
Abstract
Animal models of human disease are commonly utilized to gain insight into the potential efficacy and mode of action of novel pharmaceuticals. However, conventional (healthy) rodent and nonrodent models are generally utilized in nonclinical safety testing. Animal models of human disease may be helpful in understanding safety risks of compounds in nonclinical or clinical development, with their greatest value being in targeted or hypothesis-driven studies to help understand the mechanism of a particular toxicity. Limitations of animal models of disease in nonclinical safety testing include a lack of historical control, heterogeneity in disease expression, a limited life span, and confounding effects of the disease. In most instances, animal models of human disease should not be utilized to supplant testing in conventional animal models. While of potential benefit, testing in an animal model of human disease should only be taken after adequate consideration of relevance along with benefits and limitations of the proposed model.
Keywords
Introduction
Animal models of human disease are commonly utilized to gain insight into the pathogenesis of diseases or to evaluate the efficacy of potential therapeutic regimens. While such models are not frequently utilized in the safety assessment of compounds in clinical development, their potential use is increasingly discussed as new therapeutic modalities and targets are being identified and developed. The intent of this article is to outline recommendations for the use of animal models of human disease in drug safety assessment. A detailed discussion of a wide variety of animal models that may or may not be suitable for use in drug safety assessment is beyond the scope of this article. However, selected animal models will be discussed briefly to illustrate the decision-making process for their use in drug safety assessment. Animal models may be very selective (e.g., targeted gene knockouts) or more general to as closely as possible mimic complex human disorders with multiple contributory factors including complex genetics, nutrition, environmental factors, and infectious agents.
Several guidance and regulatory documents outline the utilization of conventional nonclinical models to evaluate the safety risk of new chemical entities (NCEs) (European Medicines Evaluation Agency [EMEA] 2009, 2011). Retrospective analysis indicates that toxicity evaluation in healthy rodent and nonrodent species results in prediction of human risk in approximately 71% of instances (Olson et al. 2000). While predictability depends on multiple factors, one of the most important determinants is the organ system involved. Nonclinical evaluation of small molecule NCEs is most predictive of hematologic, gastrointestinal, and cardiovascular toxicities and least predictive of cutaneous, neurological, and hepatobiliary toxicities (Olson et al. 2000). The reason for the low predictability for cutaneous changes in nonclinical studies is not completely understood. While cutaneous changes may be significant enough to result in discontinuation of dosing in some patients or even failure to support marketing of the compound, they are readily monitored and typically reversible. For neurological findings, there may be multiple explanations for low predictability, including: (1) inability to accurately assess clinical signs such as dizziness, hallucinations, etc. in nonclinical species; and (2) differences in blood–brain transport between healthy nonclinical species and human subjects that have a disorder compromising the blood–brain barrier. The third toxicity with relatively low predictability, hepatobiliary toxicity, has achieved particular attention in recent years as drug-induced liver injury has been noted to be a frequent cause of clinical attrition and post-approval withdrawal (U.S. Food and Drug Administration [FDA] 2009b). There are likely a multitude of reasons for the low predictability of hepatic toxicities, including but not limited to (1) differences in hepatic transporters between nonclinical species and humans, (2) differences in elimination (e.g., renal vs. hepatic) between nonclinical species and humans, and (3) the relatively high incidence of idiosyncratic (immune-mediated) hepatotoxicity in a clinical setting (Thiele 2007).
Understandably, there is a desire to enhance predictability of toxicities that may be encountered in human subjects. Animal models of human disease may result in improved assessment of human relevance in some instances, primarily by helping to understand the mechanism of toxicity once a particular toxicity has been identified. Unfortunately, even the optimal use of animal models of disease in nonclinical testing will not result in a 100% predictability or understanding of toxicities that may be encountered in a clinical setting, in large part because of limitations of any animal model as outlined in the paragraph above.
The use of animal models of disease should be seen as an adjunct to safety testing in conventional animals, with the focus of targeted animal models being a hypothesis-based approach. The authors envision that animal disease models will be most appropriate for investigative studies into specific toxicities. This article will outline general principles and considerations of the appropriate use of animal models of disease in drug development and safety evaluation. While the emphasis of this article will be on small molecules, many of the principles are applicable to biopharmaceutical agents as well.
Are There Any Specific Regulatory Guidance Documents That Pertain to Use of Animal Models of Disease?
When considering the use of animal models of human disease, many of the relevant challenges are highlighted by the FDA Draft Guidance for Industry: Animal models—essential elements to address efficacy under the animal rule (U.S. FDA 2009a). In this draft Guidance document, the FDA promulgated rules that, for the first time, permit the Agency to approve drugs based upon efficacy data obtained from animal studies in the absence of human clinical trials establishing efficacy. This new regulation, 21 CRF 314 Subpart 1, commonly called the Animal Rule, allows reliance on adequate and well-controlled animal studies to support approval of new drugs or biological products when human efficacy studies are neither ethical nor feasible. It should be noted that this guidance provides information on use of animal models of disease to support filing from an efficacy but not a nonclinical toxicology perspective. However, several of the principles of this guidance are indeed applicable to consideration of animal model use in general and will be briefly outlined below.
Under the Animal Rule Guidance, the animal model to be utilized for determination of efficacy should be supported by natural history studies that establish the similarity to humans in pathophysiology of disease, including progression (i.e., outcome) as well as manifestation of symptomatology and pathology (i.e., extent and type of organ involvement). Inconsistencies of a given model in disease manifestation can have negative safety and efficacy implications and may suggest that use of the model is not justified. The natural history studies must be followed by a pivotal efficacy study in the established animal model conducted in compliance with Good Laboratory Practices and analytically validated and biologically qualified assays. These studies must demonstrate that the hallmarks of human disease occur in the animal model and are mitigated by therapy (using a humanized dose regimen) through similar response pathways (e.g., immune response) and at the appropriate timing to demonstrate a true treatment effect. Any limitations must be taken into consideration and a solution provided to effectively bridge the discrepancy. Finally, early and regular regulatory interaction is encouraged to facilitate success of such a nontraditional approach.
While the “Animal Rule” guidance provides information on the use and development of animal models to support efficacy in humans, there are no currently available guidance that describe the use and development of animal models in nonclinical general toxicology studies. However, materials published on the subject of hepatobiliary toxicity (EMEA 2010; U.S. FDA 2009b; Health Canada 2004) may be instructive in developing an understanding of a tiered approach to nonclinical evaluation of toxicities. Tier I studies are standard studies typically involve testing in conventional nonclinical species (e.g., Sprague-Dawley® rat and beagle dog). They are required for all novel pharmaceuticals unless there is a specific reason why they would not be appropriate. Providing a sufficient safety margin exists, it is typically not necessary to progress past Tier I studies, particularly if the toxicity in question is capable of being monitored in the clinics. However, if additional information is considered helpful to more clearly define human risks (e.g., investigation of a specific pathology finding, seriousness of the finding, dose dependency, safety margins in the different species, ability to use a premonitory clinical biomarker). Also, even if a sufficient safety margin exists and the toxicity can be monitored in the clinics, it still may be advantageous to perform additional studies in order to understand the mechanism of the toxicity and to determine the feasibility of avoiding the toxicity with backup compounds. Tier II studies are specialized studies undertaken to further characterize a change or address a specific nonclinical or clinical issue. Tier III studies may be initiated if there is need for more intense research efforts, depending on the type of toxicity identified in Tier I and II studies, in conjunction with an unfavorable risk/benefit assessment and an unknown mechanism. Tier II and III approaches thus provide supportive data in addition to traditional safety evaluation studies. However, most of the Tier II and Tier III assays are not validated, and, in general, their predictive value for toxicity in longer term nonclinical studies and in clinical studies is not known. Some basic considerations of the tiered approach, as outlined in the hepatic toxicity guidance, are presented in Table 1 below.
Tier I testing is most frequently conducted in conventional healthy animal models (i.e., Sprague-Dawley® rat, CD-1 mouse, beagle dog, cynomolgus monkey, minipig), at least for small molecules. Conventional healthy animal models are ideal for Tier I testing of small molecules based on the homogeneity of the models with low incidence and extensive knowledge of background changes. In contrast, biopharmaceuticals may exhibit a high degree of species specificity, requiring use of specific although healthy animal models (i.e., genetically modified animals or surrogate molecules) rather than conventional animal models in order to evaluate on-target activity for Tier I testing. Thus, the use of genetically modified animals or surrogate molecules may be required for the core Tier I evaluation of these compounds. Tier II or Tier III testing may be conducted in either conventional healthy animal models or animal models of disease and will depend upon the hypothesis being tested and the methodology designed to investigate that hypothesis. Regarding biopharmaceuticals, recommendations have been provided in a recent manuscript focused on the use of animal disease models, genetically modified rodents, and surrogate molecules in safety evaluation (Bussiere et al. 2009).
Non-clinical evaluation via tiered approach.

ADME = Absorption, Distribution, Metabolism, Elimination.
When Are Animal Models of Disease Useful or Appropriate?
Animal Model and Mechanism of Toxicity/Hypothesis Testing
Animal models are best used in the testing of a specific hypothesis in order to understand the pathogenesis and/or relevance of toxicities noted in conventional animals or to understand the pathogenesis of a new toxicity noted in a clinical setting. Considerations for the use of animal models of disease in these settings will be discussed briefly below.
Toxicities that emerge in standard toxicology studies may trigger the use of animal disease models. Animal models of disease may be helpful in providing an understanding of the mechanism of a particular toxicity in a conventional animal in order to predict whether the toxicity is relevant in a particular disease state. This is of particular value in cases where the pharmacodynamic effect of drugs under healthy physiologic conditions is different from a drug’s effect in disease states. Examples include adverse effects caused by glucose-lowering agents in normoglycemic animals, antihypertensive agents in normal animals, and others. Demonstrating that adverse effects observed in standard toxicology species do not occur in an appropriate animal disease model supports a lower human safety risk with a similar disease state.
In cases where toxicity is detected in human patients but not in conventional animal studies, one may consider whether study of the compound in an animal model of disease could provide useful context. Repeated nonclinical studies in the same conventional animal model evaluated previously are unlikely to provide additional insight into either the significance or the pathogenesis of the human clinical finding. Assuming that a suitable nonclinical study species (e.g., with similar metabolite profile, receptor interactions, pharmacodynamic effects, etc.) was used previously, careful evaluation of an appropriate animal model of disease as part of a Tier II or Tier III approach could be considered. For example, potential treatment-related but minimal increases in the incidence of pancreatitis in type 2 diabetics treated with glucagon-like peptide-1 (GLP-1) analogs (Cure, Pileggi, and Alejandro 2008) have been investigated in several rodent models of diabetes and pancreatitis (Tatarkiewicz et al. 2010). These studies have helped clarify the potential link between NCEs and adverse clinical findings.
A given hypothesis may be evaluated retrospectively, followed by prospective evaluation of additional or follow-up compounds in targeted models. An example of this approach is the elucidation of the mechanism of an unpredicted hyperbilirubinemia in human subjects receiving selected protease inhibitors to treat human immunodeficiency virus (HIV) with the subsequent ability to predict which compounds might result in this effect. The mutation of uridine 5″-diphosphoglucuronosyltransferase (UGT1A1) in the Gunn rat rendered this species more susceptible than conventional rats to bilirubin elevations (Iyanagi, Watanable, and Uchiyama 1989) for compounds that had an interaction with UGT1A1. Retrospective analysis provided support for the hypothesis. Prospective evaluation was then conducted in an attempt to screen and rank order compounds in a nonclinical setting (Kempf et al. 2006) that may result in increases in indirect bilirubin without concomitant alterations in hepatobiliary histopathology.
Another example of a specific hypothesis evaluated retrospectively in an animal model of disease involves unexpected hepatotoxicity evident in clinical investigations. Certain compounds administered to subjects with underlying inflammatory conditions, such as rheumatoid arthritis, tend to have a substantially increased incidence of adverse hepatic events compared to subjects without such an underlying disease condition. Thus, use of an animal disease model of inflammation may provide insight into the pathogenesis of hepatic changes and/or provide a better understanding of the role of the inherent pro-inflammatory status of the subjects. An example of this is the bacterial lipopolysaccharide (LPS) model in which rats receive a low dose (nontoxic) injection of LPS prior to dosing of a test compound. Utilization of the LPS model has provided insight into the potential pathogenesis of compounds causing idiosyncratic hepatic toxicity, particularly in subjects in a pro-inflammatory state (Buchweitz et al. 2003; Luyendyk et al. 2003; Roth et al. 1997). Thus, use of such an animal model may provide assistance in helping to understand the pathogenesis of a hepatic change noted in a particular clinical setting. If the compound in question is intended to be administered to humans with a pro-inflammatory state, then the results of such investigative studies may aid in prioritizing or deprioritizing compounds.
It should be emphasized that a well-planned and targeted approach will provide the most reliable and robust data and predictive interpretation. For example, a mouse model humanized for PPARα has been used in determination of the mechanism of hepatocarcinogenesis for peroxisome proliferator agonists and to aid in the assessment of human risk of carcinogenesis (Cheung et al. 2004). Similarly, mice humanized for hepatic expression of the constitutive androstane receptor (CAR) have been used to evaluate CAR-induced hepatocarcinogenicity, which is not considered to be human relevant. While potentially valuable for addressing specific questions, the use of these models for general toxicology assessment is problematic due to differences in receptor expression pattern among tissues and other potential secondary phenotypic effects. Use of such models in a general toxicology study would require thorough understanding of the distribution of mouse versus human receptor for a wide range of tissues in order to fully interpret toxicity findings.
Animal Model and Proof-of-Concept/Efficacy Studies
The most common use of animal models in drug discovery is to help establish nonclinical proof-of-concept evidence for the target of interest and efficacy for a particular molecular entity. In addition to determining whether changes in primary efficacy markers occur, other endpoints that support target engagement and downstream pharmacodynamic effects are commonly evaluated.
A partial listing of select disease models, commercially available for use in proof-of-concept validation, is listed in tabular format below (Table 2). The vast majority of currently available models are for hypertension, endocrine/metabolic disease, or immunodeficiency. The use of a variety of cancer models has also proven to be a valuable resource for selection of therapeutic candidates prior to progression to nonclinical safety evaluation. While the listing below primarily focuses on rodent models, it should be noted that other species are also available. For example, nonhuman primate (NHP) species such as macaques have been utilized in studies of bioenergetics, obesity, cardiovascular and reproductive health, central nervous system function, and cognitive and social behavior, among others (Shively and Clarkson 2009). A wide variety of other animal species are also available (dog, guinea pig, rabbit, prairie dog, woodchuck) for specific models of human disease (Quimby 2002).
Examples of animal models of disease.

Animal Model and Early Discovery Safety Information
An animal model of disease may be of assistance in providing early evidence of safety risk through incorporation of a targeted set of safety endpoints and biomarkers into the design of an efficacy study. The goal of this approach is to aid in identification of target organ toxicities or consequences of exaggerated pharmacology. For example, the effect of target blockade by novel therapeutic candidates can be estimated in knockout mice as compared to wild-type mice in order to gain understanding of the potential for either beneficial (pertaining to efficacy) or adverse (on-target toxicity) effects (Bussiere et al. 2009).
Animal Model and Therapeutic Safety Margin versus Hazard Identification
Animal models of disease are more valuable for hazard identification rather than for determining exposure multiples for selection of first-in-human dosages and exposure limits. For example, a lower safety margin or unique toxicities may occur in an animal model of disease (as compared to a conventional animal model) with reduced hepatic or renal clearance. Thorough comparison of pharmacokinetics/toxicokinetics in the animal model of disease with a conventional animal model is of paramount importance to discern whether unique toxicities in the animal model are a manifestation of a mechanistic difference or are secondary to higher systemic exposures.
Animal Model and Target Engagement When Standard Species Lack the Target
In some cases, the use of animal models for safety evaluation is desirable for prediction of on-target adverse drug reactions. For example, animal models may be of use to evaluate the on-target toxicity of pharmaceutical agents intended to treat inborn errors of metabolism, such as lysosomal storage diseases. Some lysosomal storage diseases are more commonly reported as naturally occurring models noted in larger species (Haskins 2007). However, from a practical standpoint, large animal models of specific diseases are less desirable for use in toxicology studies, due to the necessity of establishing breeding colonies and the comparatively long gestation period and small litters per birth. While several mouse models of LSDs do not exhibit satisfactory similarity to the human counterpart (Ohshima et al. 1997; Xu et al. 2003), others such as Niemann–Pick disease and Pompe’s disease have been successfully modeled in rodents (Bijvoet et al. 1999; Horinouchi et al. 1995). Because of the substrate buildup in these animals, they are particularly appropriate for determining the outcome of treatment in actual patients, including the effect of abrupt metabolism of intracellular materials and subsequent introduction of breakdown products to the affected cells and distant tissues (Gieselmann 2007; Smith and Schuchman 2008). Investigators should consider the possible biological effects that could result from a bolus of potential breakdown products when such metabolic diseases are corrected by enzyme replacement. The use of an appropriate animal model may allow for adjustment of treatment regimens to avoid potential negative outcomes (Vitsky et al. 2006). Nonclinical studies utilizing the α-glucosidase knockout mouse have been utilized to support filings from a pharmacology and safety perspective (e.g., Myozyme, therapy for Pompe’s disease). Studies conducted from a pharmacology perspective involved demonstration of clearance of tissue glycogen load under varying conditions (varying dose levels, treatment regimens, ages of animal, product preparation). In contrast, in a safety study, a knockout mouse was utilized to investigate the nature of the proposed hypersensitivity reaction in the CD-1 mouse.
Mouse models of Alzheimer’s disease (AD) provide another example of the potential use of an animal model of human disease for investigation of efficacy and safety hypothesis testing. Several models currently exist, including the PDGF promoter expressing amyloid precursor protein (PDAPP) mouse (Johnson-Wood et al. 1997). The PDAPP transgenic mouse overexpresses human amyloid precursor protein (APP717VàF). The use of the PDAPP mouse (or other models) for efficacy testing (prevention or reduction in amyloidosis and plaque burden) has tremendous potential to guide selection of efficacious therapeutic agents. While dedicated safety testing in such a model may not be necessary, if adverse clinical effects are noted in the animal model of disease but not in conventional animal models, strong consideration should be given to investigation of the toxicity in the animal disease model prior to clinical trials.
General Considerations for Selection of the Animal Model
Various factors come into play in evaluating the predictive value of an animal model for a particular human disease. Critical factors include determination of the on-target potency and species–specific metabolism properties. Some of the species differences can be addressed through the use of humanized transgenic animals that express the human target of interest, modeling of a particular of metabolic pathway, or, in some instances, the use of species–specific surrogates (Bussiere et al. 2009).
Can the Animal Model of Disease Help to Differentiate Between Off-target and On-target Toxicity?
The use of animal models of disease may facilitate exploration of toxicity when dose-limiting pharmacologic effects occur in non-diseased animal models at relatively low multiples of anticipated therapeutic exposure. For example, dose-limiting hypoglycemia or polycythemia would be expected to be evident at relatively low exposure multiples in non-diseased animals treated with a therapeutic agent designed to lower blood glucose in hyperglycemic patients or increase red blood cells in anemic patients, respectively. Utilizing an appropriate animal model of disease may allow for more robust evaluation of toxicity, particularly off-target toxicity.
The use of an animal model of disease may also be helpful in differentiation of an on-target compared to an off-target effect. A classic example of exaggerated pharmacology resulting in complication of safety assessment in normal animals is hypoglycemia associated with certain antidiabetic agents. It is well documented that intermittent insulin-induced hypoglycemia can result in a variety of histopathology changes, including peripheral neuropathy in rats and mice (Tabata 2000; Ozaki et al. 2010). Unfortunately, nerve fiber degeneration, as characterized by axonal atrophy and myelin sheath disruption, is not specific for intermittent hypoglycemia and such changes may be an off-target manifestation of the hypoglycemic agent. However, selective safety assessment of such agents in naturally hyperglycemic or diabetic animals can be more relevant to assessment of risk. While pharmacologically induced acute hypoglycemia is relatively straightforward to monitor in nonclinical studies, a diabetic model in spontaneously diabetic (Wistar Bonn/Kobori) WBN/Kob rats has been utilized to help determine the on-target effects (secondary to hypoglycemia) versus. off-target effects of insulin. In one study, rats were treated with insulin and severe peripheral neuropathy was noted only in those rats with serum glucose concentrations less than 50 mg/dl, substantiating the on-target (hypoglycemia-related) effect (Ozaki et al. 2010).
Can the Animal Model of Disease More Closely Predict Human Toxicity (As Compared to Conventional Animals)?
Frequently, there are differences in toxicity profiles between species. When there are interspecies differences in metabolites, an obvious question is whether these differences in metabolites are responsible for the toxicity or if they are relevant to human risk. Animal models may be of benefit in elucidating the role of a particular metabolite in the toxicity of an NCE. This may involve testing of the toxicant in question either in mice genetically deficient in specific cytochrome P450 (CYP) enzymes (Valentine et al. 1996) or through coadministration of a blocking agent.
What Are Some Challenges or Limitations in Utilizing an Animal Model of Disease?
For an animal disease model to have human relevance, particularly for safety testing, it is critical that the animal model exhibits considerable homogeneity with respect to the severity of the disease phenotype. It is also imperative to have an understanding of background changes and unintended manifestations of the animal model of disease. If the animal model of disease is intended to answer a targeted question, then establishing that there is homogeneity between animals and no confounding unintended adverse manifestations may be sufficient. However, if there is consideration of utilizing the animal model of disease for more general toxicology evaluation, then a more in-depth understanding to include evaluation of incidence, severity, and homogeneity of background changes is needed in order to determine whether the animal model of disease will be satisfactory.
Analogy to the Human Disease
An animal model of disease should reliably exhibit all the critical manifestations of the disease relevant to the investigation. An example of incomplete manifestation in an animal model was apparent when attempts were made to develop a mouse model of cystic fibrosis (mice deficient in CFTR, cystic fibrosis transmembrane conductance regulatory gene). While the mice failed to develop pulmonary effects that were consistent with cystic fibrosis, they did exhibit severe gastrointestinal obstruction, which can be another manifestation of cystic fibrosis in humans (Davidson and Rolfe 2001). Subsequent work suggested that the lack of pulmonary findings may be a manifestation of differences in tissue-specific gene regulation between different species (Odom et al. 2007).
Homogeneity with Respect to Disease
Consistency with respect to disease manifestation is important to ensure reliable evaluation of the potential for exacerbation of the disease phenotype or target, especially in groups that achieve high-dose pharmacologic exposures. For example, in the investigation of histopathologic effects in a hypertensive model, use of a heterogeneous population of animals may result in exclusion of potentially beneficial compounds as it may appear that the compound exhibits no benefit or, alternatively, exacerbates hypertension-associated histopathologic changes. In the latter case, the apparent histopathologic changes may simply be secondary to preexisting heterogeneity in manifestation of the disease. The inability to develop a model with sufficient homogeneity with respect to the disease may hinder the use of the model for both drug efficacy and toxicity studies.
Background Changes
An in-depth discussion of the specifics for validation of a model including evaluation of background changes is beyond the scope of this article, as the needs will vary depending upon the model and the hypothesis/question. However, several recent publications provide detailed recommendations in respect to the use of genetically engineered models, as it pertains to study design (Brayton, Treuting, and Ward 2012), the effect of microflora on phenotype (Treuting et al. 2012), and the design of phenotyping studies (Zeiss, Ward, and Allore 2012).
The number of animals to be evaluated will depend on the phenotypic homogeneity of the model as well as variability encountered in early screening evaluations. Additionally, the robustness of the evaluation will depend upon the questions to be answered. Prior to use of an animal model of disease in general toxicology testing, robust evaluation of background changes should be conducted. While less extensive evaluation of background changes may be sufficient when considering an animal model to address a targeted question, the testing should still address critical targets that could influence the ability to answer the question of interest.
Unintended Manifestations
It is also important to determine whether a given group of animals modeling a specific disease can be maintained over the intended duration of efficacy and safety studies. Many genetically manipulated models may not have sufficient long-term survivability, which may substantially limit their use, particularly for safety studies. Similar considerations would be apparent for animal models with naturally occurring or implanted tumors as control and low-dose animals may have survival issues, precluding long-term nonclinical safety testing.
Specific Considerations for Surgically Created Models
When considering use of a surgically modified model to mimic a human disease, additional issues should be taken into account. Sufficient animal numbers must undergo surgical modification in order to guarantee healthy animals have been successfully modified and to provide enough statistical power to mitigate impact of individual differences in response to surgical conditions. For example, rodents that are less hydrated during ischemia/reperfusion kidney studies are more susceptible to kidney injury, which confounds the surgical manipulation parameters (Racusen, Prozialeck, and Solez 1984). The surgical procedure must be followed by adequate time to allow for complete healing postoperatively and to permit complete washout of any drugs used for anesthesia, analgesia, and supportive care (i.e., antibiotics). Postoperative healing and scarring from fibrosis may result in marked differences in functional parameters in ensuing toxicity studies and in histopathologic scoring. Finally, successful execution of the surgical protocol, in order to provide the same surgical manipulation across all animals in all dose groups, must be followed. This often requires surgical modification of sham animals to control for variables introduced as a result of the operative procedures.
All of these variables can be difficult to control for and are undesirable when using a surgical model of disease for toxicity studies. Generation of sufficient numbers of healthy surgically modified animals is resource-intensive and time-consuming. Most importantly, a greater number of animals are required to undergo surgical modification in order to guarantee a sufficient number of viable, successfully modified animals for study.
When Are Animal Disease Models Not Useful or Appropriate?
Model Does Not Favorably Mimic Pathophysiology
Although animal disease models are widely used for efficacy, critical issues may arise in drug toxicity studies due to insufficient knowledge of model-specific background pathology and sensitivity and functional/anatomical differences among species. Several examples are provided below to illustrate issues to consider when evaluating the appropriate use of targeted animal models.
Animal models of Alzheimer’s disease
Alzheimer’s disease (AD) research is an area in which available animal models vary widely in their similarity to human AD. Most AD models demonstrate a sexual dimorphism not representative of the human disease (Lewis et al. 2001), and few models demonstrate a range of clinical signs and characteristic pathologic lesions that are comparable to human AD (Oddo et al. 2003). Use of mice that are transgenic for multiple proteins that exhibit abnormalities in humans has resulted in more appropriate modeling (Jay et al. 2011). While even the best animal models for AD do not recapitulate all the features of the human counterpart, these models may still have value as instruments for efficacy evaluation (Wong et al. 2002). From a safety standpoint, mitochondrial dysfunction and dysregulation of inflammation are theorized to be central to neurodegenerative disease pathogenesis (Chen, Stern, and Yan 2006; Rojo et al. 2008). For this reason, certain AD models have been used for specialized evaluation of inflammation and/or hemorrhage in response to treatment, such as changes associated with the removal of amyloid-β peptide plaques (Jay et al. 2011; Nicholl et al. 2003). However, given the heterogeneity of the animal models available, they are unlikely to yield reliable safety information that parallels human disease and are best utilized in efficacy screens.
Aged rat model for evaluating sensitivity to potential renal toxicants
Responses to nephrotoxicants are greatly affected by age, as renal function and morphology undergo dramatic age-related changes in rodents. Rats are commonly affected by chronic progressive nephropathy (CPN), a rodent-specific, age-related renal disease first detected at 2 to 3 months of age. Deaths from renal failure due to CPN may commence at <18 months in Sprague-Dawley rats (Hard and Khan 2004; Palm 1998; Travlos, Betz, and Hard 2007). CPN does not mimic key hallmarks of kidney senescence in humans, and the key morphologic features in the basement membrane of tubules in rats with CPN do not recapitulate similar features in humans with chronic renal failure (Trevisan, Nicolli, and Chiara 2010). Reduced cortical mass as observed in CPN has been shown to enhance renal injury by increasing delivery of nephrotoxin to the remaining functioning nephrons. Underlying kidney disease also affects baseline clinical pathology parameters and may obscure a biomarker indicative of kidney injury that otherwise would be identified in a healthy kidney from a young rodent. The hugely variable severity of CPN between individuals of the same age prevents standardization between groups. Thus, an aged rat with CPN is not considered a suitable model for renal senescence in humans due to differing histopathologic features and confounding variability that obscures to ability to reliably detect a renal change (Hard, Johnson, and Cohen 2009).
Obese rodent models of diabetes
In efficacy evaluations of antidiabetic agents, Obese Zucker Diabetic Fatty (ZDF) rats are widely used as an experimental model of genetic, obesity-induced type 2 diabetes mellitus (T2D). Development and pathology of the disease in the ZDF rat shares many features with obesity-induced human T2D. These rats are obese and develop progressive insulin resistance, glucose intolerance, hyperinsulinemia, and hyperlipidemia in a relatively predictable, age-dependent fashion (Ionescu, Sauter, and Jeanrenaud 1985; Kasiske, O’Donnell, and Keane 1992; Kurtz, Morris, and Pershadsingh 1989). ZDF rats are euglycemic at 5 to 6 weeks but become increasingly hyperglycemic and hypoinsulinemic, developing diabetes at about 10 weeks with progressive islet failure. These rats eventually die at 30+ weeks of age due to diabetes-related complications that include pancreatic islet fibrosis, hepatic steatosis, and renal damage (Clark, Palmer, and Shaw 1983; Janssen et al. 1999; Tokuyama et al. 1995). Given the range of preexisting islet pathophysiology, resultant complications in several organ systems, and increased mortality, it is expected that the expression of drug-induced effects in obese ZDF rats will be markedly altered (particularly with an advanced diabetic state) compared to healthy animals. Conducting studies evaluating drug toxicity in obese ZDF rats requires rigorous understanding of the model, its pathophysiology, and its morphologic characteristics. The difficulty in separating direct effects (drug-induced) from indirect effects (diabetes-related complications) limits the utilization of obese ZDF rats in nonclinical toxicity studies. It can also be extremely difficult to detect target organ toxicity in organs that are directly or indirectly affected by hyperglycemia and hyperlipidemia, such as liver, kidney, and pancreas. Other factors that complicate or limit the use of this model in general nonclinical toxicity testing include potentially confounding low long-term survival of control animals and variation in severity of disease between animals.
Recommendations for Use of Animal Disease Models in Safety Testing
General Considerations for Safety Evaluation in Nonclinical Toxicology Studies Used for Regulatory Filing for Entry into Human Subjects
Use of a targeted animal disease model to assess pharmacological relevance, evaluate pharmacodynamic effects, elucidate mechanisms of toxicity, or assess disease relevance of an identified safety study finding in nonclinical or clinical studies are examples from a Tier II–III approach. Using animal disease models in this manner potentially allows for concurrent efficacy and safety endpoint evaluation and also for direct monitoring of improvements in the therapeutic index (Boelsterli 2003) and assessment of on-target safety when healthy nonclinical toxicology species lack the target of interest. Nonetheless, the use of animal disease models in complete safety evaluations in human-enabling nonclinical safety toxicology studies requires a rigorous risk:benefit assessment in the context of drug development. Several considerations of animal disease models in nonclinical safety toxicology studies should be included in determining whether the anticipated benefits substantially outweigh the risks: (1) establish a clearly defined scientific rationale for using a targeted animal disease model, (2) determine whether the model will substitute for or complement traditional safety evaluations, (3) understand the impact of safety findings on development of the compound and target, (4) determine whether the endpoints are clearly defined and the associated tests are validated and/or qualified in that species, and (5) determine whether the safety endpoint of interest can be alternatively evaluated appropriately in an efficacy study to rank-order compounds and understand response to therapy in states of target over/underexpression. These concepts apply regardless of whether the model is being utilized in the development of small molecules, biotherapeutics (Bussiere et al. 2009), or stem cell therapies (Cabral et al. 2011; Chen 2010). Risks in this context may include increased timelines, lack of concordance to the human population of interest, and the potential impact of ill-understood or even irrelevant safety findings that do not translate clinically or do not provide information that can be used for clinical decision making.
From a pathology perspective, the evaluation of animal disease models is challenging. Animal disease models are in species or strains that are generally poorly characterized and that lack historical information on spontaneous background findings. In addition, the considerable inter-individual variation in primary or secondary disease-associated pathology poses additional challenges in interpretation. Thus, discerning whether clinical and anatomic pathology findings are attributable to incidental age-related or background changes, anticipated primary or secondary disease manifestations, test article–related adverse effects, test article–related nonadverse effects, or resolution of disease phenotype will require additional experience and data accumulation specifically with that model system.
A decision tree outlining the salient features to consider when evaluating the potential utility of an animal disease model for use in safety testing is presented below (Figure 1).
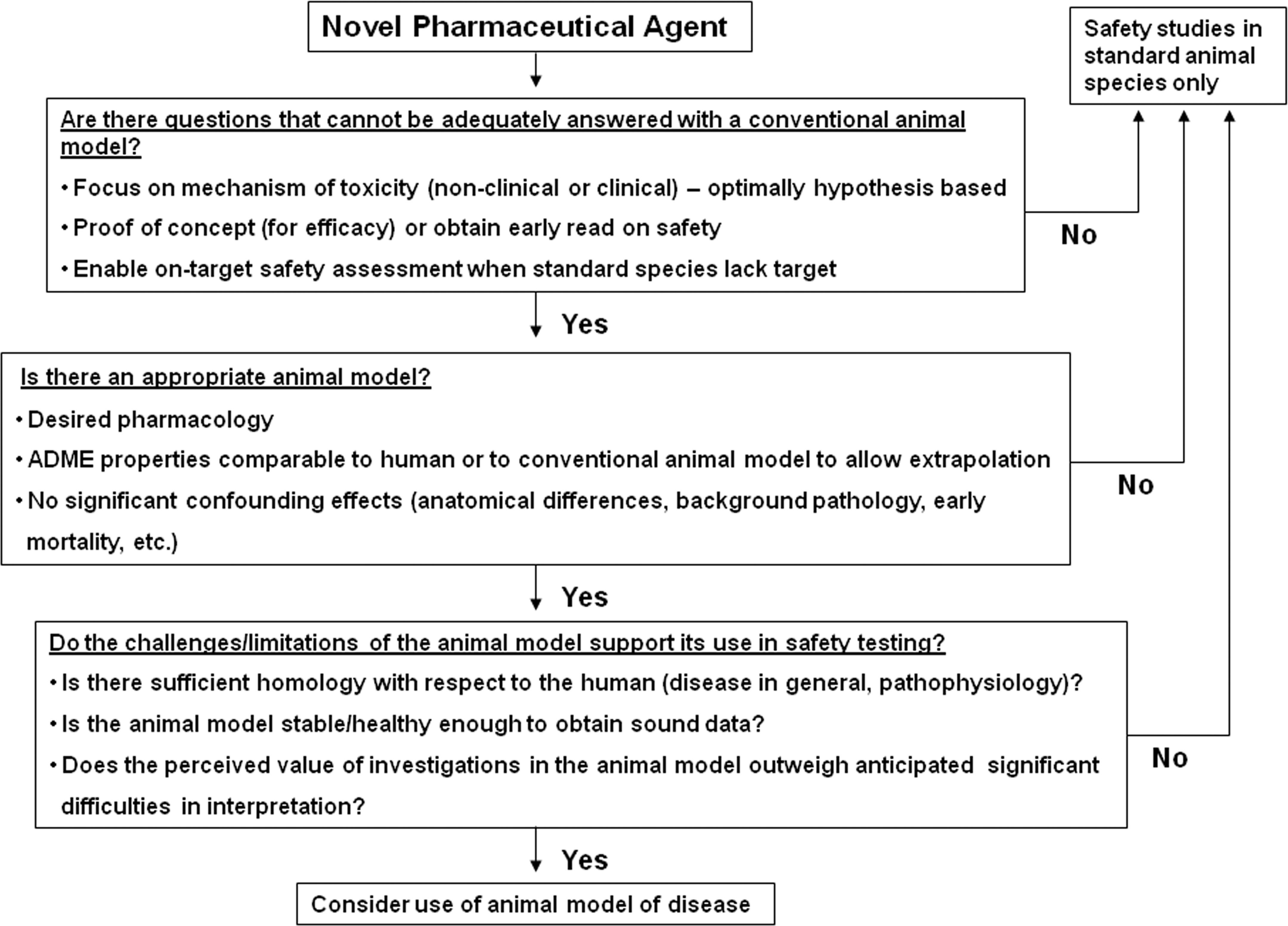
Decision tree—Selection of animal disease models for conducting nonclinical safety studies of small-molecule pharmaceuticals.
Summary
Establishing knowledge of efficacy and safety parameters is essential for drug discovery, development, and approval. Animal models of human disease are valuable research tools for elucidating pathogenic mechanisms and testing the efficacy of prophylactic or therapeutic interventions. In contrast, conventional animal models such as the Sprague-Dawley rat® and beagle dog are utilized nearly exclusively to evaluate the safety of compounds, and studies evaluating on-target toxicities in animal models of human disease should not be expected to replace traditional nonclinical safety studies in these well-characterized species.
The decision to utilize an animal model of human disease as a part of a nonclinical safety package is usually driven by a need to test a specific hypothesis. Typically, such hypotheses are generated after target organs have been identified in standard nonclinical toxicity studies in healthy animals. Animal models of disease rarely, if ever, accurately recapitulate all the key aspects of the corresponding human disease, may have inconsistent disease manifestations, and are less well-characterized toxicology models than standard models utilizing healthy animals (Bolon and Galbreith 2002; Cordaro 1989). Thus, targeted animal models are typically inappropriate to use in general toxicity studies that include complete evaluation of clinical and anatomic pathology endpoints. Instead, animal models should be utilized in a second or third tier approach to elucidate safety risks that were identified in the first tier of traditional nonclinical safety studies.
Recommendations for the use of animal models of disease are as follows: Standard animal models should continue to be utilized in a second or third tier approach to further elucidate safety risks that were identified in the first tier of traditional nonclinical safety studies. The use of animal models of disease should be reserved as an adjunct to answer specific hypothesis-driven questions as it pertains to the safety assessment. The use of animal models of disease in safety testing should be focused on hazard identification/understanding rather than safety margin calculation. A rigorous risk:benefit assessment of the appropriateness of the animal of disease and its intended use in preclinical drug development is paramount to success. Critical steps prior to consideration of the animal model of disease include (1) determination of degree of homogeneity with respect to the human disease and (2) rigorous characterization of the animal model.
Examples of particularly useful applications may include (1) allowing for more robust evaluation of toxicity, particularly off-target toxicity; (2) differentiation of an on-target versus off-target effect; and (3) investigation of unexpected toxicity evident in clinical investigations not found in conventional animal models.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
Authors’ Note
The recommendations contained in this article have been endorsed by the Society of Toxicologic Pathology.