
Abstract
Animal models of human disease are a critical tool in both basic research and drug development. The results of preclinical efficacy studies often inform progression of therapeutic candidates through the drug development pipeline; however, the extent to which results in inflammatory bowel disease (IBD) models predict human drug response is an ongoing concern. This review discusses how murine models are currently being used in IBD research. We focus on the considerations and caveats for commonly used models in preclinical efficacy studies and discuss the value of models that utilize specific pathogenic pathways of interest rather than model all aspects of human disease.
Introduction
Inflammatory bowel disease (IBD) is a complex multigenic inflammatory disorder affecting approximately 1 to 1.5 million patients in the United States (Herrinton et al. 2007; Kappelman et al. 2007; Loftus et al. 2007). IBD incidence has traditionally been highest in North America and Europe (Gollop et al. 1988); however, there has been an increase in the global incidence documented beginning approximately in the middle of the 20th century (Molodecky et al. 2012).
While the introduction of biologic inflammatory mediators such as tumor necrosis factor (TNF) inhibitors has improved the IBD therapeutic landscape, there remains considerable unmet medical need particularly in patients with severe or complicated clinical manifestations and in pediatric populations (Yang, Alex, and Catto-Smith 2012; Assaa et al. 2013; Mehta, Silver, Lindsay 2013). This need continues to drive IBD research efforts at universities, research foundations, and biotechnology and pharmaceutical companies spanning the spectrum from basic science to translational medicine. A common thread in most of these research efforts is the need for appropriate animal models. Animal models provide a means of characterizing physiologic interactions when our understanding of such processes is insufficient to allow replacement with in vitro systems.
Utilization of Animal Models in IBD Research
Animal models in IBD research are used in studies that fall in 5 broad categories: addressing scientific questions, evaluating preclinical efficacy, evaluating pharmacokinetics (PK), safety testing, and biomarker studies. There is considerable variability in how frequently IBD animal models are used in these categories. For instance, IBD models are rarely used for safety studies but very frequently used to address scientific questions and for preclinical efficacy testing. IBD models are also commonly used for PK studies; however, use of existing IBD models has raised relatively few concerns in this area when compared to issues with model use in efficacy, scientific, and biomarker studies. While this acceptance of current models for PK studies may need to be revisited at some future date, we will not discuss PK testing further in this review.
It is challenging to assess the full scope of use of IBD animal models to address scientific questions. A quick PubMed search shows greater than 600 IBD publications per year for the past 3 years that were flagged as animal models studies. This undoubtedly underrepresents the extent of animal model use in IBD-related studies but does illustrate that this use is both common and important to scientific research. Animal models are typically used in these studies as a means of hypothesis testing but are also utilized in exploratory or hypothesis generating experiments. For these experiments, scientists may modify standard models or create new models to enable the analysis. A typical approach might involve creating a new genetically modified mouse or using existing lines and subjecting these lines to a chemical challenge (Ramirez-Carrozzi et al. 2011; Cox et al. 2012).
Another major category of animal model use in IBD research is for preclinical efficacy testing. This is of considerable importance to pharmaceutical and biotechnology companies, and results of preclinical efficacy studies frequently serve as gating criteria that determine whether a therapeutic candidate will advance in the drug development pipeline. Preclinical efficacy studies most commonly utilize well-established chemically induced or genetic models of intestinal inflammation (Table 1). It is uncommon for new IBD models to be developed specifically for preclinical efficacy testing of a new therapeutic candidate, although models developed in academic labs are sometimes adopted for testing of specific therapeutic candidates. The predictive value of results from preclinical efficacy studies is a major concern with existing IBD models and will be covered in more depth later in this review.
Commonly used models of inflammatory bowel disease.
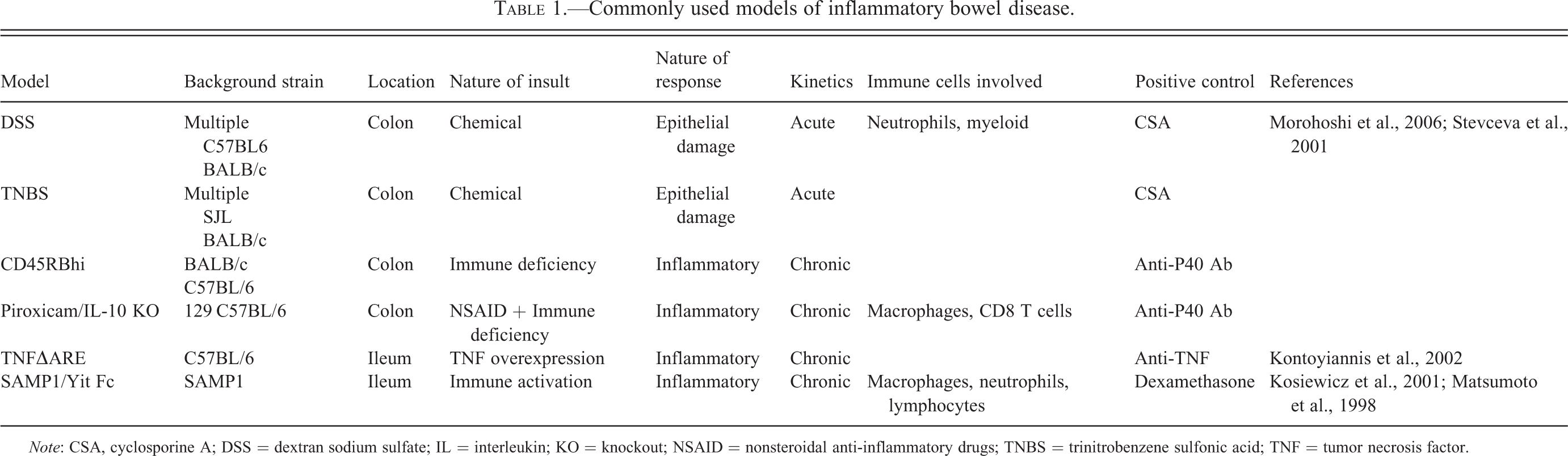
Note: CSA, cyclosporine A; DSS = dextran sodium sulfate; IL = interleukin; KO = knockout; NSAID = nonsteroidal anti-inflammatory drugs; TNBS = trinitrobenzene sulfonic acid; TNF = tumor necrosis factor.
An emerging area for animal model use is biomarker development. Biomarkers are objective measures of normal or pathologic processes or, in the case of pharmacodynamics (PD) markers, of response to therapeutic intervention (Biomarkers Definition Working Group 2001; Strimbu and Travel 2010; Goodsaid 2012). Human IBD drug trials, particularly those in Crohn’s disease, are complicated by both uncertainty about current disease activity and also by the relapsing and remitting clinical course. These variables are believed to be significant contributors to high placebo rates, which can obscure therapeutic benefits of drug candidates (Su et al. 2004; Sands et al. 2005; Sands 2009; D’Haens et al. 2012). The need to reduce placebo risk in IBD trials has led to increasing use of expensive and sometimes invasive imaging modalities to compensate for the lack of highly reliable indicators of disease activity (Solem et al. 2008; Loftus et al. 2007; Buisson et al. 2013), so development of reliable and noninvasive disease activity biomarkers is an ongoing area of interest. In addition to disease activity biomarkers, predictive biomarkers are an important area of IBD-related research. Predictive biomarkers are used to identify individuals within the larger IBD patient population who are likely to have a positive response to treatment with a specific therapeutic entity (de Castro et al. 2013; Hanania et al. 2013). A successful predictive biomarker used to enroll or interpret trial results can make the difference between a successful or failed trial. In addition, a robust biomarker can reduce risk to patients by eliminating those unlikely to derive clinical benefit from exposure to a drug that is still in the testing phase (Becker Jr. and Mansfield 2010).
While much IBD biomarker work is done in human patient populations, the effectiveness of this is limited because it is often difficult to do hypothesis testing until relatively late in drug development. If a strong predictive biomarker hypothesis can be generated from in vitro data, then initial testing can be done in phase II patients and the results confirmed in phase III. But, more commonly in IBD trials, biomarker candidates are not sufficiently robust to be incorporated in phase II study design and, instead, phase II trial samples are frequently used to generate a biomarker hypothesis that may get initial testing in phase III. This longer time line creates challenges for creation and launch of companion diagnostics (Moore, Babu, Cotter 2012). These issues underscore the need for animal models that share pathogenic features and/or utilize molecular pathways of interest to enable hypothesis testing or even to do preliminary hypothesis generating studies.
Ideally, a disease model should closely parallel the human disease in clinical manifestations, pathophysiology, and response to existing therapeutic reagents. However, this is rarely possible with complex human diseases such as IBD where multiple genetic and environmental influences determine disease onset and clinical course. This creates ongoing challenges for scientists involved in IBD model development and analysis. Planning for success in IBD animal model development may require that we consider Voltaire’s comment that “the perfect is the enemy of the good” (Voltaire 1772) and focus our efforts on identifying the specific features we need to model rather than trying to recapitulate the entirety of human disease in an experimental animal.
In this review, we discuss some examples of needs that arise in a drug development environment and the current state of model development to address those needs. Our intention, rather than to provide a comprehensive review of every model and how it may or not meet these needs, is to encourage our readers to consider the needs specific to their own programs and pathways of interest with the goal of optimizing IBD models to address their most important questions. Colitis models exist in rats, nonhuman primates, and dogs, but, in this review, we will limit our discussion to only murine IBD models (Mansfield et al. 2001; Holst et al. 2012; Wadie et al. 2012). The use of mouse models enables relative ease in incorporating animals with specific genetic modifications, use of larger cohort sizes, and decreases the quantity of therapeutic reagents required.
Characteristics of Human IBD
IBD encompasses a heterogeneous group of disorders that have 2 major phenotypic forms, Crohn’s disease and ulcerative colitis, both with a pattern of chronic relapsing and remitting intestinal inflammation (Abraham and Cho 2009; Kaser, Zeissig, and Blumberg 2010; Levine et al. 2011). Ulcerative colitis, the most common of the IBDs (Danese and Fiocchi 2011), is characterized by continuous mucosal inflammation involving the rectum and progressing up the colon with increasing disease severity. Inflammatory infiltrates are confined to the mucosa and consist primarily of lymphocytes and plasma cells plus granulocytes in crypt abscesses and present in the mucosa during disease flares. Ulceration is common during active disease and colonic epithelial cell changes such as goblet cell depletion, distorted crypt architecture, and epithelial dysplasia are associated with chronic disease.
By contrast, Crohn’s disease is characterized by discontinuous inflammation that can affect any part of the gastrointestinal tract, although ileum and colon are most frequently involved (Farmer, Hawk, and Turnbull 1975). Inflammation due to Crohn’s disease can extend throughout the intestinal wall, and this transmural inflammation appears to be responsible for many of the serious complications associated with Crohn’s disease, such as fibrostenotic disease, abscesses, and fistula formation (Keighley et al. 1982; Maeda et al. 1994). Epithelioid granulomas are a characteristic of Crohn’s disease and provide diagnostic differentiation from ulcerative colitis when identified histologically. However, these granulomas are found in only about 20 to 40% of biopsies and 60% of surgical resection specimens (Scholmerich and Warren 2003).
Current models of IBD pathogenesis hypothesize that disease arises from interactions between genetically susceptible hosts and the gut microflora (Xavier and Podolsky 2007). It is clear that host genetic susceptibility plays an important role in IBD pathogenesis. The first Crohn’s disease susceptibility gene, nucleotide oligomerization domain 2 (NOD2), which is encoded by the caspase recruitment domain-containing protein 15 (CARD15) gene, was identified in 2001 (Hugot et al. 2001). Subsequent genome-wide linkage and association studies have revealed many additional genetic polymorphisms, and there are currently more than 160 IBD susceptibility genes or loci recognized (Jostins et al. 2012). A number of these susceptibility genes are shared between ulcerative colitis and Crohn’s disease, which suggests the existence of shared pathways in IBD pathogenesis despite differences in clinical phenotype (Budarf et al. 2009; Zhernakova, van Diemen, and Wijmenga 2009). While the functional role of many loci or single nucleotide polymorphisms is incompletely understood, many of the susceptibility genes are associated with aspects of mucosal immunity including immune response to microbial pathogens (Jostins et al. 2012). These genetic data are in accord with observations that development of immunologic responsiveness to commensal microorganisms is a hallmark of IBD (Arnott et al. 2004; Lodes et al. 2004; Targan et al. 2005; Danese and Fiocchi 2011) and that shifts in the bacterial makeup of intestinal microflora have been documented in IBD (Frank et al. 2007; Walker et al. 2011; Nagalingam and Lynch 2012).
While the classic phenotypic descriptions of ulcerative colitis and Crohn’s disease seem to clearly differentiate these disease conditions, there is a subset of patients in whom disease differentiation can be problematic. There is a low but persistent incidence of diagnosis change in patients from ulcerative colitis to Crohn’s disease or the reverse (Myren et al. 1988) illustrating that clinical phenotype may change over time in individual patients. In addition, approximately 10% of patients presenting with symptoms of IBD may have indeterminate features that preclude an initial diagnosis of either ulcerative colitis or Crohn’s disease (Guindi and Riddell 2004). This diagnostic uncertainty primarily arises from the lack of a specific test that can differentiate ulcerative colitis from Crohn’s disease resulting in heavy reliance on clinical phenotype which can be heterogeneous. This phenotypic heterogeneity creates obvious problems for clinical patient management, but it should also serve as a reminder to those of us engaged in animal model development of just how complex are the diseases under the broad category of IBD.
General Guidelines for IBD Models in Preclinical Efficacy Studies
Once the decision has been made to initiate studies within a preclinical model system, a series of thought questions and tests should be employed to ensure that the model is operating in a reproducible and consistent manner. Below, we describe a series of factors that must be thoughtfully considered prior to experimentation. These factors are arranged in ascending order according to how easy they are to manage, from easily controllable (e.g., study size and powering of groups) to more challenging (e.g., microflora).
Reproducibility is a key component of any scientific experiment, whether it means being able to generate consistent data repeatedly within the same laboratory, or being able to repeat published work performed at other sites. Typically, the former is more easily constrained than the latter. However, recent publications have highlighted several major pharmaceutical companies’ struggles in attempting to reproduce published literature (Arrowsmith 2011; Prinz, Schlange, and Asadullah 2011; Begley and Ellis 2012). While this review cannot address all of the issues involved in data reproducibility, we will try to highlight those factors that critically influence preclinical IBD models such as study size, mouse strain and gender, study reagents including positive and negative treatment controls, and study group randomization.
Study Size
Preclinical efficacy studies should be of sufficient power to draw a meaningful conclusion. Ideally, this would be determined through review of the study design and past model results with a biostatistician. While our typical group size is approximately 10 mice, it is not possible to make a blanket recommendation that will be applicable to all institutions even for a single model. The number of mice needed is largely determined by the variability typically observed within a group. This varies widely between institutions and by specific model so must be determined locally. In general, the fewer animals per group, the more likely subtle outliers will influence the interpretation of an experiment and the lower the likelihood of generating reproducible data. Despite the fact that inbred strains of mice are typically used in preclinical models of IBD, variability in the response is frequently observed and must be taken into account. In our experience, models of chemically induced colitis have a higher degree of variability than genetic models. This may be a reflection of the relatively short disease course in acute chemical models, which can highlight early kinetic differences in disease onset that might equilibrate over time. In contrast, chronic or progressive models of disease often develop, over sufficient periods of time, a rather homogeneous disease burden.
Gender
Gender can also provide a source of variability, and gender-specific effects have been documented for dextran sodium sulfate (DSS) colitis (Mahler et al. 1998), 2,4,6-trinitrobenzene sulfonic acid (TNBS) colitis (Bouma, Kaushiva, and Strober 2002), and inflammatory disease in senescence accelerated mouse prone 1 (SAMP1)/Yit Fc mice (Pizarro et al. 2011). Regardless of whether the induction of disease is possible in both genders, we would recommend using female animals whenever possible. Male animals are more prone to fighting and the inflammation and stress associated with such behavior often has a negative impact on studies. In long-term or chronic studies, fight wounds may also fester and require euthanizing the animals in accordance with Institutional Animal Care and Use Committee (IACUC) protocols. While single housing of experimental animals is an option, it becomes challenging as study sizes increase.
Reagent Validation
For proper interpretation of a scientific study, the reagents and materials used in the study should be rigorously validated. For genetically engineered animals, this should include confirmation that the genetic targeting is correct and complete (e.g., gene and protein expression for knockout animals). In chemically induced models of disease, such as DSS and TNBS colitis, each new lot of chemical inducer should be titrated prior to the study to understand the dose–response relationship. If the amount of DSS is too little, the overall histological burden will be insufficient to provide a window to see differences between treatments. If the amount of DSS is too high, the dose may be fatal or may result in damage too extreme to reasonably expect a detectable protective effect, regardless of treatment reagents.
Mouse Strain
Rodents, in particular mice, have been the model of choice for immunologists, and inbred mice are available in a wide range of strains. However, each of these strains has a unique genome that makes it susceptible or resistant to intestinal inflammation. DSS colitis has been attempted using many different mouse strains with varying degrees of success. The current hypothesis of how colitis-inducing injury occurs is that DSS is directly toxic to the gut epithelium (Laroui et al. 2012). The loss of epithelial integrity and subsequent migration of bacteria into the crypt results in inflammation and triggers a pathologic loss of structure. Despite this rather broad postulated mechanism of action, BALB/c mice and C57BL/6 mice require significantly different doses of DSS to induce disease. At present, the reasons behind this difference are unknown, but it must be taken into account when choosing an animal model. In contrast, induction of colitis with TNBS results in a similar degree of acute toxicity to the epithelium and subsequent inflammation; however, SJL and BALB/c strains are susceptible to disease induction while C57BL6 are resistant (Scheiffele and Fuss 2002).
Another aspect to consider in choosing a mouse strain and preclinical model is the availability of genetically engineered animals for the gene of interest. The majority of genetically engineered animals are constructed on the C57BL6 background. Thus, if an investigator wishes to determine the effects of their particular genetically engineered animal on a particular model system, they must either choose a model compatible with this genetic background or spend significant time and resources backcrossing their animals to a different strain.
Treatment Controls
In order to draw meaningful conclusions from any scientific experiment, positive and negative controls must be incorporated. In preclinical models of IBD, this can be challenging as the number of clinically validated therapies is relatively low and the number of models that respond to these therapies is also low. Although they are not typically used in the clinic due to safety issues, high-dose immunosuppressants can be used in most inflammation-mediated models as a positive control (Hoshino et al. 1992; Melgar et al. 2008; Hirano and Kudo 2009; McNamee et al. 2010, 2013). In Table 1, we summarize the standard controls used in various animal models and their respective effects on disease.
Randomization
Once the model has been chosen and the number of animals is decided, animals should be randomized into experimental groups based on factors identified as important to the model in question. This process, termed initial balancing or randomizing, ensures that subtle differences across the cohort will be less likely to influence the experimental outcome. For IBD models, this typically consists of genotype, date of birth or age, body weight, and if animals are to be treated after the onset of disease, some accounting for disease burden prior to start of treatment. Understanding the degree of pathological change without histology is technically challenging, however. In our experience, the pathological changes that occur in colitis models can be positively correlated with body weight (Figure 1). In contrast, disease activity scores such as diarrhea and presence of occult blood are typically not quantitative enough to be meaningfully correlated with pathology (i.e., these are binary readouts) and are not used in balancing cohorts. When available, more sophisticated techniques such as endoscopy can be employed to assess colon damage and group animals (Laroui et al. 2012). In contrast to colitis models, ileitis models do not positively correlate with body weight loss, and visualization of the mouse ileum is not possible using endoscopy methods. For this instance, we are investigating the use of disease biomarkers as a means of balancing cohorts. However, the number of predictive biomarkers in preclinical models of IBD is limiting, although recent studies have been published looking at fecal biomarkers such as calprotectin and lipocalin (Chassaing et al. 2012; Cury et al. 2013).
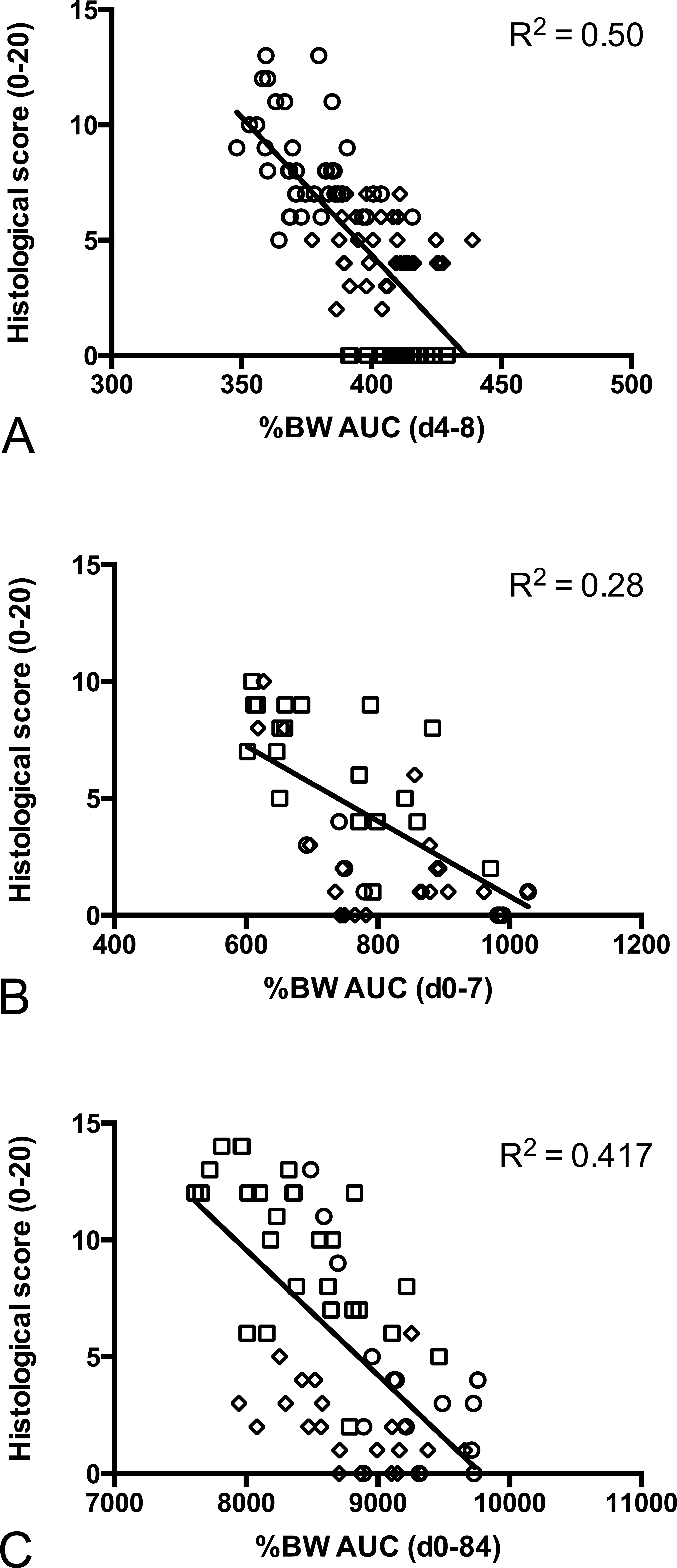
Correlations between body weight and histology in various IBD models are shown for various preclinical models of IBD. Body weight loss is reflected as percentage body weight (relative to baseline) area under the curve (AUC) for the portion of the study indicated. (A) DSS; results are a meta-analysis of 5 studies; diamond = cyclosporine A (CSA), circle = no DSS control, square = isotype control antibody. (B) TNBS; results are a meta-analysis of 2 studies; diamond = CSA, circle =
Animal Source
Despite efforts to properly control studies, variability may derive from a myriad of other external sources. The gut microflora, a collective term for the various species of bacteria inhabiting the intestinal tract, has been demonstrated in multiple model systems to be important in disease. Antibiotics decrease the severity of DSS- and TNBS-induced colitis (Aranda et al. 1997; Hans et al. 2000; Fiorucci et al. 2002), and the immune response in CD45RBhi transfer colitis is believed to be largely targeting endogenous bacteria (Stepankova et al. 2007). Similarly, the emergence of intestinal disease in the SAMP1/Yit mouse corresponded with the transfer of the colony from a native flora facility to a specific pathogen free facility (Matsumoto et al. 1998), and gnotobiotic animals are often resistant to disease induction. Since different vendors, and even different facilities of the same vendor, are known to harbor slightly different microflora (Ivanov et al. 2009), the same source of animals should be used across experiments. However, other recent studies have suggested that the microflora of genetically deficient animals may be unique but also transferrable to wild-type littermates, suggesting such effects may mask phenotypes observed within a colony (Elinav et al. 2011). Further complicating this matter, there is also an emerging role for nonmicrobial pathogens, such as norovirus (Cadwell et al. 2010), and the fact that the microflora diversity and frequency changes in response to the induction of colitis (Okayasu et al. 1990). Despite these challenges, we still believe that wild-type littermate controls should be used in experiments whenever possible, since differences in disease induction can be observed within “wild-type” animals of different colonies but the same strain (Figure 2).
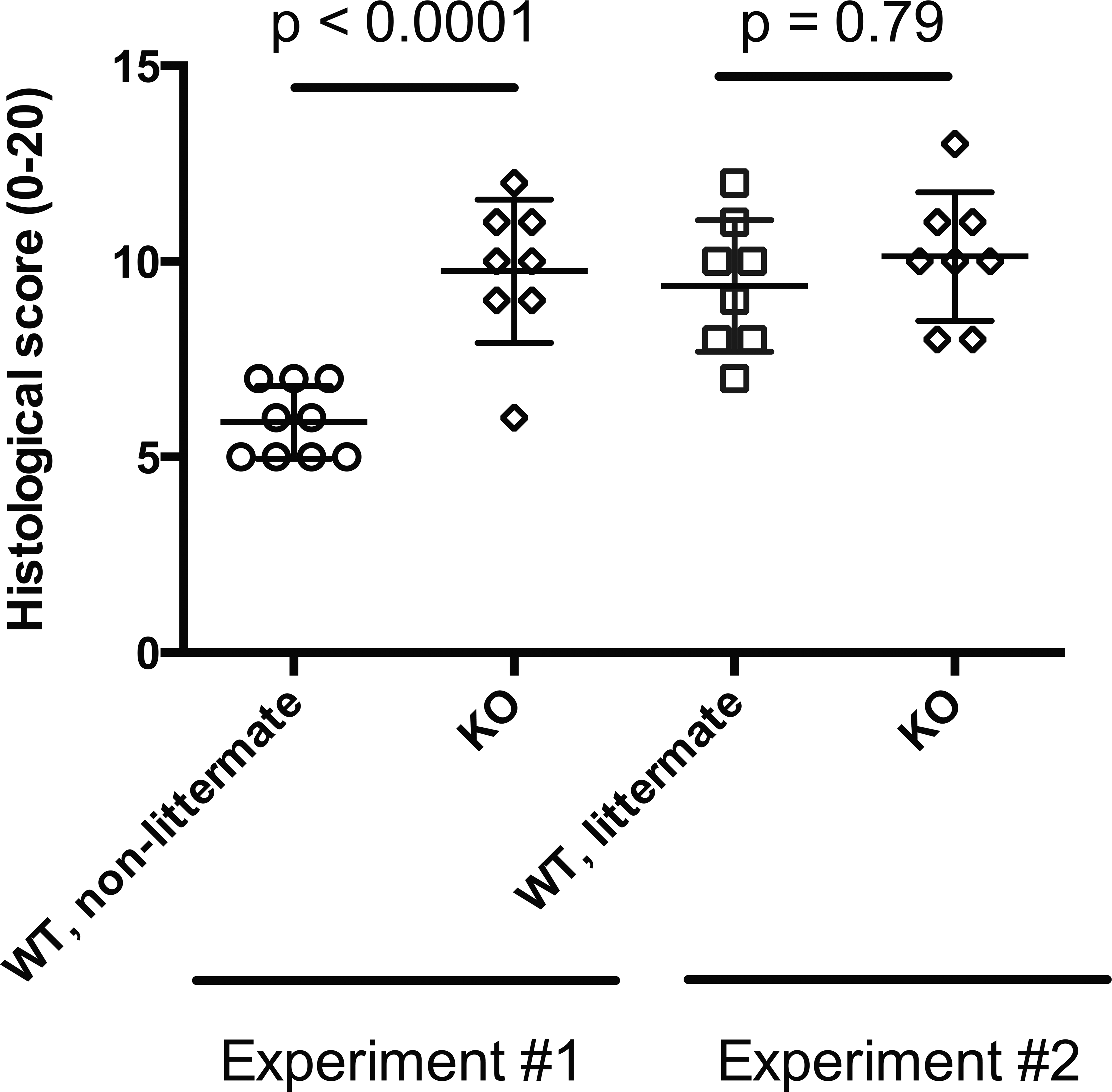
Disparity in disease induction in wild-type (WT) and knockout (KO) animals. In experiment 1, wild-type animals from another colony were used. In experiment 2, littermate controls were used. Symbols indicate individual animals within the experiment. Bars are mean histology score +/− standard deviation.
Histologic Processing and Analysis
Finally, because histologic analysis is still the gold standard for evaluating the extent of intestinal lesions and protective effect of therapeutic candidates, maintaining quality and consistency during preparation of histologic specimens is important. In our opinion, there is not a single “perfect” system for preparing tissue samples for histologic analysis. Instead, we find that maintaining internal consistency in how intestinal tissues are collected and prepared allows for best results. At our facility, we fix either colon or small intestine held in a Swiss roll configuration by a standard plastic histology cassette using 10% buffered formalin. Our technique is a variation of that described in the Moolenbeek which we have modified by not making the longitudinal opening made in the intestinal wall and instead removing fecal material using a saline flush (Moolenbeek and Ruitenberg 1981). This allows for rapid evaluation of the entire colon or small intestine by the pathologist or using an automated algorithm (Kozlowski et al. 2013).
Reported histologic scoring systems vary considerably between institutions, pathologists, and the various disease models. While it might be useful to develop some “best practices” in regard to scoring common preclinical efficacy models, that task is beyond the scope of this review. At our institution, we use a robust and rapid histologic scoring system that we apply to all colitis studies with some model-specific adjustments. The scoring system is designed to capture both extent and severity of lesions by scoring 4 anatomic regions of colon (proximal, middle, distal colon, and rectum) on a severity scale from 0 to 5. Criteria for scoring severity are model-specific. For instance, DSS colitis is primarily scored based on loss of colon crypts, while transfer colitis is scored based on inflammatory infiltrate and change in intercryptal distance. But the scoring output is consistent and, as such, is readily interpretable by a variety of collaborating investigators. Examples of scoring output can be seen in Figures 1 and 2.
Considerations for Choosing a Preclinical Model
The above guidelines apply to all preclinical IBD models. In this section, we will discuss strategies for determining the model of greatest relevance to the particular scientific question to be addressed. It would be outside of the scope of this article to discuss every model published to date. To simplify, we will focus on a set of standard preclinical models frequently used in pharmaceutical companies to assess therapeutic efficacy. These models span a diverse landscape of biology and are summarized in Table 1.
As described previously, Crohn’s disease and ulcerative colitis can occur in different anatomic regions of the gastrointestinal tract. For many years, the majority of animal models published focused on the large intestine, with DSS, TNBS, CD45RBhi cell transfer, and interleukin-10 (IL-10) gene deficiency induced inflammation all largely affecting the colon (Neurath, Fuss, and Strober 2000; Read and Powrie 2001; Perse and Cerar 2012). The colon and the small intestine, however, differ in anatomical and cellular structure as well as bacterial diversity and burden (Eckburg et al. 2005). Thus, it is reasonable to postulate that the mechanisms governing disease in these distinct locations may be similarly unique. Fortunately, the emergence of a number of models that develop inflammation in the small intestine, including the Tumor necrosis factor alpha delta AU rich element (TNFΔARE) and the SAMP1/Yit (Fc) has provided an additional series of platforms to test therapeutic candidates (see Table 1).
IBD models tend to segregate by the nature of the response that is evoked. To better determine whether a model would suit a particular scientific question, it is helpful to ask what is the dominant pathological response that is observed. For example, does the postulated therapeutic mechanism of action target prevention of damage to the epithelium or intestinal architecture, inflammation caused by innate immune cells, or inflammation mediated by an adaptive immune response?
Chemically induced models of colitis, such as DSS and TNBS, induce acute damage to the epithelium. These models are useful to explore pathways or therapeutics that are thought to protect intestinal epithelial cells from damage or stress response as well as pathways that maintain gut integrity in the presence of a biological insult. Animals that have genetic deficiencies in permeability through a variety of mechanisms (Laukoetter et al. 2007; Kong et al. 2008; Chogle et al. 2011; Williams et al. 2012) have been shown to be more susceptible to DSS colitis. Consistent with this, therapeutics that maintain or increase gut permeability are protective in DSS colitis (Ukena, Singh, and Westendorf 2007; Chogle et al. 2011). However, damage to the epithelium is only the opening salvo of DSS colitis. Upon loss of barrier integrity, bacteria rapidly penetrate the epithelium and mediate both direct and inflammation-associated toxicity (Johansson et al. 2010; Johansson et al. 2013). Thus, therapeutic agents that moderate the gut flora or modify subsequent inflammatory responses to gut flora also have the potential to demonstrate efficacy in the DSS colitis model. Illustrating this point, treatment of specific pathogen-free colonized animals with antibiotics improves colitis (Hans et al. 2000; Rath, Schultz, and Sartor 2001). Surprisingly, however, the administration of DSS to gnotobiotic animals results in severe disease—suggesting that normal flora is required to establish those mechanisms that protect the epithelium from insult (Kitajima et al. 2001; Hudcovic et al. 2007). Consistent with this hypothesis, germ-free animals have significantly decreased mucus layer thickness in the lumen of the colon, and animals with a genetic deficiency in mucus production have exacerbated responses to DSS (Petersson et al. 2011). From this standpoint, DSS colitis and other acute biological insult models represent a meaningful means of assessing gut integrity, host responses to microflora mediated by intestinal epithelial cells, and innate immune cells.
Innate immune cell response dominates in acute inflammation models when there is insufficient time to develop an adaptive immune response. This is also true of intestinal inflammation models that have been performed in commonly used immunodeficient mouse strains. These models have been used to ascertain the contribution of innate immune cells to the maintenance of immune tolerance and responses to inflammation in the gut. While several studies have suggested a role for T cells in the DSS response, disease induction is similar in mice homozygous for the severe combined immunodeficiency (SCID) mutation or the Rag1 mutation which is devoid of B and T cells, clearly demonstrating that these cells are neither necessary nor sufficient for disease development (Axelsson et al. 1996; Kim et al. 2006). Within the damaged epithelium, neutrophils and macrophages are the most common immune cells present. Therapies that target the downstream mediators of these cells, such as reactive oxygen species, are therefore protective (Krieglstein et al. 2001; Amrouche-Mekkioui and Djerdjouri 2012; Vong et al. 2012; Yasukawa et al. 2012).
While the acute chemical-induced colitis models typically only incorporate features of innate biology (both epithelial and immune), the chronic and progressive models typically illustrate a complex interplay between innate biology and adaptive immune responses. The TNFΔARE mouse model, in which loss of translational repressors result in chronic TNFα overexpression (Kontoyiannis et al. 1999, 2002), provides an example of this complexity and the resulting opportunities for testing therapeutic entities that target various aspects of immune function. In normal animals, the regulation of TNFα expression is tightly controlled, including regulation through mRNA destabilization mediated by a 3′ adenylate-uridylate (AU)-rich region (Anderson 2000). When the AU-rich region is removed from the gene, TNFα overexpression is observed in all cell types normally capable of expressing TNFα and these TNFΔARE mice develop spontaneous arthritis and ileitis (Kontoyiannis et al. 1999). A series of genetic crosses suggest that ileitis observed in this model is dependent on CD8+ T cells and the effector cytokines IL-12/23 p40 and interferon-γ (Kontoyiannis et al. 2002). However, while disease was greatly diminished in animals that lack the β2-microglobulin gene, a necessary component of major histocompatibility class 1, and subsequently display greatly diminished CD8+ T cells, lymphocyte-specific overexpression of TNFα resulted in only mild ileitis. Thus, while CD8 T cells are effector cells necessary for disease, they are not sufficient. In contrast, driving TNFα overexpression in the myeloid compartment using LysM-cre was sufficient to cause ileitis comparable to a TNFΔARE animal indicating that myeloid immune cells are an important mediator of disease (Kontoyiannis et al. 2002). Finally, an additional study suggested that TNF receptor expression solely via the collagen VI promoter, restricting the ability to respond to TNF to the epithelial layer of the gut and other stromal tissues, was sufficient to induce disease (Armaka et al. 2008). This model highlights the complex interplay between the various facets of an immune response—myeloid production, epithelial sensing, and an effector response mediated by CD8 T cells and the production of interferon-γ all contribute to induce a disease process controlled by TNFα.
The TNFΔARE model example illustrates the complexity of an intact immune response, highlighting the difficulty in modeling such a system in an in vitro fashion. It also demonstrates how detailed knowledge of the mechanistic basis of disease can help to determine what therapeutics may be active in a disease context. Neutralization of TNFα in the TNFΔARE model is very efficacious (McNamee et al. 2013); however, targeting of downstream effector functions allows the interrogation of the events set in motion by this single cytokine.
Interpreting Preclinical Data from IBD Models
The previous sections have discussed decision making during IBD model development and the use of knowledge about the underlying biology to determine when to employ such a model. In this section, we will discuss the interpretation of data generated from these models and recommend how to weigh the predictive value of study data. In this context, it is useful to consider whether a model is intended to serve as a translatable model or a pathway model.
A truly translatable preclinical model of human disease would incorporate several features including similar pathological hallmarks of disease, shared underlying mechanisms and biology, a response to standard of care therapeutics that mirrors what is observed in human patients, and accurate prediction of response to experimental therapeutics in clinical trials. Unfortunately, there are no models that currently exist which fulfill these criteria. We choose to view preclinical models of disease as pathway models that will inform whether or not modulation of a specific pathway in the disease state will have an effect on the overall process. To the extent that the mechanism of action for a given therapeutic is known, the downstream effect of modulation can be queried. This information can then be linked to developing the knowledge of human disease biology and genetics from which therapeutic candidates arise. When a specific pathway is determined to be activated or repressed in human disease, we then determine whether or not it is similarly modulated in an animal model using many of the principles articulated earlier in this review.
If a given pathway is modulated in both human and mouse disease and therapeutic targeting of that pathway in a relevant preclinical model results in disease amelioration, how much does this inform our understanding? Does this need to be demonstrated in a single model or multiple models? To what extent should a preclinical model be used for deciding whether or not to advance to a clinical trial? The answers to these questions are complex and will likely vary depending on goals of the individual scientist posing the question as well as the current goals of the sponsoring organization. Given that we have already addressed the limitations of preclinical animal models and do not consider them as truly translatable, we view preclinical efficacy as a criterion for advancing candidate therapeutic agents to the next stage of development. However, preclinical efficacy cannot serve as the only criteria.
If an experimental therapeutic is advanced into clinical trials, does preclinical data accurately predict success? In a recent review of 108 phase II clinical trials, over half of the trials failed due to efficacy (Arrowsmith 2011). Similarly, a review of 83 phase III clinical trials noted that 66% of the trials failed due to efficacy (Arrowsmith 2011). It is reasonable to assume that some form of preclinical data was generated to support these studies, suggesting that either the model was not translatable to human disease or the effect to which target was modulated in mouse and human was not comparable. The latter hypothesis, that a given drug fails due to insufficient target coverage, can be addressed in both rodents and humans provided a PD marker is identified. PD markers provide a context in which to understand relationships between the dose of drug administered and the biological effect that the dose produces. Ideally, the same PD marker is informative in both the preclinical model and the human model, although distinct PD markers can be used if they both faithfully report on the downstream biology in each species. In IBD, however, the most relevant organ in which to collect samples for PD interrogation is the gut and frequent biopsies may not be desirable to all IBD patients. A blood-based PD marker, therefore, is desirable but extensive validation studies must be performed to ensure that the PD readout in the blood is reflective of target modulation in the end organ.
Other factors that some investigators choose to assess within a preclinical model include immunogenicity and safety. However, both of these are difficult to interrogate within the context of an IBD model. The predictive value of preclinical models for immunogenicity is a controversial subject and many animal models show limited predictive values (Norlander, Gotthard, and Ström 1990; Brinks et al. 2013; Thway et al. 2013), owing to differences in treatment regimens, dosing routes, and host immune response. In addition, human subjects may be on immunosuppressant regimens that will further complicate the anticipation of antidrug responses. Safety assessment within an animal model is similarly contentious, especially if baseline values typically assessed in a safety study are significantly affected by the disease process. For this reason, we choose to assess immunogenicity and safety in separate studies performed in healthy animals.
We view the current landscape of IBD model development for scientific as well as preclinical efficacy testing as challenging but one in which careful decision making can enhance the opportunities to derive meaningful data from these studies. Both the increasing understanding of factors contributing to human disease and the expanding repertoire of mouse models of intestinal inflammation provide opportunity for investigators to dissect mechanistic pathways and response of those pathways to experimental intervention. While no single mouse model recapitulates all of the features of human IBD, it is worth remembering that there is considerable genotypic and phenotypic variation even within humans with a similar diagnosis of either Crohn’s disease or ulcerative colitis. Therefore, it is unlikely and possibly not even desirable to be able to develop a single prototypic model of disease. Careful utilization of pathway models to query specific scientific or efficacy questions is likely to be the more successful approach to existing needs.
Footnotes
Abbreviations
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.