
Abstract
3,3′,4,4′-tetrachloroazobenzene (TCAB) is a contaminant formed during manufacture of various herbicide compounds. A recent National Toxicology Program study showed B6C3F1 mice exposed to TCAB developed a treatment-related increase in lung carcinomas in the high-dose group, and urethral carcinomas, an extremely rare lesion in rodents, in all dose groups. As the potential for environmental exposure to TCAB is widespread, and the mechanisms of urethral carcinogenesis are unknown, TCAB-induced urethral and pulmonary tumors were evaluated for alterations in critical human cancer genes, Kras and Tp53. Uroplakin III, CK20, and CK7 immunohistochemistry was performed to confirm the urothelial origin of urethral tumors. TCAB-induced urethral carcinomas harbored transforming point mutations in K-ras (38%) and Tp53 (63%), and 71% displayed nuclear TP53 expression, consistent with formation of mutant protein. Transition mutations accounted for 88% of Tp53 mutations in urethral carcinomas, suggesting that TCAB or its metabolites target guanine or cytosine bases and that these mutations are involved in urethral carcinogenesis. Pulmonary carcinomas in TCAB-exposed animals harbored similar rates of Tp53 (55%) and Kras (36%) mutations as urethral carcinomas, suggesting that TCAB may induce mutations at multiple sites by a common mechanism. In conclusion, TCAB is carcinogenic at multiple sites in male and female B6C3F1 mice through mechanisms involving Tp53 and Kras mutation.
Keywords
Introduction
3,3′,4,4′-tetrachloroazobenzene (TCAB) is generated as a synthetic contaminant and/or degradation product during the manufacture of 3,4-dichloroaniline and its herbicidal derivatives Propanil®, Linuron®, Diuron®, and Neburon® (Poland et al. 1976; Miller, Zisook, and Zepp 1980; Hill et al. 1981). Exposure of humans to TCAB may potentially occur through several routes including the manufacture and application of 3,4-dichloroaniline and its herbicidal derivatives, consumption of crops contaminated with 3,4-dichloroaniline-derived herbicides, and the use of 3,4-dichloroaniline in other products such as dyes (Taylor et al. 1977; U.S. Environment Protection Agency 1985). Environmental contamination by TCAB can occur from chloroanilide herbicide (acylanilides, phenylcarbamates, and phenylureas) degradation by peroxide-producing soil microorganisms (Bartha, Linke, and Pramer 1968) and providing another route of potential exposure. TCAB produces toxic effects in rodents similar to dioxins since it shares structural similarity to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD or dioxin), and possesses aryl hydrocarbon receptor-binding activity (van Birgelen et al. 1999) and may be subject to enterohepatic cycling (Pillai et al. 1996). Based on this similarity and the possibility for human exposure, toxicity and carcinogenicity testing of TCAB is of high importance.
There is a limited body of mutagenicity data for TCAB in the literature including conflicting outcomes of assays, thus making definitive conclusions difficult. Convincing evidence that TCAB or its metabolites are genotoxic has not been forthcoming. In Salmonella typhimurium reversion assays and Chinese hamster ovary/hypoxanthine–guanine phosphoribosyltransferase assays (with and without S9 metabolic activation), Propanil® and 3,4-dichloroaniline derivatives failed to show any genotoxic effects (McMillan et al. 1988). Weak mutagenicity has been detected over a narrow range of concentrations in the S. typhimurium fluctuation test and in the TA97 strain when accompanied by rat liver S9 activation (van Birgelen 1998; van Birgelen et al. 1999; Witt et al. 2000). Further evidence that TCAB has the potential to act as a genotoxic carcinogen was shown by in vivo mouse micronucleus tests, in which significant increases in micronuclei were detected after subchronic exposure of male and female mice to TCAB (Witt et al. 2000).
The National Toxicology Program (NTP) performed a 2-year chronic bioassay investigating the exposure of TCAB in B6C3F1 mice and Harlan Sprague Dawley (HSD) rats (NTP 2010). In rats, significant treatment-related findings included increased incidences of cystic keratinizing epithelioma of the lung in males and females, and an increased trend of cholangiocarcinoma in male rats. In mice, exposure to TCAB resulted in the development of neoplastic lesions in the urinary tract, lung, stomach, and skin, providing clear evidence of the carcinogenic activity of this chemical (Singh et al. 2010). The primary sites of neoplasia in the urinary tract were the urethra and ureter. There was also a significantly increased incidence of pulmonary adenoma in all male dose groups and a significantly increased incidence of pulmonary carcinoma in the high-dose (30 mg/kg) female group. Pulmonary and urethral carcinomas were not observed in the concurrent HSD rat study. In other short-term (13-week) studies using a structurally related compound, 3,3′,4,4′-tetrachloroazoxybenzene in mice and rats, urethral lesions were not observed in B6C3F1 mice or F344 rats (NTP 1998).
Proliferative lesions of the lower urinary tract are extremely rare in rodents; there have been no reports of spontaneous neoplastic lesions of the urethra or ureter in the NTP historical control database reported for B6C3F1 mice or F344/N rats. In addition, there has only been one chemical (nitriloacetic acid trisodium monohydrate) tested by NTP, which has been associated with an increased incidence of ureteral carcinomas; and this occurred in female rats (Goyer et al. 1981). The effects of TCAB exposure were more pronounced in the urethra with a high incidence (98%) of transitional epithelial tumors in male mice (Singh et al. 2010). A less marked effect was observed in the ureter, where the incidence of transitional epithelial tumors was 2%. In all animals, carcinomas were invasive to surrounding tissue and/or metastatic to distant sites (Singh et al. 2010). Although a rare occurrence, bladder carcinomas in mice have been shown to arise through a sequence of morphological changes studied and described in animals treated with N-butyl-N-(4-hydroxybutyl)nitrosamines (Becci et al. 1981; Ogawa et al. 1998). The sequence of changes begins with the formation of nonpapillary dysplastic lesions, progressing to carcinoma in situ and then to invasive and metastatic carcinoma (Cohen 2002). Correlation of the morphological features of neoplasia with genetic changes has shown that mutations in the Tp53 gene occur in dysplastic and carcinomatous lesions in approximately 70% of mice (Ogawa et al. 1998; Morimura et al. 1999). Furthermore, it has been shown that the disruption or activation of various genetic pathways leading to urothelial tumorigenesis in rodents has involved other key tumor suppressor and oncogenes that may play a role in human bladder cancer. These include cell cycle genes (Tp53, Cdk1, Rb1, and Cdkn2a), apoptosis genes (Bcl2), and signal transduction genes (Hras, Kras, Fgfr3, and Erbb2; Knowles 2001; Mitra, Bartsch, and Cote 2009). In particular, TP53 genetic mutations are independent prognostic factors for poor progression-free survival in urothelial carcinomas (Ecke et al. 2008). In human pulmonary carcinomas, Kras accounts for 90% of RAS mutations, and approximately 97% of Kras mutations involve codons 12 or 13 (Forbes et al. 2006).
The potential for environmental exposure to TCAB is widespread, and the underlying tumorigenic mechanisms in TCAB-treated B6C3F1 mice are unclear. Therefore, our primary objectives in the current study were to confirm the urothelial origin of TCAB-induced urinary tract tumors and to evaluate TCAB-induced urinary tract and pulmonary tumors for the involvement of alterations in oncogenes and tumor suppressor genes (K-ras and Tp53) important in the pathogenesis of a variety of human cancers, including lung and urinary tract neoplasia (Berggren et al. 2001; Lin et al. 2005; Ecke et al. 2008).
Materials and Methods
Histopathology and Immunohistochemistry (IHC)
Histopathologic lesions observed and tissue handling in this study were described in detail by Singh and colleagues (2010). Serial sections of 6 µm and 8 µm were cut for IHC and laser capture microdissection (LCM), respectively. Proliferative urothelial and pulmonary lesions induced by TCAB in B6C3F1 mice in the NTP 2-year bioassay included 26 urethral hyperplasias, 129 urethral carcinomas, 1 ureteral carcinoma, 1 renal (pelvic) carcinoma, 40 pulmonary adenomas, and 14 pulmonary carcinomas (NTP 2010). Based on their relatively large size, 21 urothelial samples (8 normal vehicle controls, 3 urethral hyperplasias, and 10 carcinomas [8 urethral, 1 ureteral, and 1 renal]) were chosen for IHC and mutation analysis. There were 9 pulmonary adenomas (8 TCAB treated and 1 spontaneous) and 6 pulmonary carcinomas (3 TCAB treated and 3 spontaneous) analyzed.
Laser Capture Microdissection (LCM)
Due to their small size, urethral carcinomas and normal urothelium were isolated by LCM using the MMI® CellCut Plus® Laser Microdissection System (Molecular Machines, MI, USA). In general, 500 hits with a laser pulse were used to obtain approximately 1,000 cells or 2 × 106 μm2 of tissue. The cap with the attached transfer film and captured tissue was then fitted onto a 0.5-ml microtube containing 50-μl lysis buffer (0.5% Tween-20, 1 mM ethylene diamine tetraacetic acid pH 8.0, 50-μM Tris pH 8.5, 0.5-μg/μl Proteinase K). DNA was extracted from 19 LCM samples and 20 whole lysate samples using the Arcturus® PicoPure® DNA Extraction Kit (Life Technologies, Foster City, CA).
Mutation Analysis
Ten 10-µm serial sections of each lung tumor were scraped from noncharged slides using a scalpel blade for DNA isolation. DNA was isolated from urethral carcinomas and associated normal tissues using LCM. DNA amplification reactions were carried out using seminested polymerase chain reaction (PCR) using the primer sets for exons 5 to 8 of the mouse Tp53 gene and exons 1 to 2 of the mouse K-ras gene, corresponding to the hot spot mutation sites in humans. Controls lacking DNA were run with all sets of reactions. PCR products were purified using a QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). The purified PCR products were cycled with Terminal Ready Reaction Mix-Big Dye (Perkin Elmer, Foster City, CA), and the extension products were purified with the DyeEx 2.0 Spin Kit (Qiagen, Valencia, CA). The lyophilized PCR products were sequenced with an automatic sequencer (Perkin-Elmer ABI model 3100). Comparisons were made of electropherograms from vehicle control and TCAB-treated group samples.
IHC
IHC was performed on TP53, uroplakin III (UPIII), CK7, and CK20. CK7 is a 54-kDa intermediate filament protein showing cytoplasmic localization that recognizes simple epithelium in most glandular and transitional epithelium but not stratified squamous epithelium. CK20 is a type I keratin primarily expressed in gastric and intestinal epithelium, urothelium showing cytoplasmic localization. For IHC, unstained, paraffin-embedded tissue sections were deparaffinized, rehydrated, and subjected to antigen retrieval using 1× citrate buffer in a Decloaking Chamber (Biocare Medical, Walnut Creek, CA) for 5 min at 120°C. Following nonspecific blocking steps, polyclonal and monoclonal primary antibodies were applied at the following dilutions: polyclonal rabbit anti-cytokeratin 20 (Abcam, Cambridge, MA) at 1:50; monoclonal mouse anti-cytokeratin 7 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:50; monoclonal mouse anti-UPIII antibody (American Research Products, Waltham, MA) at 1:100; and polyclonal rabbit anti-p53 (Vision Biosystems Inc, Norwell, MA) at 1:500. Negative controls substituted normal serum from the same species the primary antibody was made in. Positive controls included male reproductive and gastrointestinal tract, tissues known to express the proteins of interest. The avidin–biotin enzyme complex was used for primary antibody-binding amplification, and 3,3′-diaminobenzidine was used for visualization. Sections were then dehydrated in graded alcohols and coverslipped. Detailed protocols can be found at http://www.niehs.nih.gov/research/atniehs/labs/lep/path-support/core-support/immuno/protocols/immunohistochemistry/index.cfm
Results
Urethral Carcinoma Protein Expression
Since the tumors seemed to arise from the urothelial surface in some cases while others appeared to arise from the underlying urethral glands, characterization of these tumors as to the cell of origin and differentiation was essential. Exposure of B6C3F1 mice to TCAB resulted in a dose-dependent increase in urethral carcinomas (Table 1). The location and morphologic features of many of the urethral carcinomas in this study described by Singh et al. (2010) were unusual; and for this reason, we used IHC to support the urothelial origin of these tumors. A total of 19 samples from male mice (2 normal, 5 hyperplasias, and 12 carcinomas from different animals ([3-, 10-, and 30-mg/kg doses]) and 4 female mice (2 normal and 2 carcinomas) were labeled with UPIII, CK7, and CK20 antibodies. In all, 11 of the 14 (79%) neoplastic lesions (all urethral carcinomas; 12 males and 2 females) and 3 of the 4 (75%) of urethral hyperplasias expressed UPIII (Table 2; Figure 1). Histologically normal membraneous urethra and collecting ducts from tumor-bearing mice and 2 control mice were negative for UPIII. Immunoreactive cells showed positive cytoplasmic staining specific to urothelial cells (Figure 1). Elevated TP53 expression was detected in 71% (10/14) of carcinomas and 20% (1/5) of hyperplasias; no immunoreactivity to TP53 was detected in normal urothelium (0/4; Table 2; Figure 2). TP53 immunoreactive carcinomas included a single ureteral carcinoma and a renal urothelial carcinoma (Table 2). All urethral hyperplasias in males were immunoreactive for CK7 and CK20. Of the 11 urethral carcinomas in males, 9 (82%) were immunoreactive for CK7 and 10 (91%) were immunoreactive for CK20 (Table 2). Both carcinomas found in females were immunoreactive for CK20, and 1 was immunoreactive for CK7.
Incidences of TCAB-induced urethral carcinomas in the National Toxicology Program (NTP) study.a
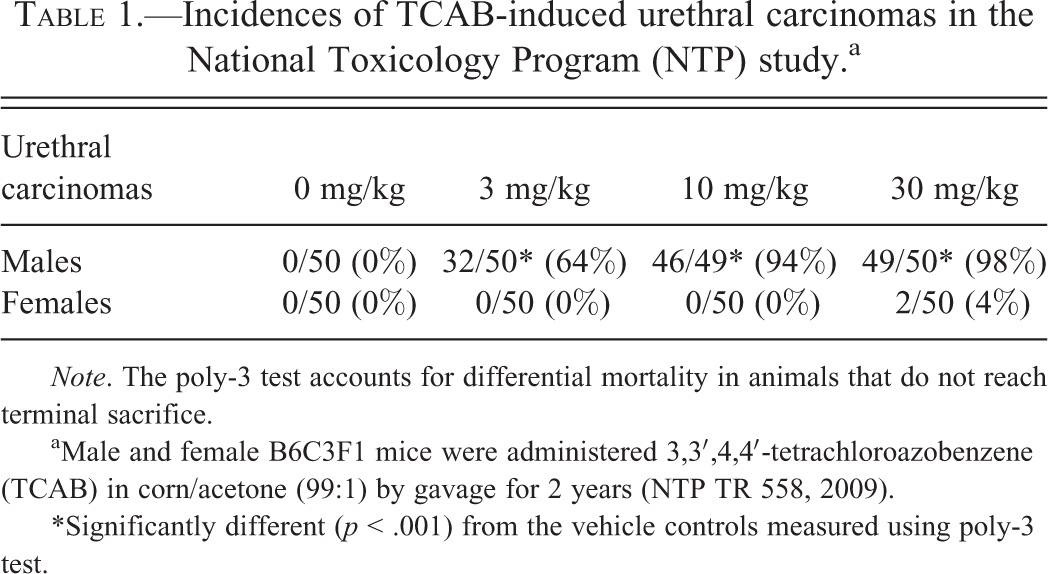
Note. The poly-3 test accounts for differential mortality in animals that do not reach terminal sacrifice.
aMale and female B6C3F1 mice were administered 3,3′,4,4′-tetrachloroazobenzene (TCAB) in corn/acetone (99:1) by gavage for 2 years (NTP TR 558, 2009).
*Significantly different (p < .001) from the vehicle controls measured using poly-3 test.
Immunohistochemical detection of p53, uroplakin III, CK7, and CK20 in urothelial carcinomas and hyperplasias in TCAB-treated male and female mice.

Note. Two control animals and normal appearing proximal urothelium, collecting ducts from tumor-bearing animals were negative for uroplakin III, p53, and positive for CK7 and CK20.
a Male and female B6C3F1 mice were exposed to 0-, 3-, 10-, or 30-mg 3,3′,4,4′-tetrachloroazobenzene (TCAB)/kg body weight daily in corn/acetone by gavage for 2 years (NTP TR 558, 2009).
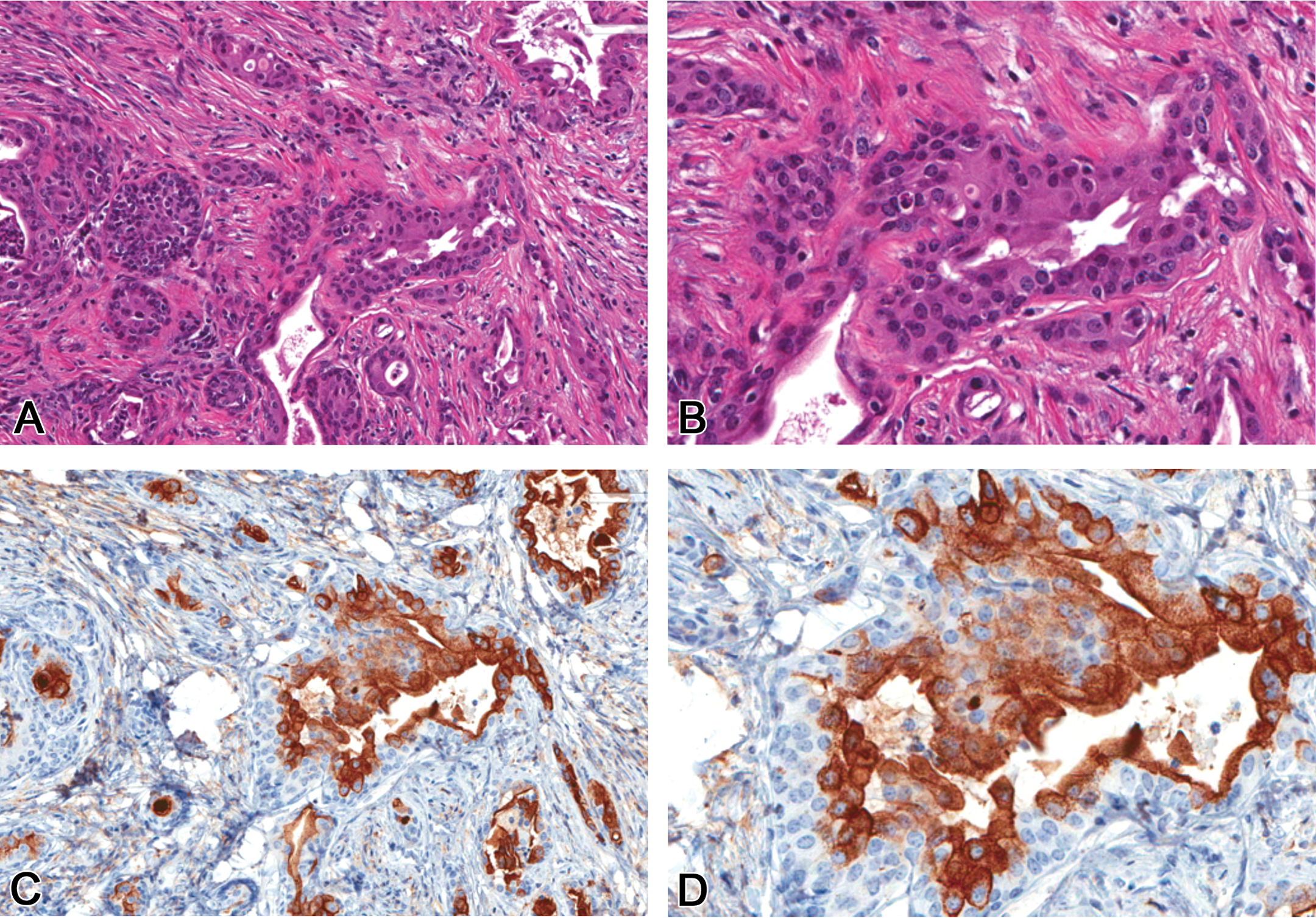
Showing an invasive urethral carcinoma, 200× and 400× original magnification, H&E (A&B) and corresponding positive immunostaining of a urethral carcinoma of Uroplakin III, 200× and 400× original magnification (C & D) from a B6C3F1 male mouse treated with TCAB. H&E = hematoxylin and eosin; TCAB = 3,3′,4,4′-tetrachloroazobenzene.
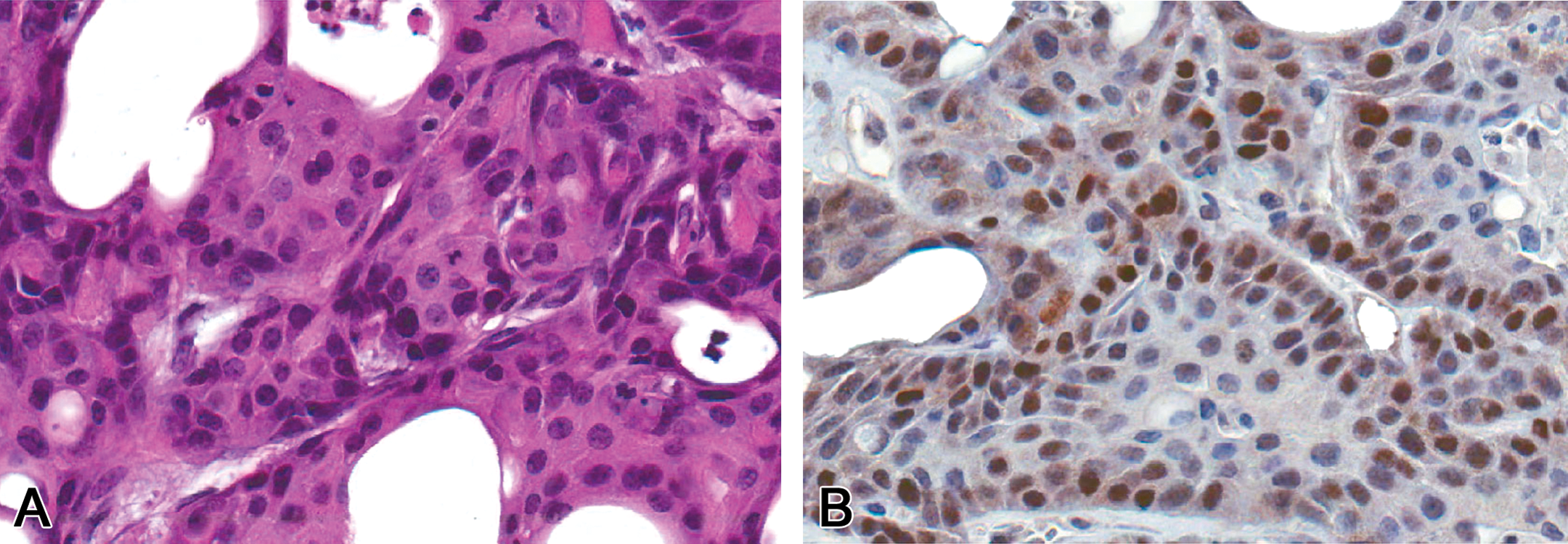
Showing H&E (A) and p53 immunohistochemistry positivity of a urethral carcinoma from a B6C3F1 male mouse treated with TCAB (B). 400× original magnification. H&E = hematoxylin and eosin; TCAB = 3,3′,4,4′-tetrachloroazobenzene.
Genetic Alterations in Urethral Carcinomas
To assess for mutations in the Kras oncogene and Tp53 tumor suppressor gene, DNA was extracted from 21 paraffin-embedded LCM samples (8 vehicle control normal urothelium, 3 hyperplasias, 8 urethral carcinomas, 1 ureteral carcinoma, and 1 renal urothelial carcinoma). Sequence analysis was performed on the highly conserved domains of exons 5 to 8 of the Tp53 gene and exons 1 to 2 of the Kras gene. In urethral carcinomas, Tp53 mutations were detected in 5 of the 8 samples (63%). Tp53 mutations were not detected in the ureteral or renal carcinomas, urethral hyperplasias, or normal urothelium (Table 3). The mutations were exclusively point mutations of the missense type resulting in nonsynonymous amino acid changes in exons 7 and 8 (Figure 3). Mutations were not detected in exons 5 and 6. Of the 5 tumors that harbored Tp53 mutations, 3 had double point mutations leading to G241E and G263R-, C239S and G263R-, P247S and G263R-predicted amino acid changes. Of these 8 mutations, 7 (87.5%) were transitions (GC à AT) and 1 (12.5%) was a transversion (TA à AT). Mutations predominantly occurred in codons 263 and 264 (63%). All mutation-positive tumors also demonstrated the highest level of nuclear TP53 by IHC. However, the concordance of nuclear TP53 protein expression and gene mutation was 71% for all samples analyzed (Table 3). Further, in 1 urethral hyperplasia, 1 urethral carcinoma, and the single ureteral and renal carcinomas, nuclear TP53 staining was observed, but Tp53 mutations were not detected.
Summary of Tp53 and K-ras mutations in mouse urethral carcinomas following TCAB exposure.a
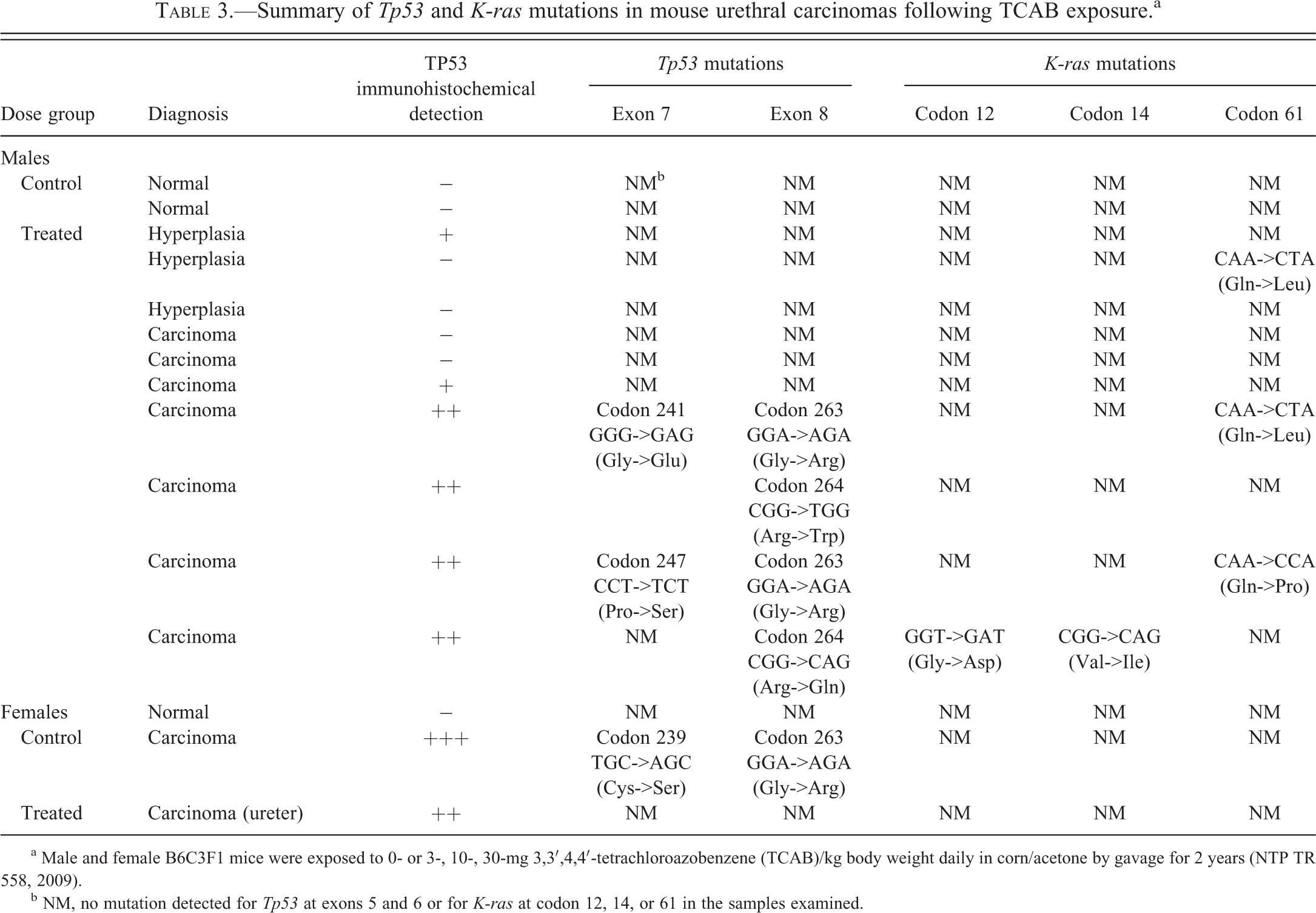
a Male and female B6C3F1 mice were exposed to 0- or 3-, 10-, 30-mg 3,3′,4,4′-tetrachloroazobenzene (TCAB)/kg body weight daily in corn/acetone by gavage for 2 years (NTP TR 558, 2009).
b NM, no mutation detected for Tp53 at exons 5 and 6 or for K-ras at codon 12, 14, or 61 in the samples examined.
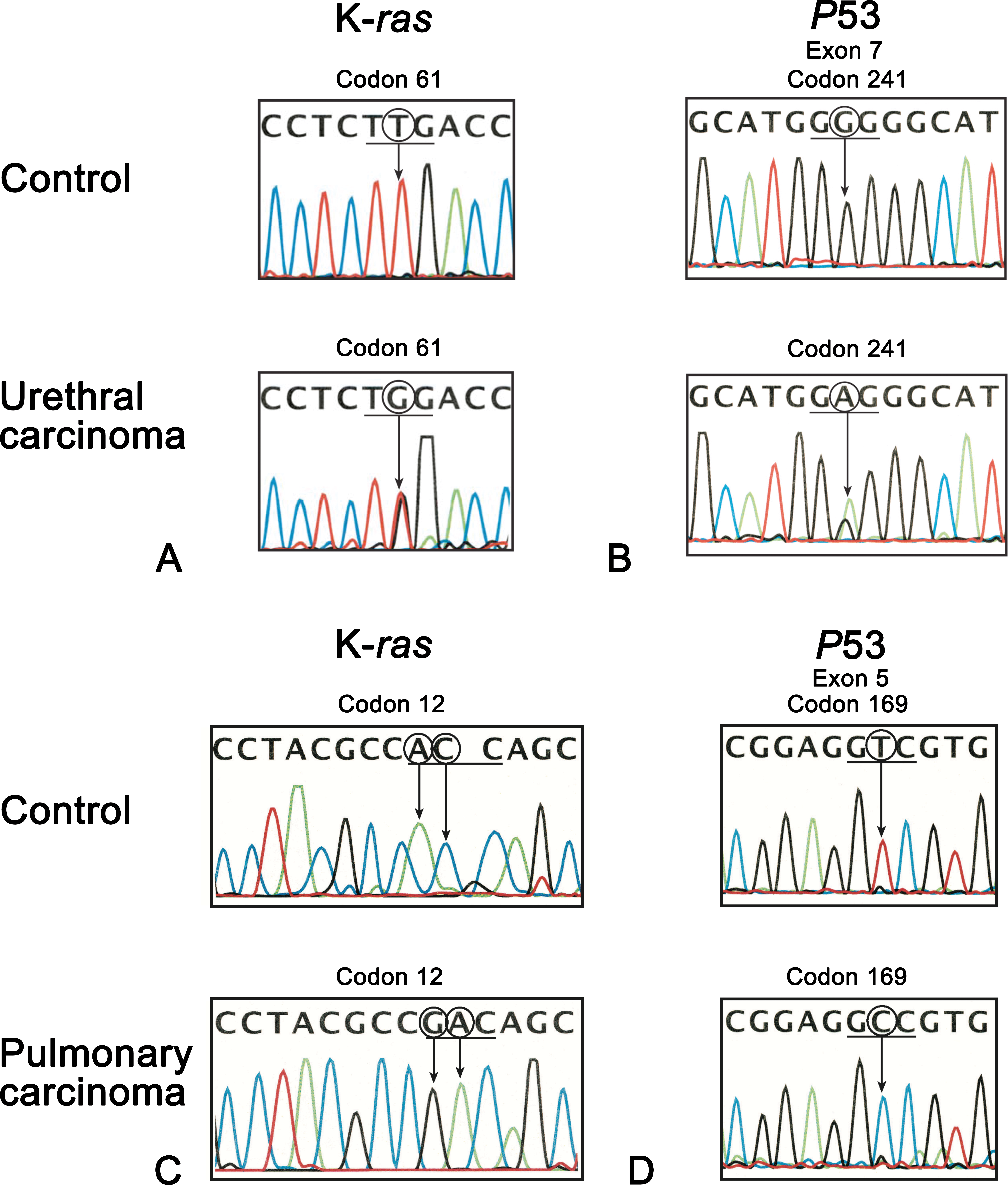
Electrophoretograms indicating point mutations within K-ras and Tp53 genes amplified from urethral carcinomas and pulmonary carcinomas of B6C3F1 mice chronically exposed to TCAB for 2 years. (A) Identification of point mutations in normal K-ras gene codon 61 (CAA) and urethral carcinoma with mutated codon 61 (CCA; B) urethral carcinoma with mutated Tp53 gene codon 241 (GGG > GAG; C) pulmonary carcinoma with mutated K-ras gene codon 12 (GGT > GTC). (D) Pulmonary carcinoma with mutated Tp53 gene codon 169 (GTC > GCC). TCAB = 3,3′,4,4′-tetrachloroazobenzene.
Mutations in Kras were detected in 3 of the 8 (38%) urethral carcinomas (Table 3). A mutation was also detected in 1 of the 3 (33%) urethral hyperplasias. Kras mutations were not detected in the ureteral or renal carcinoma, or in normal urothelium (Table 3). Kras mutations were exclusively point mutations of the missense type resulting in a nonsynonymous amino acid change in both exons 1 and 2 (Figure 3). Of the 4 samples that exhibited Kras mutations, 1 carcinoma displayed a double point mutation resulting in G12D- and V14I-predicted amino acid change (Table 3). In the remaining 3 samples, mutations were detected exclusively in codon 61 and were predicted to produce Q61L, Q61P amino acid alterations (Table 3). Of the 5 mutations observed, 3 were transversions and 2 were transitions. No consistency in the type of base substitutions that occurred was detected; however, mutations occurred predominantly (80%) in the second position of the affected codons. Overall, mutations of either Tp53 or Kras were present in 75% (6/8) of urethral carcinomas with comutation of both p53 and Kras genes occurring in 25% (2/8) of samples.
Genetic Alterations in Pulmonary Lesions
Mutation analysis of Tp53 and Kras was performed on DNA isolated from paraffin-embedded lung tumors (9 adenomas and 6 carcinomas). Spontaneous lung tumors (1 adenoma and 3 carcinomas) from concurrent vehicle control mice did not have Tp53 or Kras mutations. The incidence of Tp53 mutations in pulmonary tumors from TCAB-treated mice was 55% (6/11; 5 adenomas and 1 carcinoma), similar to that seen in urethral carcinomas (63%). Point mutations in Tp53 were of the missense type, which led to a predicted amino acid change in 3 of the 6 samples (Table 4). Missense mutations were predicted to cause V169A, V169A, and M166V amino acid alterations and all occurred in adenomas (Table 4). In the remaining 3 samples, 1 adenoma harbored a double mutation that resulted in a synonymous amino acid change. Of the 7 Tp53 substitutions that occurred (missense and silent), all were transitions. The incidence of Tp53 mutations, excluding silent alterations, in pulmonary adenomas was 38% (3/8); carcinoma samples did not harbor missense Tp53 mutations.
Summary of Tp53 and K-ras mutations in mouse lung tumors following TCAB exposure.a
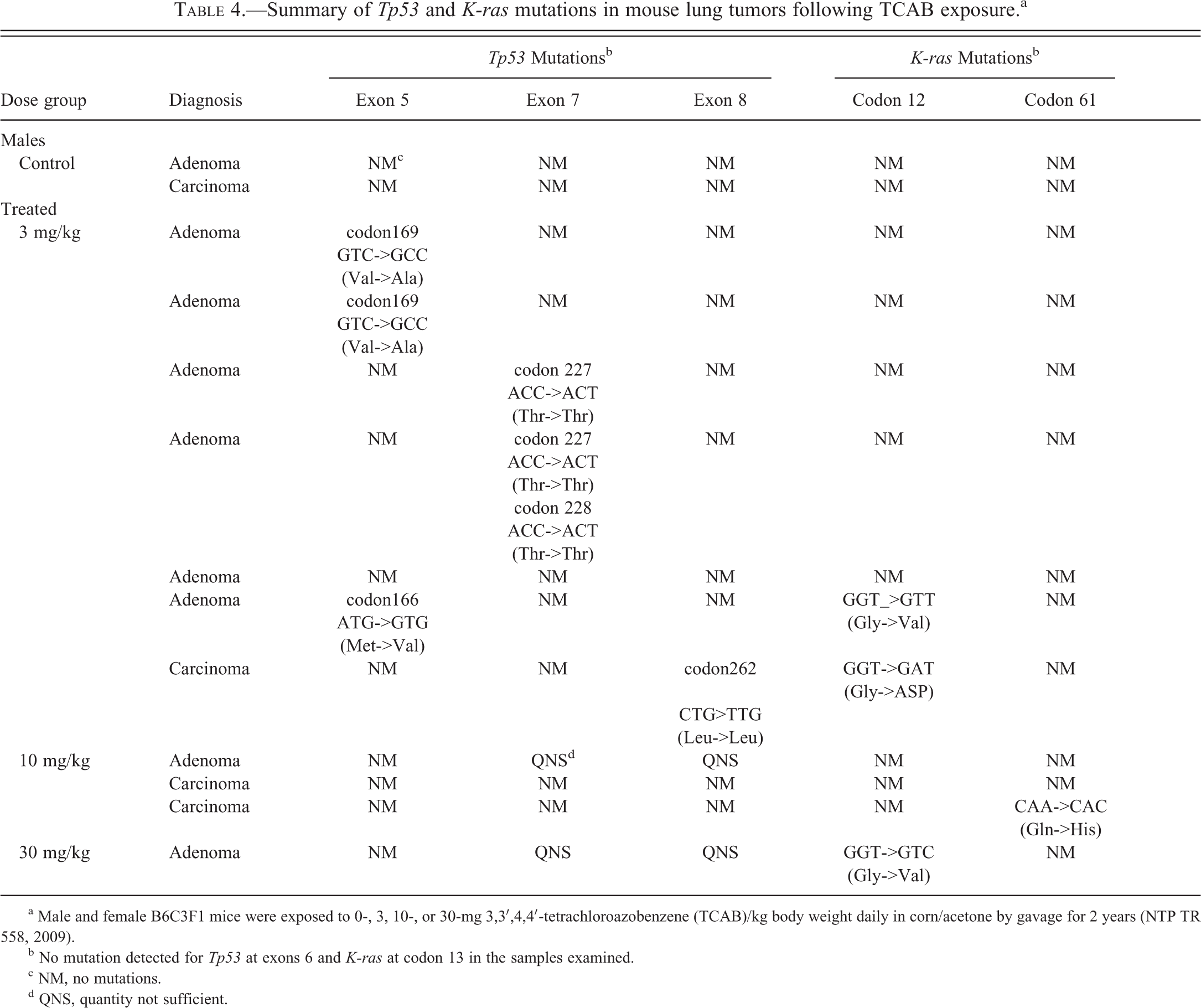
a Male and female B6C3F1 mice were exposed to 0-, 3, 10-, or 30-mg 3,3′,4,4′-tetrachloroazobenzene (TCAB)/kg body weight daily in corn/acetone by gavage for 2 years (NTP TR 558, 2009).
b No mutation detected for Tp53 at exons 6 and K-ras at codon 13 in the samples examined.
c NM, no mutations.
d QNS, quantity not sufficient.
The overall frequency of Kras gene mutations in lung tumors (4/11; 2 adenomas, 2 carcinomas, 36%) was similar to that observed in urethral carcinomas (Table 5). Mutations detected were all single nucleotide substitutions that led to missense mutations predicted to produce an amino acid change. The incidence of Kras alterations was higher in pulmonary carcinomas (67%, 2/3) than in pulmonary adenomas (25%, 2/8). Substitutions were predominantly transversions, present in 3 of the 4 mutations; transitions were less common, occurring in only 1 tumor (Table 4). Kras mutations were not detected in spontaneous pulmonary adenomas or carcinomas from vehicle control animals. Overall, the frequency of either Tp53 (missense and silent) or Kras mutation was 73% (8/11; Table 5) in pulmonary tumors, with 1 sample harboring double mutations in both Tp53 and Kras genes.
Mutation profile of p53 and K-ras genes in mouse urethral and pulmonary tumors following TCAB exposure.a

Note. No mutation detected in 4 control urethral epithelia and glands; no mutation detected in 4 spontaneous alveolar/bronchiolar adenomas and carcinomas.
a Male and female B6C3F1 mice were exposed to 0-, 3-, 10-, or 30-mg 3, 3′, 4, 4′-tetrachloroazobenzene (TCAB)/kg body weight daily in corn/acetone by gavage for 2 years (NTP TR 558, 2009).
b No mutation detected for p53 at exon 6 in the samples examined.
Discussion
Human urethral cancer is rare and is most often associated with human papillomavirus (HPV) infection (HPV 16 or 18; Swartz et al. 2006). Common histologic types of human urethral cancer are transitional cell (55%), squamous cell (21.5%), and adenocarcinoma (16.4%; Swartz et al. 2006). As the occurrence of proliferative lesions in the lower urinary tract of rodents is very rare, particularly in the mouse (Singh et al. 2010), the assessment of tumorigenesis mechanism resulting from TCAB exposure, and the relationship to human cancer, is of particular importance. In addition, since TCAB acted as a multisite carcinogen, determining whether a similar mechanism of tumorigenesis was occurring in multiple organs was of interest. Further, since some of the urethral tumors arose from the surface epithelium, while others appeared to arise from urethral glands, characterization of these tumors as to the cell of origin was important. Studies have shown that UPs are expressed abundantly in the urothelium but are undetectable in nonurothelial tissues (Moll 1995; Kaufmann, Volmerig, and Dietel 2000). Moreover, their expression has also been proposed as an objective marker of terminally differentiated cells (Olsburgh et al. 2003). Importantly, tissue specificity of UPIII expression is fairly well maintained throughout the process of oncogenic transformation in the urothelium, and it can be used for identifying carcinomas of urothelial origin (Lobban et al. 1998; Olsburghet al. 2003; Wu et al. 2009). Of the urethral carcinomas in this study, 79% were UPIII positive, confirming the urothelium as probable tissue of origin (Figure 1). The epithelium of the membranous urethra and collecting ducts were negative for UPIII. Generally, urothelium expresses UPs. However, the proximal membranous urethra is lined by a single to bilayered flattened to short cuboidal epithelium, which is morphologically different than the stratified transitional morphology of the urothelium. Therefore, the differences in immunostaining for UPIII may represent differences in subtopography from which they arose and/or degree of differentiation.
Since there was a treatment-related increase in the incidence of urethral and pulmonary tumors, we next investigated whether genes relevant for human cancer were similarly altered in tumors at both sites, which would suggest a common mechanism for tumorigenesis of TCAB. Nuclear accumulation of mutant TP53 is a common finding in transitional cell carcinomas of the bladder in humans (Yamamoto et al. 1997; Ogawa et al. 1998; Morimuraet al. 1999); however, attempts to utilize accumulation as a prognostic marker, or as an indicator of more aggressive tumors, has produced varying conclusions (Sidransky et al. 1991; Vet et al. 1995; Gao et al. 2000). In this study, we observed TP53 protein nuclear accumulation in a majority (71%) of urethral carcinomas, which correlates well with previous studies of bladder cancer in humans; for example, Kuczyk et al. (1995) reported significant TP53 immunoreactivity in 70% of human bladder cancer cases (Kuczyk et al. 1995). Although good concordance was observed between TP53 nuclear accumulation and the presence of Tp53 gene mutations in urethral carcinomas, some lesions analyzed showed accumulation of TP53 without gene mutations. These included single cases of urethral hyperplasia, urethral carcinoma, and ureteral and renal carcinomas that expressed higher levels of protein by IHC. These types of discordant results have been reported previously and are likely either the result of gene mutations occurring in unsequenced exons or a reflection of a low percentage of mutation-positive tumor cells (Bian et al. 2001; Kropveld et al. 1996; George et al. 2007).
Tp53 mutations were detected in a majority (63%) of exons 5 to 8 in TCAB-induced urethral tumors. Exons 5 to 8 of Tp53 represent the “hot-spot” mutation site frequently altered in human cancers. Mutations in this study were primarily clustered within exons 7 and 8, which contrasts somewhat with the findings from other studies in rat and mouse urothelial tumors, in which the most frequently mutated exons were 5, 6, and 7 (Pavletich, Chambers, and Pabo 1993; Jackson et al. 2006). Since exons 5 to 8 also represent the DNA-binding region of Tp53, it is possible that mutations arising from TCAB exposure have the potential to affect protein function. This is consistent with the accumulation of mutant TP53 protein seen by IHC in these lesions.
The spectrum of Tp53 mutations in pulmonary tumors was similar to that of urethral carcinomas, but the overall frequency of Tp53 mutations occurred at a lower rate. Furthermore, Tp53 mutation occurred only in pulmonary adenomas and not in carcinomas. Considering it is believed that adenomas progress to carcinomas, one might expect the incidence of Tp53 mutation in carcinomas to be similar to or increased over that of adenomas; however, the absence of Tp53 mutations in pulmonary carcinomas may indicate that these tumors in TCAB-treated mice do not develop through a Tp53-dependent mechanism, but rather that other genomic alterations may be required for further tumor progression to carcinoma. Further investigation of additional mutational events involving other common tumor suppressors or oncogenes in TCAB-related proliferative lesions may be warranted.
The incidence of Kras mutation was similar in both urethral and pulmonary tumors. In addition, a Kras mutation was detected in a single hyperplastic urethral lesion, suggesting that Kras mutation may be an early event in the process of urethral carcinogenesis in TCAB-exposed animals. In fact, Kras mutations have been observed in preneoplastic lesions in other types of tumors, including aberrant crypt foci in the colon, and even in histologically normal mucosal epithelium adjacent to neoplasms in chemically induced rodent models and human colorectal cancer patients (Jacoby 1991; Hu et al. 2009). In the lung, Kras mutations were detected with greater frequency in pulmonary carcinomas (67%) than in adenomas (25%), which may support an increased mutation rate with progression of malignancy. Mutations in this gene are a common finding in mouse lung tumors; a survey of the literature indicates that approximately 60% of carcinomas and 70% of adenomas harbor Kras mutations (Jacksonet al. 2006).
While the genotoxic activity of TCAB is still not completely understood, mutations in Kras and Tp53 following TCAB exposure imply that this chemical or its metabolites may cause DNA damage, and suggest a possible genotoxic mode of action. However, given the conflicting genotoxicity data and the known AhR activity (Poland et al. 1976), it is possible that TCAB induces indirect DNA damage through nongenotoxic mechanisms such as chronic xenobiotic or oxidative stress, cytotoxicity, or other mechanisms, although no such evidence is currently available. Determining the genetic fingerprint of exposure has shown that transition substitutions were the most common type of mutations, occurring in guanine or cytosine residues. In urethral carcinomas, Tp53 mutations were predominantly observed in codons 263 and 264, which may represent TCAB-specific target sites in the urethra; however, mutations at this site were not identified in any pulmonary tumors examined. Urethral carcinomas and pulmonary tumors shared similar frequency (36–38%) and spectra (codons 12 or 61) of Kras mutations, suggesting that these sites may be targets for TCAB or its reactive metabolites. However, as the number of lesions with mutations was fairly low, a more extensive study of a larger group of tumors may further define the fingerprint of TCAB exposure. Likewise, the lack of Kras/Tp53 mutations in the single renal or ureteral carcinomas might indicate divergent pathways of carcinogenesis distinct from those involved in the urethral carcinomas. Further studies would be needed to assess this possibility.
In conclusion, exposure of B6C3F1 mice over 2 years to TCAB resulted in the development of rare urethral carcinomas and pulmonary tumors associated with mutations in Kras and Tp53, critical genes often altered in human cancer. It is significant that these urethral and pulmonary tumors were characterized by transition mutations in the Tp53 gene; this suggests that TCAB or its metabolites target guanine or cytosine bases in the Tp53 gene and that these mutations may play a role in carcinogenesis in B6C3F1 mice exposed to TCAB.
Footnotes
Acknowledgments
The authors would like to thank the CMPB Histology, Immunohistochemistry, and Special Techniques Core Laboratories, and the NIEHS DNA Sequencing Laboratory for their excellent technical assistance.
Authors’ Note
This article may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH); however, the statements, opinions, or conclusions contained therein do not necessarily represent the statements, opinions, or conclusions of the NIEHS, NIH, or the U.S. government.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), and The Division of the National Toxicology Program (DNTP).