
Abstract
Neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases, have olfaction impairment. These pathologies have also been linked to environmental pollutants. Vanadium is a pollutant, and its toxic mechanisms are related to the production of oxidative stress. In this study, we evaluated the effects of inhaled vanadium on olfaction, the olfactory bulb antioxidant, through histological and ultrastructural changes in granule cells. Mice in control group were made to inhale saline; the experimental group inhaled 0.02-M vanadium pentoxide (V2O5) for 1 hr twice a week for 4 weeks. Animals were sacrificed at 1, 2, 3, and 4 weeks after inhalation. Olfactory function was evaluated by the odorant test. The activity of glutathione peroxidase (GPx) and glutathione reductase (GR) was assayed in olfactory bulbs and processed for rapid Golgi method and ultrastructural analysis. Results show that olfactory function decreased at 4-week vanadium exposure; granule cells showed a decrease in dendritic spine density and increased lipofuscin, Golgi apparatus vacuolation, apoptosis, and necrosis. The activity of GPx and GR in the olfactory bulb was increased compared to that of the controls. Our results demonstrate that vanadium inhalation disturbs olfaction, histology, and the ultrastructure of the granule cells that might be associated with oxidative stress, a risk factor in neurodegenerative diseases.
Introduction
Environmental exposure to particulate matter (PM2.5) has been associated with airway diseases. Metals such as vanadium are the major components of PM2.5, and it has been suggested that it plays an important role in PM-induced mucin production and may result in the direct access of these pollutants into the central nervous system (CNS;Yu et al. 2011; Genc et al. 2012; Shafer et al. 2012). Workers exposed to vanadium exhibited neurobehavioral alterations in attention, cognition, and short-term memory (Li et al. 2013). Exposure to air pollutants causes alterations in the blood-brain barrier integrity, and degeneration of cortical neurons in dogs, and increase in prefrontal cortex tau hyperphosphorylation with pre-tangle material in children and young adults from Mexico City (Calderón-Garcidueñas et al. 2011, 2002). Human interaction with environmental pollutants is recognized as a significant contributing factor in the development of neurodegenerative diseases such as Parkinson’s disease (PD) and Alzheimer’s disease (AD; Caudle et al. 2012; Moulton and Yang 2012; Guglielmotto et al. 2010).
Data obtained from the residents of Mexico City show that air pollution appears to have a substantial impact on olfactory dysfunction and an upregulation of inflammation biomarkers such as cyclooxygenase 2 and interleukin 1 β in the olfactory bulb and frontal cortex (Hudson et al. 2006; Kovács 2004; Calderón-Garcidueñas et al. 2010, 2013). Early-stage olfactory dysfunction in neurodegenerative diseases and aging is associated with significant cell loss in the olfactory bulb layers (Doty 2009; Haehner, Hummel, and Reichmann 2011; Kovács, Cairns, and Lantos 2001). In PD, the olfactory dysfunction is present approximately in 90% of early-stage cases and may precede motor symptoms (Doty 2012).
Environmental pollutants may enter the brain through the nasal olfactory epithelium to the olfactory bulb. In the olfactory bulb, the agent is carried by the axons of mitral and tufted cells. In addition, the pollutant may enter the cortex because it is carried by granule cells that modulate the output pathway to piriform cortex and to the superficial layers of the rostral entorhinal cortex (Zou, Li, and Buck 2005).
In rodent’s brain, some metals are transported through the olfactory nerve and some reports mention their accumulation in the olfactory bulb. These structures tend to accumulate certain metals such as Al, Cu, Mn, and Zn (Suderman 2001).
Vanadium, a trace element, is widespread in the environment (Zhang et al. 2001). The main source of vanadium in urban areas is the combustion of gasoline that causes an increase in its concentration in the air, and consequently, its increase in the lung tissue (Fortoul et al. 2002). Vanadium compounds have been related to the pathogenesis of some human diseases (Environmental Protection Agency [EPA] 2011; Mukherjee et al. 2004). Vanadium pentoxide (V2O5) is the most toxic and common form of this element (International Agency for Research on Cancer [IARC] 2006). An exposure of 1.1-mg vanadium/m3 in 2-year-old mice causes nasal inflammation and olfactory epithelium atrophy. Likewise, 0.56-mg vanadium/m3 causes hyaline degeneration of olfactory bulb and respiratory epithelium as reported previously (National Toxicology Program [NTP] 2002). Soluble vanadium compounds in the lung are well absorbed and induce pulmonary inflammation (Barceloux 1999; Rondini, Walters, and Bauer 2010).
In rat neonates (Soazo and Garcia 2007), exposure to sodium metavanadate causes behavioral alterations and myelin deficit. Furthermore, V2O5 exposure (1.4 mg/m3) in male mice causes neurotoxic changes, as well as a loss of tyrosine hydroxylase immunoreactive in substantia nigra and a decrease in dendritic spine density in medium size striatal spiny neurons. In pyramidal neurons, vanadium inhalation causes necrosis of the hippocampus CA1 with spatial memory impairment (Avila-Costa et al. 2003, 2006). Blood-brain barrier disruption (Avila-Costa et al. 2005) as well as increased activity of metalloproteinase 9 (MMP-9) in striatum, hippocampus, and olfactory bulb has also been reported (Colín-Barenque et al. 2008).
A potential mechanism of injury to human tissues after exposures to PM2.5 is through metal-catalyzed oxidant generation (Ghio et al. 2002). Several studies have demonstrated that vanadium toxicity is mediated by the generation of free radicals and oxidative stress (Crans et al. 2004).
Experimental studies have shown that vanadate mediated generation of reactive oxygen species (ROS) and that oxidative stress and lipid peroxidation are involved in adverse biological effects (Huang et al. 2003; Cortizo et al. 2000; García et al. 2004). It is known that in vivo vanadium increases glutathione peroxidase (GPx) and glutathione reductase (GR) activities in the lungs (Wennig and Kirsch 1988). In addition, the accumulation of ROS induces cellular damage that is an important cause of neurodegenerative diseases (Gibson and Huang 2002; Practico and Delanty 2000; Smith et al. 2000), and as we previously mentioned, olfactory dysfunction is an early symptom in AD and PD (Doty 2001; Hawkes 2003; Haehner, Hummel, and Reichmann 2011). Moreover, a cytological study showed that oxidative stress generated by ozone caused olfactory bulb alterations (Colín-Barenque et al. 1999). The aim of this study was to investigate the relationship between a simple olfactory function test, GPx and GR activities, and morphological changes in granule cells from the olfactory bulb as a consequence of vanadium inhalation.
Materials and Methods
Forty-five CD-1 male mice weighing 33 g were housed in hanging plastic cages under controlled lighting conditions (12 hr light/dark regime) and were fed with Purina rat chow and water ad libitum. The experimental protocol was conducted in compliance with the Animal Act of 1986 for Scientific Procedures and the Mexican Guideline Animal Welfare (NOM-062-200-1999). All efforts were made to minimize the number of animals used and their suffering.
Buried Food Test
The buried food test measures the ability to recognize odors. Twenty mice were trained before the inhalation. Food and water were removed from the cages 12 hr before the beginning of both stages in which the test was divided. In the first stage, a chocolate chip was placed inside the clean cage so that chocolate scent was associated with its flavor during a period of 10 to 12 hr. During this time, the animals consumed the chocolate chip. Afterward, food and water were restored. Two days later, a night before the buried food test began, food and water were removed again.
The buried food test was performed at baseline (before exposure) and at 2 and 4 weeks after vanadium exposure. Food and water were removed a night before buried food test was performed. The test began when we transferred each mouse from its home cage to a clean cage, in which a chocolate chip was buried beneath the surface by 2 cm of the bedding cage. The latency to locate the chocolate was measured up to 120 sec. This procedure was repeated 3 times with the chocolate chip buried in different locations of the cage. Chocolate localization testing was performed based on a method previously described (Ferguson et al. 2000).
Vanadium Inhalation Exposure
Inhalations were performed as described by Fortoul et al. (2008). Knowing that the half-life of vanadium is about 48 hr, we planned a twice a week exposure. Forty animals were placed in an acrylic chamber and were made to inhale 0.02-M V2O5 1 hr twice each week for a 4-week period.
Ten control mice inhaled only the vehicle (saline) for the same period. Inhalations were performed in closed acrylic boxes (35-cm wide × 45-cm long and 20-cm high) connected to an ultranebulizer (Yue Hua, WH-802, Taiwan) that was used to nebulize the vanadium solution, maintaining a constant flow of 10 L/min. Based on the manufacturer’s instructions, about 80% of the aerosolized particles reaching the mice would be expected to have a mass median aerodynamic diameter (MMAD) of 0.5- to 5-μm range. Concentrations of V2O5 in the chamber were quantified as follows: a filter was positioned at the outlet of the ultranebulizer during the entire inhalation time. After each exposure, the filters were removed and weighed; the element was quantified following the same protocol as with tissue samples. Six filters for each inhalation were evaluated (Fortoul et al. 2002).
The experimental group was exposed to V2O5 in the same experimental conditions as that of the controls. Both groups were sacrificed at 1, 2, 3, and 4 weeks after vanadium exposure.
Histological and Ultrastructural Method
After the scent detection evaluation, 20 mice were anesthetized with sodium pentobarbital and perfused via aorta with saline (sodium chloride [NaCl] 0.9%), 2% paraformaldehyde, and 2% glutaraldehyde (pH 7.4) in 0.1-M phosphate buffer. Olfactory bulbs were removed and placed in a fixative solution for 2 hr at 4°C and processed according to the rapid Golgi method (Valverde 1970). All the samples were processed in the same experimental conditions and at the same time. Blocks were cut into 90-μm sections. In the cytological analysis of control and exposed mice, the major and minor axes of 25 granule cells were measured directly in a light microscope, which has a micrometric ocular, and the dendritic spines in 20 μm of 50 secondary dendrites were counted per mouse from each group (4 animals), which gives a total of 100 neurons per group.
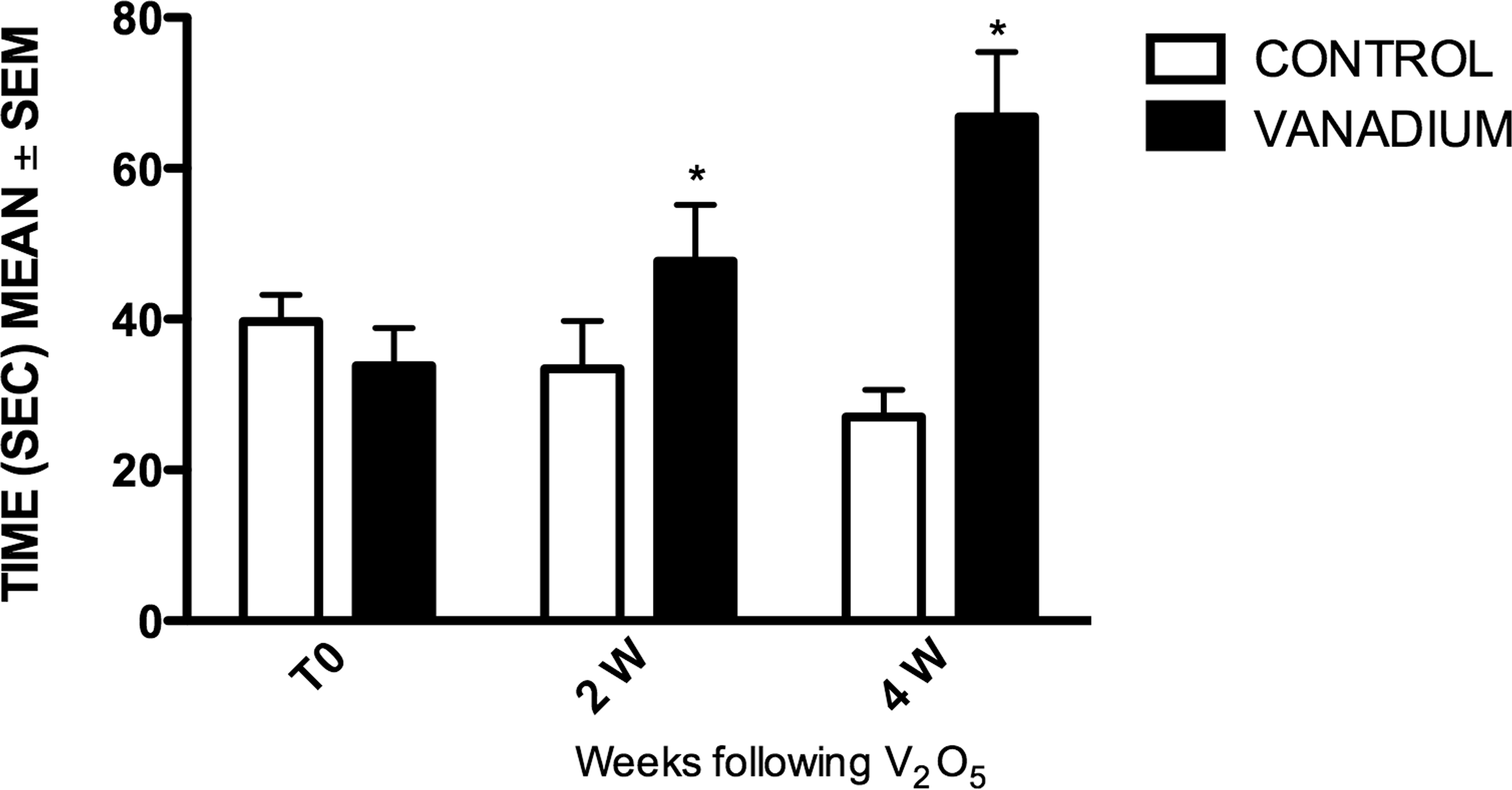
Graph shows the average (+/− standard error of mean) olfactory test results. Mice exposed to vanadium at 2 and 4 weeks had significantly increased times compared with controls. Statistical significance *p < .05 analysis of variance (ANOVA).
For ultrastructural evaluation with conventional transmission electron microscopy, the olfactory bulbs were postfixed in 1% osmium tetroxide (pH 7.4), buffered with sodium cacodylate, dehydrated in graded alcohols, and embedded in Araldite 6005 for 1 hr. Sections were obtained with a Reichert-Jung microtome placed on grids, contrasted with uranyl acetate and lead citrate, and analyzed in a Zeiss electron microscope (EM) 10 and Jeol 100× EM (Dykstra and Reuss 2003). The study also included analysis of granule neurons from each sample of all groups.
GPx and GR Activities
Twenty-five mice were anesthetized with sodium pentobarbital and perfused via aorta with saline solution (NaCl 0.9%). The olfactory bulbs were quickly removed and rinsed in cold 0.9% NaCl, weighed and homogenized (10% w/v) with a sonic dismembrator (Model 100, Fisher Scientific International Inc. Hampton, NH) for 3 sec in cold 50-mM potassium phosphate, 0.1% Triton X-100, pH 7.0 (Pedraza-Chaverri et al. 2001). The homogenate was centrifuged at 19,000×g and 4°C for 30 min and then the supernatant was separated to measure total protein content and the activities of GR and GPx.
GPx
GPx activity was assayed by a method previously described (Pedraza-Chaverri et al. 1995). The reaction mixture consisted of 50-mM potassium phosphate pH 7.0, 1-mM EDTA, 1-mM sodium azide, 0.2-mM nicotinamide adenine dinucleotide phosphate-oxidase (NADPH), 1 U/ml of GR, and 1-mM reduced glutathione (GSH). Appropriate dilution (19 Vl) of tissue homogenates was added to 0.15 ml of the mixture and allowed to incubate for 5 min at room temperature before initiation of the reaction by adding 19 μl of 0.18-mM H2O2 solution. Absorbance at 340 nM was recorded for 3 min, and the activity was calculated from the slope of these lines as micromoles of NADPH oxidized per minute taking into account that the mM absorption coefficient for NADPH is 6.22. Blank reactions with homogenates replaced by distilled water were subtracted from each assay. The results were expressed as U/mg protein.
GR
Tissue GR activity was assayed using oxidized glutathione (GSSG) as substrate, and the disappearance of NADPH was measured at 340 nM (Barrera et al. 2003). The reaction mixture contained 0.1-M potassium phosphate pH 7.6, 0.5-mM EDTA, 1.25-mM GSSG, and 0.1-mM NADPH. A reaction mixture (200 μl) was mixed with 10 μl of homogenate at appropriate dilution and the optical density at 340 nM was read for 3 min. One unit of GR was defined as the amount of enzyme that oxidizes 1 μmol of NADPH/min. Data were expressed as U/mg protein.
Statistical Analysis
Results are expressed as mean ± standard error of mean (SEM). One-way ANOVA with Dunnett test was performed to identify differences through exposure times. p < .05 was considered significant.
Results
Olfactory Function
Differences in olfactory function between the control and exposed groups were statistically significant. After a 4-week inhalation period of V2O5, the exposed group increased odorant localization significantly, compared with controls. Interestingly, the control group diminished the time of locating the chocolate chip throughout the 4-week period, compared with its initial record and their predose values.
Histological Analysis
The analysis showed a decrease in the size of the granule cells soma in vanadium-exposed groups when compared with the control group (Figure 2). The major and minor axes were significantly reduced by the exposure to vanadium during the experiment.
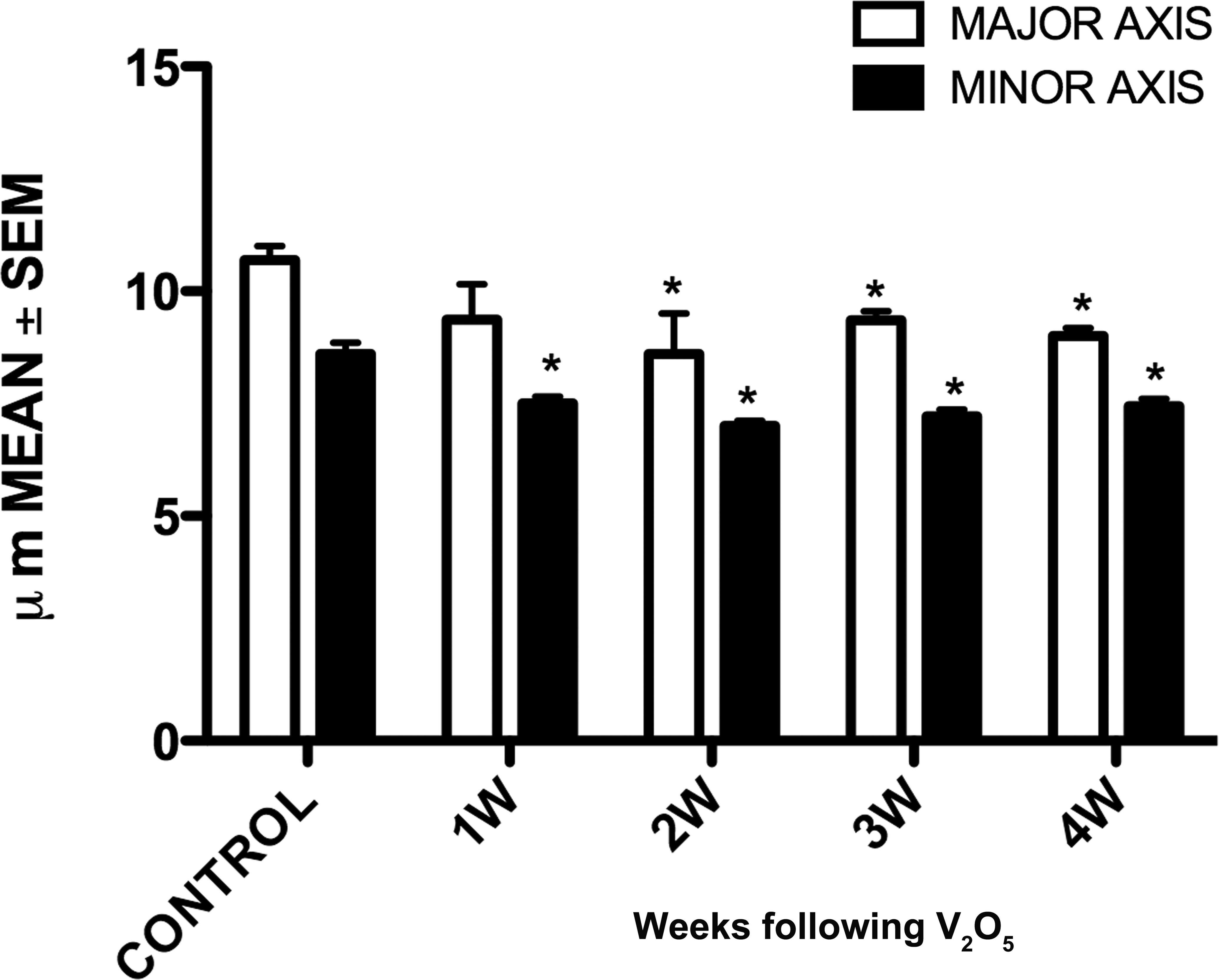
Graph shows that vanadium exposure induced decrease in major and minor axis in granule cells since 1 week in minor axis and 2 weeks in major axis until the 4-week exposure. *p < .05 versus control, one-way analysis of variance.
Dendritic spine loss in the granule cells of olfactory bulb in exposed mice was observed in all exposure times compared with controls (Figure 3). By the 4th week of exposure, dendritic spine density had decreased dramatically (5.09 ± 0.12; Figure 4).
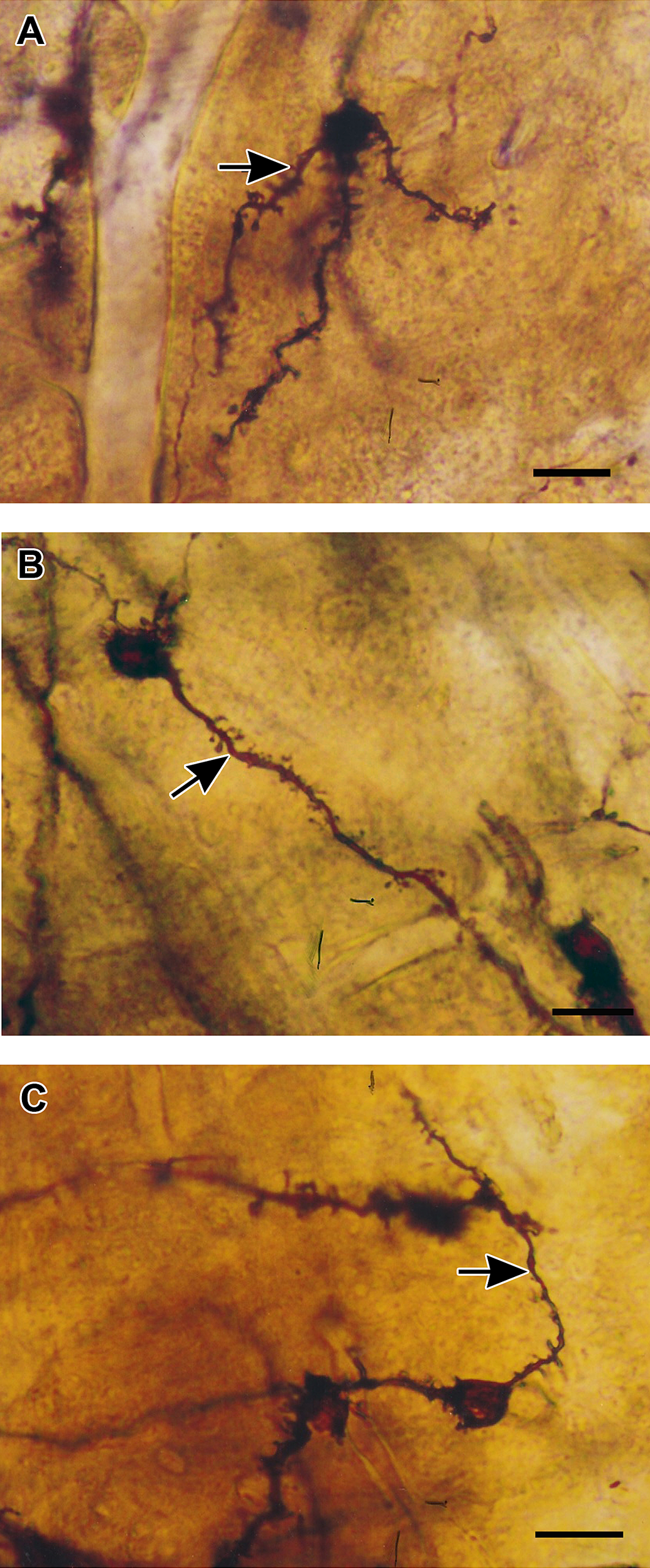
Golgi silver impregnation of the olfactory bulb granule cells. (A) Note the presence of spines on the soma and dendrites (arrow) in the granule cell from a control mouse. Figures (B) and (C) are examples of 2 and 4 weeks of vanadium exposure, respectively. Note the prominent loss of dendritic spines in the secondary dendrites (arrows).
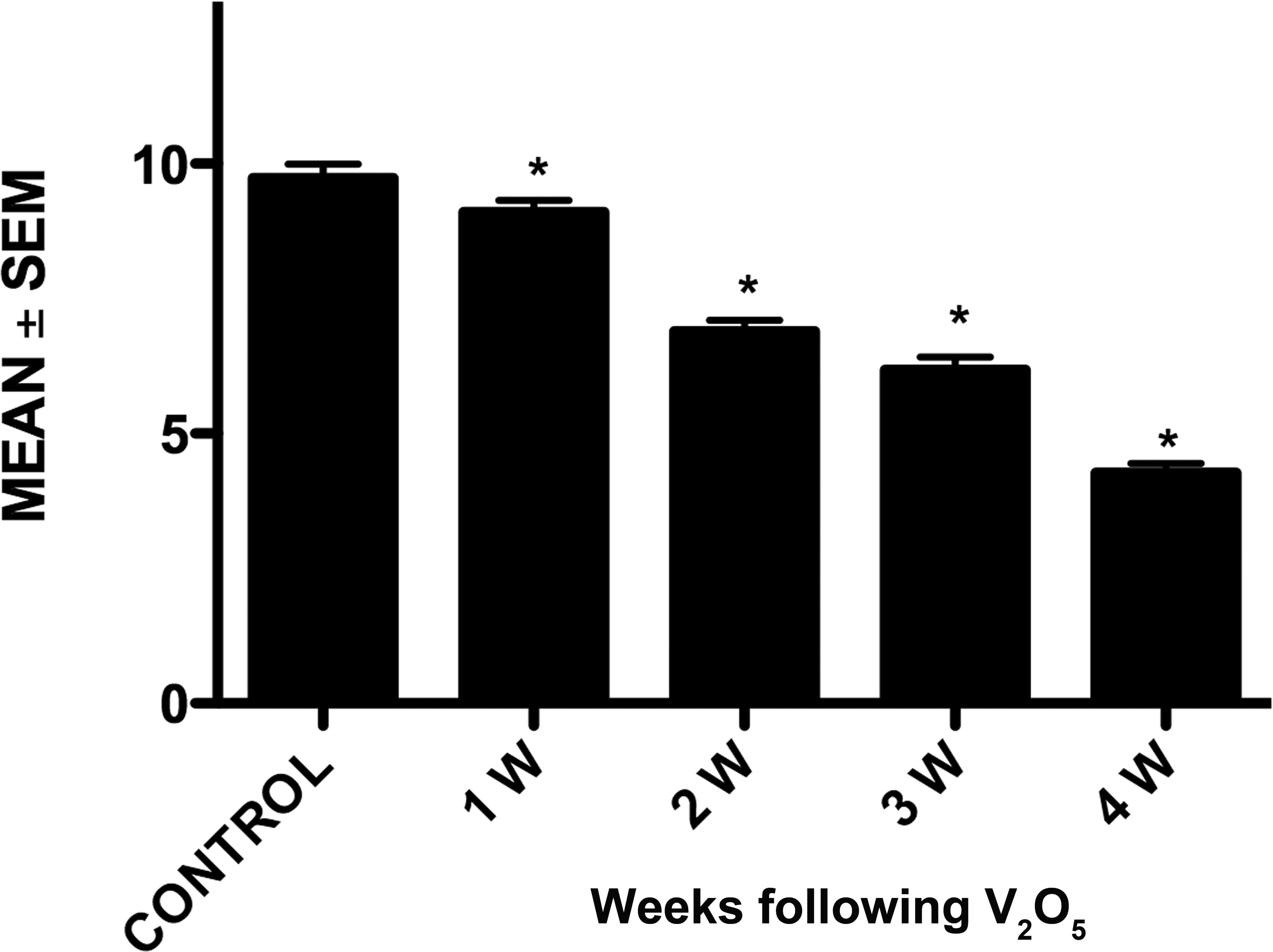
Quantitation of dendritic spine density in control and vanadium exposed mice. The bars show a decrease in dendritic spine density with increasing vanadium exposure. Mean values ± standard error of mean (n = 5). *p < 0.05 (Dunnett’s test: comparison with control).
Ultrastructural Analysis
Granule cells from the olfactory bulb in controls had normal organelles (Figure 5). However, ultrastructural alterations were observed at 1, 2, 3, and 4 weeks after vanadium exposure (Figure 6). The changes observed were apoptotic cells with condensation and margination of chromatin (Figure 7), dark cells with shrunken soma, pyknotic nucleus, and irregularly clumped chromatin. This last modification is compatible with a variant of necrotic neuronal death (Figure 8). Lipofuscin granules increased (Figure 9); swelling of the organelles and vacuolation and condensation of the cytoplasm were observed. Disrupted mitochondria with dissolution of their crista and a dilated Golgi apparatus were also seen (Figure 9).
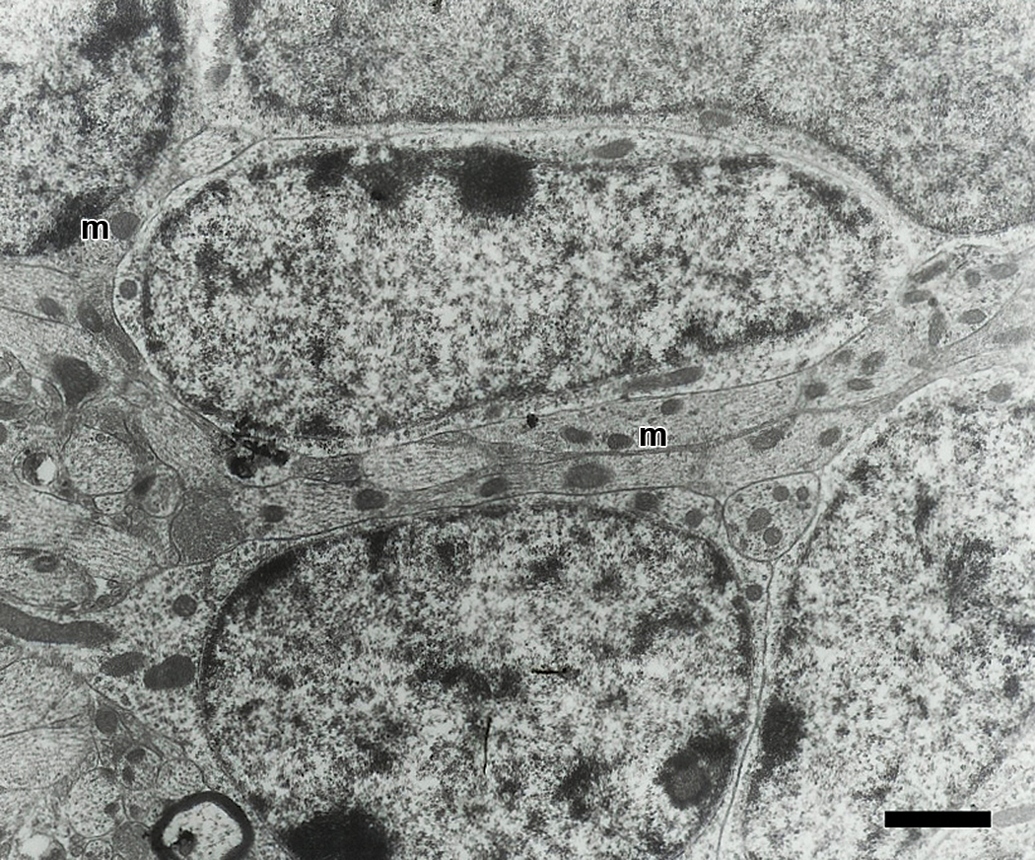
Electron micrograph of granule cells from olfactory bulb of a control mouse. The cells have a prominent nucleus and scarce cytoplasm with mitochondria (m). Scale bar = 1.5 μm.
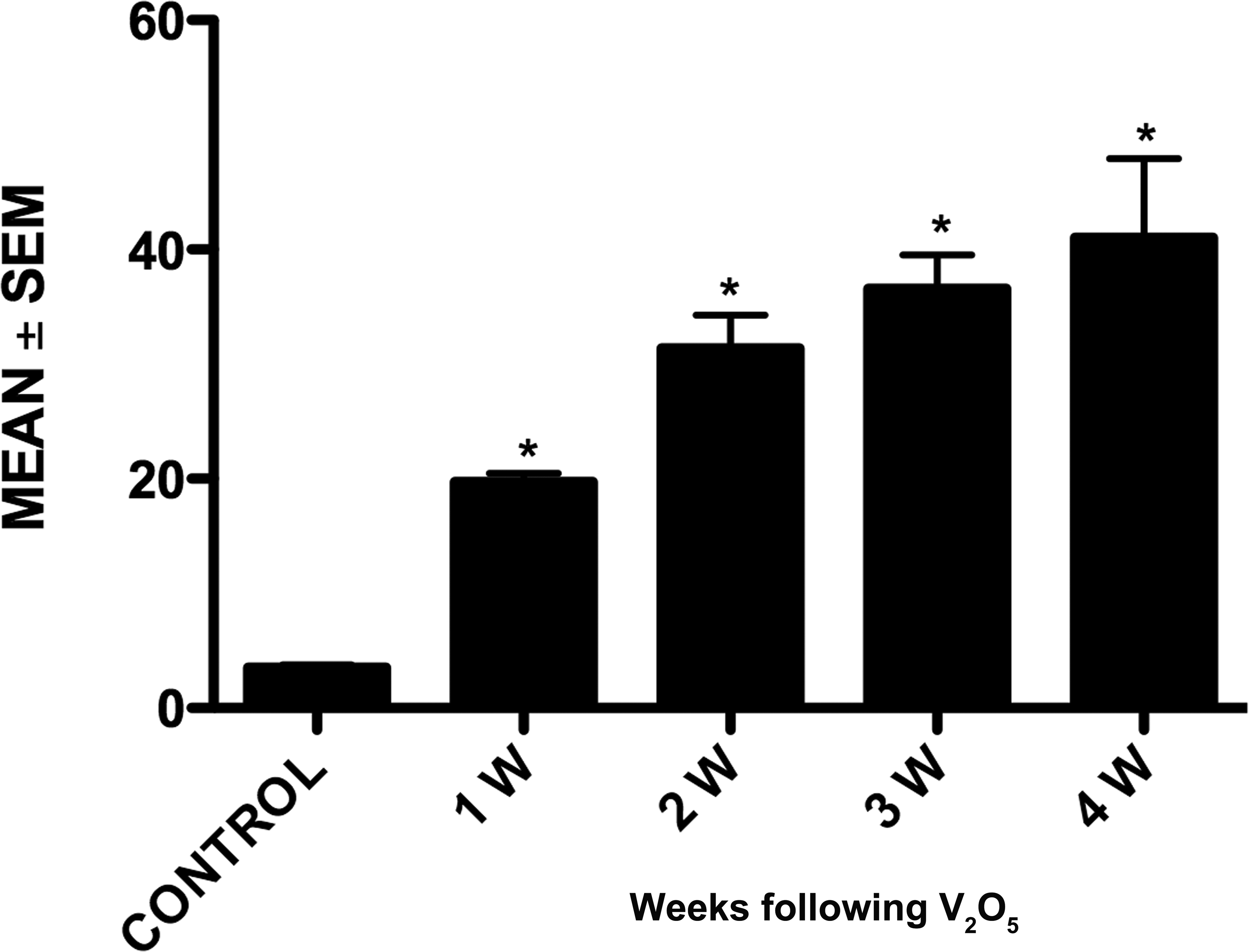
Graph shows ultrastructural alterations in the olfactory bulb from vanadium exposure groups, including necrosis, apoptosis, and mitochondrial edema. *p < 0.05 (Dunnett’s test: comparison with control).
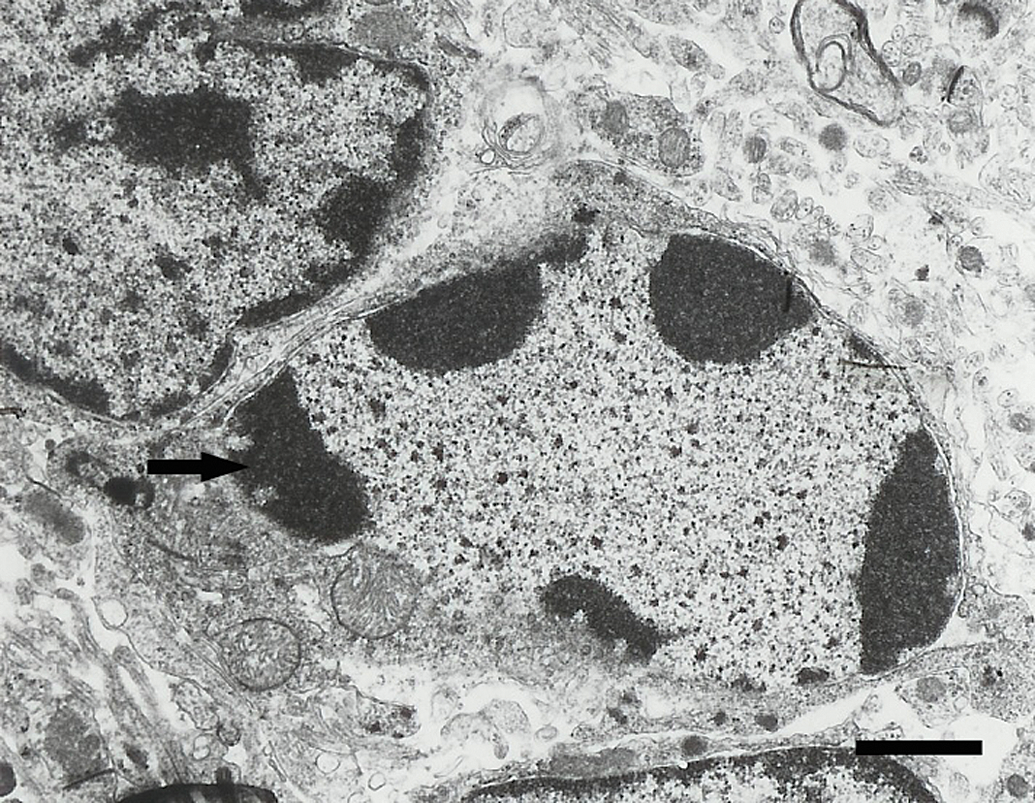
Electron micrograph of an apoptotic granule cell from a mouse in the 3 week exposure group. Note the condensed and marginated chromatin (arrow). Scale bar = 1.5 μm.
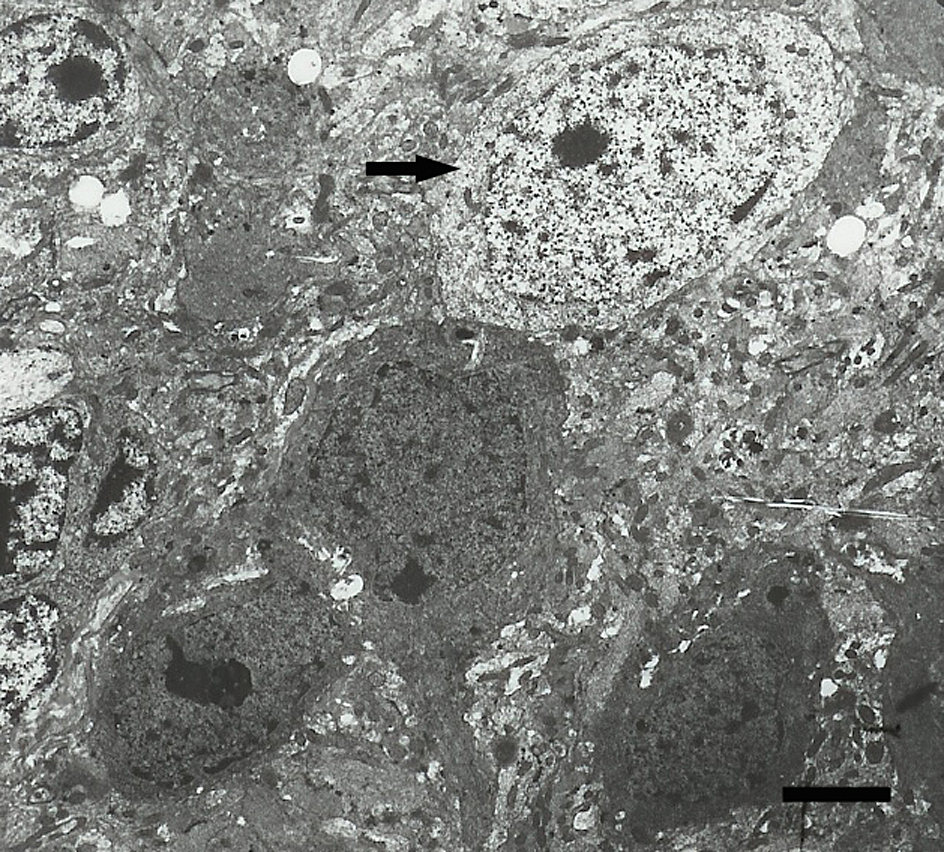
Three necrotic granule cells next to a normal one (arrow) in a 4-week vanadium-exposed mouse. Scale bar = 4 μm.
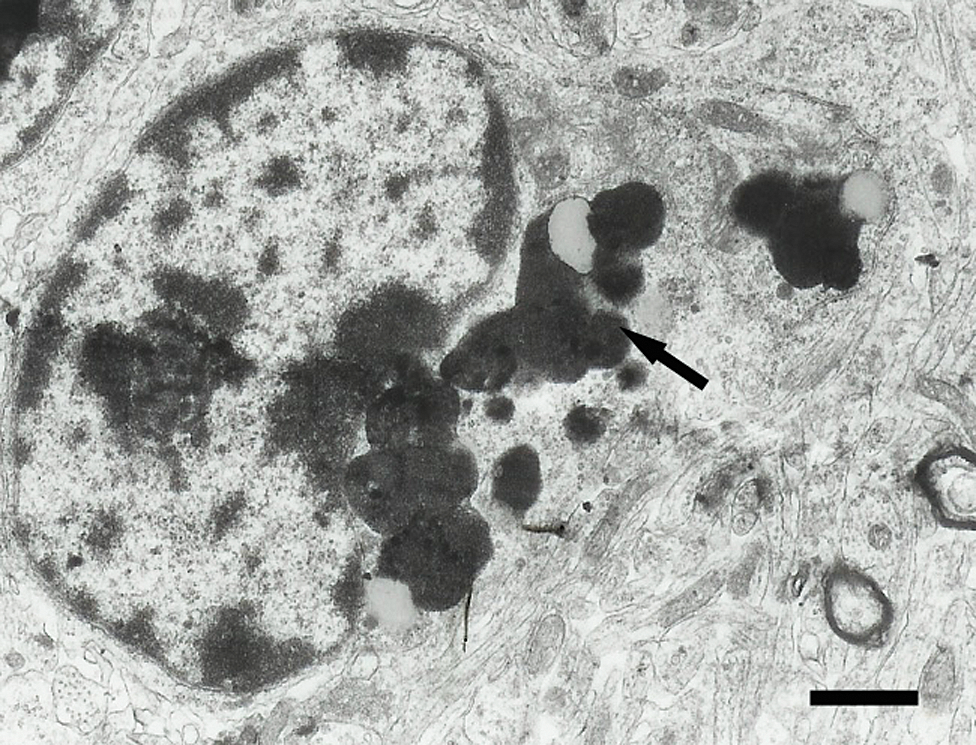
Olfactory bulb granule cell from a 4-week vanadium exposed mouse that contains many electron dense lipofuscin granules in the cytoplasm (arrow). Scale bar = 1.5 μm.
Activity of Antioxidant Enzymes
The enzyme GPx in olfactory bulb showed a significant increase in its activity during the 2-week exposure to vanadium compared with controls. GR enzyme showed high activity at 2 and 4 weeks after exposure when compared with controls (p = .001; Figure 10A and B).
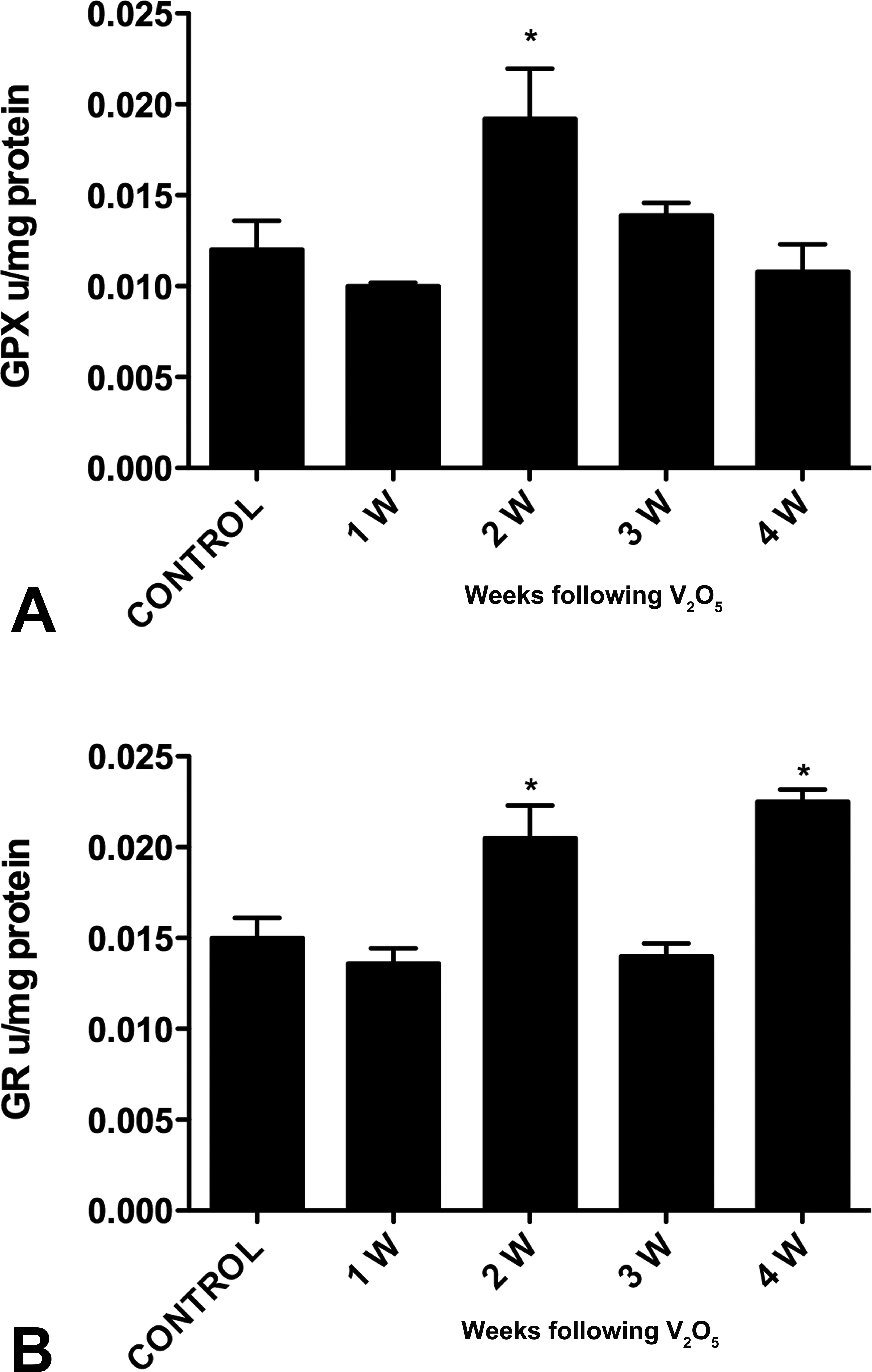
Effect of vanadium on olfactory bulb GPx (A) and GR (B) activities. Data represent average ± standard error of mean of U/mg proteins. Abbreviations: GPx, glutathione; GR, glutathione reductase. *p < 0.05 (Dunnett’s test: comparison with control).
Discussion
Vanadium inhalation resulted in olfactory impairment and alterations in histological, ultrastructural, and enzymatic activities. The exposure caused an olfaction deficit at 2- and 4-week exposure that might be explained by the loss of dendritic spine and the death of granule cells observed at 4-week exposure. These effects implicate that changes in tufted-granule cell circuit might mediate low threshold perception of odorants (Nagayama et al. 2004). In addition, direct damage to the olfactory epithelium might be modifying the functional results because in our mice model, we have identified necrosis and picnosis of this epithelium (data not shown).
Exposure to vanadium causes neurobehavioral alterations and pathophysiological manifestations in humans (World Health Organization [WHO] 2001; Li et al. 2013). The direct input of metals is through nasal–brain connection pathway, and vanadium is present in a gradient from olfactory mucosa–olfactory bulb and frontal cortex as described in a study by Calderón-Garcidueñas et al. (2003) in Mexico City residents. In this study, we show that vanadium causes cytological alterations in the size of olfactory bulb granule cells soma in all groups after the exposure that might be associated with modifications in homeostasis (Aureliano 2009) that could result in cell retraction. In fact, vanadium acts as a phosphate analog inhibiting protein tyrosine phosphatases and activating kinases inducing phosphorylation that precede at cell retraction and also acts on Rho GTPases that change cell morphology through their regulation of the cellular cytoskeleton (Mukherjee et al. 2004; Zeng et al. 2000; Govek, Newey, and Aelst 2005).
The remarkably reduced number of dendritic spines in granule cells in vanadium-exposed mice is similar to our previous findings in the medium spiny striatal neurons (Avila-Costa et al. 2003). However, in the olfactory bulb, the loss was earlier than in the striatum. Dendritic spines are known to be temporal structures due to their constant transformation (Segal 2005; Yuste and Bonhoeffer 2001). Changes in the number of spines occur under pathological conditions such as in excitotoxicity, during oxidative stress, and in response to neural activity under physiological circumstances (Smart and Halpain 2000). Pathologies of spine distribution affect many of these structures in the neuronal surface including the quantity of spines (Fiala, Spacek, and Harris 2002).
It has been demonstrated that the spine loss caused by vanadate is related to ROS generation, among which includes hydroxyl radicals (Cortizo et al. 2000) that may occur as a consequence of Ca2+ (calcium) influx (Segal, Korkotian, and Murphy 2000; Mattson 2003). These results agree with the changes described in experimental models and neurodegenerative diseases that have been associated with oxidative stress (Colín-Barenque et al. 1999; Avila-Costa et al. 2001; Smith et al. 2000; Practico and Delanty 2000; Yu and Lu 2012).
In addition, the structural spiny modifications are modulated by changes in the extracellular microenvironment (Howell and Gottschall 2012; Kasai et al. 2010), such as activation of metalloproteinases (MMPs) observed in some CNS pathologies (Yong et al. 2001), which induce spiny remodeling. In a previous study, we reported an increased MMP-9 activity in the mice olfactory bulb after a 2- and 4-week exposure period to vanadium. The MMP-increased activity was associated with dendritic spine loss as well as ultrastructural alterations involved in synaptic remodeling (Colín-Barenque et al. 2008; Michaluk et al. 2011).
Another possible explanation for our findings is the direct effect of vanadium on the spine’s cytoskeleton that is mainly constituted by actin (Svitkina et al. 2010), and because in our experimental model, we have demonstrated in mice testes the effect of vanadium on this microfilament (Rodriguez-Lara et al. 2013). Some reports indicated that decavanadate and vandyl inhibit G-actin polymerization leaving actin filaments (F-actin) and producing different perturbations of F-actin structure (Ramos, Moura, and Aureliano 2012; Yang et al. 2004). Actin filaments are thought to be responsible for the spine changes (Hotulainen and Hoogenraad 2010). The spine loss might represent a retraction of the spine neck, due to an effect on spine cytoskeleton that alters the coupling between spine and dendrite (Majewska, Tashiro, and Yuste 2000).
Ultrastructural alterations induced by vanadium exposure might be explained by oxidative damage that was confirmed by 4-hydroxynonenal presences in cortex pyramidal neurons in our model (Fortoul et al. 2011). Oxidative stress causes abnormalities that precede clinical and pathological manifestations in neurodegenerative diseases; these are the earliest pathological changes along with lipid peroxidation in olfactory neurons associated with olfactory dysfunction in AD and PD (Ghanbari et al. 2004; Perry et al. 2003; Babizhavev, Devev, and Yegorov 2011). Our results are supported by a report in the olfactory bulb of elderly humans that oxidative stress was evidenced by high levels of lipofuscin and pathological changes in olfactory bulb of children exposed to urban air pollution (Vaishnav et al. 2007; Calderón-Garcidueñas et al. 2010).
Neuronal death is an important process in neurodegenerative diseases that is associated with oxidative stress. In this study, oxidative stress was generated by vanadium exposure, and the changes we observed were neuronal death by apoptosis and necrosis. Vanadium increased apoptotic proteins such as p53. It also induced mitochondrial membrane permeability disruption of mitochondrial membrane potential, release of cytochrome c, and activation of proteases such as caspase 9 and 3. These changes resulted in evident ultrastructural mitochondrial damage (Chakraborty et al. 2005; Caicedo et al. 2008; Roos and Kaina 2006; Mattson 2003; Zhao et al. 2010).
Necrosis observed in granule cells could be attributed to hydrogen peroxide and hydroxyl radical generated by vanadium that induced transient hyperpolarization of the mitochondrial membrane potential, ROS generation from complexes I and II (Cuesta, Frances, and García 2011; Shi, Liu, and Yang 2011), and cell death (Choi et al. 2009). These ROS can react with cellular macromolecules through oxidation, inducing cells into necrosis and apoptosis (Loh et al. 2006; Higgins, Beart, and Nagley 2012).
Studies on various cell lines reveal that vanadium exerts its antitumor effects through inhibition of cellular tyrosine phosphatases and/or activation of tyrosine phosphorylases. Both effects activate signal transduction pathways leading to either apoptosis or necrosis (Evangelou 2002; García et al. 2004). These results also support such neurotoxic effects of vanadium in dopaminergic neuronal cells via caspase-3-dependent protein kinase C (PKC), demonstrating that PKC is a key element in the apoptotic process (Afeseh et al. 2009).
On the other hand, it has been reported that intracellular oxidation of vanadate to vanadyl may be mediated by GSH and NADH (Valko, Morris, and Cronin 2005; Aureliano 2009).
Previous studies have shown that 1 hr after intraperitoneal injection of sodium vanadate in mice, there is a decrease in GSH and NADPH, that is, indicative of oxidative stress (Mahmoud et al. 2011). In addition, rats treated with vanadium showed an increase in plasma GSH levels (Francik et al. 2011).
We found that vanadium inhalation increased the activity of GPx and GR in the olfactory bulb, in which granule cells are the main cell population. GR catalyzes the regeneration of GSH, from GSSG, that is used by GPx to reduce hydrogen peroxide and lipoperoxides induced by vanadium exposure.
Finally, our data suggest that vanadium inhalation increased the activity of GR and GPx that could be associated with neuronal death, decreased number of dendritic spine, and olfactory impairment likely mediated by oxidative stress.
Conclusion
The results presented in this study demonstrate that inhalation of vanadium causes significant functional and morphological alterations that might be explained to be a consequence of the oxidant/antioxidant imbalance that could increase oxidative damage that is a risk factor in neurodegenerative diseases. Further studies to analyze the functional and molecular consequences of vanadium inhalation in olfactory system should be done in order to understand this phenomenon.
Footnotes
Acknowledgments
The authors thank Armando Zepeda, Francisco Pasos, and Jesus Espinosa-Villanueva for their photographic artwork and thank Patricia Aley for the technical assistance.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: DGAPA-UNAM (IN-220211 and IN210713) supported this work.