standards for nonclinical development of pharmaceutical products and to promote high-quality and effective nonclinical safety testing that can enable human risk assessment. DruSafe is creating an industry-wide database to determine the accuracy with which the interpretation of nonclinical safety assessments in animal models correctly predicts human risk in the early clinical development of biopharmaceuticals. This initiative aligns with the 2011 Food and Drug Administration strategic plan to advance regulatory science and modernize toxicology to enhance product safety. Although similar in concept to the initial industry-wide concordance data set conducted by International Life Sciences Institute’s Health and Environmental Sciences Institute (HESI/ILSI), the DruSafe database will proactively track concordance, include exposure data and large and small molecules, and will continue to expand with longer duration nonclinical and clinical study comparisons. The output from this work will help identify actual human and animal adverse event data to define both the reliability and the potential limitations of nonclinical data and testing paradigms in predicting human safety in phase 1 clinical trials.
Keywords
Introduction
Nonclinical safety assessment, utilizing animal toxicology studies, plays an important role in drug development. Data from animal toxicology studies are used to characterize potential safety risks for humans (identify target organs of toxicity) and to help determine a safe starting dose for first-in-human (FIH) clinical trials. The drug development process is highly controlled, based on specific regulatory agency criteria. Drug candidates must be evaluated in animal toxicology studies to support their first entry into the clinic, longer duration clinical trials, and marketing approval. The International Conference on Harmonization (ICH) provides guidance on the selection of animal models and the conduct of the animal toxicology studies for both small and large molecules (ICH M3 (R2) 2009; ICH S6 1997; ICH S6 (R1) 2011). The majority of relevant data from animal toxicology studies, for example, the identification of target organs of toxicity and establishing the no observed adverse effect level (NOAEL), is determined by histopathology results. Thus, the toxicologic pathologist is a key contributor to nonclinical drug safety data generation, interpretation, and risk assessment.
The typical testing paradigm, based on the ICH, is to evaluate drug candidates in both a rodent and a nonrodent animal model. While the mouse is often favored for nonclinical pharmacology and efficacy studies, it is not routinely used for toxicology studies. The rat is the rodent model generally preferred for toxicology studies because of its larger size that provides ease of handling and blood sampling. The beagle dog is the typical nonrodent animal model because of the long history in utilizing purpose-bred beagle dogs in pharmaceutical development (large historical database) and because of their amicable human interactions (Tomaszewski 2004). Many large molecules (e.g., monoclonal antibodies), however, are highly specific for only the human target, usually resulting in the selection of the nonhuman primate (NHP) as the relevant (e.g., target homology, similar pharmacodynamic response as humans) nonrodent species for safety assessment (Chapman et al. 2007, 2009). More recently, the minipig is being evaluated as an alternative nonrodent species to the NHP and dog (Forster et al. 2010).
Current Testing Paradigm: Based on Science or Tradition?
It is noteworthy to chronologically review how regulatory agencies and the pharmaceutical industry arrived at the current nonclinical safety testing paradigm that resulted in evaluating new drug candidates in both a rodent and a nonrodent animal model (Zbinden 1993). Early pharmacologists did most of their work with mice but oftentimes followed up with studies in additional animal models, such as the rat and guinea pig (Trevan 1927). So, for pharmacologists branching into toxicology, it was becoming clear that acute toxicity testing should be completed in several species. By 1935, a prominent pharmacologist reported that acute toxicity testing should be conducted in mice and rats followed by small numbers of guinea pigs, rabbits, cats, or dogs (Zbinden 1993).
Moving forward to the early 1940s, an official paper was issued by the Food and Drug Administration (FDA) based on a working group (WG) consisting of experts from the Council of Pharmacy and Chemistry of the American Medical Association (Woodward and Calvery 1943; Van Winkle et al. 1944). The recommendation, in regard to species selection, was based on length of the toxicology study. Three or more species were needed for acute and chronic studies, while 1 or more species were needed for subacute testing. The definitions of acute, subacute, and chronic study duration were not provided. It was stated, however, that the actual species to be used in the studies was at the discretion of the toxicologist.
During the next 2 decades, efforts to provide formal guidelines for nonclinical safety testing were never achieved through joint efforts of the government and private industry, so the regulatory agencies (both in the United States and in the ex-United States) took the lead on their own. In a scientific presentation in 1963, the FDA presented their views on animal species and toxicology testing (Lehman 1963): “acute” testing—4 species and 1 being a nonrodent and “subacute” or “chronic” testing—2 species and 1 a nonrodent. Within the United States, these “guidelines” became more or less formalized over time in regard to what species should be used.
The next document, issued by the Pharmaceutical Manufacturers Association (PMA 1977) in the 1970s, became the unofficial but generally followed guideline for nonclinical safety testing for pharmaceuticals. This guideline reaffirmed that both the rodent and the nonrodent were the preferred combination, suggesting that tradition, rather than science, dictated this paradigm. Purpose-bred beagle dogs of good quality also became available in sufficient numbers during this time period (Zbinden 1993).
The European Society for the Study of Drug Toxicity, a nongovernmental organization in Europe, also had agreed on the preferred combination of the rat and dog (Zbinden 1993). The European Society for the Study of Drug Toxicity recommended that drug absorption, distribution, metabolism, and excretion (ADME) data of the test substance were needed from the animal models in order to compare them with the respective human data to help determine the most appropriate nonclinical animal model. However, at the time, ADME data were often not readily available (Zbinden 1993) and toxicology testing in the rodent and nonrodent was in reality, just following the standard approach. Current default nonclinical safety testing of both the rodent and the nonrodent and the rationale of multispecies testing in industrial toxicology is discussed by Zbinden (1993) and the reader is encouraged to review his article.
Review of Animal to Human Toxicology Concordance Literature
The major goals of nonclinical safety testing in animals are to ensure human safety in the clinic, a safe starting dose, and the identification of potential target organs of toxicity that can be monitored in the clinic. Currently, the conduct of toxicology studies is based on historical precedence and ICH recommendations. This process is based on the assumption that the current choice of animal models and the design of the toxicology studies are truly predictive of possible human hazard (Olson et al. 2000). There are, however, limited publications that scientifically address correlations between observed toxicities in animal models to adverse events (AEs) observed in the clinic following their respective administration of novel biopharmaceutical agents. One of the first published analyses of animal-toxicity data for pharmaceuticals was made by Litchfield (1962), based on a very small number of compounds, in which he concluded that toxicities in rats were only rarely observed in humans and that toxicities observed in dogs were not much better in their correlation to humans. However, toxicities occurring in either the rat or dog resulted in a 70% concordance with humans (Litchfield 1962).
A more robust and scientifically rigorous approach to evaluate nonclinical to clinical correlations was undertaken by the International Life Sciences Institute’s Health and Environmental Sciences Institute (ILSI-HESI), comprising 12 pharmaceutical companies contributing data from 150 compounds (Olson et al. 2000). The approach taken by ILSI-HESI was to create a database from a survey of the participating companies in which significant human toxicities (HTs) were identified during any stage of clinical development. HTs were included if the following criteria were met: HT was responsible for termination of drug development, HT limited the dose escalation, HT required dose level monitoring, or HT restricted the target patient population (Olson et al. 2000). Then in a subsequent step, the pharmaceutical company toxicologists retrospectively reevaluated the animal toxicology study reports, including clinical signs, hematology, clinical chemistry, and histopathology data from the rodent and nonrodent toxicology studies and the physiological measurements from safety pharmacology studies. A “toxicology correlation” was considered to be positive if the same target organ was involved in humans and in animals in the judgment of the company clinicians and toxicologists (Olson et al. 2000).
The results of the ILSI-HESI initiative, which are still widely referenced today, demonstrated a true positive HT concordance rate of 71% for rodent and nonrodent species combined, with nonrodents alone being predictive of 63% of HTs and rodents alone of 43% (Olson et al. 2000).
Data from a cross-company data sharing initiative in cardiovascular safety pharmacology evaluated the concordance between preclinical (conscious telemetry dog studies) and phase I cardiovascular assessments; results indicated a good concordance in identifying potential QT interval changes (Ewart et al. 2014). Interestingly, if the ILSI-HESI data (Olson et al. 2000) are corrected for the positive correlations made in the cardiovascular HT category based on safety pharmacology results, the rodent and nonrodent would be equivalent as to “target organ” predictively based on the nonclinical safety studies (Monticello 2009).
Martin and Bugelski (2012) reported on the concordance of preclinical to clinical pharmacology and toxicology data of 14 approved monoclonal antibodies and fusion proteins targeted to soluble targets. In this retrospective review, the preclinical and clinical data were obtained from either the U.S. FDA product reviews and Prescribing Information (USPI, product label) or the European Medicines Agency (EMA) European Public Assessment Reports scientific discussions. The authors concluded that a good concordance existed between the NHP and human for pharmacodynamics end points but not for predicting human adverse effects.
More recently, a review of 142 approved drugs in Japan evaluated the advantages and limitations of the nonclinical safety assessment paradigm for predicting human adverse drug reactions (ADRs; Tamaki et al. 2013). Similar to the Olson approach, this evaluation was retrospective in that ADRs (at an incidence rate of
WG 1 (WG1): Nonclinical to Clinical Translational Safety Database
The International Consortium for Innovation and Quality (IQ) in Pharmaceutical Development is a technically focused organization of more than 40 pharmaceutical or biotechnology companies with a mission of advancing science-based and scientifically driven standards and regulations for pharmaceutical and biotechnology products worldwide (Figure 1). The IQ provides a sustained forum for the exchange of ideas within and across technical disciplines in these industries. The Consortium is leading initiatives in the areas of chemistry, manufacturing, and control; preclinical safety; drug metabolism; clinical pharmacology; quality; and the reduction, refinement, and replacement of animal testing. As part of these initiatives, which are managed by Leadership Groups and Working Groups, members lead and participate in collaborative research, industry surveys, and benchmarking exercises.

Mission of the Innovation and Quality (IQ) consortium.
The Preclinical Safety (DruSafe) Leadership Group is one of the “biological” groups of the IQ. The mission of DruSafe is to advance the science of nonclinical safety and influence the global regulatory environment through their collective experiences and sharing of preclinical noncompetitive data, in order to help accelerate the delivery of safe medicine to patients. The DruSafe Leadership has created WG1, a Nonclinical to Clinical Translational Safety Database, to help achieve their mission (Figure 2).
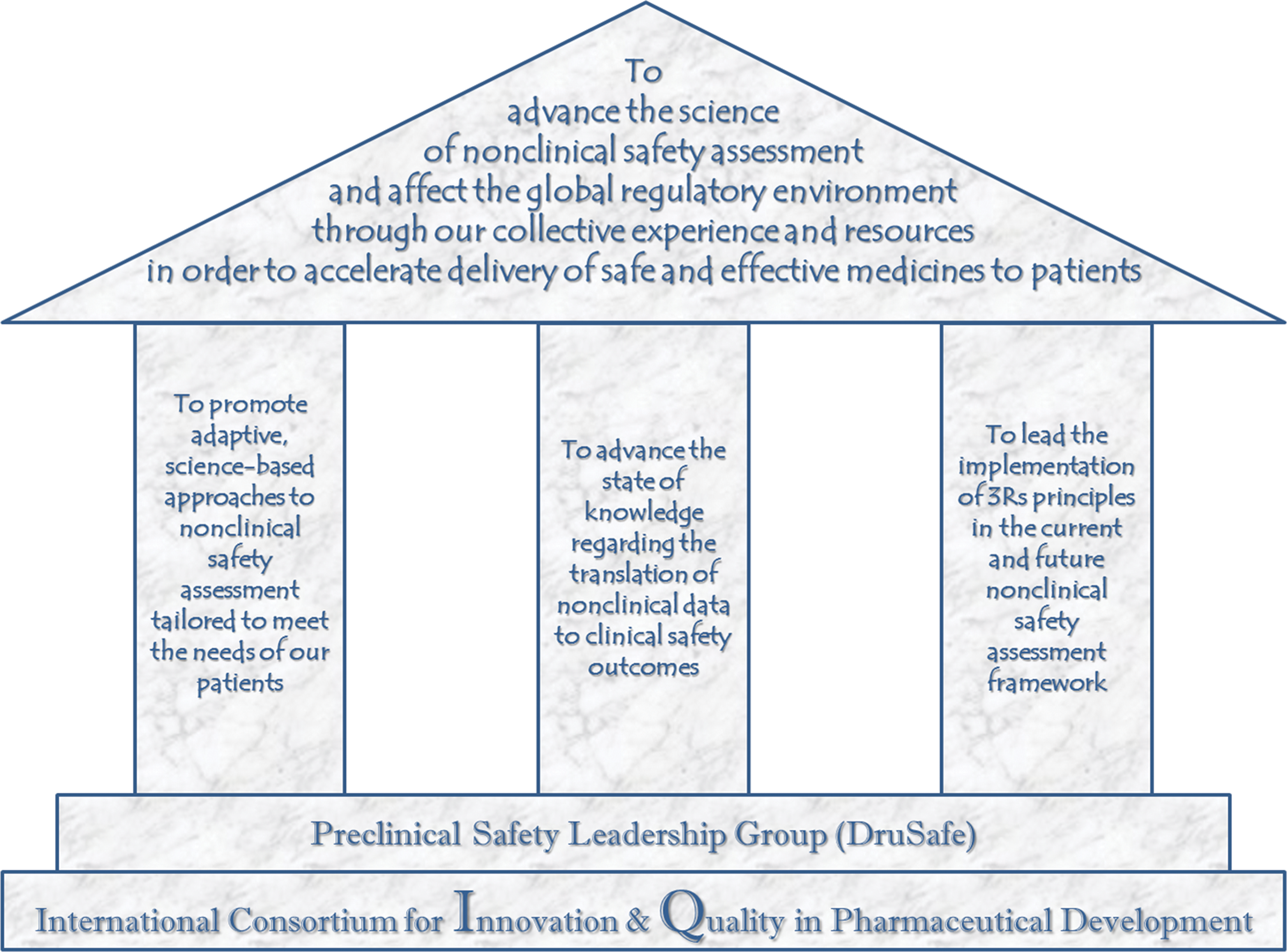
Mission of the Innovation and Quality (IQ) DruSafe leadership group. Working group 1, “Nonclinical to clinical translational database initiative,” supports each “Foundational Pillar.”
The basis of WG1 is to create and analyze a cross-industry database to clarify the accuracy with which the interpretation of nonclinical safety animal models correctly predicts potential human risk. The database will use actual human and animal AE data from the Investigator’s Brochure (IB) of new drug candidates entering the clinic to define the reliability and possible limitations of the nonclinical data and to evaluate the performance and interpretation of conventional biomarkers of toxicity across different organ systems to define if gaps exist. Currently, approximately 25 biopharmaceutical companies are contributing to this noncompetitive data sharing exercise.
WG1 is in direct support of the FDA’s strategic plan of advancing regulatory science. One of their priorities is to modernize toxicology to enhance product safety. Statistical evaluation of the WG1 database in response to specific questions will help address the FDA’s concern of determining true predictive accuracy of current toxicology models and safety assays. The FDA states that there is a need for a more rigorous validation against actual human and animal AE data to better define the reliability and possible limitations of the current nonclinical safety assessment paradigms (U.S. FDA 2011).
Unlike earlier published concordance evaluations, the WG1 foundational database for the nonclinical interpretation of potential clinical safety liabilities will be obtained from the IB from FIH packages, which identifies potential safety risks based on the results of the animal toxicology studies. The database will be “prospective” in that the potential safety risks based on the animal data, identified in the IB, will then be followed in the clinic. The initial output from this WG will determine how well FIH-enabling animal safety assessment studies guide and predict clinical safety for phase 1 trials. The objectives of the phase 1 human studies (usually up to 1 month in duration) are to establish pharmacokinetics, pharmacodynamics, and possible AEs (maximum tolerated dose), usually at somewhat higher doses than in subsequent longer term clinical trials. Thus, in the original database for WG1, we are comparing the results of nonclinical and clinical studies of similar durations and with similar end points. In addition to being a prospective database, other different aspects, as compared with to previous concordance databases, include the addition of exposure data, clinical pathology (biomarker) data, and the evaluation of both large and small molecules. The WG1 database will be “living” in that it will continue to be updated and populated with later stage molecules (longer duration toxicology studies) and longer duration clinical trials.
For the initial evaluation of the database being limited to FIH packages that have completed phase 1, concordance will be determined based on the following parameters: true positive (the identified nonclinical safety liability is observed in the clinic), true negative (lack of identified nonclinical safety liability translates to no clinical safety liability), false positive (an identified nonclinical safety liability is not observed in the clinic), and false negative (no identified nonclinical safety liability but a safety liability is identified in the clinic). Other additional end points will also be queried from the database such as variables that are based on species selection, therapeutic indication, exposure margins, or modality (e.g., small vs. large molecule).
Caveats exist that must be recognized when establishing a nonclinical to clinical correlative database. For example, the database is inherently biased in that many compounds fail in exploratory safety studies because of unacceptable toxicity and therefore are never progressed into investigative new drug (IND)-enabling toxicology studies or the clinic.
The initiation of this database is limited to FIH-enabling studies and clinical data from only phase 1. The goals of FIH-enabling studies are to identify and characterize target organs of toxicity (even if at unrealistically high exposures) and to determine an NOAEL (which translates into a safe starting dose). Therefore, the doses in the clinic, based on the animal toxicology study NOAEL, may never reach exposures where nonclinical toxicity was observed. Moreover, the incidence of clinical AEs will be limited to the phase I human clinical trials.
In summary, the IQ is creating a noncompetitive cross-industry database to clarify the accuracy with which the interpretation of nonclinical safety animal data correctly predicts potential human adverse effects. The database will help evaluate the performance and interpretation of conventional biomarkers of toxicity across different organ systems and define possible gaps. The initial output from this WG will determine how well animal safety assessment studies guide and predict clinical safety for phase 1 trials. The database will continue to expand with longer duration nonclinical and clinical study data.
Footnotes
Author Contributions
Thomas M. Monticello contributed to conception or design, data acquisition, analysis, or interpretation, drafting the manuscript, and critically revising the manuscript. Thomas M. Monticello gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.