
Abstract
The past several decades have seen great effort devoted to mimicking the key features of pancreatic ductal adenocarcinoma (PDAC) in animals and have produced 2 robust models of this deadly cancer. Carcinogen-treated Syrian hamsters develop PDAC with genetic lesions, which reproduce those of human, including activation of the Kras oncogene, and early studies in this species validated nongenetic risk factors for PDAC including pancreatitis, obesity, and diabetes. More recently, PDAC research has been invigorated by the development of genetically engineered mouse models based on tissue-specific Kras activation and deletion of tumor suppressor genes. Surprisingly, mouse PDAC appears to arise from exocrine acinar rather than ductal cells, via a process of phenotypic reprogramming that is accelerated by inflammation. Studies in both models have uncovered molecular mechanisms by which inflammation promotes and sustains PDAC and identified targets for chemoprevention to suppress PDAC in high-risk individuals. The mouse model, in particular, has also been instrumental in developing new approaches to early detection as well as treatment of advanced disease. Together, animal models enable diverse approaches to basic and preclinical research on pancreatic cancer, the results of which will accelerate progress against this currently intractable cancer.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a relatively rare cancer: at approximately 45,000 new cases per year in the United States, it currently ranks as the 11th most common cancer overall. Despite this overall low incidence, pancreatic cancer is the fourth leading cause of cancer death in both men and women, with approximately 38,000 deaths per year and an overall 5-year survival rate of only 6% (Siegel, Naishadham, and Jemal 2013). This rate increases only to 20% for the rare patients (approximately 1 in 10) who present with apparently local disease and thus qualify for surgical resection (Schneider et al. 2005). For those patients ineligible for surgery, conventional chemotherapy or radiotherapy approach appears to extend life span by only a few months. The overall cure rate of PDAC has been estimated at less than 1%, and even this number is likely inflated by misdiagnosis.
The fact that PDAC commonly recurs even after total pancreatectomy indicates that it is diagnosed only after already having seeded metastases in the liver, lung, and elsewhere. Other solid cancers are similarly deadly following metastasis; pancreatic cancer is remarkable in never being caught early enough to cure. Nonetheless, a recent study estimates that >10 years pass between the first genetic “hit” of PDAC and the formation of invasive cancer, and >6 years more before metastatic lesions are established (Yachida et al. 2010). In theory, this provides time for intervention; yet, the relative scarcity of PDAC patients—let alone those with precancerous lesions detected prior to death—hinders the study of disease initiation and progression. This motivates efforts to develop animal models of autochthonous (i.e., originating in the place where it is found) PDAC, which might also serve as preclinical models to test new therapies for advanced cancer. These are the focus of this review: what animal models exist for pancreatic cancer, how have they enhanced our understanding of PDAC biology, and how they inform our efforts to improve patient outcomes.
Evolving Approaches to an Animal Model of Pancreatic Cancer
Two roughly parallel tracks have been pursued to mimic human PDAC in animals: mutagenesis and transgenesis. Chemical carcinogen treatment can induce a variety of tumors in rodents, including skin cancer in mice. In this so-called multistage carcinogenesis model, tumors are induced by exposure to the mutagen 9,10-dimethyl-1,2-benzanthracene (DMBA), followed by treatment with chemical agents, such as 12-O-tetradecanoylphorbol-13-acetate (TPA), that promote their growth into self-sustaining carcinomas (Zoumpourlis et al. 2003). As discussed later in this review, a multistage initiation–promotion paradigm is likely to apply in the pancreas as well, with inflammatory stimuli playing the role of tumor promoter. Efforts to model PDAC via carcinogen treatment, however, have produced robust success only in the Syrian hamster, where a single injection of the mutagen N-nitrosobis(2-oxopropyl)amine (BOP) induces invasive PDAC in up to 90% of animals (Pour et al. 1977). Invasive tumors arise within 3 to 12 months and exhibit close histological similarity to their human counterparts. This model has been used to validate and dissect numerous conditions thought to modulate human cancer risk (Takahashi et al. 2011).
Importantly, hamster PDAC accumulates many of the same genetic alterations as human. The defining mutational event of pancreatic cancer is activation of the KRAS proto-oncogene, which occurs in >90% of all human PDAC (Almoguera et al. 1988; Maitra and Hruban 2008). KRAS encodes a member of the RAS family of small GTPase signaling proteins, and mutational activation (most commonly affecting amino acid Gly12) disables GTPase activity and traps RAS in a GTP-bound “on” state that normally requires mitogen stimulation (Pasca di Magliano and Logsdon 2013). As depicted in Figure 1, KRAS mutations are detected in the earliest precancerous lesions of the human pancreas, pancreatic intraepithelial neoplasia (PanIN)-1, while more advanced lesions accumulate additional mutations such as loss of the tumor suppressors CDKN2A (p16), TP53 (p53), SMAD4, and BRCA2 (Maitra and Hruban 2008). The hamster CDKN2A homolog is also mutated in BOP-induced pancreatic cancer (Takahashi et al. 2011), further supporting the suitability of hamster as a model for human PDAC.
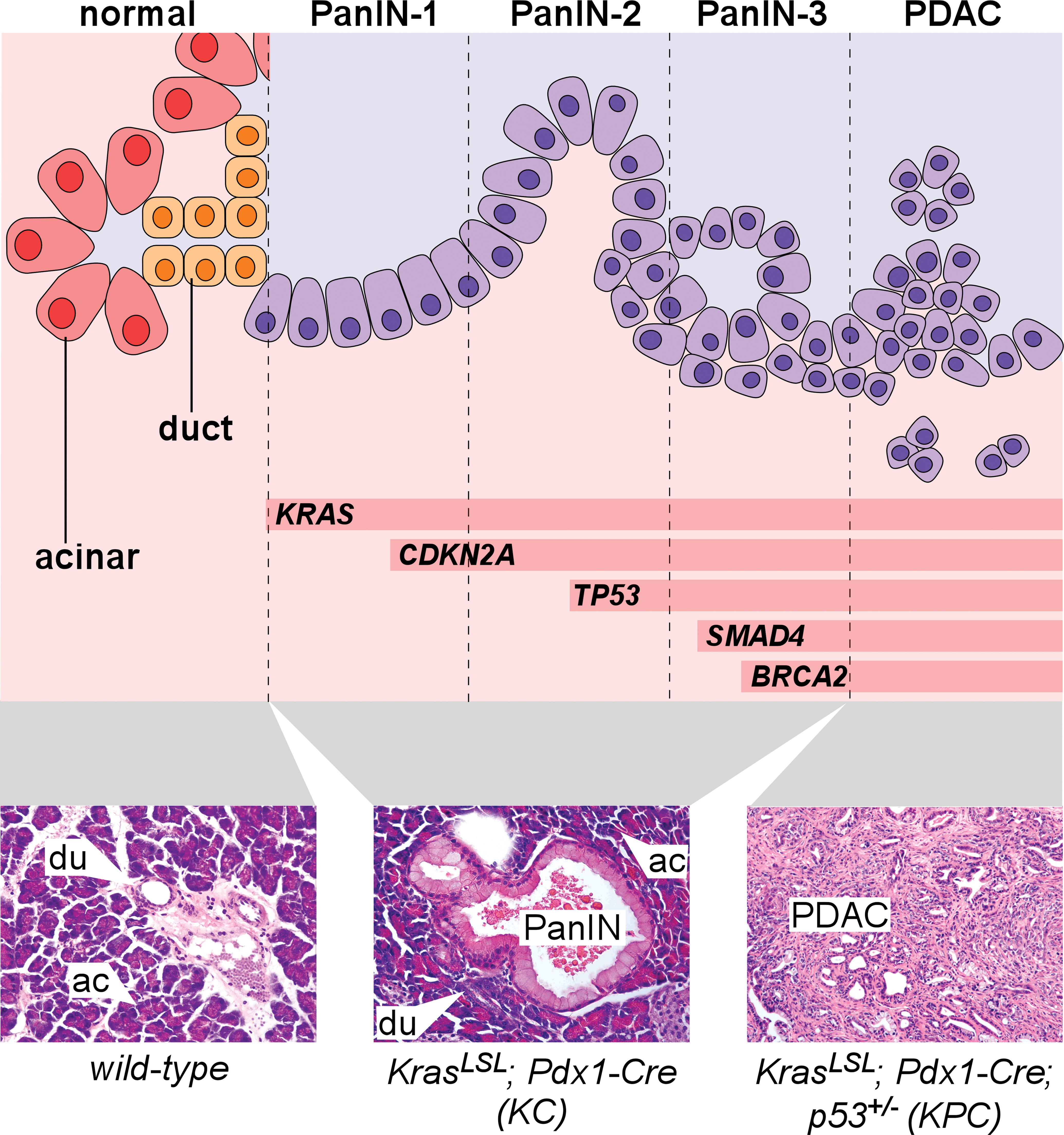
Pancreatic cancer progression in human and mouse. Top: schematic representation of normal exocrine pancreas cells, transitioning from left to right into increasingly dysplastic PanIN and PDAC. In addition to visible abnormalities including papillary morphology, loss of polarity, nuclear atypia, intraluminal budding, and stromal invasion (see Table 1 for additional details), PanIN-PDAC progression is associated with increasing accumulation of genetic lesions including activation of KRAS and loss of tumor suppressor genes including CDKN2A, TP53, SMAD4, and BRCA2. Bottom: H&E-stained sections of mouse pancreata of indicated genotypes, illustrating normal acinar (ac) and duct (du) cells as well as an early-stage PanIN lesion and advanced PDAC. PanIN = pancreatic intraepithelial neoplasia; PDAC = pancreatic ductal adenocarcinoma; H&E = hematoxylin and eosin.
The major drawback of a mutagenesis-based model is that the investigator cannot control what genes are actually mutated in a given tumor: in addition to KRAS and CDK2NA, BOP-induced cancers likely harbor additional mutations of unknown relevance to human tumors. Hamsters also lack the extensive genome manipulation toolkit of mice, which has facilitated the development of genetically engineered mouse (GEM) models of cancer (van Miltenburg and Jonkers 2012). Even studies of carcinogen-induced tumors in mice, such as the skin cancer model, have benefited from genetic manipulation to address issues such as tumor suppressor gene function (Zoumpourlis et al. 2003).
A barrier to studying PDAC in mouse, however, is that it is difficult or impossible to induce with BOP or other mutagens (Takahashi et al. 2011). Instead, mice offer the approach of directly expressing mutant KRAS in susceptible cells of the pancreas. A “first generation” of GEM models employed transgenic mice driving activated human KRAS expression with promoters of the duct-specific Cytokeratin-19 (Krt19) or acinar-specific Elastase1 (Ela1) genes (Brembeck et al. 2003; Grippo et al. 2003). Acinar cells normally produce digestive enzymes that are channeled to the gut via a network of ducts; together, acini and ducts comprise the exocrine pancreas, with anatomically scattered islets of Langerhans making up the endocrine pancreas. The resemblance of PDAC to ducts, in morphology and marker expression, has generally been taken to suggest an origin among normal duct cells. Surprisingly, however, Krt19-KRASG12V mice develop only mild inflammation without cancer or dysplasia (Brembeck et al. 2003), while Ela1-KRASG12D mice exhibit premalignant lesions of mixed ductal–acinar character (Grippo et al. 2003). Similar lesions were described more than 20 years ago, in mice overexpressing the oncogenic transcription factor Myc via the same Ela1 promoter (Sandgren et al. 1991). Although mixed acinar–ductal lesions do not have a clear human counterpart (Hruban et al. 2006), the Ela1-Myc and Ela1-KRASG12D studies were among the first to indicate that duct-like tumor precursors might arise from acinar cells.
Two shortcomings affect conventional transgenic approaches: they require a promoter element that is active in a “competent” cell of origin for the cancer, and they inevitably drive oncogene expression at nonphysiological levels. This is a particular concern for mutant RAS, which actually induces senescence when overexpressed (Serrano et al. 1997). The creation of conditional mutant alleles of the endogenous mouse Kras gene, 10 years ago, therefore constituted a major advance in GEM cancer models (Figure 2A; Guerra et al. 2003; Tuveson et al. 2004). (Here and elsewhere, we use all capital letters for human gene symbols, while capitalizing only the first letter of mouse genes.) These were immediately used to establish a “second generation” of PDAC models that have since become the major animal model of pancreatic cancer (Aguirre et al. 2003; Hingorani et al. 2003; Perez-Mancera et al. 2012).
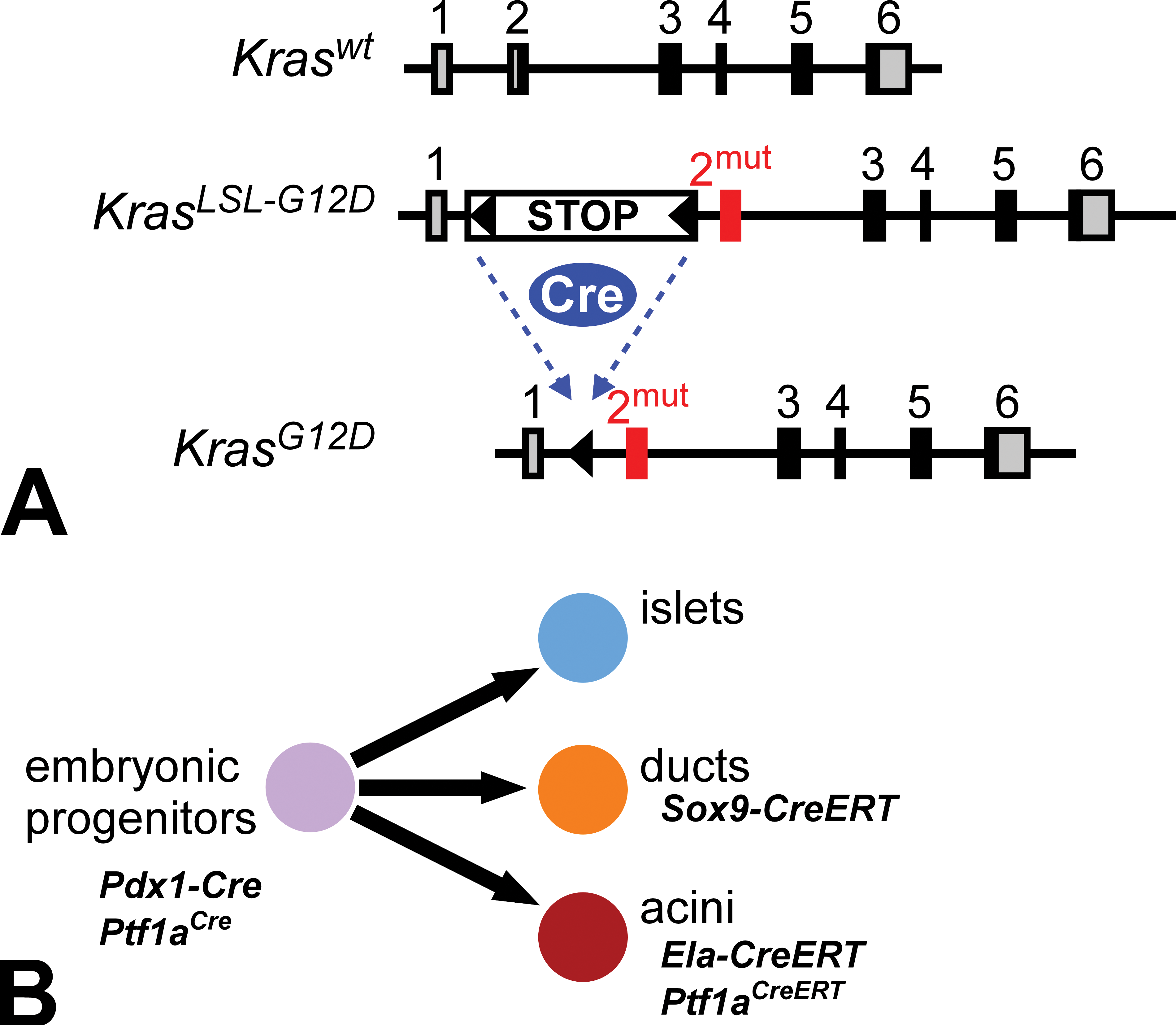
Inducible Kras mouse model of pancreatic cancer. A, Exon–intron structure of mouse Kras locus, comparing the wild-type allele to the LSL-G12D targeted allele, before and after Cre-mediated recombination. Gray boxes indicate 5′ and 3′ untranslated regions (UTRs); black boxes indicate coding regions. The oncogenic G12D mutation is targeted to exon 2, preceded by a floxed STOP cassette that prevents expression prior to Cre recombination. B, Tissue-specific Cre lines used for PDAC modeling. Pdx1-Cre and Ptf1aCre induce widespread recombination in multipotent progenitors of the embryonic pancreas, which give rise to all 3 mature pancreatic lineages. Adult ductal recombination is achieved with Sox9-CreERT, while acinar cells are labeled with Ela-CreERT and Ptf1aCreERT . PDAC = pancreatic ductal adenocarcinoma; LSL = loxP-STOP-loxP.
Two conditional mutant Kras alleles exist, with similar overall design: a transcriptional “STOP” sequence, consisting of multiple strong polyadenylation signals, is placed upstream of Kras exon 2, in which the Gly12 codon has been mutated to encode either aspartic acid (G12D) or valine (G12V), the two most prevalent mutations in human PDAC (Guerra et al. 2003; Tuveson et al. 2004). The STOP sequence prevents expression of mutant Kras, but it is flanked by loxP sites (floxed) so that it can be recognized and excised by Cre recombinase protein. Without Cre, mutant Kras is silent and the resulting mice, with one wild-type and one silent mutant allele, are phenotypically normal. When Cre is active in a specific mature or progenitor cell (i.e., by breeding conditional Kras to a Cre-expressing mouse), the STOP cassette is removed and that cell and its offspring will permanently express oncogenic Kras. Functionally, this should be identical to a spontaneous G12D or G12V point mutation, except that its prevalence potentially encompasses an entire organ rather than a single cell. Mutant Kras is expressed from its endogenous locus and does not induce senescence associated with RAS overactivity (Guerra et al. 2003; Serrano et al. 1997; Tuveson et al. 2004). The conditional G12D and G12V alleles behave generally similarly, although G12D appears slightly more transforming in the pancreas. We will refer to both alleles as KrasLSL , short for “loxP-STOP-loxP.”
How is mutant Kras targeted to the pancreas? Borrowing tools from pancreas developmental biology, the vast majority of published PDAC modeling experiments have used “deletor” strains in which Cre is expressed in multipotent progenitor cells of the early embryonic pancreas, under control of the Pdx1 or Ptf1a promoters (Figure 2B; Murtaugh and Melton 2003). In the resulting bigenic mice (e.g. KrasLSL-G12D/+ ; Pdx1-Cre), almost all pancreatic cells express oncogenic Kras from embryonic stages. (These mice are frequently referred to in the literature as KC, for “Kras/Cre”; when mutant p53/Tp53 is also present, they are called KPC.) At birth, mutant Kras-expressing pancreata are relatively normal, but within 1 to 2 months they begin to develop lesions resembling human PanIN-1 (Figure 1; Aguirre et al. 2003; Hingorani et al. 2003). PanINs become more numerous and dysplastic with age, resembling human PanIN-2 and PanIN-3 (Hruban et al. 2006). (Table 1 summarizes the key histological features of both human and mouse PanINs, as defined by pathologist working groups; Hruban et al. 2006; Hruban et al. 2001). At >1 year of age, a minority of these otherwise “wild-type” mice develop invasive PDAC, presumably due to additional somatic mutations. Mouse PDAC is dramatically accelerated when oncogenic Kras is compounded with heterozygous or homozygous mutations in tumor suppressor genes such as p16/Cdkn2a, Tp53, Smad4, and Brca2, in some cases resulting in lethal, metastatic disease within 3 months of birth (Aguirre et al. 2003; Bardeesy, Aguirre, et al. 2006; Bardeesy, Cheng, et al. 2006; Hingorani et al. 2005; Skoulidis et al. 2010). KrasLSL mice thus directly validate the relevance of human PDAC mutations and have become the major preclinical model in the field.
Classification of human and mouse PanIN progression.

Note: PanIN = pancreatic intraepithelial neoplasia. Key morphological characteristics of preinvasive pancreatic ductal lesions in the human and mouse, as defined by pathologist working groups of the Pancreatic Cancer Think Tank and Pancreatic Cancer in Mice and Man conferences, respectively (Hruban et al. 2006; Hruban et al. 2001). Progression to pancreatic ductal adenocarcinoma (PDAC) is defined by invasion of tumor epithelial cells through the basement membrane. Additional diagnostic information is available in the original publications and online at the Sol Goldman Pancreatic Cancer Research Center of Johns Hopkins Medicine: http://pathology.jhu.edu/pc/professionals/DuctLesions.php.
In the KrasLSL models, activation by Cre leaves cells in an irreversibly KRAS-active state from embryogenesis onward. Given that human PDAC affects older adults, activation in utero is probably nonphysiological. Furthermore, while the irreversibility of KrasLSL activation mimics the situation of a human cancer cell, it precludes investigation of “oncogene addiction,” that is, the hypothesis that maintenance of a cancer requires the same activated oncogenes that triggered its initiation. Although RAS molecules are considered challenging drug targets (Pasca di Magliano and Logsdon 2013), validating KRAS addiction in PDAC would provide a clinical rationale for pursuing this goal.
Two groups have recently developed “third-generation” GEM models to address these issues (Collins et al. 2012; Ying et al. 2012). These models combine 3 genetically engineered components: a pancreatic Cre deletor (Ptf1aCre ) activates expression throughout the organ of the synthetic transcription factor rtTA, which itself binds to and activates a TetO-KrasG12D transgene in a doxycycline-dependent manner. The end result is that KrasG12D can be turned on and off almost at will in these mice, which we refer to as iKras (inducible Kras). Confirming that activated KRAS can induce PDAC in adulthood, iKras mice develop PanINs when doxycycline administration is delayed until 4 to 6 weeks of age (Collins et al. 2012). More importantly, if doxycycline is subsequently withdrawn to downregulate KrasG12D , these PanINs disappear: early-stage PanINs appear to redifferentiate into normal exocrine pancreas tissue, while more advanced PanINs undergo apoptosis. And most important, when PDAC is induced by Tp53 deletion in iKras mice, doxycycline withdrawal causes regression of both local and metastatic cancer (Collins et al. 2012; Ying et al. 2012). The dramatic oncogene addiction demonstrated in these studies provides a gold standard for preclinical studies aimed at blocking oncogenic KRAS.
Where and Why Does PDAC Initiate: Insights from Animal Models
Almost 200 articles have been published using the KrasLSL models in the past 10 years, and at least as many using the Syrian hamster model since its development in the mid-1970s. A comprehensive review of these models is therefore impossible, and we will instead focus on 3 areas in which they have proved particularly useful: dissecting the initial steps of tumorigenesis, providing insights into risk factors and prevention, and serving as a preclinical test platform for detection and treatment modalities.
The first of these issues, how PDAC initiates, may seem trivial, given the abundant evidence that KRAS mutations induce pancreatic tumorigenesis. Yet, a cursory examination of the KrasLSL mouse model reveals that mutant KRAS is not sufficient for tumor initiation: almost all cells of the pancreas express activated Kras, but PanINs arise in isolation within an abundance of apparently normal tissue (Figure 1; Hingorani et al. 2003). Two questions arise from this observation: what cells within the pancreas actually give rise to PanINs and what determines the ability of KRAS to transform these cells?
The question of the cell of origin for PDAC has been controversial at least as far back as the development of the Syrian hamster model, with opposing histological evidence marshaled in support of an origin among ductal or acinar cells (Pour 1984). An analogous controversy, of even longer standing, concerns the question of where new insulin-producing β-cells come from the resting or injured pancreas (Granger and Kushner 2009). Only recently has any definitive answer been approached, using the technique of Cre-mediated lineage tracing (Kretzschmar and Watt 2012). This technique relies on Cre-activated reporter genes, such as green fluorescent protein (GFP) or β-galactosidase, the activation of which will mark the descendants of a Cre-expressing progenitor cell. In the pancreas, this approach revealed that all pancreatic lineages arise from cells expressing the transcription factors Pdx1 and Ptf1a (Murtaugh and Melton 2003); these same Cre lines are now used in the KrasLSL PDAC model. Cre recombinase can be fused to a modified form of the estrogen receptor (ER)–ligand binding domain, responsive to the drug tamoxifen; such an “inducible Cre,” referred to as CreERT, will activate recombination only after tamoxifen administration. CreERT lines have been used to follow the fates of islet, acinar, and duct cells in the adult mouse and to show that new β-cells arise from old β-cells, not via transdifferentiation of ducts or acini (Desai et al. 2007; Dor et al. 2004; Kopinke and Murtaugh 2010; Kopp et al. 2011; Solar et al. 2009).
These same lineage-specific CreERT lines have recently been used to test the susceptibility of adult cells to mutant KRAS (Figure 2B). Working independently, several groups demonstrated that activating KrasLSL in adult acinar cells produces PanIN lesions indistinguishable from those obtained in the conventional Pdx1-Cre/Ptf1aCre -based models (De La O et al. 2008; Guerra et al. 2007; Habbe et al. 2008). Acinar-derived PanINs formed more quickly and more abundantly when the mice were subjected to acute or chronic pancreatitis via treatment with the cholecystokinin analog caerulein (De La O and Murtaugh 2009; Guerra et al. 2007; Morris et al. 2010) and progressed to invasive PDAC in the context of either chronic pancreatitis or Tp53 deletion (Guerra et al. 2007; Ji et al. 2009). Remarkably, targeting KrasLSL to adult duct cells via Sox9-CreERT produced almost no PanINs, even after pancreatitis (Kopp et al. 2012). By carefully comparing the efficiency of Cre deletion between acini and ducts, it was concluded that the latter cell type was >100-fold less susceptible to KRAS transformation than the former. These results led to the provocative conclusion that pancreatic ductal cancers originate from differentiated, nonductal cells, and they highlight the unique power of mice for understanding basic tumor biology.
Three caveats apply to these studies. First, they do not exclude the existence of KRAS-sensitive duct cells that are Sox9-negative and are thus inaccessible to Sox9-CreERT. Centroacinar cells (CACs), for example, a subset of duct cells that lie at the junction of ductal and acinar epithelium, have been ascribed both progenitor-like and tumor-initiating potential (Cleveland et al. 2012; Ekholm et al. 1962; Pour 1984). Although Sox9-CreERT appears to be active in most CACs (Kopp et al. 2011; Kopp et al. 2012), it is conceivable that specialized, Sox9-negative CACs were missed in these experiments. Second, while these findings indicate that adult acinar cells can give rise to PanINs and PDAC, they do not directly address the origin of lesions that arise with embryonic KrasLSL activation (i.e., using Pdx1-Cre or Ptf1aCre ). Third, these experiments were performed in mice and, like any result obtained in an animal model, may not apply to humans. Interestingly, expression profiling defines up to a third of human PDACs as “exocrine like,” in which ductal tumor cells express detectable levels of acinar enzymes (Collisson et al. 2011). It is tempting to speculate that these tumors retain an epigenetic memory of their origin.
Although similar profiling has not been performed in mice, it is safe to say that acinar-derived PanINs in the KrasLSL model do not betray obvious acinar characteristics, implying that lesion formation requires extensive phenotypic reprogramming (De La O et al. 2008; Kopp et al. 2012). Rare but apparently sustained acinar–ductal reprogramming (ADR) also occurs in vivo after chronic pancreatitis (Strobel et al. 2007) and can be induced both in vitro and in vivo by exposure to the transforming growth factor-alpha (TGF-α; Blaine et al. 2010; Means et al. 2005), which itself activates endogenous KRAS. Recent studies address mechanisms controlling KRAS-induced reprogramming: it requires Sox9 (Kopp et al. 2012) that is upregulated at an early stage of the process, and it is inhibited by Wnt/β-catenin signaling and the acinar transcription factor Mist1, both of which are downregulated during reprogramming (Morris et al. 2010; Shi et al. 2013). In these respects, it is distinct from “acinar–ductal metaplasia” (ADM), the transient upregulation of duct markers by wild-type acinar cells in response to injury: ADM does not absolutely require Sox9, nor does the resolution of ADM absolutely require β-catenin or Mist1 activity (Keefe et al. 2012; Kopp et al. 2012; Kowalik et al. 2007). It should be noted that the distinction between ADR and ADM has only recently been drawn, with both phenomena previously termed metaplasia (Kopp et al. 2012). We reserve the term reprogramming for the complete conversion of acinar cells to a duct-like phenotype, which we propose represents a rate-limiting step of PDAC initiation (Figure 3).
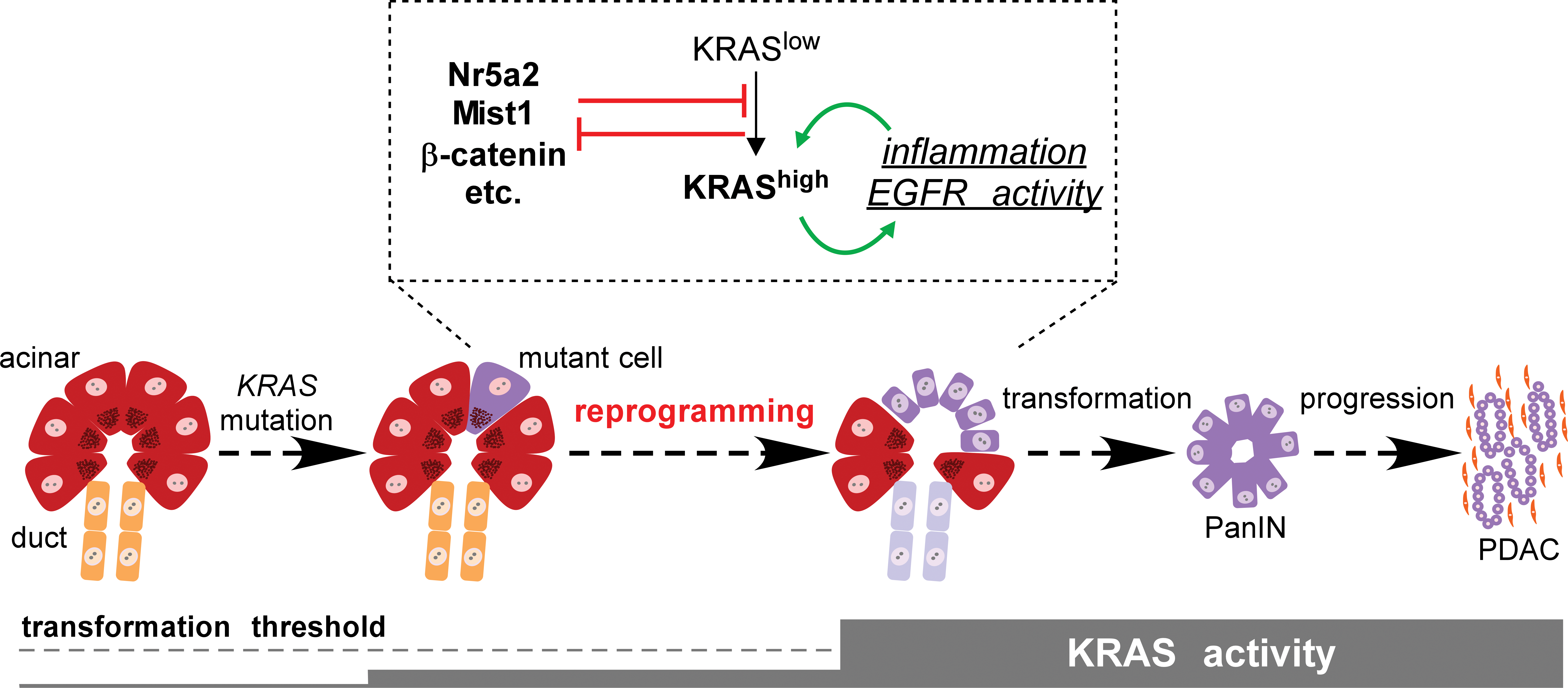
Acinar reprogramming in PDAC initiation. PDAC originates from differentiated acinar cells, in which KRAS mutation alone does not elevate total RAS activity to the levels required for transformation. Acinar–ductal reprogramming occurs when KRAS signaling increases in response to exogenous inflammatory stimuli and/or EGFR agonists; as KRAS activity increases, mutant cells produce endogenous signals that establish a positive feedback loop between KRAS and inflammation. Several genes counteract acinar reprogramming, including Nr5a2, Mist1, and β-catenin but are themselves inhibited by high levels of KRAS signaling. We hypothesize that acinar reprogramming represents a key step of PDAC initiation, which might be inhibited by anti-inflammatory agents in a chemopreventive setting. PDAC = pancreatic ductal adenocarcinoma; EGFR = epidermal growth factor receptor.
The earliest lesions of the KrasLSL model contain abnormal cells, coexpressing acinar and duct markers, that have upregulated the epidermal growth factor receptor (EGFR) but are negative for phospho-ERK (pERK), a downstream target of the EGFR/RAS/RAF/MAPK pathway (Zhu et al. 2007). These cells comingle with pERK-positive cells that express only duct markers and have PanIN-like morphology, presumably further along the reprogramming pathway. Remarkably, genetic or pharmacologic inhibition of EGFR blocks acinar reprogramming and abolishes ERK activation in cells expressing oncogenic KRAS (Ardito et al. 2012; Navas et al. 2012). Together, these results suggest that EGFR upregulation is a proximal target of mutant KRAS, but that full activation of the “RAS pathway,” including ERK phosphorylation, requires autocrine EGFR stimulation. Most acinar cells do not experience autocrine EGFR signals and therefore remain normal despite expressing mutant KRAS.
The net effect of EGFR stimulation is to amplify signaling through mutant KRAS itself, which at physiological levels has only modest effects on downstream signaling components (Guerra et al. 2007; Huang et al. 2013; Tuveson et al. 2004). Once the tumors reach the invasive PDAC stage, their RAS activity becomes elevated and independent of EGFR. Mimicking this process by KrasG12V overexpression in acinar cells is sufficient to induce widespread ADR and rapid PanIN/PDAC progression (Ardito et al. 2012; Ji et al. 2009). Thus, acinar reprogramming both unleashes and is induced by high RAS activity, creating a positive feedback loop that drives tumor initiation. Although EGFR is a major component of this loop, there is also a key requirement for inflammatory stimuli, which themselves both feed into and are upregulated by oncogenic RAS (Daniluk et al. 2012; Ji et al. 2009). As we will discuss in the next section, a variety of inflammatory conditions have been linked to human pancreatic cancer risk (Greer and Whitcomb 2009), and we hypothesize that a major impact of inflammation is to enhance ductal reprogramming of KRAS mutant acinar cells (Figure 3).
Actionable Insights into Pancreatic Cancer Risk Factors from Animal Models
Mice provide an ideal model to test genetic risk factors, of particular relevance to PDAC given that up to 10% of cases occur in familial clusters (Maitra and Hruban 2008; Petersen et al. 2006). A few major PDAC risk genes have been identified, including the tumor suppressors CDKN2A and BRCA2 that are frequently disrupted during “sporadic” PDAC progression. As described above, heterozygous mutations in these genes increase PDAC incidence in mice, validating them as familial PDAC susceptibility genes (Bardeesy, Aguirre, et al. 2006; Skoulidis et al. 2010).
These relatively high-impact risk alleles account for only a minority of familial PDAC cases, and genome-wide association studies (GWAS) have recently been used to identify additional risk alleles more widespread in the population, albeit of lower penetrance. These include common noncoding single nucleotide polymorphisms (SNPs) in the orphan nuclear hormone receptor NR5A2, which are associated with approximately 1.3-fold increased PDAC risk (Li et al. 2012; Petersen et al. 2010). Mouse NR5A2, also called liver receptor homolog-1 (LRH1), normally promotes acinar-specific gene expression in collaboration with the PTF1 transcription factor complex (Holmstrom et al. 2011). In the KrasLSL model, heterozygous loss of Nr5a2 dramatically increases PanIN formation, while cell-autonomous deletion of Nr5a2 sensitizes acinar cells to KRAS-induced reprogramming (Flandez et al. 2013; von Figura et al. 2013). While these manipulations are more severe than the presumably subtle effects of human NR5A2 SNPs, they provide the first experimental assessment of a GWAS-identified PDAC risk factor and implicate acinar reprogramming as a key step of human PDAC initiation.
In addition to sensitizing acinar cells to mutant KRAS, loss of Nr5a2 reduces their capacity for regeneration after experimentally induced pancreatitis (Flandez et al. 2013; von Figura et al. 2013). These are the latest in an abundance of studies linking pancreatitis and pancreatic inflammation to PDAC risk, including epidemiological research (Duell et al. 2012; Greer and Whitcomb 2009). In fact, one of the most penetrant PDAC risk alleles in humans is a gain-of-function mutation in the cationic trypsinogen gene, PRSS1, which causes hereditary chronic pancreatitis and increases PDAC risk >50-fold (Maitra and Hruban 2008). Studies have found that acute pancreatitis accelerates ADR and PanIN formation in KrasLSL mice, while chronic pancreatitis promotes progression to PDAC (Carriere et al. 2009; De La O and Murtaugh 2009; Guerra et al. 2007; Morris et al. 2010). Of note, the PDAC-promoting role of pancreatitis was first demonstrated in the Syrian hamster, a decade before strong epidemiological results in humans and almost 25 years before similar findings in mouse (Pour et al. 1983).
Indeed, the Syrian hamster provides independent validation of additional nongenetic risk factors for PDAC and ought not be neglected in the GEM model era (Takahashi et al. 2011). Among these epidemiologically identified risk factors are obesity and type-2 diabetes, which are themselves linked and which may promote PDAC through elevated systemic inflammation (Greer and Whitcomb 2009). Feeding KrasLSL mice a high-fat diet accelerates PanIN formation (Khasawneh et al. 2009), a result anticipated in hamster by almost 3 decades (Birt, Salmasi, and Pour 1981). Interestingly, hamsters exhibit constitutive hyperlipidemia, and treatment with the PPARγ ligand pioglitazone both lowers circulating lipids and reduces PDAC incidence after exposure to BOP (Takeuchi et al. 2007). Pioglitazone and other PPARγ ligands have both antidiabetic and anti-inflammatory effects and have been shown to inhibit pancreatitis in mice (Konturek et al. 2005; van Westerloo et al. 2005). Together, these studies highlight the intimate link between inflammation and PDAC and raise the question of whether anti-inflammatory drugs could serve as chemopreventive agents.
Autocrine and paracrine signals between epithelium, fibroblast, and immune cells establish an inflammatory microenvironment from early stages of PanIN progression (Clark et al. 2007; Perez-Mancera et al. 2012). Mechanistically, pancreatic inflammation activates STAT3 and nuclear factor kappa B (NFκB) signaling, both of which are required for tumorigenesis (Daniluk et al. 2012; Fukuda et al. 2011; Ji et al. 2009; Lesina et al. 2011; Maniati et al. 2011). As inflammation does not cause cancer in the absence of mutant KRAS, it appears to represent a “tumor promoter” analogous to TPA treatment in mouse skin cancer (Zoumpourlis et al. 2003): unable to induce tumorigenesis itself but required to expand and sustain mutant cells. NFκB appears particularly critical in early tumorigenesis, as it participates in the positive feedback loop that amplifies KRAS signaling to the levels required for ADR (Daniluk et al. 2012). Insofar as anti-inflammatory agents could inhibit this loop, they should prevent pancreatic cancer. Indeed, several such agents have proven effective at delaying PanIN progression and PDAC initiation in the hamster and KrasLSL mouse models, including cyclooxygenase inhibitors (Daniluk et al. 2012; Funahashi et al. 2007; Takahashi et al. 1990), dexamethasone (Rhim et al. 2012), and the diabetes drug metformin (Schneider et al. 2001), the last of which exhibits anti-inflammatory effects due to inhibition of NFκB (Isoda et al. 2006). Interestingly, metformin use in human diabetics has been associated with decreased risk of PDAC (Li et al. 2009), suggesting that the drug has chemopreventive properties within this patient population.
Additional PDAC risk factors remain to be assessed in animal models. For example, cigarette smoking imposes at least a 2-fold increased risk in humans (Bosetti et al. 2012), comparable to that associated with nonhereditary chronic pancreatitis (Duell et al. 2012). The fact that this not been pursued in animals may reflect the already high emphasis placed on antismoking efforts. For more innocuous-seeming risk factors, however, animal studies could establish causal relationships with pancreatic cancer. For example, individuals with periodontal disease have similarly elevated PDAC risk to smokers (Michaud et al. 2012); this could indicate a tumor-promoting role for infectious bacteria, or it could mean that both PDAC and periodontal disease occur secondary to immune system defects.
We expect that animal PDAC models will continue to validate human risk factors, identify their mechanisms of action, and help develop counteracting therapeutics. Prospective testing of chemoprevention in humans will be a challenge, however, given that only a small minority of individuals subject to any of the risk factors discussed here will ultimately develop PDAC. Chemopreventive trials will likely have to focus on high-risk patients defined by a combination of risk factors, and perhaps also exhibiting signs of incipient cancer. In the next section, we will discuss the use of animal models in developing new methods for early PDAC detection, as well as efforts to improve treatment of the advanced cancers that represent most newly diagnosed cases.
Before ending this discussion of risk factors, it is important to consider the possibility that animal models are too cancer susceptible and thus affected by risk factors of minor or no human relevance. For example, although deletion of the retinoblastoma (Rb1) tumor suppressor accelerates KrasLSL tumorigenesis (Carriere et al. 2011), the human homolog of this gene is not mutated in familial or sporadic PDAC. Similarly, studies in hamster indicate increased susceptibility to BOP-induced tumors on a diet supplemented with heterocyclic amines, by-products of meat cooking (Yoshimoto et al. 1999), yet there is scant epidemiological evidence that meat consumption increases human PDAC risk independent of its contribution to obesity (Nitsche et al. 2011).
It is with this context in mind that one must consider the recent controversy over whether glucagon-like peptide-1 (GLP-1)-based diabetes drugs, such as sitagliptin and exenatide, increase the risk of pancreatic cancer (Gale 2012). Following up on preliminary epidemiological evidence for excessive pancreatitis and PDAC cases among patients on these drugs (Elashoff et al. 2011), a preclinical study indicated that exenatide treatment increased PanIN burden and organwide inflammation in KrasLSL mice (Gier et al. 2012). However, the interaction of KrasLSL and exenatide appears less severe than that of KrasLSL and caerulein-induced pancreatitis (Carriere et al. 2009; Guerra et al. 2007; Morris et al. 2010). Given that only a small minority of pancreatitis patients develops PDAC, the risk associated with GLP-1 drugs may be quite low. On the other hand, there is little comfort in the fact that PanINs and PDAC do not arise in wild-type animals treated with GLP-1 drugs (Nyborg et al. 2012), given that rodents rarely, if ever, develop these lesions without induced or engineered Kras mutations. If further epidemiological studies confirm a link between GLP-1 drugs and human PDAC risk, animal models will undoubtedly help dissect the basis of that link.
Use of Animal Models for Improved PDAC Diagnosis and Treatment
There are currently no diagnostic tools with the specificity and sensitivity required for PDAC screening of the general population. Even restricted to high-risk individuals, current screening modalities may be of limited clinical impact (Canto et al. 2013; Poruk et al. 2013). A key advantage of animal models is that they represent the ultimate “high-risk” patients: all affected individuals will develop PanINs or PDAC, depending on the model, while all untreated or wild-type littermates will remain completely cancer free. The KrasLSL mouse has been particularly useful in this regard: one of the first studies of this model included serum proteomic analysis to detect potential circulating biomarkers of PanIN burden (Hingorani et al. 2003), and subsequent work has correlated specific circulating protein markers between PanIN/PDAC-bearing mice and human PDAC patients (Faca et al. 2008). A recent study of the KrasLSL model indicates that mutant cells themselves are shed into the bloodstream from PanIN formation onward, providing a potentially powerful mechanism to detect tumor-prone cells before they become invasive (Rhim et al. 2012).
New imaging modalities are also emerging from studies of the KrasLSL model, including targeted nanoparticles detectable by magnetic resonance imaging (MRI) imaging (Kelly et al. 2008) and optical imaging based on tumor-specific enzymatic activity (Cruz-Monserrate et al. 2012; Eser et al. 2011). While these have yet to be scaled up to the human organ, they also provide facile methods to monitor the evolution of tumor burden and metastasis in living mice. Such an approach is essential if we are to realize the most pressing goal of pancreatic cancer research, which is improving the treatment of advanced disease. A disadvantage of autochthonous cancer models, as opposed to traditional models based on xenografts or transplanted cell lines, is that tumor progression is inevitably asynchronous between individual animals. Simultaneously treating age-matched GEM mice with a particular drug can therefore yield incoherent results: some mice will be too far along to respond, while others may not have invasive disease at the onset of treatment. By applying in vivo imaging, irrespective of the traditional methods such as ultrasound or newer methods discussed above, a clinical trial-like study design can be implemented in which individual mice are “enrolled” only once they develop detectable cancer (Cook et al. 2008).
Such approaches continue to be refined but have already confirmed the particular utility of KrasLSL mice as a model to improve existing drugs and test new ones. For example, the standard-of-care chemotherapy drug gemcitabine was found to almost cure transplantation models of human and mouse PDAC but had practically no effect on tumors arising in situ in KrasLSL mice (Olive et al. 2009). In this respect, the GEM model is much closer to the human situation, where gemcitabine barely extends the life span of PDAC patients. A potential reason for the inefficacy of chemotherapy is that tumor cells are separated from blood vessels by abundant stromal cells, limiting access to circulating drugs. Inhibiting the Hedgehog signaling pathway in KrasLSL mice caused stromal regression and enhanced gemcitabine killing of tumor cells, thus extending survival (Olive et al. 2009). Further supporting the importance of the stroma is the recent finding that increasing its physical permeability, by hyaluronidase treatment, also increases gemcitabine efficacy in KrasLSL mice (Provenzano et al. 2012).
A central goal of cancer research, in the pancreas and elsewhere, is development of targeted therapies that kill cancer cells based on their unique molecular properties. EGFR inhibitors, for example, are known to inhibit the growth of human lung cancers carrying activating EGFR mutations (Laurent-Puig, Lievre, and Blons 2009). These drugs are effective at inhibiting PDAC growth in transplantation models but are generally unhelpful in human PDAC patients. Importantly, this lack of effect is reproduced in KrasLSL mice (Singh et al. 2010), providing an example of the GEM model better predicting clinical outcome than traditional xenograft methods. The inefficacy of EGFR inhibitors may reflect the fact that this pathway acts only during PDAC initiation (Ardito et al. 2012), as well as the fact that mutant KRAS signaling is thought to bypass any need for upstream EGFR activation. Interventions targeted downstream of mutant KRAS might be more effective, and a recent study indicates that genetic or pharmacological inhibition of the RAS effector phosphoinositide (PI)-3-kinase inhibits both initiation and maintenance of KrasLSL -induced PDAC (Eser et al. 2013). Clinical trials of PI-3-kinase inhibitors in PDAC are ongoing (www.clinicaltrials.gov) and should indicate whether the GEM model can predict positive as well as negative patient outcomes.
Conclusions and Future Directions
Studies of Syrian hamster and mouse have converged on a model for PDAC initiation and progression that begins with activation of the KRAS oncogene in differentiated acinar cells, establishment of a positive feedback loop to increase KRAS signaling and induce acinar reprogramming into ductal tumor precursors, and subsequent loss of tumor suppressor genes to facilitate invasion and metastasis. Several of the key risk factors for PDAC have been reproduced in these models and collectively indicate an important role for inflammation in enhancing KRAS activity and promoting acinar reprogramming. Animal models are well suited for validation and dissection of known and emerging human risk factors as well as for developing preventive interventions targeting those factors. The KrasLSL mouse is particularly suited for studies of pancreatic cancer genetics, and it has emerged in the last decade as the model of choice for most researchers. We encourage the field not to neglect the hamster model, however, because it provides independent validation of many results recently obtained in mouse.
Ultimately, progress toward the goal of reducing PDAC deaths will require improved prediction based on genetic and nongenetic risk factors, improved biomarker, and imaging modalities to screen potential high-risk individuals for incipient cancer, and pharmacological interventions to prevent PDAC progression as well as limit the growth of advanced disease. The refinement of animal models in the past several decades has driven progress in each of these areas, and these models will continue to make major contributions in the years ahead.
Footnotes
Acknowledgments
I thank Nathan Krah, Ben Stanger, and Nicole Aiello for providing images, the Murtaugh lab for helpful discussions, and the pancreatic cancer research community for its remarkable collegiality.
Author’s Note
I apologize to those colleagues whose work I could not discuss for lack of space.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grant 1-R21-CA179453 from the National Institutes of Health/National Cancer Institute.