
Abstract
Antiretroviral therapy has dramatically reduced mortality in human immunodeficiency virus (HIV) infection. In 1988, the suggestion that the first antiretroviral drug, zidovudine, was the potential cause of muscle pathology in HIV-infected persons resulted in structural and biochemical patient studies demonstrating acquired mitochondrial dysfunction. Assessment of subsequent nucleoside analog reverse transcriptase inhibitor (NRTI) antiretroviral drugs has indicated that mitochondria are a common target of NRTI toxicity in multiple tissues, leading to a wide variety of pathology ranging from lipodystrophy to neuropathy. Overwhelmingly, these complications have emerged during post-licensing human studies. Subsequent animal and in vitro studies have then elucidated the potential pathological mechanisms, suggesting that NRTI-associated mitochondrial toxicity arises principally from inhibition of the sole mitochondrial DNA (mtDNA) polymerase gamma, leading to a reduction in mtDNA content (depletion). Millions of patients have been treated with mitochondrially toxic NRTIs and these drugs remain the backbone of antiretroviral rollout in much of sub-Saharan Africa. Here we describe the 25-year history of antiretroviral associated mitochondrial pathology and critically review the strength of evidence linking clinical, histopathological, and molecular data. We discuss recently described novel mechanisms of NRTI-associated mitochondrial damage and whether or not recently licensed NRTIs may be considered free from mitochondrial toxicity.
Keywords
Introduction
Human Immunodeficiency Virus (HIV) and Antiretroviral Therapy
HIV is a member of the retrovirus family and is the causative agent for the acquired immune deficiency syndrome (AIDS). The first antiretroviral drug, azidothymidine (zidovudine [AZT]), was licensed in 1987 and paved the way to triple combination “highly active antiretroviral therapy” (HAART), which was first implemented in 1996 and remains the basis of modern anti-HIV therapy (Rodriguez-Novoa et al. 2006).
Although multiple steps in the HIV replicative cycle can now be targeted with licensed antiretroviral drugs, HAART usually consists of a combination of 2 nucleoside analog reverse transcriptase inhibitor (NRTIs) and a 3rd drug from either the protease inhibitor (PI) class or the NNRTI (non-nucleoside reverse transcriptase inhibitor) class as reviewed by Dalakas, Semino-Mora, and Leon-Monzon (2001). Shortly after entry into the host cell, HIV starts to reverse transcribe its single stranded RNA into proviral DNA using the viral enzyme reverse transcriptase. NRTIs are nucleoside analogs that incorporate into the elongating HIV proviral DNA and cause premature chain termination. The currently licensed NRTIs are AZT, didanosine (ddI), stavudine (d4T), lamivudine (3TC), emtricitabine (FTC), and abacavir (ABC). Zalcitabine (ddC) is no longer licensed, and tenofovir (TDF, formulated as tenofovir disoproxil fumarate) is a nucleotide reverse transcriptase inhibitor (N(t)RTI; Petit et al. 2003).
Mitochondrial Genetics and Disease
Mitochondria are the only cytoplasmic organelles that contain their own mitochondrial DNA (mtDNA). Human mtDNA is a 16,569 base pair (bp) circle of double stranded DNA. It is highly compact and contains 37 genes that encode for 13 polypeptides of the respiratory or electron transport chain. Replication of mtDNA occurs via the activity of the sole mtDNA polymerase, polymerase gamma (pol γ) as reviewed by Mahad, Lassmann, and Turnbull (2008). The respiratory chain is made up of 4 multimeric complexes, with adenosine triphosphate (ATP)-synthase being referred to as the unofficial 5th complex. Complex II (succinate dehydrogenase [SDH]) is the only respiratory complex that is solely encoded by nuclear DNA; all others have at least 1 gene that is mitochondrially encoded.
Patients with inherited mitochondrial disease have mutations either within the mitochondrial genome or within a nuclear gene responsible for mtDNA replication or maintenance (Schapira 2012). The result is disruption of mitochondrial oxidative function at the cellular level, leading to a wide range of clinical presentations including lactic acidosis, myopathy, neuropathy, encephalopathy, sensorineural deafness, hepatic dysfunction, diabetes and renal tubulopathy, as discussed by Schon, DiMauro, and Hirano (2012). In general terms, those tissues most affected by mitochondrial disease tend to be the ones containing cells with the highest energy (ATP) demands, in particular muscle and neurons.
Diagnosis of Mitochondrial Pathology in Tissue Specimens
There are a number of histopathological techniques used to assess mitochondria; however, the first step is to consider the possibility that mitochondrial pathology may be present. Simple hematoxylin and eosin stains will generally not give any clues to the presence of mitochondrial damage. Histologically, the stereotypical feature of mitochondrial myopathy is the presence of ragged red fibers (RRF), for example, by the use of modified Gomori trichrome or similar stains. RRFs are characteristic of mitochondrial damage in diseased muscle tissue and the accumulations of diseased mitochondria are what underlie the RRF phenomenon.
Resin histology was historically performed on noncontracted muscle samples to image mitochondria either side of the Z discs using light microscopy; however, this has been replaced by electron microscopy (EM). Although seldom performed in routine diagnostic practice, EM may be regarded as a “gold standard” in that it is the only technique that enables mitochondrial morphology to be observed directly and in great detail. Typical abnormalities include mitochondrial enlargement with vacuolation and “ballooning” of cristae (DiMauro et al. 1985).
Contemporary diagnostics for mitochondrial disease frequently employs sequential COX/SDH (cytochrome c oxidase/SDH) histochemistry and thus is performed on a frozen skeletal muscle biopsy. This histochemical technique depends on the activity of 2 complexes of the respiratory chain. COX is involved in accepting electrons from cytochrome c and generating the membrane’s electrochemical potential. COX activity is determined using an electron donor in place of cytochrome c. Upon oxidation, this forms a brown pigment and thus produces a brown stain where activity is present. If there is absent or minimal COX activity within a cell, then there will be absent or minimal brown stain. COX is encoded by both nuclear and mtDNA. Thus, if an mtDNA defect is present within a cell (at high enough levels to impair respiratory chain function), then COX activity in that cell will be lost. Therefore, staining sequentially for complex II, SDH, which is entirely nuclear DNA encoded, is used as a blue counterstain and thus clearly highlights the cells that contain mtDNA defects (Figure 1). SDH catalyzes the oxidation of succinate into fumaric acid. Therefore, histochemical demonstration of this enzyme is performed by the use of a tetrazolium compound that is reduced to a formazan dye in the presence of SDH activity. Frequently, COX-deficient skeletal muscle fibers demonstrate hyperintense SDH staining. This finding is due to proliferation of mitochondrial and mtDNA content as an attempted compensation for the respiratory chain defects: the histochemical correlate of RRF. The morphological features of RRFs are characterized by the aggregation of abnormal mitochondria, which stain an intense red (Gomori trichrome) creating the “ragged red” subsarcolemmal outline to muscle fibers. The molecular diagnostics of mitochondrial disorders is further reviewed by Rötig et al. (2004).
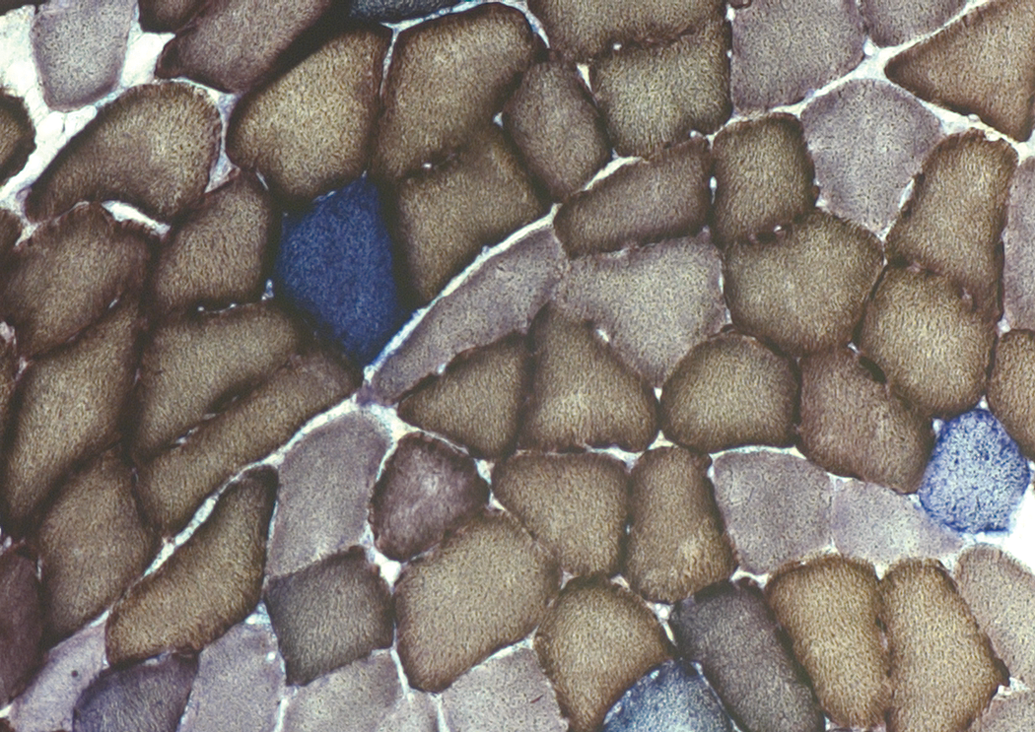
Sequential COX/SDH histochemistry of skeletal muscle from a patient with prior exposure to multiple pol γ inhibiting NRTIs, showing mosaic pattern of normal COX+ (brown) and COX-/SDH+ (blue) fibers (Payne et al. 2011) indicating mitochondrial defect due to a lack of mitochondrially encoded COX but sufficient genomic DNA encoded SDH. COX/SDH histochemistry typically exhibits a slightly granular appearance to the staining, which is thought to be due to localization of the mitochondria within the cytoplasm. COX = cytochrome c oxidase; NRTI = nucleoside analog reverse transcriptase inhibitor; SDH = succinate dehydrogenase.
Recently, techniques and new technology have been implemented to measure the respiratory chain activity in greater detail, such as the level of oxygen consumption and individual complex activity measurements. Such analyses do not provide a definitive diagnosis of mitochondrial disease, but they do complement a histochemical diagnosis. The final step in a complete mitochondrial pathological analysis is then making a molecular diagnosis. For example, sequencing of the mitochondrial genome will detect primary mtDNA disorders. Southern blot or real-time polymerase chain reaction (PCR) will detect the presence of mtDNA depletion (reduction in mtDNA content per cell); and long-range PCR will detect multiple large-scale mtDNA deletion mutations. Both these molecular abnormalities would point to the presence of an underlying mtDNA maintenance disorder due to a nuclear gene defect, which could then be screened for (Wong and Boles 2005).
NRTIs and Organ-specific Mitochondrial Pathology
Muscle Pathology
One of the defining clinical features of most inherited mitochondrial disease is myopathy. Therefore, in the late 1980s, when AZT-treated patients developed myopathy, mitochondrial abnormalities were sought. Since then, numerous other clinical and pathological adverse effects of NRTI therapy have been associated with acquired mitochondrial injury. In general terms, these toxicities have become apparent at the post-licensing stage, and the underlying histological and molecular defects have been subsequently defined through a combination of human, animal, and in vitro studies.
NRTIs, mtDNA, and the Pol γ Hypothesis
The biochemical and structural pathology of AZT myopathy was relatively well understood from the early 1990s; however, there was still a gap in the understanding of the molecular mechanism by which AZT caused such mitochondrial toxicity. Nuclear encoded disorders of mtDNA maintenance exert their effect on mitochondrial function via secondary mtDNA defects. These may take the form of depletion (reduction in cellular mtDNA content) or somatic (acquired) mtDNA mutations. The proposed molecular mechanism underlying NRTI-induced mtDNA depletion and consequent mitochondrial pathology has become known as the pol γ hypothesis (Lewis 2003). The theory states that the action of the mitochondrial polymerase is inhibited by chain termination due to the incorporation of NRTIs during mtDNA replication. Biochemical studies have demonstrated that the affinity of NRTIs for pol γ is hundreds of fold less than that for HIV-RT (HIV reverse transcriptase), but hundreds of fold greater than that for the human nuclear DNA polymerases. The affinity of NRTIs for the 4 major human DNA polymerases has thus been described γ > β > α = ∊ (Kakuda 2000). In vitro studies have demonstrated a “hierarchy” of pol γ inhibition among the NRTIs, namely ddC > ddI > d4T ≥ AZT > 3TC = ABC = TDF (Lim and Copeland 2001); this is further reviewed by Höschele (2006). One of the least inhibitory of these drugs, ABC, was highlighted to have a Ki (inhibitory constant) of 6 μM for pol γ, with stronger inhibitors such as ddC being significantly lower at 0.034 μM (Cherrington et al. 1994). In many respects, this hierarchy fitted well with emerging clinical impressions of the relative frequency of mitochondrial toxicities seen (as described in more detail later in this review). Perhaps the most striking anomaly in this hierarchy was the low relative level, compared to other NRTIs, to which AZT was seen to inhibit the polymerase function of pol γ, despite the well-described association with mitochondrial myopathy. However, this study also suggested alternative means by which AZT, at least in vitro, might detrimentally affect mtDNA, showing that AZT has a disproportionally strong adverse effect on the exonuclease function of pol γ and is particularly prone to cause base misincorporation. In a similar vein, other studies have hinted that mtDNA depletion may not be the sole pathological mtDNA defect in NRTI-induced mitochondrial toxicity. Early studies of AZT myopathy also showed the presence of large-scale mtDNA deletion mutations within affected muscle. These secondary mtDNA defects are a molecular hallmark of the myopathy seen in mtDNA maintenance disorders, such as inherited defects of polymerase γ gene–catalytic subunit, the nuclear gene encoding pol γ. However, the significance of these molecular defects was not fully explored at the time, and the role of somatic (acquired) mtDNA mutations in NRTI-induced mitochondrial toxicity has received relatively little study until recently. In our recent work, we have presented a new hypothesis linking pol γ inhibition, mtDNA depletion, and mtDNA mutations in skeletal muscle, suggesting that a temporary decrease in mtDNA content due to NRTI exposure causes an increase in the small number of preexisting mtDNA deletion mutations present within individual cells, through a clonal expansion mechanism (Payne et al. 2011). Recent investigation of NRTI-associated mtDNA molecular pathology beyond depletion, and its possible implications for patient health, is discussed further at the end of this review.
AZT Myopathy
As early as 1988, the seminal paper describing AZT myopathy was published and proposed that AZT rather than HIV was the cause of the myopathy in treated patients (Helbert et al. 1988). Helbert and colleagues described that the myopathy resolved upon AZT cessation but not on dose reduction. Subsequent animal work by Lewis et al. (1992) implicated mitochondria as the pathological target in the AZT myopathy, finding that rodents treated for 35 days with AZT presented selective changes in striated muscle which appeared to be ultrastructurally localized to the mitochondria upon EM analysis. The electron micrographs showed abnormal mitochondrial architecture and apparent lysis of mitochondria. Distortion was not only evident in the mitochondria but also within the muscle fiber morphology with thin sarcomeres present throughout the samples assessed (Figure 2). This study showed not only the morphological abnormalities of mitochondrial swelling and cristae disruption but furthermore demonstrated a reduction in mtDNA content, mtRNA, and respiratory chain protein production in AZT-treated animals compared with untreated controls (Lewis et al. 1992). Further EM work in rodent tissue revealed structural abnormalities that were in concordance with that found in human AZT subjects (Schroder et al. 1992). Despite both these studies linking AZT usage and mitochondrial dysfunction, patients at that time were treated with high doses of the drug. However, Tomelleri et al. (1992) subsequently found that abnormalities of mitochondrial EM were also seen in patients treated with lower doses of AZT (comparable with those used in contemporary practice). This study also assessed the respiratory chain activity of AZT-treated patients using COX histochemistry and found 9 of the 11 patients treated with AZT had focal COX deficiency. Furthermore, the biochemical COX assessment found that COX activity in AZT-treated patients was reduced by approximately 55% in comparison to control values, further suggesting that myopathy was mitochondrial in nature (Tomelleri et al. 1992). A more recent study noted significant histochemical COX defects in patients with AZT myopathy, but a minority of skeletal muscle fibers also showed decreased SDH expression (on immunohistochemistry). This phenomenon may be compensatory to the primary mtDNA defect that leads to the COX defect (although an increase in SDH activity due to mitochondrial proliferation would be more common in this scenario), or perhaps reflects additional abnormalities of mitochondrial protein translation. To definitively distinguish between these possibilities would require mtDNA and mtRNA copy number analysis within single muscle fibers (Yerroum et al. 2000).
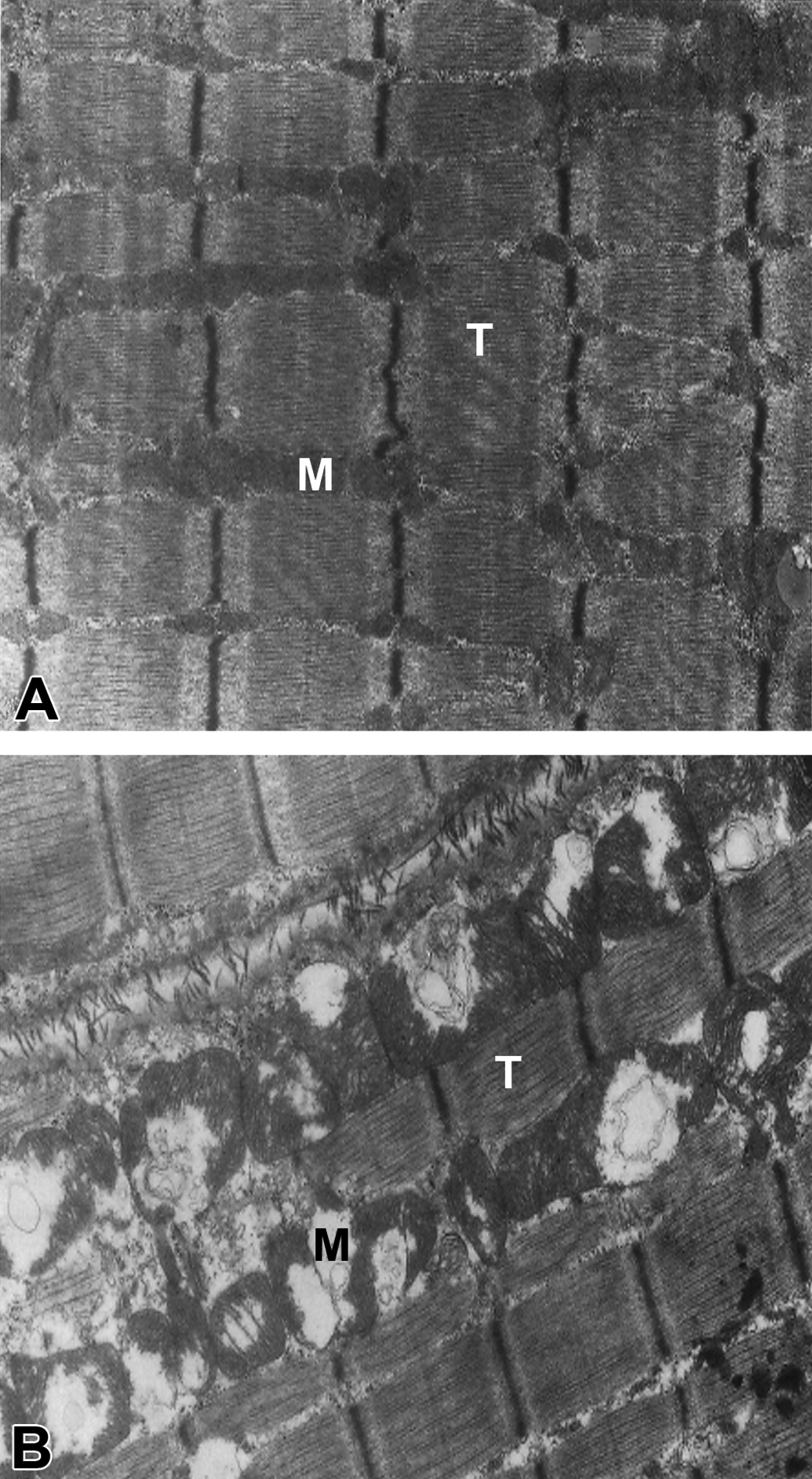
Photomicrographs of striated rat muscle (gastrocnemius) in controls (A) and AZT-treated rats (B). Image A highlights normal structure of striated muscle with no apparent mitochondrial damage (M). Image B clearly shows the damage sustained in rat muscle from AZT treatment hallmarked by the mitochondrial damage—lysis (bursting and degradation) and abnormally large morphology (M). This is coupled with apparent thinning of the sarcomere filaments (T) when compared to the untreated, image above Republished with permission of the American Society for Clinical Investigation, from the Journal of Clinical Investigation, Lewis et al. (1999) vol. 89, copyright. AZT = zidovudine.
Although much of the early data suggested that the myopathy seen in AZT-treated patients was mitochondrial, it was also previously accepted, from the pre-AZT era, that myopathy could be due to HIV infection itself. One study therefore set out to define the difference between AZT myopathy and HIV myopathy. It was concluded that histopathologically, AZT therapy can cause a toxic mitochondrial myopathy (evident by RRFs), but this appears to coexist with a T-cell–mediated inflammatory myopathy that is indistinguishable from that associated with untreated HIV infection. In patients with HIV myopathy, total cellular infiltrate was found, on average, to be almost double that of patients with AZT myopathy; there was also little histological evidence of mitochondrial damage (mean proportion of fibers which were RRFs was 0.4% in HIV myopathy, 13.86% in AZT myopathy; Dalakas et al. 1990). H&E staining of samples from patients presenting with proximal weakness due to HIV myopathy reveals degenerating basophilic myofibers and dark pink cytoplasmic bodies (Robinson-Papp and Simpson 2009). This suggests relatively distinct histopathological features between HIV and AZT myopathy. It should also be noted that HIV-associated myopathy is now a rare event and largely prevented by current HAART regimens (Robinson-Papp and Simpson 2009). We may therefore conclude that many patients presenting with clinical myopathy in the early days of the HIV epidemic, who frequently had a combination of advanced clinical disease and AZT exposure, exhibited the combined pathological effects of HIV and AZT on muscle. Although these effects appear to be clinically very difficult to distinguish, the histopathology suggests distinct processes.
ddC Myopathy
Although it is the most significantly implicated, AZT was not the only NRTI under scrutiny for being a cause of mitochondrial muscle pathology. 2′,3′-dideoxycytidine (ddC) has also been linked with myopathy that was suggested to be mitochondrially driven (Chen and Cheng 1989). However, the landmark “Delta” trial, which was the first randomized controlled trial of dual NRTI therapy (ddC/AZT), showed no excess of myopathy in the ddC/AZT arm compared with AZT alone (Darbyshire 1996). This finding is perhaps surprising if we consider the in vitro hierarchy of pol γ inhibition which implicated ddC as the most mitochondrially toxic of all NRTIs. In contrast, as we will describe below, ddC has instead been particularly implicated clinically in NRTI neuropathy. However, our recent work has considered cumulative lifetime rather than current NRTI exposure and looked at the accumulation of COX-deficient skeletal muscle fibers (Figure 1). Here we see that COX defect is indeed strongly predicted by lifetime exposure to potent inhibitors of pol γ such as ddC and ddI (Payne et al. 2011). Thus, at the histopathological level, other mitochondrially toxic NRTIs do indeed seem to have a detrimental effect on mitochondria in muscle, probably via an effect on somatic mtDNA mutations, even if such patients may not have clinically overt myopathy.
Cardiac Pathology
NRTI-associated myopathy has been most commonly studied in skeletal muscle; however, it seems likely that similar changes may be present (albeit, probably of a milder nature) in cardiac muscle tissue. Given the similarities of mitochondria in cardiac and skeletal muscle fibers, this is perhaps unsurprising. A few years after the reports of AZT-associated skeletal mitochondrial myopathy, some limited evidence emerged that patients receiving AZT were also at risk of cardiomyopathy (Herskowitz et al. 1992). Subsequently, there have been very little human data definitively linking NRTI-induced mitochondrial dysfunction and cardiomyopathy, and as such, the specific histological features are poorly characterized compared with skeletal muscle. This may partly be due to the fact that human cardiac muscle is not as easily studied, except at postmortem examination. It is also inevitable that the etiology of cardiomyopathy in HIV-infected patients is multifactorial and may include cardiovascular disease and alcohol. In contrast, however, there are significant rodent data to suggest that NRTIs can affect cardiac muscle mitochondria, so an NRTI effect on cardiac muscle in humans remains entirely plausible. When rats were treated with AZT, it was found on EM that the mitochondria of cardiac muscle were characteristically enlarged with little or no cristae present. Oxygen consumption rate was assessed and the areas with structurally abnormal mitochondria were found to have a decreased activity of mitochondrial complexes I and III (Lamperth et al. 1991). In more recent work, mice fed with either AZT or ddC for a 9-week period were found to have cardiomyopathy and enlarged mitochondria. The mtDNA content of the myocardial tissue was found to be decreased in both cases. The level of the mouse “common” (approximately 5 kb) large-scale mtDNA deletion mutation was also found to be increased in both the AZT and the ddC treatment groups (Balcarek et al. 2010).
Neuronal Pathology
Among the HIV community, there are a number of neurological complications that may develop, such as mononeuropathy, inflammatory demyelination, and autonomic neuropathy (Robinson-Papp and Simpson 2009). However, the most prevalent HIV-associated neurological complication is distal sensory polyneuropathy (DSP, also known as distal symmetric polyneuropathy). The disorder is generally characterized by painful dysesthesia that occurs spontaneously and often appeared to develop in the advanced stage of AIDS (Pardo, McArthur, and Griffin 2001). HIV sensory neuropathy (HIV-SN) therefore appeared to be related to the pathogenic effects of HIV itself perhaps driven by direct neurotoxicity of specific HIV proteins. However, almost clinically indistinguishable from HIV-SN is the antiretroviral toxic neuropathy (ATN) that occurs in the setting of NRTI treatment (Moyle 2000). Electrophysiologically, both of these are generally axonal neuropathies and are estimated to have a combined prevalence among the HIV-infected community of 30 to 60% (Kamerman et al. 2012). DSP is therefore regarded as the most prevalent neurological complication in the era of HAART (Zhou et al. 2007).
Peripheral Neuropathy
ddC and ddI were initially thought to be the cause of ATN. Dalakas, Semino-Mora, and Leon-Monzon (2001) compared three groups of patients: controls (HIV uninfected), those with ddC-neuropathy, and those with HIV-SN who had never been treated with ddC. The numbers of morphologically abnormal mitochondria were counted by the use of EM, and mtDNA content was later quantified. The results showed that across the board, morphological changes were observed, specifically axonal degeneration, with large-scale vacuolization of the mitochondria and significant cristae loss (Figure 3). However, of all mitochondria found in the axon and Schwann cells, approximately 55% were morphologically abnormal in some way (ddC-SN), compared to 9% of the untreated and HIV-SN cases. Significantly reduced mtDNA content was also detected (up to 80% depletion) in ddC-treated patients compared to the ddC-naive controls (including HIV-SN). Overall, the study concluded that the mtDNA was significantly affected by treatment with ddC and played a role in causing ddC neuropathy (Dalakas, Semino-Mora, and Leon-Monzon 2001). One of the earliest animal models used to investigate NRTI neuropathology was performed using ddC-treated rabbits. Pathological examination of the peripheral and central nervous system was performed along with electrophysiological studies. It was evident that after 16 weeks of treatment, neuropathological changes were present in the peripheral nerves, along with impaired nerve conduction. Histologically, a myelinopathy was associated with abnormal mitochondria in the Schwann cells, along with other pathologic changes including myelin splitting, intramyelinic edema, axonal degeneration, and reduction in diameter size (Anderson et al. 1994). More recently, focus has shifted to d4T as a cause of ATN (Affandi et al. 2008). d4T treatment is still widely used in developing countries and therefore numerically the neuropathological relevance of this drug remains high. Recent research in d4T-treated Malawians reported that 21% showed signs of neuropathy after 1 year of treatment (van Oosterhout et al. 2012). In reality, it seems likely that all the pol γ inhibiting NRTIs (ddI, ddC, d4T, and AZT) may to a greater or lesser extent cause an axonal neuropathy after exposure (Robinson, Li, and Nath 2007).
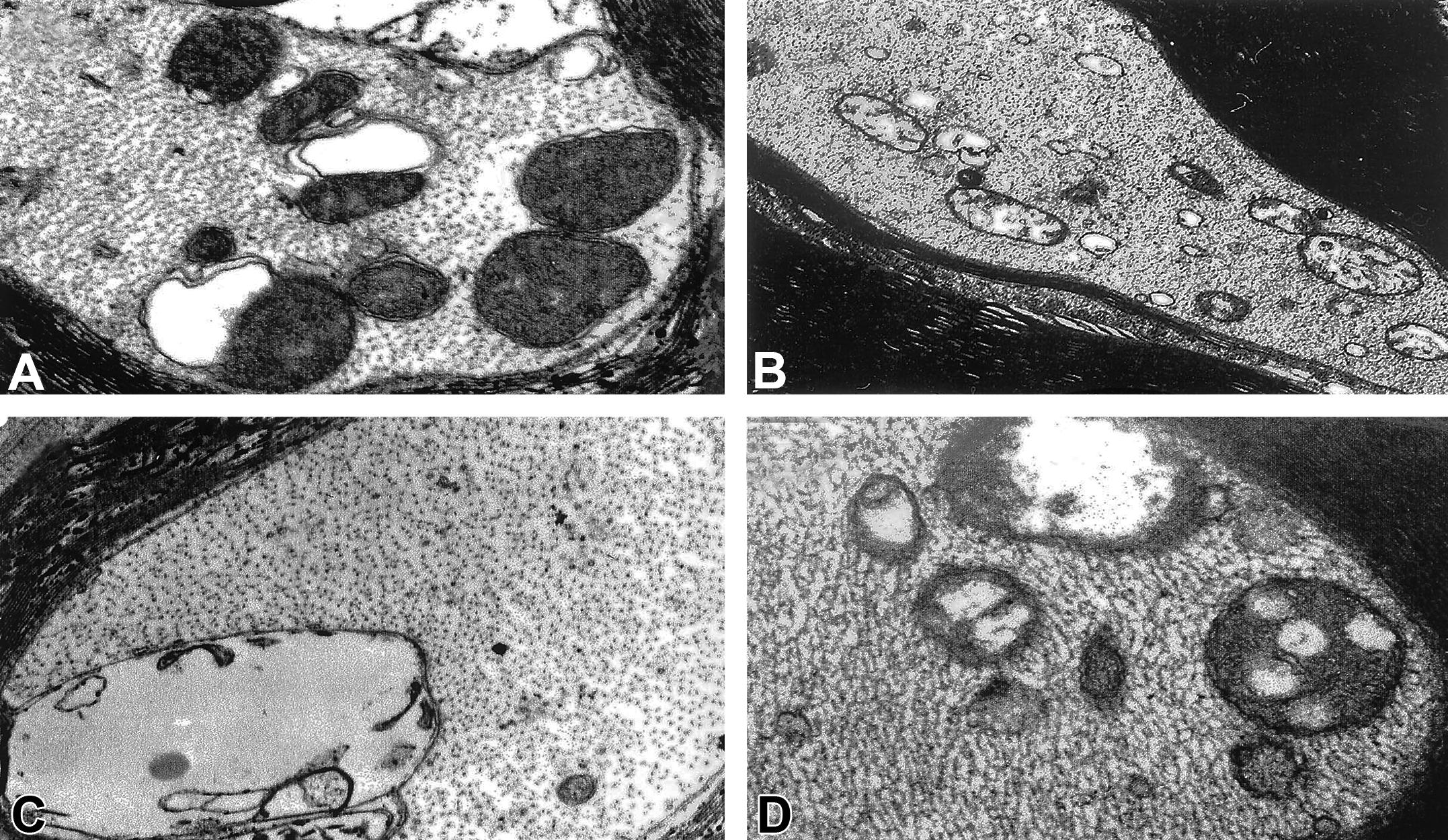
A–D illustrates electron micrographs of sural nerve biopsies (myelinated axons) taken from patients with ddC neuropathy. Large-scale vacuolization is evident in all images (prominently in C) along with disorganized matrices, swollen shape and loss of cristae (A–D). Mitochondria present in image D show signs of amorphous substance and large vacuoles. Magnification: A—37,500×; B—16,500×; C—32,500×; D—37,500×. Reprinted by permission from Macmillan Publishers Ltd.: Laboratory Investigation (Dalakas et al., 2001), copyright (2001). ddC = zalcitabine.
Lehmann and colleagues (2011) recruited patients both with and without HIV-SN and compared the levels of the 4,977 bp mtDNA “common deletion” mutation. They found that not only was there an increase in the frequency of the “common deletion” among patients with HIV-SN, but it was also more prevalent in the distal sural nerves in comparison to the dorsal root ganglia (Lehmann et al. 2011). Furthermore, this analysis was reinforced by findings from a primate model (macaques) of HIV-SN. This showed that macaques infected with a neurovirulent strain of simian immunodeficiency virus showed impaired mitochondrial function (on biochemical assays) in the mitochondria isolated from distal nerve fiber (sural nerves) when compared to the mitochondrial function in the proximal (dorsal root ganglia) nerve fibers (Lehmann et al. 2011). These findings, however, appeared to apply to HIV-SN in general, rather than delineating a specific role of NRTIs. Mitochondrial damage may therefore play a rather complex role in neuropathy, with mitochondrial abnormalities being prevalent in neuropathy per se, but exacerbated by d-drug induced mtDNA depletion.
Emphasis has been recently placed upon improved detection and monitoring of DSP among NRTI-treated patients, in part due to the high incidence rate in the developing world. Epidermal nerve fiber density (ENFD) has been validated as a reliable tool to measure nerve fiber damage (Rosenberg et al. 2005). ENFD is a technique that determines the size of the nerve fibers from a small skin biopsy; smaller fibers positively correlate with the severity of neuropathy. A recent AIDS Clinical Trials Group study has measured ENFD among 101 HIV-infected patients receiving HAART for at least 15 weeks (Zhou et al. 2007). However, the study found that although ENFD can be used to diagnose DSP, it did not correlate with any of the previously established risk factors for developing DSP (e.g., low CD4 lymphocyte count, high HIV-1 RNA plasma viral load, or exposure to specific NRTIs).
Central Nervous System Toxicity
Given the widespread occurrence of ATN, as well as many studies showing direct neurotoxicity of NRTIs such as ddI and d4T (described above), we may speculate whether mitochondrially mediated neurotoxicity also occurs in the brain. To our knowledge, relevant human data are not available; however, AZT exposure in rodents (at plasma levels equivalent to that of human dosing [Blum et al. 1988]) has been shown to cause (undefined) behavioral changes (Venerosi, Calamandrei, and Alleva 2002). These findings may be regarded as very nonspecific, and the mechanisms behind such changes require further elucidating.
Lipid Pathology
Abnormalities of fat metabolism in HIV infection and HAART treatment are widespread and are likely to be multifactorial in their pathogenesis. The common manifestations are “high-risk” serum lipid profiles and lipodystrophy (peripheral lipoatrophy and central lipohypertrophy; Carr et al. 1998, 2000).
Lipodystrophy
Lipohypertrophy (ectopic fat deposition, such as the “buffalo hump,” and central adiposity) was initially attributed to the PIs (Martínez 1998). It subsequently became clear that patients who were PI naive but receiving thymidine analog NRTIs (d4T or AZT) presented with peripheral fat wasting (facial and limb lipoatrophy; Moreno and Martínez 2000). A number of studies have shown that where lipoatrophy is present in those treated with NRTIs, mtDNA levels and respiratory chain function were decreased in comparison to those without any form of lipoatrophy, including within adipose tissue (Walker et al. 2002; Sievers et al. 2009). Histopathological features include smaller and more angular adipocytes and an increase in lipogranulomas (inflammatory lipid deposits). Data have suggested that the cause of lipoatrophy at the cellular level may be adipocyte apoptosis mediated through the mitochondrial pathway; however, some believe that inflammation due to cytokine dysregulation may be of more importance (Kotler et al. 2003; Hooker et al. 2009). It is interesting to note that lipodystrophy is not well associated with the most potent pol γ inhibitors, ddI and ddC, suggesting that there are likely to be additional adverse effects of the thymidine analogs within adipose tissue beyond mtDNA depletion.
Tissue-specific Pathology Associated with NRTIs
Lactic Acidosis
Although myopathy was the first pathology to be described as an NRTI side effect, lactic acidosis was also very prevalent among such patients. However, the earliest case of HIV-associated lactic acidosis (another hallmark of inherited mitochondrial disease) was described in 1993 and then was quickly linked with both AZT and ddI monotherapy (Baram and Cooke 1993). It was later found that all of the dideoxynucleoside analogs (“d-drugs”) were implicated in lactic acidosis. Not all NRTI-associated hyperlactemia results in symptomatic lactic acidosis; however, serum lactate levels of 2 to 5 mmol/L are often considered as significant and when such a level is persistent, may indicate an increase in mitochondrial damage and abnormal mitochondrial function (Gerschenson and Brinkman 2004).
Pancreatitis
Pancreatitis is a rare but serious complication described with ddC and ddI, presenting at a rate of 1.27 per 1,000 person-years for ddI treatment (Smith et al. 2008). Pancreatitis in patients receiving ddI very frequently coexists with lactic acidosis that is very suggestive of a mitochondrial etiology (Gerschenson and Brinkman 2004). A definitive mitochondrial pathway has not been clearly defined, but this may be due to the relatively small number of cases.
Hepatic Pathology
Hepatotoxicity as measured biochemically by modest elevations in hepatic transaminases appears to occur at a rate of 5 to 15% in HIV patients treated with NRTIs (Dolin et al., 1995). The pathophysiology is likely to be multifactorial; however, hepatosteatosis appears to account for at least some of the liver dysfunction seen in HAART-treated patients (Duong Van Huyen et al. 2003). The extent to which NRTI-associated mitochondrial dysfunction may contribute to hepatosteatosis remains unclear; however, it has been implicated in the non-HIV setting (Sunny et al. 2011). In terms of more severe hepatic injury, both animal and human studies have illustrated a link between NRTI treatment and hepatic mitochondrial toxicity. Human-equivalent doses of d4T were given to Erythrocebus patas monkeys for 80 days. They developed mtDNA depletion in hepatic mitochondria along with subnormal mitochondrial oxidative activity and elevated serum lactate levels (Dagan et al. 2002). Similarly, in the early days of NRTI treatment, occasional patients developed severe, even fatal, hepatic injury associated with lactic acidosis, and mtDNA depletion and large-scale deletion mutations in liver have been reported and reviewed (Senise, Castelo, and Martinez 2011; Montessori, Harris, and Montaner 2003).
Peripheral Blood
Owing principally to convenience of sampling, mitochondrial toxicity of NRTIs has frequently been assessed in peripheral blood mononuclear cells (PBMCs), particularly with respect to mtDNA depletion. In general terms, mtDNA depletion in PBMCs has been consistently documented in the setting of exposure to specific NRTIs, and the severity of depletion shows reasonable correlation with in vitro data on mtDNA depletion and the potency of pol γ inhibition (Lim and Copeland 2001). However, it is clear that there is not any well-defined clinical toxicity in blood that arises due to mtDNA depletion. One study has shown some correlation between PBMC mtDNA depletion and hyperlactemia; however, this finding has not been consistent (Cote et al. 2002). Furthermore, the correlation between mtDNA depletion in PBMCs and mitochondrial toxicity in clinically relevant tissues is debatable. Upon simultaneous sampling of PBMCs and subcutaneous fat, it was found that in ddI or d4T treatment, the presence of lipoatrophy did not correlate significantly with lower mtDNA level in PBMCs, but a significant correlation with the mtDNA level in fat was seen (Cherry et al. 2006). Finally, several studies have shown that untreated HIV infection may cause a severe depletion of mtDNA in PBMCs in the absence of NRTI treatment (Casula et al. 2005; Maagaard et al. 2006). Maagaard and colleagues state that due to the lack of good correlation in mtDNA content between tissues, and difficulties in differentiating mtDNA depletion due to HIV and NRTI treatment, PBMCs should be excluded as a potentially relevant biomarker of mitochondrial toxicity and the focus should remain with a more tissue-specific approach (Maagaard et al. 2006).
Nephropathology
TDF is a prodrug for the active metabolite tenofovir and is the most commonly used N(t)RTI in industrialized countries. In general terms, it is considered to have little toxicity; however, it has been suggested to cause renal dysfunction, and in particular tubulopathy. From a mitochondrial perspective, in vitro studies have demonstrated that TDF appears to have a negligible effect on pol γ function (Birkus, Hitchcock, and Cihlar 2002) and the typical clinical mitochondrial toxicities are not observed (Nelson et al. 2007). However, renal tubulopathy is a feature of some inherited mitochondrial disorders, and the question of whether TDF may exert specific and unexpected effects on renal tubular mitochondria is the subject of ongoing research. There is pathological evidence from humans to suggest that TDF has a tissue-specific effect on the proximal tubules, which is associated with morphological changes in mitochondria (Woodward et al. 2009). It has been suggested that such changes may be due to the nature of the proximal tubules lacking the ability to perform anaerobic glycolysis, relying heavily on the localized mitochondria to function efficiently (Hall 2012). Hall and colleagues present a case series of 22 patients receiving TDF all presenting with nephrotoxicity which represents 1.5% of all patients from the referring center. Kidney biopsies of the patients showed acute tubular damage and interstitial edema. Misshapen mitochondria and giant, swollen mitochondria located in the proximal tubules were seen on EM (Woodward et al. 2009).
Current Controversies
Are Current HAART Regimens Free from Mitochondrial Toxicity?
As described above, newer N(t)RTIs such as ABC and TDF appear to show a low affinity for the mitochondrial polymerase and clinically have little effect on mitochondria. However, it is possible that lower level mitochondrial toxicity may build up over time, or new organ-specific mitochondrial pathologies may emerge as has been hypothesized for TDF-induced renal tubulopathy.
Recently, the use of NRTI-free HAART regimens has been proposed, in an attempt to reduce the morbidity of NRTI-associated side effects (Riddler et al. 2008). Another method of reducing morbidity would be a more personalized approach to therapy based on increased knowledge of susceptibility determinants for NRTI toxicity, including genetic predisposition (Maggiolo, Ripamonti, and Suter 2005; Hulgan et al. 2005; Kallianpur and Hulgan 2009; Canter et al. 2008).
Preclinical Determination of Mitochondrial Toxicity
The pre-licensing pharmacology reviews (accessed from http://www.fda.gov/; on March 20, 2013) of three NRTI drugs (AZT, ABC, and FTC) were reviewed to assess at which stage (if any) mitochondrially mediated side effects were suspected, and whether any specific mitochondrial toxicology investigations were performed. The three drugs were chosen due to large time gap (16 years) of approval between them: AZT—1987, ABC—1998, and FTC—2003.
Throughout the pre-licensing preclinical studies, safety testing revealed no data indicating mitochondrial toxicity from the NRTI in question; in all cases, this was first highlighted only in post-licensing studies. There were no specific mitochondrial toxicological assessments until the most recent NRTI licensed, FTC, in 2003, despite the issue of mitochondrial toxicity first being reported in 1988. Within the pharmacology review of this drug, there was a specific aim to determine the affinity to pol γ and whether there were any effects seen on the quantity of mtDNA in an in vitro setting (MOLT-4 cells—Human T lymphocyte cells). However, due to the long development time of drugs, it may be reasonable to assume that until recently the knowledge of mitochondria and NRTI toxicity was limited. Therefore, although no specific pathology in the pre-licensing studies suggested mitochondrial toxicity, there were no specific tests performed to assess whether this would be the case, either for AZT or ABC. A useful initial assessment that would indicate potential mitochondrial damage within the preclinical safety package would be EM examination of likely target organs (muscle, nerve, fat, etc.). This EM examination would reveal any abnormal mitochondrial morphology associated with the NRTI treatment. Used in this way, EM should be highly sensitive; however, it will have poor specificity for the presence of a specific NRTI-induced mtDNA defect. While COX histochemistry is very useful and highly specific in the diagnostic evaluation of patient samples, defects might not reliably appear within the time scales of typical preclinical studies. It is also suggested that in vitro studies become routine practice in the preclinical assessment of new antiretroviral compounds as an “early warning” of mitochondrial dysfunction. Determination of cellular mtDNA quantity would be a useful indication of how toxic the NRTI in question could potentially be and would be a useful assessment to be implemented in any future NRTI licensing studies, such as that performed with FTC. The overall “health” of mitochondria could be further assessed by measurement of the respiratory rate from cells in vitro. We suggest that the combination of these two measures in vitro, along with EM on rodent samples, is likely to give the best sensitivity and moderate specificity. This would also provide the Food and Drug Administration with sufficient data to decide whether any form of mitochondrial toxicity is an issue at the time and thus, issue a request for further safety data investigations.
Beyond the Pol γ Hypothesis
It will be clear from the evidence presented above, that a simple linear relationship between pol γ inhibition, mtDNA depletion, and mitochondrial dysfunction at the tissue level is insufficient to fully explain the mitochondrially mediated toxicity of NRTIs. In addition to their role in mtDNA maintenance disorders, somatic mtDNA mutations are found within cells during the “normal” human aging process, as well as with the degenerative diseases of older age.
It has recently been suggested that HIV-infected patients, who are receiving successful (virally suppressive) antiretroviral therapy, show signs of an accelerated senescence and aging process (Effros et al. 2008), including increased frailty and age-associated diseases. Furthermore, recent studies have examined whether somatic mtDNA mutations arise or may be increased in the setting of long-term NRTI exposure. Although there are some conflicting results, it certainly appears that mtDNA mutations are increased, at least in postmitotic (nondividing) tissues, such as skeletal muscle, as has been described briefly above. In contrast to mtDNA depletion, mtDNA mutations will not be expected to be reversible once the “culprit” NRTI is switched to a cleaner agent. Recent data support this concept, suggesting that mtDNA defects and cellular mitochondrial dysfunction (COX-deficient cells) appear to persist long after exposure to the causative NRTI has ceased (Payne et al. 2011; Figure 4). These observations require further delineation as to what exactly the functional consequences of this persistent damage may be and to what extent it interacts with the natural aging process and frailty. The various molecular processes that may lead to cellular mitochondrial defects in the setting of NRTI therapy are summarized in Figure 5.
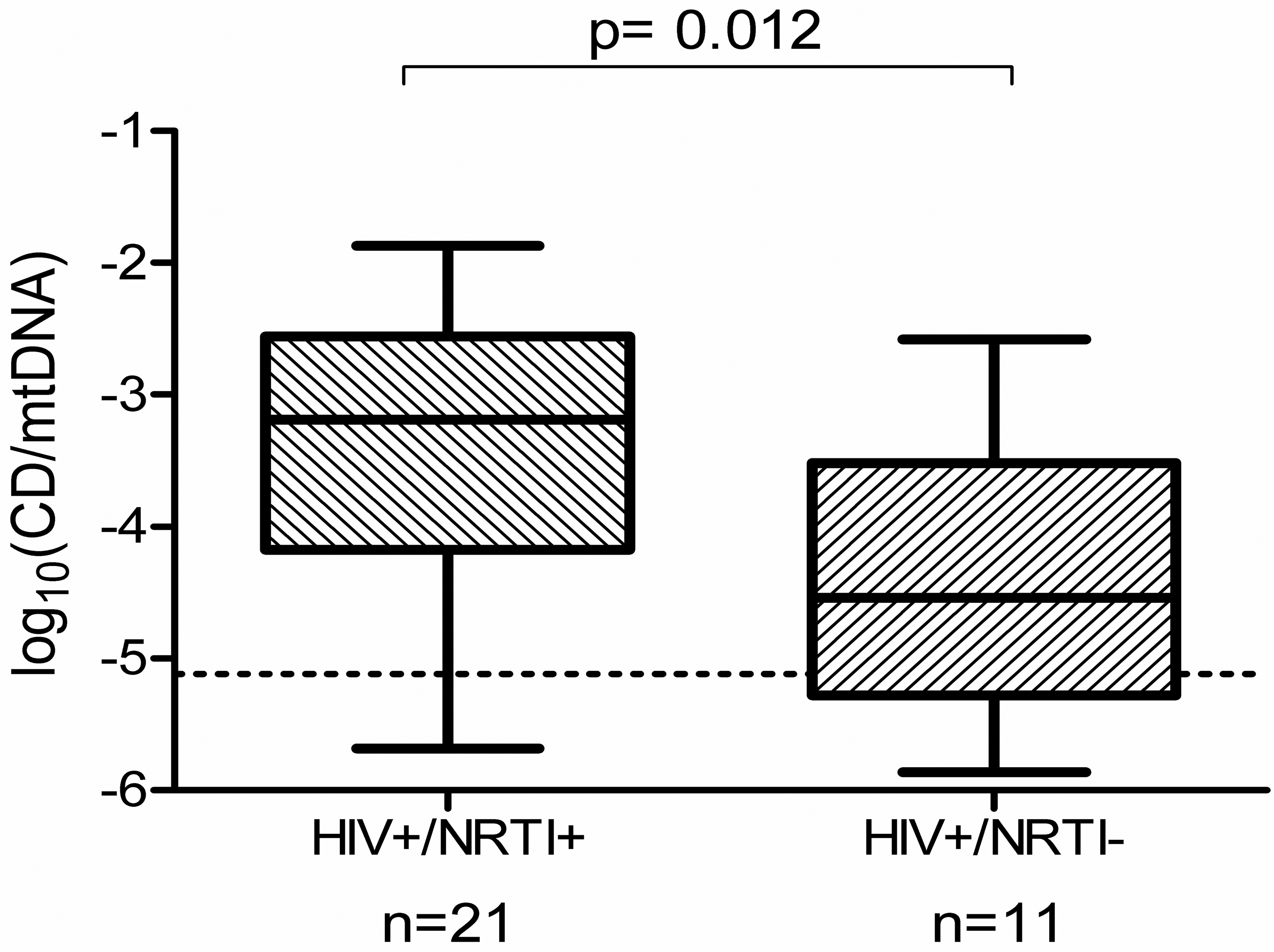
Box and whisker plot showing significantly higher proportional levels of the δ4,977 bp mtDNA “common deletion” (CD) mutation within homogenized lower limb skeletal muscle of HIV-infected patients, with (NRTI+) and without (NRTI−) exposure to pol γ inhibiting NRTIs. Dotted line indicates the lower limit of sensitivity of the real-time PCR assay (Payne et al. 2011). mtDNA = mitochondrial DNA; NRTI = nucleoside analog reverse transcriptase inhibitor; PCR = polymerase chain reaction.
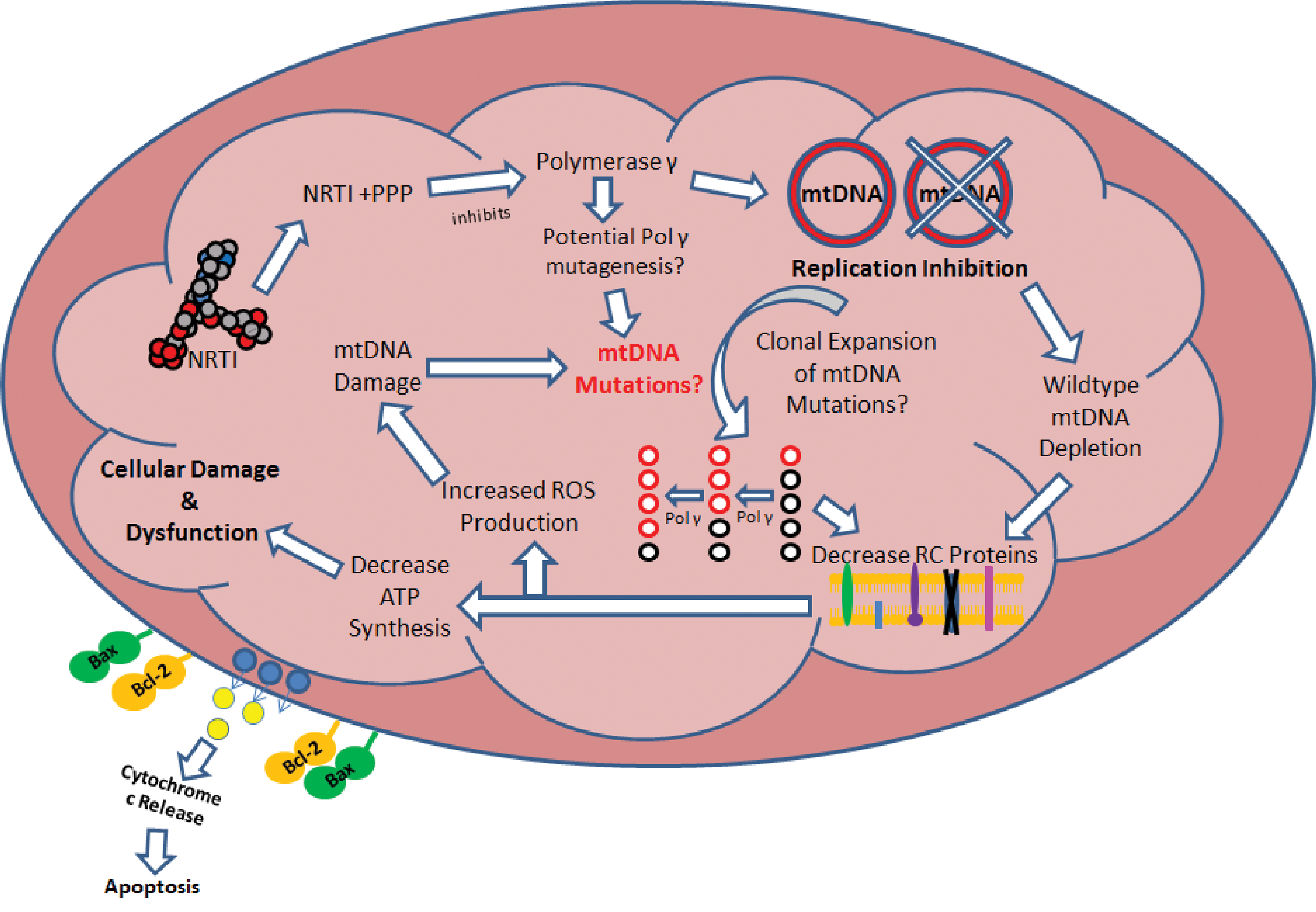
Mechanisms leading to cellular defects due to NRTI incorporation into polymerase γ. Once the NRTI has been transported into the mitochondria, it initially undergoes phosphorylation by either thymidine kinase-2 or deoxyguanosine kinase, followed by further phosphorylation to a triphosphate form of the drug (NRTI + PPP). This active metabolite is then incorporated into the replication of mtDNA by pol γ, inhibiting the full replication and therefore causing a decrease in the quantity of mtDNA. Disruption of the mtDNA level will eventually cause a decrease in the amount of respiratory chain (RC) proteins synthesized, ultimately leading to respiratory defects and cellular damage (illustrated by apoptosis and COX proteins). The process as a whole ultimately leads to the question as to whether mtDNA mutations are the consequence of the toxicological interaction and whether clonal expansion plays a part in the accumulation of mtDNA mutations. COX = cytochrome c oxidase; mtDNA = mitochondrial DNA; NRTI = nucleoside analog reverse transcriptase inhibitor.
Conclusions
The pattern of use of the older NRTIs has changed significantly over the last decade. Despite being the original antiretroviral, AZT remains widely used in the developing world and is still in use in specific situations in industrialized countries. d4T has received extensive use in the developing world antiretroviral “rollout” programs; however, its use is now being scaled back in the light of World Health Organization (2007) guidance around toxicity. ddI is still very occasionally used in industrialized countries, and ddC is no longer licensed owing to its toxicity. In the modern era, the two main concerns are therefore whether there is a persistent burden of mitochondrial damage in the many patients in industrialized countries with prior exposure to the older and what the clinical effects of recent AZT and d4T use on a very large scale (millions of patients) will be in developing countries (van Oosterhout et al. 2012).
The use of NRTIs over the last 25 years has helped transform mortality rates among HIV-infected patients. As we have used these drugs clinically, we have become aware of their multifaceted mitochondrial toxicity (Table 1). As with the inherited mitochondrial diseases, the correlation between molecular pathology, histopathology, and clinical phenotype has not always been clear-cut. Although the use of NRTIs has changed over the last quarter century, and the clinical treatment priorities have evolved, understanding drug-induced mitochondrial damage remains relevant to modern HIV care. Animal and in vitro studies continue to increase our knowledge of the precise molecular mechanisms involved. However, a greater effort may be required to specifically assess any potential mitochondrial damage during future novel NRTI drug development.
Summary of NRTIs and their putative mitochondrial pathology.
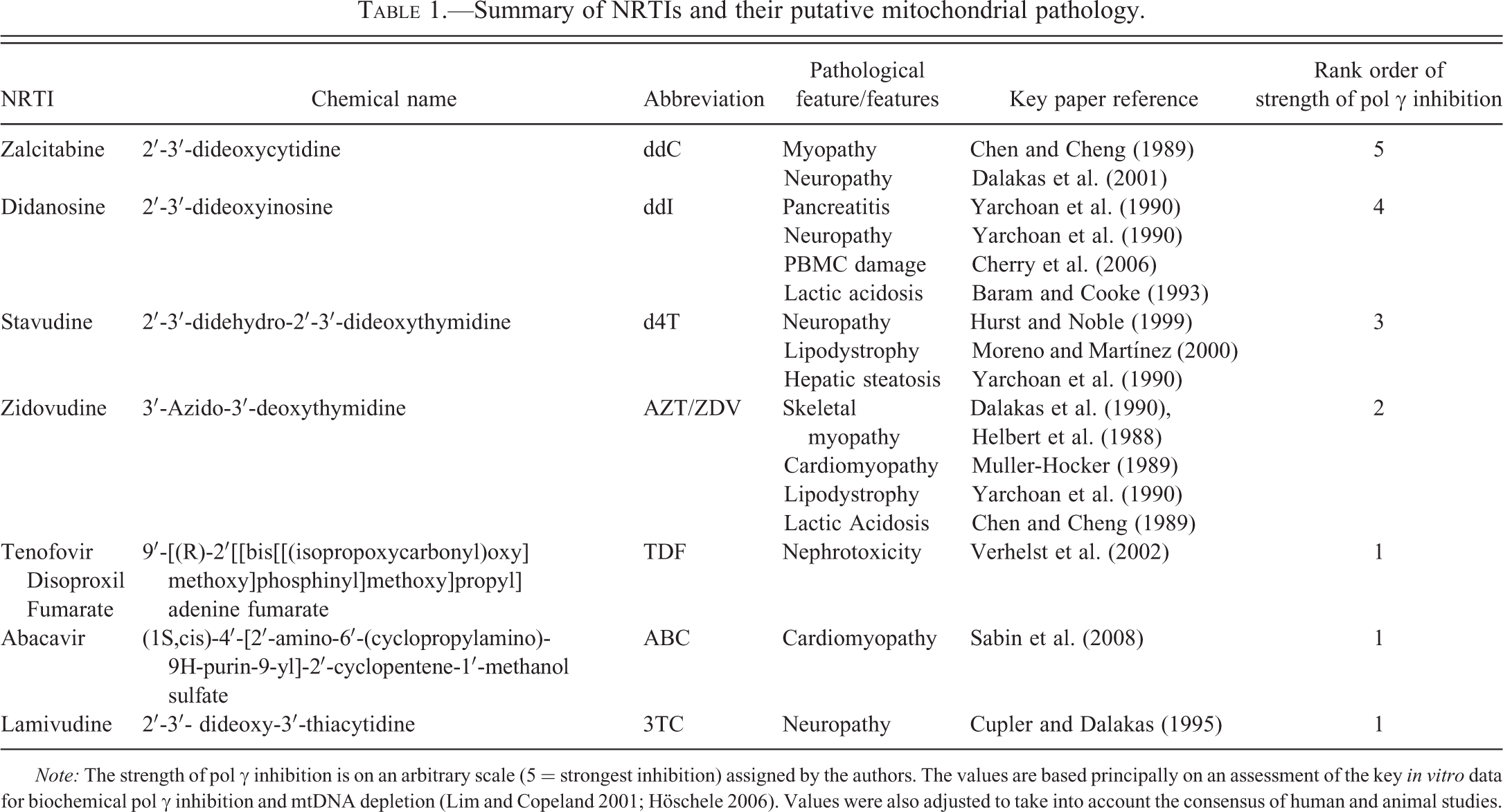
Note: The strength of pol γ inhibition is on an arbitrary scale (5 = strongest inhibition) assigned by the authors. The values are based principally on an assessment of the key in vitro data for biochemical pol γ inhibition and mtDNA depletion (Lim and Copeland 2001; Höschele 2006). Values were also adjusted to take into account the consensus of human and animal studies.
Finally, as NRTI treatment presents a unique situation of iatrogenic mitochondrial damage, it offers the potential to also further our understanding of mitochondrial biology in human health and disease.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.