
Abstract
Malignant glial tumors are the most aggressive and difficult to treat neoplasms arising in the brain. More than 22,000 people in the United States are diagnosed with a malignant glioma annually, and most will die within the first two years from diagnosis. Traditionally, gliomas have been categorized based solely on tumor histological features. However, expression studies have found that molecular signatures can be used to categorize these tumors into subclasses that more effectively predict patient outcome. The heterogeneity between tumors as well as within individual tumors makes understanding the molecular aspects of tumorigenesis extremely important. Several genetically engineered mouse models (GEMMs) of glioma have been developed that recapitulate the molecular alterations observed in the human disease. GEMMs of glioma have allowed researchers to more closely study the role of cancer stem cells (CSC) in gliomagenesis as well as the relevance of signaling within the CSC microenvironment. Knowledge of the underlying molecular signatures of malignant glial tumors coupled with the existence of a variety of human disease-relevant GEMMs of this tumor type provide researchers and clinicians with valuable resources for the discovery of new drug targets.
Introduction/Relevance
Gliomas (a primary tumor of glial cell origin) are the most common intracranial neoplasm with astrocytomas, glioblastomas, and oligodendrogliomas accounting for more than 80% (Central Brain Tumor Registry of the United States [CBTRUS] 2010; Pfister, Hartmann, and Korshunov 2009; Merchant, Pollack, and Loeffler 2010) (Figure 1 ). More than 22,000 people are diagnosed with a malignant glioma (also referred to as high-grade gliomas) in the United States each year (CBTRUS 2010; Wen and Kesari 2008), and most will die within the first two years from diagnosis despite aggressive therapy. The World Health Organization (WHO) classifies malignant gliomas as grades III and IV based on tumor histological and immunohistochemical features (Louis et al. 2007). Anaplastic astrocytomas, anaplastic oligodendrogliomas, and anaplastic oligodendroastrocytomas are classified as grade III; and glioblastoma multiforme (GBM) and other less common variants are classified as grade IV. GBMs, the most aggressive and deadly of these tumors, are the most common cancer of the central nervous system (CNS) accounting for 53% of gliomas overall (Figure 1) (CBTRUS 2010). By contrast, low-grade gliomas (WHO grades I and II) are relatively benign and afford a much better prognosis as compared to their higher-grade counterparts; however, grade II gliomas have the propensity to progress to grade IV GBMs over time, especially in the younger population (Huse and Holland 2010).
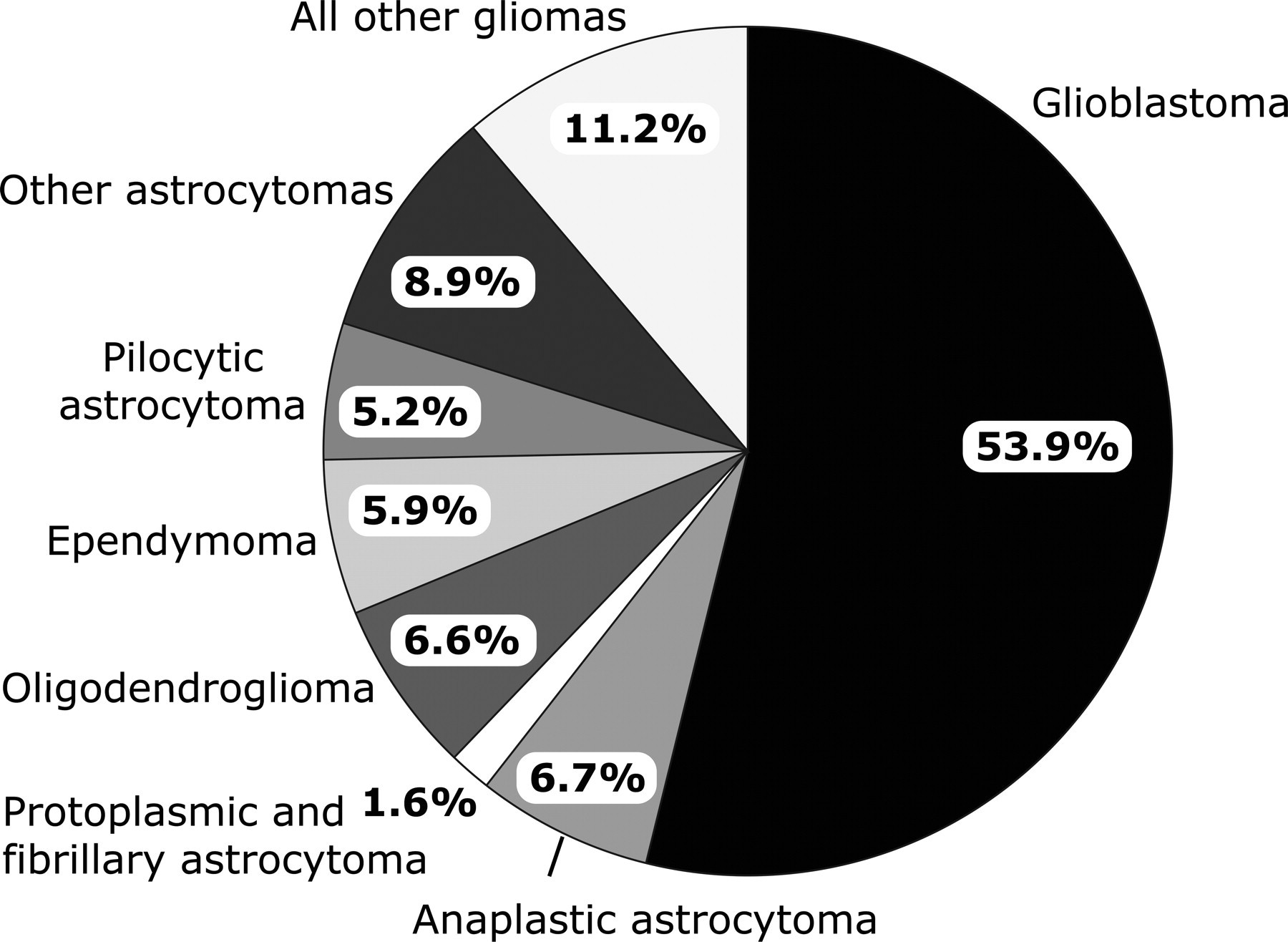
Frequency of primary CNS gliomas. Pie chart showing the frequency of primary glial tumors by histology group diagnosed in the United States from 2004 to 2006 (CBTRUS 2010).
Despite more than 40 years of research, little improvement in mortality rates has been made for patients diagnosed with malignant glioma owing to the intra- and inter-tumoral heterogeneity and the propensity of the cancerous cells to infiltrate into the normal brain parenchyma. Further complicating therapy, the blood-brain barrier prevents chemotherapeutic agents from adequately penetrating into the tumor mass and leads to suboptimal therapeutic response. The challenge in providing effective therapies for the treatment of malignant gliomas is evidenced in a 5-year survival rate (less than 5% for GBM) that has remained practically unchanged over the past several decades. Undoubtedly, the key to developing effective therapies is to obtain an in-depth understanding of the biological mechanisms important to gliomagenesis.
Histological and Molecular Pathology of Malignant Gliomas
Histological Pathology
In 1979, the WHO formalized the first classification system for tumors of the CNS. This system set criteria to group CNS tumors by grade (WHO grades I through IV) (Table 1 ), with increasing grade reflecting an increase in tumor aggressiveness and a worse prognosis. Grade I glial tumors are benign, slow-growing, and well-circumscribed tumors that are typically curable with resection. Grade II glial tumors infiltrate into normal brain tissue and exhibit moderate proliferative features. These tumors have the potential to undergo malignant transformation (~70%) within 5 to 10 years from diagnosis (Furnari et al. 2007). Grades III and IV tumors are classified as malignant gliomas, with grade IV being extremely aggressive with hallmarks of vascular and uncontrolled cellular proliferation, diffuse infiltration, and necrosis. Since the inception of this system, three updates have been made and described in subsequent editions. Most recently (2007), the WHO published its fourth edition that reflects changes in tumor grade, new entities, and variants of some CNS tumors (Louis et al. 2007; Brat et al. 2008). This classification system has been used by clinicians, pathologists, and scientists to collectively propel the advancement of epidemiologic and clinical studies of brain tumors.
World Health Organization (WHO) classification of common glial tumors in humans.

Histological features and subclasses of WHO grades I through IV gliomas and corresponding molecular subclasses. Grade IV gliomas subdivide into three distinct subclasses (Brennan et al. 2009; Verhaak et al. 2010; Phillips et al. 2006).
Molecular Pathology
Although most gliomas are sporadic, genetics plays some role in gliomagenesis. Patients with a family history of glioma can have a 5% chance of developing a glial tumor (Wen and Kesari 2008; Yanhong, Lixin, and Huaiyin 2010). Some of the familial cases have already been linked to rare inherited genetic abnormalities (i.e., neurofibromatosis [involving mutations in NF1], Turcot syndrome [abnormalities in mismatch repair genes], Li-Fraumeni syndrome [involving mutations in TP53], and melanoma-brain tumor [P16/CDKN2A mutations]), but for most the etiology remains unknown (Yanhong, Lixin, and Huaiyin 2010).
Most of the molecular alterations that have been shown to be causal in mouse models of glioma have successfully predicted the genetics in the human disease. Here, the focus will primarily be on the molecular alterations that have been identified through the use of a variety of genetically engineered glioma mouse models (Table 2 ). One example is the functional loss of the tumor suppressors RB and P53. These proteins function as G1-S phase cell cycle regulators and are common targets of inactivating mutations in malignant gliomas. The RB pathway can be targeted through the direct inactivation of RB by gene mutation or deletion (loss of 13q) or through functional inactivation by amplification of its negative regulators CDK4 (12q13-14) or, less commonly, CDK6. Inactivation of p16Ink4a prevents negative regulation of CDK4 and CDK6 and thus represents another mechanism of RB loss of function.
Molecular subclasses of glial tumors and relevant genetically engineered mouse models (GEMMs).

Molecular subclasses based on the stage of neurogenesis and defining lesion. The subclasses were identified in studies that analyzed molecular aberrations of WHO grades II, III, and IV human primary and secondary gliomas (Brennan et al. 2009; Verhaak et al. 2010; Phillips et al. 2006). Mouse models have been developed that closely mimic the glioma subclasses in humans.
Similarly, TP53 can be inactivated by direct gene mutations (or 17p loss) or by amplification of its negative regulator MDM2. P14ARF inhibits MDM2 and represents another gene that, when inactivated, can cause P53 functional loss. P16INK4a and P14ARF are both encoded at the CDKN2A locus, and hypermethylation or deletion of this locus can affect both RB and TP53 expression and lead to glioma formation. Interestingly, genome-wide association studies have found that single nucleotide polymorphisms in CDKN2A and CDKN2B are associated with the risk for developing gliomas (Shete et al. 2009; Wrensch et al. 2009).
Phosphatidylinositol 3-kinase (P13K) and mitogen-activated protein kinase (MAPK) signaling pathways are important to the promotion of cell growth and survival. PTEN (phosphotase and tensin homolog) is a tumor suppressor that functions as one of the primary negative regulators of P13K-AKT-mTOR and Ras-MAPK signaling (Knobbe and Reifenberger 2003; Ohgaki et al. 2004). Analyses of the Cancer Genome Atlas (TCGA) Research Network data found that PTEN functional loss in human GBM is commonly due to monoallelic gene mutations or deletions (chromosome 10q deletion). Functional loss of PTEN has also been correlated with overexpression of miR-26a (a microRNA negative regulator of PTEN, RB, and MAP3K2) (Huse and Holland 2009). PTEN is inactivated in 50% of malignant gliomas, and loss of function is considered as a negative prognostic factor (Chakravarti et al. 2004; Tada et al. 2001).
Mouse models of glioma have been developed by overexpressing components of signal transduction pathways that promote cell proliferation and survival (Skeen et al. 2006). AKT and KRAS are protein kinases and downstream effectors of the P13K pathway and MAPK pathways, respectively. However, mutated isoforms of AKT and KRAS resulting in constitutive activation are only observed in a small percentage of glioblastomas. Nonetheless, a convincing argument can be made for the relevance of these two tumor promoters, especially when considering the ability for these lesions in generating glioma mouse models.
Receptor tyrosine kinases (i.e., epidermal growth factor receptor [EGFR] and platelet derived growth factor [PDGFR]) and their ligands (epidermal growth factor [EGF] and platelet-derived growth factor [PDGF]) can activate the P13K-AKT-mTOR and Ras-MAPK pathways. The EGF/EGFR and PDGF/PDGFR growth factor axes have been suggested to stimulate autocrine and paracrine loops when both the ligand and the receptor are overexpressed; however, mutation or genomic amplification of the receptor tyrosine kinases alone can lead to constitutive activation of the receptor. The most common EGF mutant receptor (EGFRvIII; exon 2-7 deletion) is observed in 20% to 30% of glioblastomas (Frederick et al. 2000). In younger patients, the presence of EGFRvIII equates to a less favorable prognosis (Brennan et al. 2009). Increased expression of PDGFRα and its ligands (PDGF-A and PDGF-B) are commonly observed in both low- and high-grade tumors. However, amplifying mutations are less common as compared to EGFR (TCGA Research Network 2008; Parsons et al. 2008).
Genes important to metabolic processes have also been linked to gliomagenesis. Mutations in isocitrate dehydrogenase 1 (IDH1) were shown to be present in a large percentage (~80%) of human grade II and III astrocytic and oligodendroglial tumors as well as secondary glioblastomas (Dang et al. 2009). The IDH1 mutation results in a gain-of-function of enzymatic activity to convert α-ketoglutarate to R(-)-2-hydroxyglutartate (2HG). 2HG is an oncogenic metabolite associated with an increased risk of malignant brain tumors in patients with an inborn defect in 2HG metabolism (Kolker et al. 2002; Wajner et al. 2004; Aghili, Zahedi, and Rafiee 2009). It has also been found at increased levels in human malignant gliomas that harbor IDH1 mutations and thus has been suggested as a contributor to glioma formation and malignant progression (Dang et al. 2009). Another molecular alteration that has been observed in 60% to 90% of oligodendroglial tumors is deletion of the 1p and 19q chromosome arms (Wen and Kesari 2008). This deletion has been found to predict a less aggressive tumor behavior and a more favorable response to chemotherapy (Wen and Kesari 2008). It is worth mentioning that the identity of the presumed tumor suppressors located within the 1p and 19q loci remains unknown. This has presented a challenge in the development of mouse models that recapitulate this disease in humans.
Molecular Subclasses of Glioma
Several expression studies of malignant gliomas have been conducted and have found that patterns of somatic mutations, copy number alterations, and gene and protein expression can be used to categorize these tumors into subclasses (Verhaak et al. 2010; Brennan et al. 2009; Phillips et al. 2006). These studies have linked the molecular signatures of gliomas with stages of neuroglial development (Phillips et al. 2006). In agreement with previous studies (Phillips et al. 2006; Verhaak et al. 2010), Brennan et al. (2009) found that GBMs could be categorized into subclasses based on the levels of expression and activity of core proteins of signal transduction pathways (i.e. PDGF, EGFR, and NF1). Interestingly, the molecular signatures of these subclasses overlap with the classical, proneural, and mesenchymal transcriptomal glioma subtypes (Verhaak et al. 2010; Phillips et al. 2006).
The proneural subclass is primarily defined by PDGF expression. Other genes that are elevated in this subclass include genes important to oligodendrocyte development such as OLIG2 and PDGFRA (Brennan et al. 2009). This subclass consisted primarily of WHO grade III tumors and some GBMs occurring in younger patients (Brennan et al. 2009). The classical subclass most commonly contains molecular alterations in EGFR. INK4a/ARF loss as well as RB and Notch pathway activation is also observed more commonly in these tumors. And finally, the mesenchymal subclass is defined by NF1 alterations as well as increased expression of mesenchymal genes such as YKL40 and CH13L1 (Phillips et al. 2006; Verhaak et al. 2010). Interestingly, these subclasses were shown to be more effective at predicting clinical outcome than was histological classification (Phillips et al. 2006). The heterogeneity between tumors, as well as within individual tumors, makes understanding the molecular aspects of tumorigenesis essential to finding effective therapies.
Mouse Models of Malignant Glioma
The heterogeneous nature of glial tumors dictates the need for creating a variety of genetically engineered mouse models (GEMMs). Several models have been developed over the years, each representing a variety of molecular defects and morphological characteristics as seen in the human disease (Tables 2 and 3 ). Discussed below are mouse models of malignant gliomas that represent the classical, proneural, and mesenchymal subclasses. These models are believed to be close recapitulations of the human disease because they represent a combination of distinct genetic events, some of which are introduced as somatically acquired events, similar to the presumed evolution of malignant gliomas. To date, these models have been essential to increasing our understanding of glioma biology and play an important role in preclinical testing of novel therapies both now and in the future.
Mouse models of malignant gliomas (astrocytomas, glioblastomas, and oligoastrocytomas).
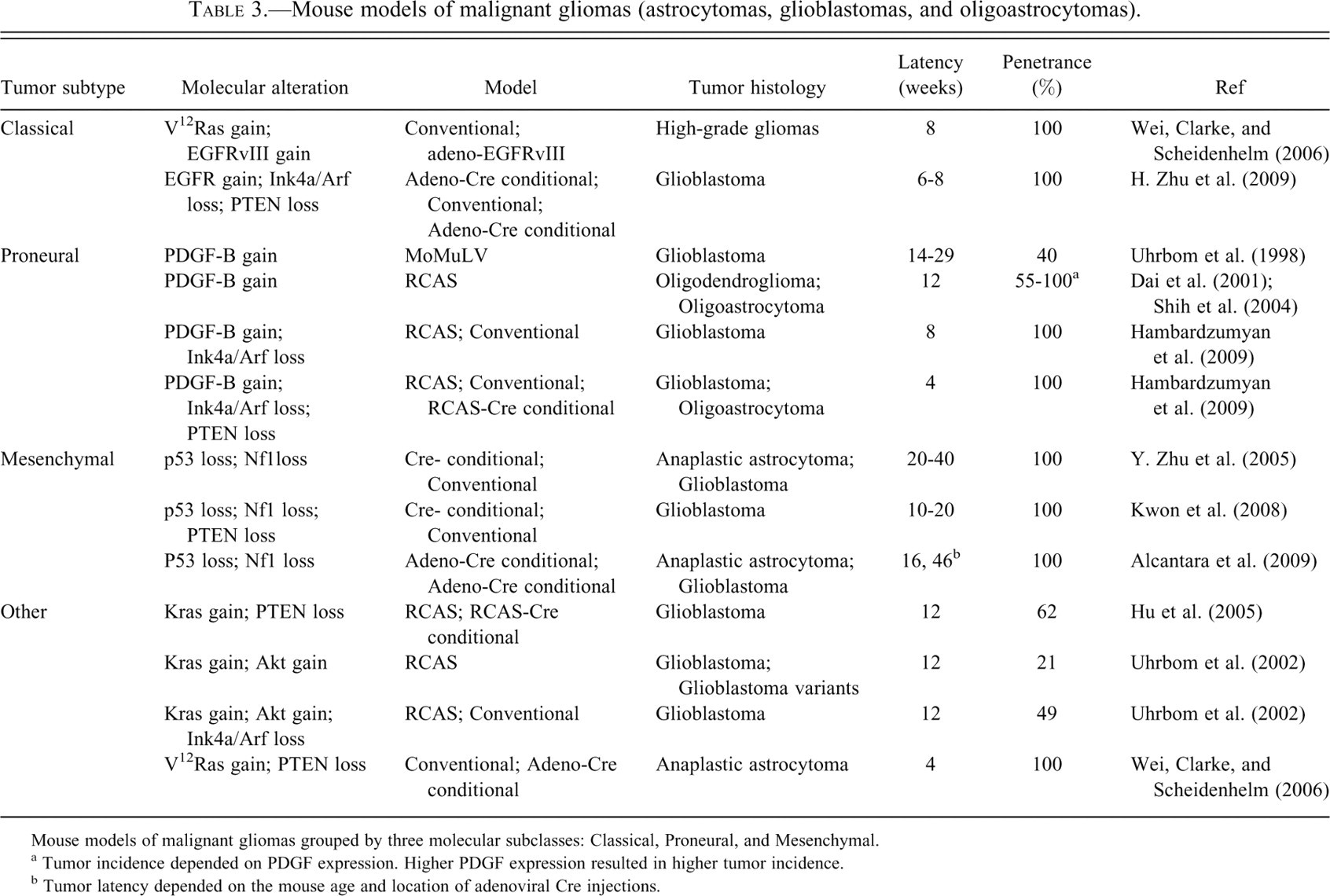
Mouse models of malignant gliomas grouped by three molecular subclasses: Classical, Proneural, and Mesenchymal.
a Tumor incidence depended on PDGF expression. Higher PDGF expression resulted in higher tumor incidence.
b Tumor latency depended on the mouse age and location of adenoviral Cre injections.
Models of “Classical” Glial Tumors
Several mouse models of glioma have been developed that harbor aberrations in receptor tyrosine kinases (i.e., EGFR, PDGFR) or their ligands. The classical molecular subclass is defined by EGFR overexpression and represents 25% to 30% of malignant glial tumors in humans. The most commonly observed EGFR variant is EGFRvIII (constitutively active variant of EGFR). GEMMs have been created to evaluate the role of EGFRvIII in gliomagenesis (Table 3). Although aberrant expression of EGFRvIII in glial cells alone does not generate gliomas in mice, when combined with other oncogenic genetic lesions, high-grade glial tumors form (Xiao et al. 2005; Wei, Clarke, and Scheidenhelm 2006). For example, when glioma-prone GFAP:V12-Ha-Ras adult mice are injected with adeno-EGFRvIII viral vector (Ad:EGFRvIII) malignant gliomas form within 8 weeks post injection (Wei, Clarke, and Scheidenhelm 2006). Another model that investigates the role of EGFR overexpression in adult gliomagenesis uses the Cre-Lox system to somatically introduce EGFRvIII expression by injecting adeno-Cre viral vector into the brain parenchyma. Ink4a/Arf null;Pten fl°xed adult mice harboring conditional EGFRvIII alleles (expressed upon Cre-mediated deletion of a floxed stop cassette) develop glioblastomas with high penetrance within 6 to 8 weeks post orthotopic injections of adeno-Cre into the brain (H. Zhu et al. 2009). Hence, EGFR expression combined with the loss of tumor suppressors is required for glioma formation, and the loss of multiple tumor suppressors shortens tumor latency (H. Zhu et al. 2009; Wei, Clarke, and Scheidenhelm 2006).
Models of “Proneural” Glial Tumors
The proneural subclass is defined by a subset of malignant gliomas with evidence of PDGF pathway activation (30% of human tumors) (Phillips et al. 2006; Brennan et al. 2009). GEMMs have been developed to ascertain what role PDGF and its receptors play in the development of glial tumors (Table 3). Uhrbom et al. (1998) developed a pediatric model of malignant glioma using a murine retrovirus, MoMuLV, to deliver PDGF-B via orthotopic injection into the frontal lobes of newborn mice. This model results in glioblastoma formation with a 40% penetrance after 14 to 29 weeks of age. Resulting tumors coexpressed PDGFRα supporting the concept of an autocrine mechanism of transformation involved in gliomagenesis.
The RCAS/tva system has been used to somatically deliver PDGF-B into glial cells. The RCAS/tva model system allows for somatic gene transfer of the oncogene of interest into targeted brain cells by driving expression of TVA (the RCAS receptor) under an astrocytic promoter, GFAP (G-tva), or a astrocyte progenitor promoter, Nestin (N-tva). This system was used to create another model of pediatric PDGF-induced glioma by orthotopically injecting RCAS-PDGF-B into newborn N-tva or G-tva mice (Dai et al. 2001). This model produces oligodendroglial or mixed oligoastrocytic tumors with a higher penetrance and shorter tumor latency as compared to PDGF-B delivery by MoMuLV (Table 3). Additionally, it was determined that higher gene dose correlated with higher tumor grade (Shih et al. 2004).
More recently, the RCAS/tva system has been used to generate adult models of PDGF-B induced glioma. Transgenic N-tva;Ink4a/Arf null;Pten fl°xed mice developed glioblastomas within 8 weeks post injection of the RCAS-PDGFB viral vector with 100% penetrance (Hambardzumyan et al. 2009). Not surprisingly, when the RCAS-PDGFB vector and RCAS-Cre vectors (conditional deletion of PTEN) were injected, malignant glial tumors formed with equal penetrance and shorter latency (4 weeks). Transgenic G-tva;Ink4a/Arf null, G-tva;Trp53, and G-tva;Arf null mice all develop glioblastomas with regions of oliogastrocytoma and mixed astrocytoma histology.
Models of “Mesenchymal” Glial Tumors
The mesenchymal subclass is defined by malignant glial tumors harboring Nf1 alterations, which is generally characterized by low NF1 expression (Verhaak et al. 2010; Brennan et al. 2009). A mouse model of this subclass was generated by crossing mice bearing a germ-line Trp53 mutation with Nf1 floxed alleles with GFAP-Cre mice (Table 3) (Y. Zhu et al. 2005). This model generated tumors with histological features consistent with high-grade gliomas such as microvascular proliferation and pseudopalisading necrosis with a 100% penetrance and a 10-month tumor latency. Another model was developed to assess the role of Akt activation in high-grade glioma formation using the Nf1/Trp53 model (Kwon et al. 2008). Not surprisingly, when PTEN haploinsufficiency was added to this model, tumor latency decreased and tumor aggressiveness increased (Table 3). However, these models of spontaneous tumor development may be more reminiscent of an inherited tumor syndrome (i.e., neurofibromatosis) since the molecular alterations were generated as germ-line mutations. A somatic cell version of the mesenchymal model for gliomas has also been generated using adenoviral CRE delivery to mice harboring floxed versions of Nf1 and p53 (Alcantara et al. 2009 ). These mice develop gliomas very closely mimicking human glioma histology. Of note, these tumors arise only if the virus is delivered to the subventricular zone (SVZ), implying that at least in this case the cell of origin is limited to CNS stem cells in this region (Llaguno et al. 2009).
Other GEMMs of Gliomas
There are other models of glioma tumors that do not specifically fall into one of the three aforementioned subclasses or that may be less representative of the accumulation of somatic mutations that occur during gliomagenesis. However, models that utilize downstream effectors of the EGFR and PDGFR signaling pathways such as constitutive expression of AKT and KRAS are still useful tools for understanding glioma biology. For example, in a pediatric model of glioma combining Akt and Kras expression generates malignant gliomas (Uhrbom et al. 2002). In this model, newborn N-tva mice injected with RCAS-Kras and RCAS-Akt viral vectors develop malignant glial tumors with a 21% tumor incidence within 12 weeks post injection (Uhrbom et al. 2002). However, when N-tva;Ink4a/Arf null mice are injected with RCAS-Kras and RCAS-Akt viral vectors, glioblastomas form at a higher tumor penetrance (Table 3) (Uhrbom et al. 2002). Glioblastomas also formed in G-tva;Ink4a/Arf null mice (incidence 42%) with histological hallmarks of the glioblastoma variant, giant cell glioblastoma.
A similar model was developed to investigate the role of Akt downstream effectors using Ntv-a;Pten fl°xed mice. When Ntv-a;Pten fl°xed mice are injected with RCAS-Kras and RCAS-Cre viral vectors, glioblastomas develop with greater penetrance (62%) at 12 weeks of age (Hu et al. 2005) as compared to the N-tva;Ink4a/Arf null mice (Table 3). Finally, an adult mouse model of malignant glioma was generated by using double transgenic mice bearing constitutively expressed V12-H-Ras and floxed Pten alleles. This model resulted in malignant astrocytoma formation within 4 weeks post injection of the adenoviral-Cre viral vector (Ad:GFAP-Cre) (Wei, Clarke, and Scheidenhelm 2006).
Brain Cancer Stem Cells and Nitric Oxide–Induced Notch Signaling
There has been a long-standing debate over the role of stem cells in brain cancer. It has been proposed that carcinogenesis is dependent on a small population of cells termed “cancer stem cells” (CSCs) that are pluripotent and capable of self-renewal (Ignatova et al. 2002; Shen et al. 2004 ). Normal neural stem cells are located in regions of the brain (subventricular and hippocampal subgranular zones) that have capillary networks capable of providing the necessary nourishment and signaling factors required for stem cell self-renewal and survival (Doetsch 2003; Shen et al. 2004, 2008; Ramirez-Catillejo et al. 2006). Evidence has suggested that brain CSCs require a similar specialized microenvironment to maintain their “stemness” as well (Calabrese et al. 2007; Gilbertson and Rich 2007). Brain CSCs migrate toward this specialized microenvironment, which is closely positioned near areas of microvasculature proliferation referred to as the “perivascular niche” (Calabrese et al. 2007; Gilbertson and Rich 2007; Riquelme, Drapeau, and Doetsch 2008). The pathological designation of the abnormal vasculature in gliomas that correspond to the perivascular niche is “microvascular proliferation”; however, these structures are composed of many cell types including endothelial cells, reactive astrocytes, microglia, smooth muscle-actin expressing pericytes, and tumor cells. There is evidence that these structures correlate with regions of blood-brain barrier breakdown and contrast enhancement on MRI scans.
Brain CSCs have been shown to express multidrug resistance proteins (e.g., ABCG2) and to be extremely resistant to conventional genotoxic therapy (Bleau et al. 2009; B. Wang et al. 2010). In fact, treatment with temozolomide in culture increases the CSC rich side population (Bleau et al. 2009), ultimately resulting in more aggressive and treatment resistant tumors upon reoccurrence. In radiation-treated medulloblastoma, it was shown that the surviving cells were located in the perivascular niche and express stem cell markers such as Nestin (Hambardzumyan et al. 2008). The perivascular niche contains a diverse group of cell types and signaling factors that are required for sustaining the cancer cells' stem-like phenotype. Identifying signaling factors important to the maintenance of the stem cell phenotype and deciphering how they contribute to gliomagenesis is important to understanding the biology of brain CSC and the development of targeted therapies against them.
Nitric Oxide–Induced Notch Signaling
The Notch pathway is important to cell differentiation and has been linked to both normal stem cells and glioma CSC biology (Baron 2003; Z. Wang et al. 2008). It has also been shown to activate the Nestin promoter in gliomas (Shih and Holland 2006). Notch receptor-ligand binding results in the release of the Notch intracellular domain (NICD), which is subsequently translocated to the nucleus where it activates target genes. In vivo studies of NICD expression in mice showed an induction of Nestin expression and an expansion of the SVZ via cooperation with Kras (Shih and Holland 2006). Additionally, Notch signaling can also influence chemoresistance by increasing drug efflux through ABCG2, a drug efflux transporter (Bhattacharya et al. 2007; Fan et al. 2006). To further support the role of the Notch pathway in brain CSC biology, when NICD is overexpressed the ratio of stem-like cells (side population) to bulk tumor cells (main population) increases, and the opposite effect is observed when the Notch pathway is inhibited (Fan et al. 2006; Bleau et al. 2009).
Endothelial nitric oxide synthase (eNOS), a member of the nitric oxide synthase family, is responsible for producing nitric oxide (NO) in the vascular endothelium (Fukumura, Kashiwagi, and Jain 2006). NO is responsible for regulating physiological processes through the NO/cGMP/Protein kinase G pathway (Fukumura, Kashiwagi, and Jain 2006). Elevated levels of eNOS expression have been observed in several cancers including pancreatic cancer and gliomas (Bakshi et al. 1998; Broholm et al. 2003; Fukumura, Kashiwagi, and Jain 2006; Lim et al. 2008). Recently, Charles et al. (2010) conducted a study to test the hypothesis that NO in endothelial cells functions to activate signaling pathways that promote a stem-cell-like phenotype in glioma cells located in the perivascular niche. They found that when human glioma cultured cells were exposed to the NO donor GSNO (S-nitrosoglutathione), the Notch pathway was activated, while other pathways important to the stem cell phenotype (i.e., Wnt and Shh) were not. Additionally, they observed a 2-fold increase in Nestin expression as compared to controls, suggesting NO can indeed activate the Notch pathway and promote stemness as measured by side population analysis.
Because the RCAS/tva PDGF-induced mouse model of glioma closely recapitulates the microenvironment of the perivascular niche and the histological character of human gliomas (Holland 2004), this model was used to ascertain the role of eNOS and NO in gliomagenesis. It was demonstrated that increased eNOS expression was specifically localized to the tumor vascular endothelium; and cells coexpressing Nestin (a stem cell marker) and NICD were in close proximity to the tumor endothelial cells (Charles et al. 2010). The NO receptor, soluble guanylate cyclase (sGC), was also found to be expressed in the stem-like cell population. Furthermore, eNOS deficient mice had a decreased tumor initiation and a longer survival as compared to eNOS wild-type mice.
Primary cultures generated from the PDGF-induced gliomas and exposed to GSNO exhibited a dose-dependent increase in NICD, confirming a collaboration between NO and Notch signaling in these tumors (Charles et al. 2010). Additionally, GSNO treatment resulted in a 2- to 5-fold increase in the stem cell side population and increased the expression of the chemoresistance gene, ABCG2. These data support the importance of NO signaling and notch pathway activation in the maintenance of the CSC phenotype in the perivascular niche during gliomagenesis.
Discussion
The WHO classification of tumors of the central nervous system has been used for decades as the gold standard for categorizing brain tumors. The most recent revisions to the WHO classification system included changes in tumor grades and the addition of new tumor variants and entities. However, the benefit of using molecular signatures over histological characterization is beginning to be realized. Although tumor histological features have historically been used as the primary consideration for therapy selection and clinical course prediction, more recently molecular profiling has resulted in a more effective subgrouping of these tumors and has shed more light on the molecular pathways involved in gliomagenesis.
Genomics has proven to be extremely useful to understanding glioma biology. It is now well understood that the molecular signatures of adult and pediatric malignant gliomas are distinctly different, even though histologically these tumors are indistinguishable. Molecular subclasses have recently been identified that stratify malignant gliomas based on expression levels of three core proteins of signaling pathways: PDGF, EGFR, and NF1. These core proteins define subclasses that represent stages of neurogenesis (proneural, classical, and mesenchymal, respectively). Interestingly, these molecularly defined subclasses have proven to be better prognostic factors than the traditional histological groups. This further supports the importance of utilizing molecular signatures to better stratify patients to ensure the most optimal treatment approach is employed.
In vivo models of cancer are essential for addressing complex questions that require a disease-host interaction to more accurately mimic the disease in humans. Several GEMMs of malignant glioma have been developed that harbor distinct molecular defects that recapitulate the biological environment of human brain tumors. Models of pediatric and adult malignant gliomas provide a tool for investigating the molecular basis for the distinct genomic differences observed between these two groups, even though they can be histologically indistinguishable. Importantly, GEMMs have been created that reflect the molecular signatures of the classical, proneural, and mesenchymal molecular subclasses (Table 3). Genetic manipulation of murine models has allowed researchers to identify genes important to disease predisposition and drug response, as well as the discovery of novel therapeutic targets. Additionally, these models have provided a tool for investigating the role of CSCs in gliomagenesis.
It has been proposed that CSCs are the cells of origin for tumorigenesis. Studies have found that CSCs are resistant to genotoxic therapy and that treatment increases the CSC population. Mouse models of cancer have aided researchers in understanding the biology of CSC. The CSC perivascular niche is a microvascular-rich microenvironment that promotes the CSC character. Identifying the diverse cell population and signaling factors involved in maintaining the CSC phenotype within this niche can help identify novel therapeutic targets. For example, a recent study found that eNOS is responsible for NO production within the perivascular niche, and elevated levels of NO activates the Notch pathway. Additionally, NO was found to promote stem cell character and increase the stem cell population within the perivascular niche. Indeed, the Notch pathway has already shown some promise in brain tumor preclinical studies (Fan et al. 2006). However, whether the Notch signaling pathway will be a fruitful target for drug development will depend on the CSC population’s response in preclinical studies.