
Abstract
The proper folding, assembly, and maintenance of cellular proteins is a highly regulated process and is critical for cellular homeostasis. Multiple cellular compartments have adapted their own systems to ensure proper protein folding, and quality control mechanisms are in place to manage stress due to the accumulation of unfolded proteins. When the accumulation of unfolded proteins exceeds the capacity to restore homeostasis, these systems can result in a cell death response. Unfolded protein accumulation in the endoplasmic reticulum (ER) leads to ER stress with activation of the unfolded protein response (UPR) governed by the activating transcription factor 6 (ATF6), inositol requiring enzyme-1 (IRE1), and PKR-like endoplasmic reticulum kinase (PERK) signaling pathways. Many xenobiotics have been shown to influence ER stress and UPR signaling with either pro-survival or pro-death features. The ultimate outcome is dependent on many factors including the mechanism of action of the xenobiotic, concentration of xenobiotic, duration of exposure (acute vs. chronic), cell type affected, nutrient levels, oxidative stress, state of differentiation, and others. Assessing perturbations in activation or inhibition of ER stress and UPR signaling pathways are likely to be informative parameters to measure when analyzing mechanisms of action of xenobiotic-induced toxicity.
Overview of Protein Folding Mechanisms in Different Cellular Compartments
The proper assembly and maintenance of cellular proteins is critical for their proper function and the process is thus highly regulated. Eukaryotic cells have assembled a complex network of multiple chaperones, foldases, and cofactors to orchestrate the proper folding and maturation of these proteins (Boelens et al. 2007). Multiple cellular compartments have adapted their own systems to monitor protein folding status and manage proteotoxic stress due to accumulation of unfolded proteins. When proteotoxic stress exceeds the capacity to adapt and restore homeostasis, these systems can trigger a cell death response involving apoptosis or necrosis as a last resort (reviewed in Austin 2009; Buchberger, Bukau, and Sommer 2010; Ellgaard, Molinari, and Helenius 1999; Haynes and Ron 2010; Malhi and Kaufman 2011; Schroder and Kaufman 2005).
In eukaryotic cells, secreted and transmembrane proteins represent approximately one-third of cellular proteins that enter the secretory pathway by translocating in the endoplasmic reticulum (ER) co-translationally where they are properly folded and posttranslationally modified (Figure 1; Kaufman 1999). In addition to protein folding, the ER also functions in lipid and steroid biosynthesis, Ca2+ homeostasis, and xenobiotic metabolism. The ER is the second major protein-folding compartment after the cytosol within eukaryotic cells. Perturbations in ER homeostasis can result in the accumulation of unfolded proteins in the lumen of the ER, leading to a pathological response referred to ER stress. The evolutionary conserved unfolded protein response (UPR) is triggered upon sensing the unfolded proteins in the ER (Xu, Bailly-Maitre, and Reed 2005). The UPR is an adaptive response with the goal of reducing the accumulation of unfolded proteins in the ER by increasing the protein folding capacity of the ER, reducing nascent protein influx in the ER, and increasing the clearance of misfolded proteins by the induction of ER-associated degradation (ERAD). ERAD is a series of pathways whereby unfolded proteins destined for destruction are translocated to the cytoplasm for ubiquitination and proteasomal-mediated degradation. The ERAD machinery is composed of several different components, such as E3 ubiquitin ligases and Derlins. The E3 ubiquitin ligases are at the center of the ERAD pathways and catalyze substrate ubiquitination and organize complexes on both sides of the ER membrane for proper ERAD coordination. Other proteins such as EDEMs (ER degradation-enhancing α-mannosidase-like protein) help target misfolded glycoproteins for ERAD (reviewed in Smith, Ploegh, and Weissman 2011). Signaling pathways that are also associated with different forms of cellular stresses such as mitogen-activated protein kinases (MAPKs), Jun N-terminal kinase (JNK), p38 MAPK, and NF-kappaB can also be activated. Apoptosis can occur if the adaptive functions of the UPR are not capable of resolving the burden of accumulated unfolded proteins (Malhotra and Kaufman 2007; Egger et al. 2003).
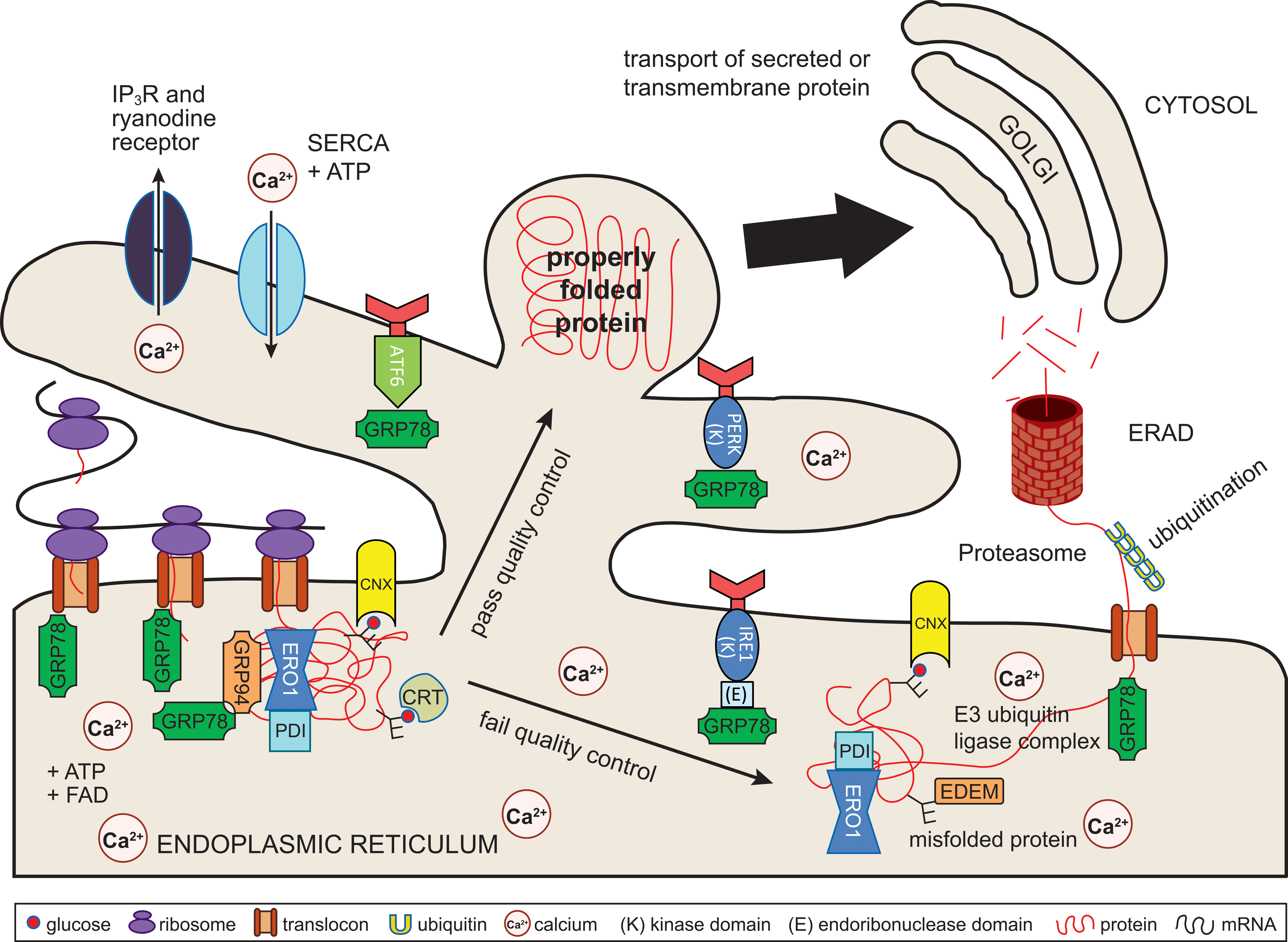
Protein folding in the endoplasmic reticulum. Secreted or transmembrane proteins enter the ER co-translationally via the translocon and interact with calcium-binding, ATP-dependent protein chaperones such as GRP78 and GRP94 that help mediate protein folding. Disulfide bond formation is driven by a protein relay involving Ero1 and members of the PDI family using FAD as a cofactor. Quality control mechanisms are in place that involve addition and processing of N-linked saccharides recognized by lectin-binding proteins CNX and CRT. Properly folded proteins that pass the quality control mechanisms are trafficked to their final destination via the Golgi. Protein chaperones as well as other proteins such as the EDEMs help target misfolded glycoproteins for ERAD, which is a series of pathways whereby unfolded proteins destined for destruction are retrotranslocated to the cytoplasm for ubiquitination and proteasomal-mediated degradation. ER Ca2+ homeostasis is regulated by ER Ca2+ release channels such as IP3R and the ryanodine receptor and Ca2+ uptake transporters such as SERCA. The 3 UPR sensors (ATF6, IRE1 and PERK) are kept in an inactive state during non-ER stress periods. Adapted from Boelens et al. (2007) and Kaufman et al. (2002). ER = endoplasmic reticulum; ATP = adenosine triphosphate; GRP78 = glucose regulated protein; Ero1 = ER oxidoreductin 1; FAD = flavin adenine dinucleotide; CNX = calnexin; CRT = calreticulin; EDEM = ER degradation-enhancing α-mannosidase-like protein; ERAD = ER-associated degradation; SERCA = sarco/endoplasmic reticulum calcium ATPase; UPR = unfolded protein response; ATF6 = activating transcription factor 6; IRE1 = inositol requiring enzyme-1; PERK = PKR-like endoplasmic reticulum kinase.
ER stress can be induced by a number of stimuli that disrupt or overwhelm the protein folding machinery or ERAD including redox disruption, hypoxia, hypoglycemia, aberrant Ca2+ regulation, energy balance disruption, viral infections, overproduction of secreted/transmembrane proteins, certain genetically mutated secretory proteins that are folding-incompetent and a number of xenobiotics (Kaplowitz et al. 2007; Kim, Xu, and Reed 2008). Cell fate between survival and death depends on the severity and duration of the stress response, with mild and/or acute ER stress generally resulting in adaptation and severe and/or chronic ER stress resulting in cellular dysfunction and/or cell death. Many factors can influence the amount and types of secreted or transmembrane proteins a given cell will produce including changes in the cellular environment and the natural life cycle of the cell. Thus, cells are frequently confronted by conditions that can transiently overwhelm the ER folding capacity leading to ER stress and therefore should not necessarily be thought of as a negative outcome unless the cell cannot overcome the increased burden on the ER.
The stress response system that manages cytosolic unfolded proteins comprises chaperones including the heat shock proteins (HSP90, HSP70, etc.) and the heat shock factors (HSF1, HSF2, HSF3, HSF4) to sense unfolded protein levels and execute a process to relieve the proteotoxic stress through induction of specific regulatory genes such as HSP90, HSP70, HSP40, HSP110, and others (reviewed in Anckar and Sistonen 2011; Fujimoto and Nakai 2010; Figure 2). The HSFs exist normally in an inactive state in monomeric form. Activation causes the assembly of a trimeric state that allows them to bind specific response elements in the nucleus and activate heat shock responsive genes (Sandqvist et al. 2009). The most studied heat shock sensor, HSF1, has several proposed sensor mechanisms including intrinsic response to heat, chaperone displacement, and the RNA thermometer mechanism. During intrinsic response to heat, HSF1, normally in an inactive monomer state, refolds in response to heat or unfolded protein stress and forms the active trimer (Mosser et al. 1990). During the chaperone displacement mechanism, HSF1 is normally complexed with the chaperone protein HSP90 in the inactive state (Guo et al. 2001). When the unfolded protein levels become high enough, HSP90 is displaced from HSF1 as it attempts to refold the unfolded proteins. When enough HSF1 is liberated, it forms a trimeric complex that activates heat shock responsive genes. During the RNA thermometer mechanism, heat shock RNA-1 (HSR1) forms a complex with eEF1A during heat shock which stimulates the activation of HSF1 from a monomeric state to a trimeric state (Shamovsky et al. 2006). This latter process can link the heat shock response with inhibition of translation.
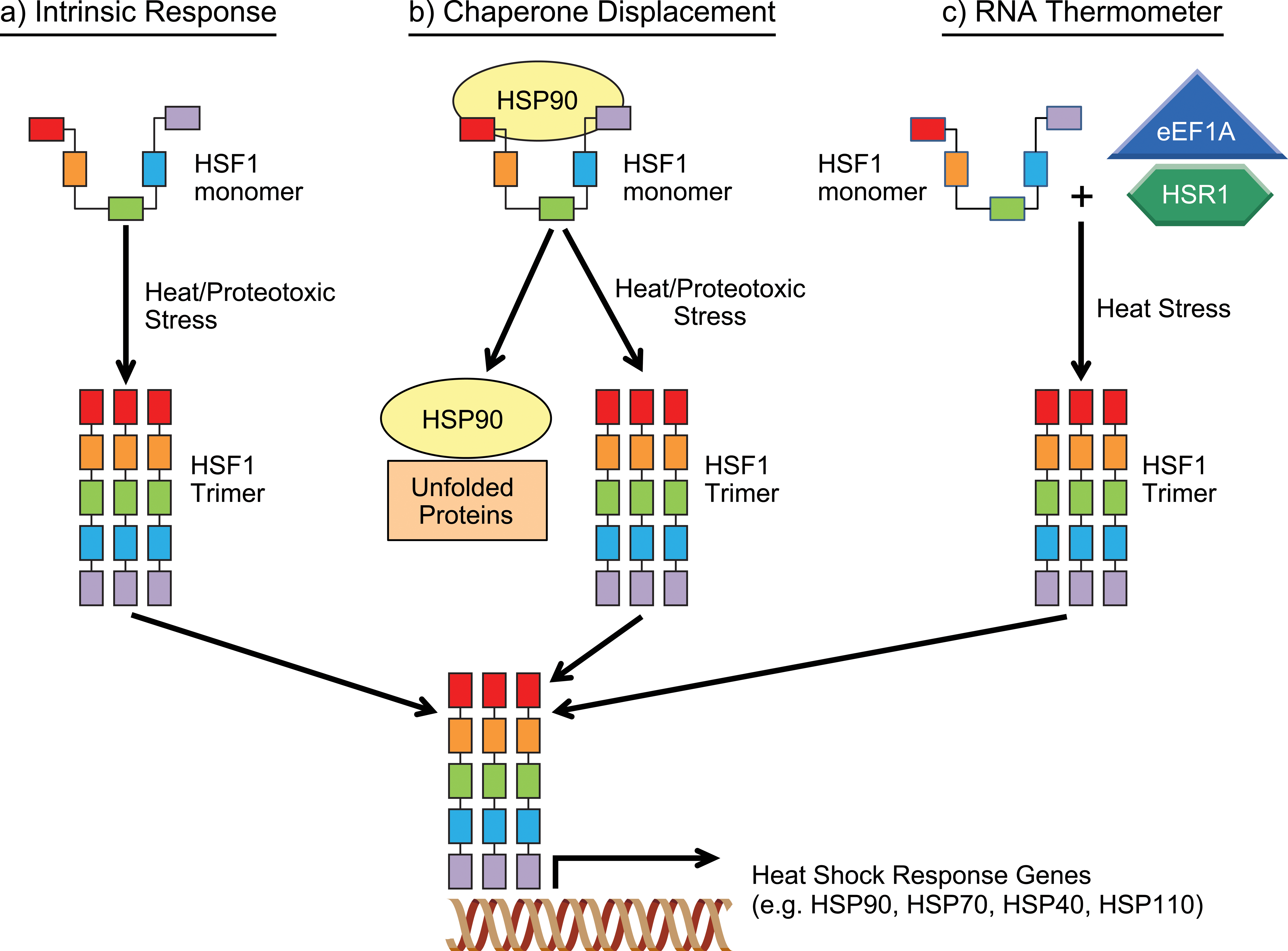
Cytoplasmic stress response. The stress response system in the cytosol that manages unfolded proteins comprises chaperones including the HSPs and HSFs to sense unfolded protein levels and execute a process to relieve the proteotoxic stress. HSF1 exists normally in an inactive monomeric state. Activation causes the assembly of a trimeric state that allows HSF1 to bind heat shock elements to activate specific stress response genes. Several activation mechanisms for HSF1 have been proposed including the intrinsic response, chaperone displacement, and the RNA thermometer mechanism. A, Intrinsic response: HSF1 is normally in an inactive monomer state; however, it can refold intrinsically in response to heat or unfolded protein stimuli to form the active trimer. B, Chaperone displacement: HSF1 is normally complexed with HSP90 in the inactive state. During heat or unfolded protein stress, HSP90 is liberated from HSF1 as it attempts to refold the unfolded proteins. When enough HSF1 is liberated, it forms the active trimeric complex. C, RNA thermometer: heat shock RNA-1 (HSR1) forms a complex with eEF1A during heat shock which stimulates the activation of HSF1 from a monomeric state to a trimeric state. Adapted from Anckar and Sistonen (2011). HSP = heat shock protein; HSF = heat shock factor.
Mitochondria also have their own UPR system (Reviewed in Haynes and Ron 2010; Figure 3). Within the mitochondrial matrix exists chaperone proteins (e.g., mtHSP70, HSP60, etc.) that are imported into the matrix and sense unfolded proteins (Tatsuta and Langer 2008; Neupert 1997; Neupert and Herrmann 2007). When the unfolded protein load exceeds the chaperone levels, an increase in the expression of mitochondrial chaperones (e.g. HSP60) and mitochondrial protease complex (ClpXP) occurs (Haynes et al. 2007). The increase in ClpXP complex degrades excess unfolded proteins into peptides that are exported out of the mitochondria into the cytosol by the transmembrane transporter HAF-1 (Haynes et al. 2010). The increase in peptides in the cytoplasm is a signal for a basic leucine zipper (bZIP) transcription factor (ZC376.7) that increases transcription of the mitochondrial chaperones such as HSP60 (Haynes and Ron 2010).
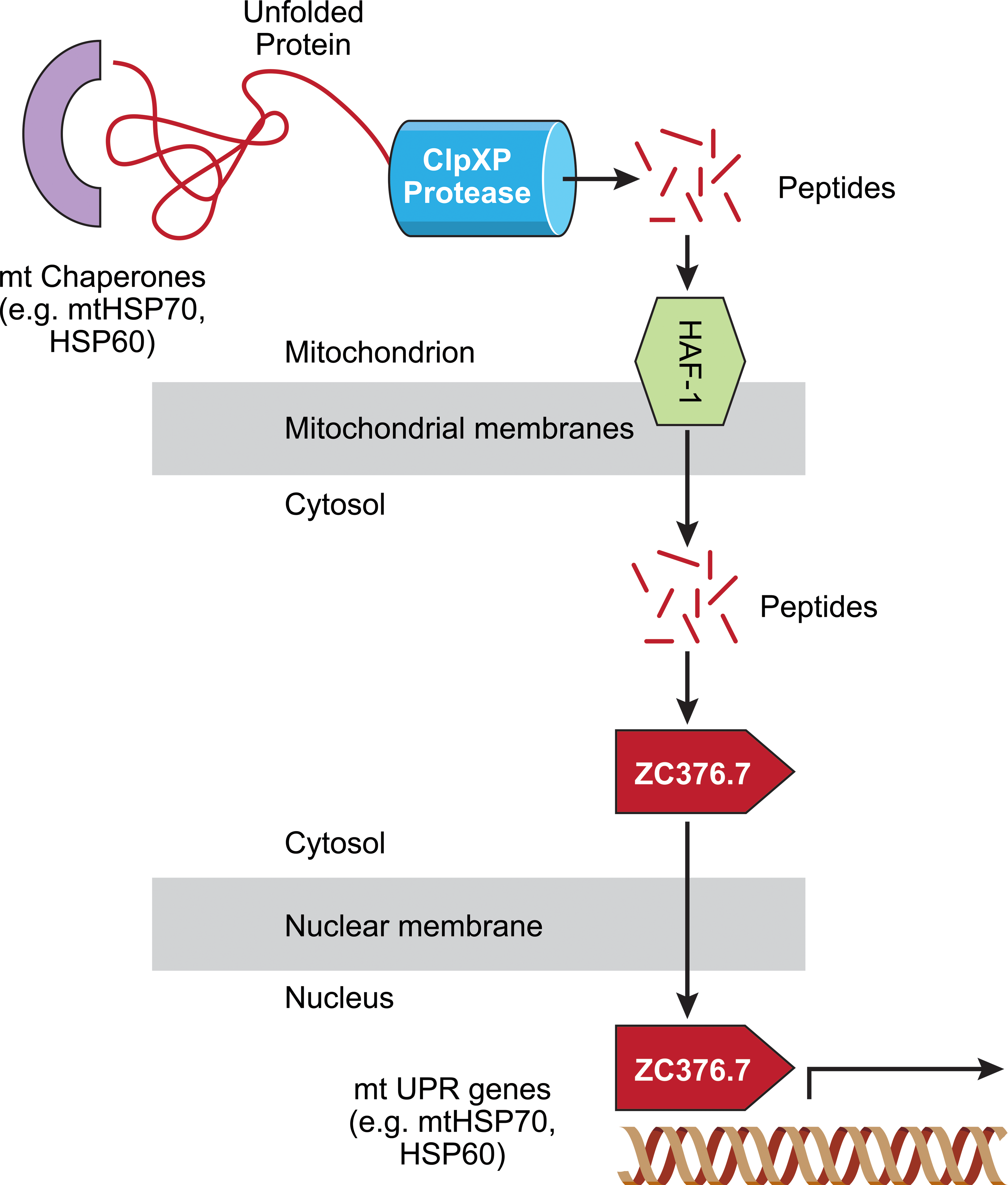
Mitochondrial unfolded protein response. Mitochondrial UPR signaling is activated when the unfolded protein load exceeds the capacity of the mitochondrial chaperones (e.g., mtHSP70, HSP60). This results in degradation of unfolded proteins into peptides by the ClpXP protease complex that is exported into the inner mitochondrial membrane by the transmembrane transporter HAF-1, and then migrates across the outer mitochondrial membrane into the cytosol. The increase in peptides in the cytoplasm leads to activation of the bZIP transcription factor ZC376.7 that translocates to the nucleus to increase transcription of mitochondrial chaperones. Adapted from Haynes and Ron (2010).
These different compartment pathways are not mutually exclusive when responding to a stress event. For example, it is known that hyperthermia, which classically induces a heat shock response, can also induce an ER stress response (Xu et al. 2011). HSP72 has been shown to protect insulin secreting cells from LPS-induced ER stress (Hagiwara et al. 2009). In another study, HSP72 protected cells from ER stress–induced apoptosis via IRE1-Xbox-binding protein 1 (XBP1) signaling through a physical interaction (Gupta et al. 2010). Inhibition of HSP90 can increase IRE1 activation in INS-1 pancreatic cells (Ota and Wang 2012). It also appears that prior ER stress can induce an enhanced heat shock response (Asmellash, Stevens, and Ichimura 2005). Thus, the cell appears to integrate proteotoxic responses between cellular compartments that permit the cell to develop adaptive resistance to subsequent or mixed insults.
Endoplasmic Reticulum UPR Signaling Pathways (PERK, IRE1, and ATF6)
Activation of the UPR involves signaling cascades from three different pathways originating from the ER: the PKR-like endoplasmic reticulum kinase (PERK or eukaryotic translation initiation factor 2α [eIF2]AK3), inositol-requiring enzyme 1 (IRE1 or ERN1), and activating transcription factor 6 (ATF6) pathways (Figure 4). PERK, IRE1, and ATF6 are transmembrane ER resident proteins with luminal sensor domains that relay information about the protein folding status of the ER to the cytosol and nucleus via changes in transcriptional activity and effects on mRNA translation or degradation. The early phase of the UPR consists of a rapid response to reduce the influx of proteins into the ER to allow the cell to regain ER homeostasis. General protein translation is inhibited via the PERK pathway to reduce increased protein burden in the ER (Harding, Zhang, and Ron 1999). In addition, certain mRNAs coding for secreted/transmembrane proteins may be degraded by IRE1 (Hollien et al. 2009). ER translocation of secretory and membrane proteins is rapidly attenuated with rerouting to the cytosol for degradation (Kang et al. 2006; Oyadomari et al. 2006). The later phase of the UPR response regulated by the 3 UPR arms involves the differential expression of genes that promote increased ER protein folding capacity as well as genes that regulate apoptosis or attenuate UPR signaling. The three arms are interconnected with feedback mechanisms and cross talk with other signaling pathways to help the cell commit to survival or death. The identification of several unique regulatory binding proteins to each of the three UPR arms (see below) may also help fine-tune and integrate signals within the cell as to the type and intensity of stress stimulus within the ER. These interactions may differ between cell types and differentiation states to allow differential responses to ER stress stimuli.
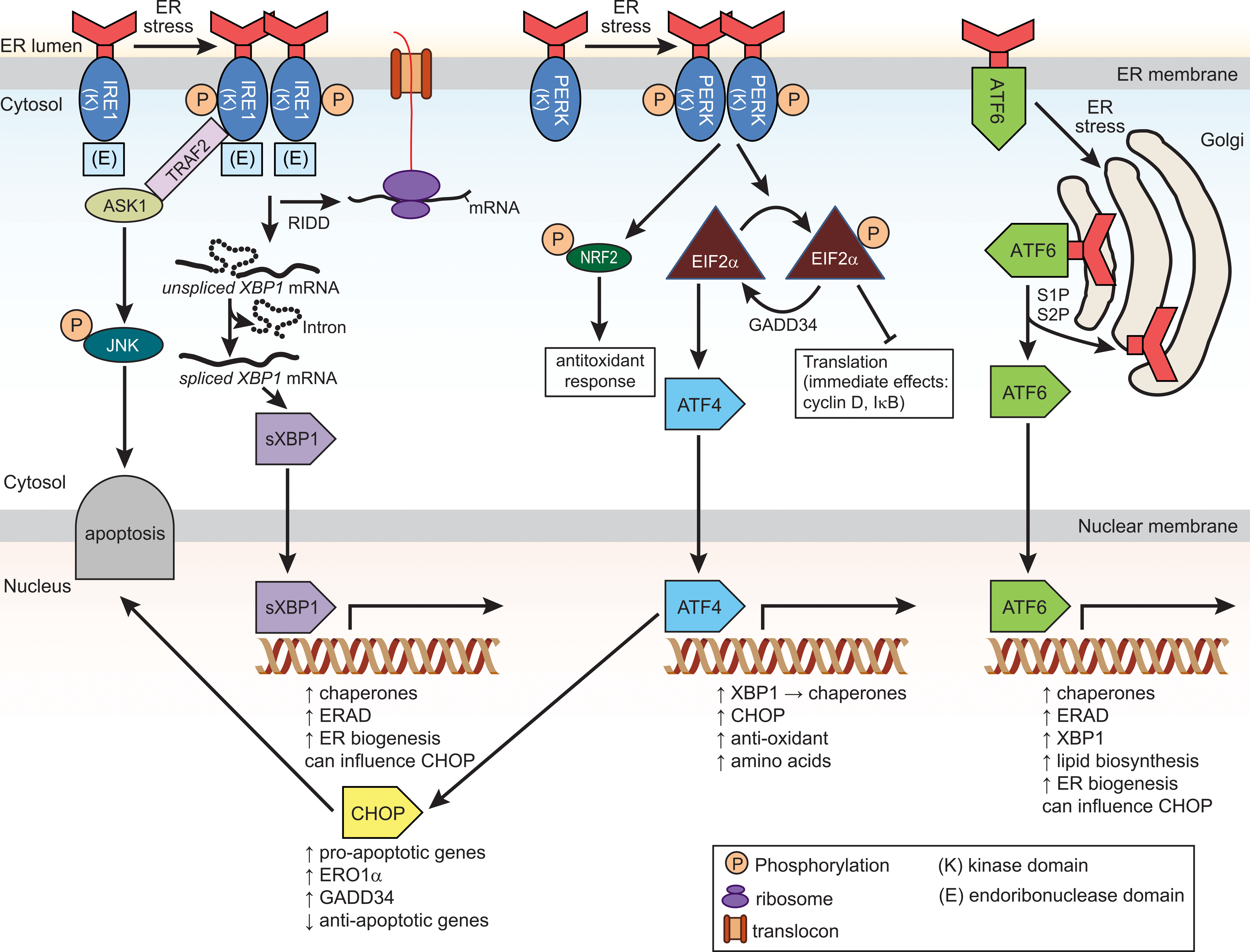
The unfolded protein response. Accumulation of unfolded or misfolded proteins in the ER leads to ER stress and activation of the 3 UPR sensors ATF6, IRE1, and PERK. IRE1 activation leads to oligomerization and trans-autophosphorylation resulting in activation of its endoribonuclease domain. The later removes an intron within the XBP1 mRNA generating the spliced XBP1 transcription factor which translocates to the nucleus to regulate expression of UPR target genes. Activated IRE1 can also degrade cytosolic mRNAs via RIDD. The adapter protein TRAF2 can bind activated IRE1 to recruit ASK1 which leads to JNK phosphorylation that can influence signaling pathways involved in apoptosis and autophagy. PERK activation leads to oligomerization and trans-autophosphorylation. Activated PERK can phosphorylate downstream targets such as eIF2α and Nrf2. Phospho-eIF2α leads to global translation attenuation with immediate effects on proteins with short half-lives such as cyclin D and IκB having effects on proliferation and NF-κB signaling. Certain genes are preferentially translated during these conditions such as the transcription factor ATF4 which activates a number of UPR target genes as well as genes involved in amino acid biosynthesis and antioxidative stress genes. CHOP expression is increased by ATF4 which activates multiple pro-apoptotic genes and suppresses antiapoptotic genes pushing the balance toward cell death. CHOP can also increase the expression of GADD34 which acts to dephosphorylate eIF2α to turn off PERK-mediated signaling during periods of adaptation. Upon activation, ATF6 translocates to the Golgi where it is cleaved by S1P and S2P proteases to liberate the transcription regulating domain into the cytosol. This fragment then migrates to the nucleus to increase the expression of UPR target genes. Adapted from Walter and Ron (2011) and Hetz (2012). UPR = unfolded protein response; ATF6 = activating transcription factor 6; IRE1 = inositol requiring enzyme-1; PERK = PKR-like endoplasmic reticulum kinase; XBP1 = Xbox-binding protein 1; RIDD = regulated IRE1-dependent decay; ASK1 = apoptosis signal-regulating kinase 1; JNK = Jun N-terminal kinase; eIF2α = eukaryotic translation initiation factor 2α; Nrf2 = nuclear factor (erythroid-derived 2)-like 2; NF-κB = nuclear factor-κB; CHOP = C/EBP homologous protein; GADD34 = growth arrest and DNA damage-inducible protein 34; S1P = site-1 protease; S2P = site-2 protease.
PERK
PERK is a type I transmembrane protein with a serine/threonine protein kinase domain that upon activation undergoes oligomerization, trans-autophosphorylation with activation of its kinase domain. Activated PERK phosphorylates the eIF2α at serine 51, resulting in inactivation of eIF2α, preventing assembly of the preinitiation ribosome complex. This attenuates general mRNA translation, reducing the net influx of proteins into the ER (Harding, Zhang, and Ron 1999). However, under conditions of eIF2α phosphorylation, a subset of mRNAs with small upstream open reading frames (such as activating transcription factor 4 [ATF4]) or internal ribosome entry sites are preferentially translated (Fernandez et al. 2002; Miller and Hinnebusch 1990; Harding, Novoa, et al. 2000). ATF4 is a bZIP transcription factor that controls the regulation of expression of pro-survival UPR-target genes (e.g., the chaperones glucose regulated protein [GRP]78 and GRP94) as well as genes involved in other functions such as redox homeostasis, amino acid metabolism, apoptosis, and autophagy (Ameri and Harris 2008; Rzymski et al. 2010; Harding et al. 2003). The PERK arm can therefore transduce both pro-survival and pro-death signals depending on the characteristics of the stress response.
A direct consequence of translational attenuation is the rapid decline in proteins with short half-lives such as cyclin D1 which results in immediate cell cycle arrest (Brewer et al. 1999). A rapid decline in the NF-κB inhibitory protein IκB is also seen resulting in NF-κB activation with effects on immune, inflammatory, and antioxidant responses (Deng et al. 2004). PERK can also phosphorylate the bZIP transcription factor Nrf2 leading to dissociation of the Keap1-Nrf2 interaction, causing Nrf2 to migrate to the nucleus and activate a number of antioxidant genes (Cullinan et al. 2003).
The UPR can be turned off via a negative feedback mechanism involving a variety of proteins such as growth arrest and DNA damage-inducible protein 34 (GADD34) whose expression is increased by ATF4 late in the stress response. GADD34 associates with protein phosphatase 1 (PP1) to dephosphorylate eIF2α, thereby acting as an off switch for the translational attenuation during adaptation (Ma and Hendershot 2003). If the cell has not successfully resolved its unfolded protein burden, premature dephosphorylation of eIF2α can worsen ER stress and further promote apoptosis (Boyce et al. 2005). Other regulatory proteins that can influence the dephosphorylation of eIF2α include Nck-1 (Latreille and Larose 2006), CReP (Jousse et al. 2003), and SIRT1 (Ghosh, Reizis, and Robbins 2011). p58IPK, an HSP40 co-chaperone thought to be upregulated by IRE1 and/or ATF6 signaling, can also turn off the UPR by inhibiting PERK activity (Yan et al. 2002). HSP90 interacts with the cytosolic domains of both PERK and IRE1 and can promote their stabilization (Marcu et al. 2002).
The critical role of PERK signaling as an adaptive response to ER stress is demonstrated in PERK-null cells and genetically modified cells that express a nonphosphorylable serine residue at position 51 in eIF2α. These cells show survival impairment when confronted with ER stress due to a lack of the protective PERK signaling (Harding, Zhang, et al. 2000; Scheuner et al. 2001). No gross developmental defects were apparent in PERK−/− mice; however, postnatally, the pancreatic ER was distended and activation of IRE1α was observed accompanied by increased cell death with progressive diabetes and exocrine pancreatic insufficiency (Harding et al. 2001). PERK gene mutations have also been found in humans with the autosomal recessive disorder Wolcott–Rallison syndrome. These patients exhibit similar pathologies in PERK-null mice, with infant-onset diabetes due to massive β-cell loss and pancreatic exocrine insufficiency (Julier and Nicolino 2010). Other pathologies associated with PERK deficiency include acute liver failure, cardiovascular disease, osteopenia, neutropenia, renal dysfunction, and intellectual deficits (Julier and Nicolino 2010).
IRE1
IRE1 is a type I transmembrane protein with a serine/threonine protein kinase domain and an endoribonuclease domain. Upon activation, IRE1 oligomerizes with subsequent trans-autophosphorylation resulting in a conformational change that activates its endoribonuclease domain. The endoribonuclease domain of IRE1 has 2 functions: (a) the unconventional splicing of an intron (approximately 26 nucleotides depending on the species) within the mRNA encoding the bZIP transcription factor XBP1. Removal of this intron leads to a translational frame shift producing a qualitatively different and more stable protein (Calfon et al. 2002). Spliced XBP1 translocates to the nucleus where it can form heterodimers with other transcription factors such as NF-Y and binds to the ER stress-response element (ERSE) to control the regulation of a subset of UPR-related genes that function in protein quality control, protein folding, ERAD, and ER biogenesis (Lee, Iwakoshi, and Glimcher 2003). The unspliced XBP1 protein has been shown to associate with both ER membranes and its own mRNA, thereby tethering the XBP1 mRNA in close proximity to the IRE1 endoribonuclease domain for efficient splicing upon IRE1 activation (Yanagitani et al. 2009). In addition, although unspliced XBP1 has a short half-life, it can complex with spliced XBP1 in the cytosol, sequestering it from the nucleus and enhancing its proteasome-mediated destruction. This mechanism may help attenuate IRE1 signaling during the recovery phase of ER stress (Yoshida et al. 2006). The regulatory subunits of PI3K (p85α and p85β) can also interact with XBP1 to increase its nuclear translocation (Park et al. 2010). Posttranslational modifications of XBP1 can modulate its activity; p38 MAPK phosphorylates spliced XBP1 resulting in enhanced nuclear translocation (Lee et al. 2011). Acetylation (mediated by p300) of XBP1 increases its protein stability and transcriptional activity (Wang, Chen, and Ouyang 2011), whereas sumoylation of spliced XBP1 (mediated by PIAS2) reduces its transcriptional activity (Chen and Qi 2010). The endoribonuclease domain of IRE1 can also degrade mRNAs from secreted and transmembrane proteins in the cytoplasm in a process known as regulated IRE1-dependent decay (RIDD), thereby reducing the influx of new proteins in the ER and ameliorating ER stress (Hollien et al. 2009; Hollien and Weissman 2006). Alternatively, IRE1 can degrade mRNAs encoding chaperones, thus worsening ER stress leading to apoptosis (Han et al. 2009). IRE1 regulation may be governed by a timer; if ER stress has not been mitigated, IRE1 signaling turns off (IRE1 oligomers disassemble with dephosphorylation) and remains refractory to further activation. However, if ER stress is resolved, IRE1 resets so it can be reactivated to future episodes of ER stress (Li et al. 2010).
A multitude of both positive and negative regulatory proteins interact with IRE1, indicating that a complex signaling platform is assembled at the level of IRE1 to modulate its activity (Hetz and Glimcher 2009). IRE1 can bind to the adaptor protein TNF receptor-associated factor 2 (TRAF2) to initiate the activation of apoptosis signal-regulating kinase 1 (ASK1) and JNK signaling pathways (Nishitoh et al. 2002; Urano et al. 2000). JNK can then phosphorylate BCL-2 to reduce its antiapoptotic activity (Yamamoto, Ichijo, and Korsmeyer 1999) and phosphorylate the pro-apoptotic protein BIM (BCL2-interacting mediator of cell death) resulting in BCL-2-associated X protein (BAX)-dependent apoptosis (Putcha et al. 2003). BAX and BCL-2 antagonist/killer (BAK) can directly interact with the cytosolic domain of IRE1, thereby modulating its activation (Hetz et al. 2006). Conversely, BAX inhibitor 1 (BI-1) that acts to suppress cell death has also been shown to bind IRE1 and inhibit its endoribonuclease activity (Lisbona et al. 2009). The scaffold protein receptor for activated C-kinase-1 (RACK1) interacts with IRE1α and protein phosphatase 2A (PP2A) to regulate glucose-stimulated IRE1α signaling in pancreatic β cells (Qiu et al. 2010). Other proteins reported to associate with IRE1 (but not PERK) and/or modulate its activity include HSP72 (Gupta et al. 2010), protein-tyrosine phosphatase 1B (PTP-1B; Gu et al. 2004), ASK1-interacting protein 1 (AIP1) (Luo et al. 2008), Jun activation domain-binding protein-1 (JAB1; Oono et al. 2004), and JNK-inhibitory kinase (JIK; Yoneda et al. 2001). The majority of these regulatory proteins act to enhance IRE1 signaling.
There are two forms of IRE1 in mammals, IRE1α and IRE1β. IRE1α is expressed ubiquitously, whereas IRE1β is more restricted to epithelial cells of the gastrointestinal tract (Bertolotti et al. 2001; Tirasophon, Welihinda, and Kaufman 1998). IRE1α-null mice die in utero at E9.5 (Urano, Bertolotti, and Ron 2000), demonstrating the importance of this pathway in development. However fibroblasts generated from IRE1α−/− embryos show defects in UPR activation using reporter genes (Lee et al. 2002). IRE1β-null mice do not show overt developmental defects; however, hyperlipidemia is observed in IRE1β-null mice (Iqbal et al. 2008), and they show increased susceptibility to experimentally induced colitis using dextran sodium sulfate (Bertolotti et al. 2001). XBP1−/− mice also die in utero at E12.5/E13.5 with severe liver hypoplasia correlated with abnormal red blood cell production leading to anemia with subsequent embryonic death (Iwakoshi, Lee, and Glimcher 2003). Reintroduction of XBP1 expression specifically in the liver of XBP1−/− mice died postnatally from severe impairment of pancreatic digestive enzyme production leading to hypoglycemia and eventually death (Lee et al. 2005).
ATF6
ATF6 α and β are type II transmembrane proteins that upon activation translocate to the Golgi from the ER where they undergo regulated intramembrane proteolysis (RIP) by site-1 and site-2 proteases (S1P and S2P). This releases the ATF6 bZIP transactivation domain into the cytosol that translocates to the nucleus to regulate transcription. ATF6α regulates a number of UPR pro-survival genes such as chaperones (e.g. GRP78, GRP94, and protein disulfide isomerase [PDI]), ERAD-related genes and XBP1 (Haze et al. 1999; Yamamoto et al. 2007). ATF6α can homodimerize or heterodimerize with other bZIP transcription factors such as NF-Y and XBP1 and bind to the ERSE (Yoshida et al. 2000). Other ATF6 family members include BBF2H7 (CREB3L2), CREB4, CREB-H, Luman (CREB3), and Oasis (CREB3L1; Zhang et al. 2006; Kondo et al. 2005, 2007; Stirling and O’Hare 2006; DenBoer et al. 2005). Unlike ATF6α, these family members tend to have a more limited tissue distribution and our understanding of their role in ER stress is more limited.
The Wolfram syndrome 1 protein (WFS1) negatively regulates ATF6α signaling, through proteasomal degradation of ATF6α (Fonseca et al. 2010). It has also been proposed that termination of ATF6α signaling may be mediated by unspliced XBP1. Although unspliced XBP1 has a short half-life, the increased expression of XBP1 during UPR activation may lead to accumulation of the unspliced form during the recovery phase which can physically associate with activated ATF6α and spliced XBP1 for degradation via the proteasome, thereby contributing to attenuation of ATF6α signaling (Yoshida, Uemura, and Mori 2009).
Studies in ATF6α−/− mice show that ATF6α is dispensable for embryonic and postnatal development and is not required for basal expression of ER chaperones such as GRP78, GRP94, and PDI. ATF6α is, however, required for optimal protein folding, secretion and degradation during periods of ER stress, assisting recovery from acute ER stress and tolerance to chronic ER stress (Wu et al. 2007). Mice deficient in ATF6β had no embryonic or postnatal developmental defects and cells from these mice showed minimal effects on UPR responses. However, knockout of both ATF6α and ATF6β is embryonic lethal (Yamamoto et al. 2007).
Major ER Stress Regulated Genes and Their Function
Chaperones
Although many protein-folding principles are shared within the ER and the cytosol, protein folding in the ER is more complex due to posttranslational modifications such as glycosylation and lipidation for proper protein function (Ma and Hendershot 2004). Protein-folding reactions must fulfill thermodynamic and kinetic requirements and are aided in the ER by the actions of protein chaperones such as GRP78 and GRP94. The high Ca2+ concentrations in the ER maintained by Ca2+ ATPases (e.g. SERCA) allow for the proper functioning of the calcium- and ATP-dependent GRP78 and GRP94 chaperones of the HSP70 and HSP90 classes, respectively. Transcription of these chaperones is increased in response to several stimuli that disrupt ER function.
Protein chaperones help maintain proteins in a folding-competent state (i.e., binding and shielding hydrophobic sequences), prevent protein aggregation, buffer ER Ca2+, and perform quality control functions such as recognition of surface hydrophobic regions to ensure misfolded proteins are retro-translocated to the cytoplasm for degradation (Quinones, de Ridder, and Pizzo 2008). The quality control mechanisms that report on the folding status of a protein also involve addition and processing of N-linked saccharides recognized by lectin-binding proteins such as calnexin (CNX) and calreticulin (CRT; Ruddock and Molinari 2006). The ER lumen is an oxidative environment that is crucial for the formation of disulfide bonds. Disulfide bond formation is driven by a protein relay involving ER oxidoreductin 1 (Ero1) and members of the PDI family. Ero1, using flavin adenine dinucleotide (FAD) as a cofactor, is oxidized by molecular oxygen and in turn acts as a specific oxidant of PDI, which then directly oxidizes disulfide bonds in folding proteins. Using molecular oxygen as the terminal electron acceptor leads to oxidative stress through the production of reactive oxygen species (ROS; H2O2; Tu and Weissman 2004). Normally the ROS are kept in check via intracellular antioxidants such as glutathione, however excessive demand on protein folding can overwhelm the antioxidant response. Glutathione depletion can also result as the cell tries to reduce improper disulfide bonds leading to a futile cycle of disulfide bond formation and breakage, further generating ROS. In support of this theory, antioxidants have been shown to improve protein folding and reduce apoptosis in response to ER stress (Malhotra et al. 2008).
C/EBP homologous protein (CHOP)
ER-stress-mediated apoptosis is largely mediated by C/EBP homologous protein (CHOP; DDIT3 [DNA damage inducible transcript 3] or GADD153), a transcription factor normally expressed at low levels that is induced by a variety of adverse conditions including amino acid starvation and ER stress (Oyadomari and Mori 2004). It operates mainly downstream of the PERK pathway during UPR signaling, although IRE1 and ATF6 can also contribute to its induction (Ma et al. 2002; Scheuner et al. 2001; Harding et al. 2003). The IRE1-ASK1-p38 MAPK pathway can phosphorylate CHOP, thereby enhancing its transcriptional activity (Wang and Ron 1996). CHOP can function either as a transcriptional activator or repressor. CHOP can sensitize cells to ER stress through mechanisms involving downregulation of antiapoptotic proteins such as BCL-2 and BCL-XL and enhanced oxidative injury by depletion of glutathione and exaggerated production of ROS by increasing expression of Ero1α (McCullough et al. 2001; Marciniak et al. 2004). In addition, CHOP can increase the transcription of pro-apoptotic proteins such as BIM (Puthalakath et al. 2007) further exacerbating cell injury and apoptosis. CHOP can directly activate GADD34 (Marciniak et al. 2004; Ma and Hendershot 2003) and modulate gene expression by interactions with members of the AP-1 transcription factor family (Ubeda, Vallejo, and Habener 1999). CHOP can also increase the expression of TRB3, a protein that inhibits AKT, thereby blocking its cytoprotective actions. GSK-3β, a target of AKT, is thought to target mitochondria promoting permeabilization and further promoting apoptosis (Song, De Sarno, and Jope 2002). Additionally, CHOP can increase the expression of the pro-apoptotic death receptor-5 (DR-5; Yamaguchi and Wang 2004).
CHOP−/− cells are often found to be resistant to apoptosis by ER stress induction compared to the wild-type cells. This is seen in pancreatic β cells as CHOP is a major player in pancreatic β cell apoptosis under conditions of increased insulin demand as several genetically induced or high-fat diet induced diabetes mouse models show preservation of β-cell mass, reduced apoptosis, and improved glycemic control in CHOP−/− mice compared to wild-type mice (Song et al. 2008). This may be due to the ability of β cells to better tolerate oxidative stress in CHOP-deficient mice (Song et al. 2008). Also, mice deficient in CHOP show reduced renal injury and apoptosis after treatment with tunicamycin (Zinszner et al. 1998). Although several lines of evidence suggest a clear involvement for CHOP in ER-stress-induced apoptosis, CHOP-independent apoptosis induction mechanisms exist, as PERK–/– and EIF2α (Ser51Ala) knock-in cells are hypersensitive to ER-stress-induced apoptosis and do not induce CHOP expression (Harding et al. 2003).
How Are Unfolded Proteins Recognized?
Exactly how unfolded proteins are sensed in the ER to activate the UPR is still unresolved. Several models have been proposed as not all findings are supported by one model (reviewed in Parmar and Schroder 2012). Mechanistic insights have largely been reported for yeast and mammalian IRE1 activation, with less data on PERK and ATF6 activation. However, the predominant model involves ER resident chaperones (e.g., GRP78) as master regulators of the UPR. The ER luminal domains of the UPR sensors (PERK, IRE1, and ATF6) and unfolded proteins compete for binding sites on GRP78. When the UPR sensors are bound to GRP78, they are kept in an inactive state. Accumulation of unfolded proteins overwhelms the protein folding chaperones and sequesters them away from the UPR sensors unmasking oligomerization sequences in IRE1 and PERK or Golgi localization sequences in ATF6 (Bertolotti et al. 2000), resulting in their activation.
ER Stress Links with Calcium, Mitochondria, and Oxidative Stress
The concentration of Ca2+ within the ER is several thousand fold higher than in the cytosol and is regulated by ER Ca2+ release channels such as inositol-1,4,5-triphosphate receptor (IP3R) and the ryanodine receptor and Ca2+ uptake transporters such as sarco/endoplasmic reticulum calcium ATPase (SERCA). These channels and transporters can be modulated by a variety of factors including cytokines, hormones, neurotransmitters, lipids, and certain pharmaceutical agents. Decreased ER Ca2+ levels lead to inhibition of proper protein folding resulting in induction of ER stress and can activate several pro-apoptotic cytosolic molecules (Hajnoczky et al. 2000; Michelangeli and East 2011; Lanner et al. 2010).
Intracellular Ca2+ and free radicals such as ROS or reactive nitrogen species (RNS) are key messengers mediating interactions between mitochondria, ER stress, oxidative stress, and inflammation. Although ROS and RNS can act as signaling messengers, they also can modify or damage DNA, lipids, and proteins; therefore, their production is tightly regulated. Multiple mechanisms can lead to the production of ROS including mitochondrial oxidative phosphorylation or excessive ER oxidative protein folding which in turn affects ER localized Ca2+ channels and chaperones resulting in Ca2+ release into the cytosol. This excessive cytosolic Ca2+ can be taken up by mitochondria, causing opening of the mitochondrial permeability transition pore (MPTP), depolarization of the inner mitochondrial membrane, generating further ROS exacerbating Ca2+ release from the ER, and triggering oxidative stress, ER stress, cytochrome c release, and apoptosis (Gorlach, Klappa, and Kietzmann 2006; Zhang 2010; Simmen et al. 2010; Rizzuto et al. 2009). The increase in cytosolic Ca2+ can also result in uncontrolled calpain (calcium-dependent cysteine protease) activation that can lead to proteolysis of intracellular targets contributing to apoptosis (Nakagawa and Yuan 2000). These interactions lead to a vicious cycle involving ROS production from the ER or mitochondria, depletion of ER Ca2+, and induction of ER stress (Figure 5).
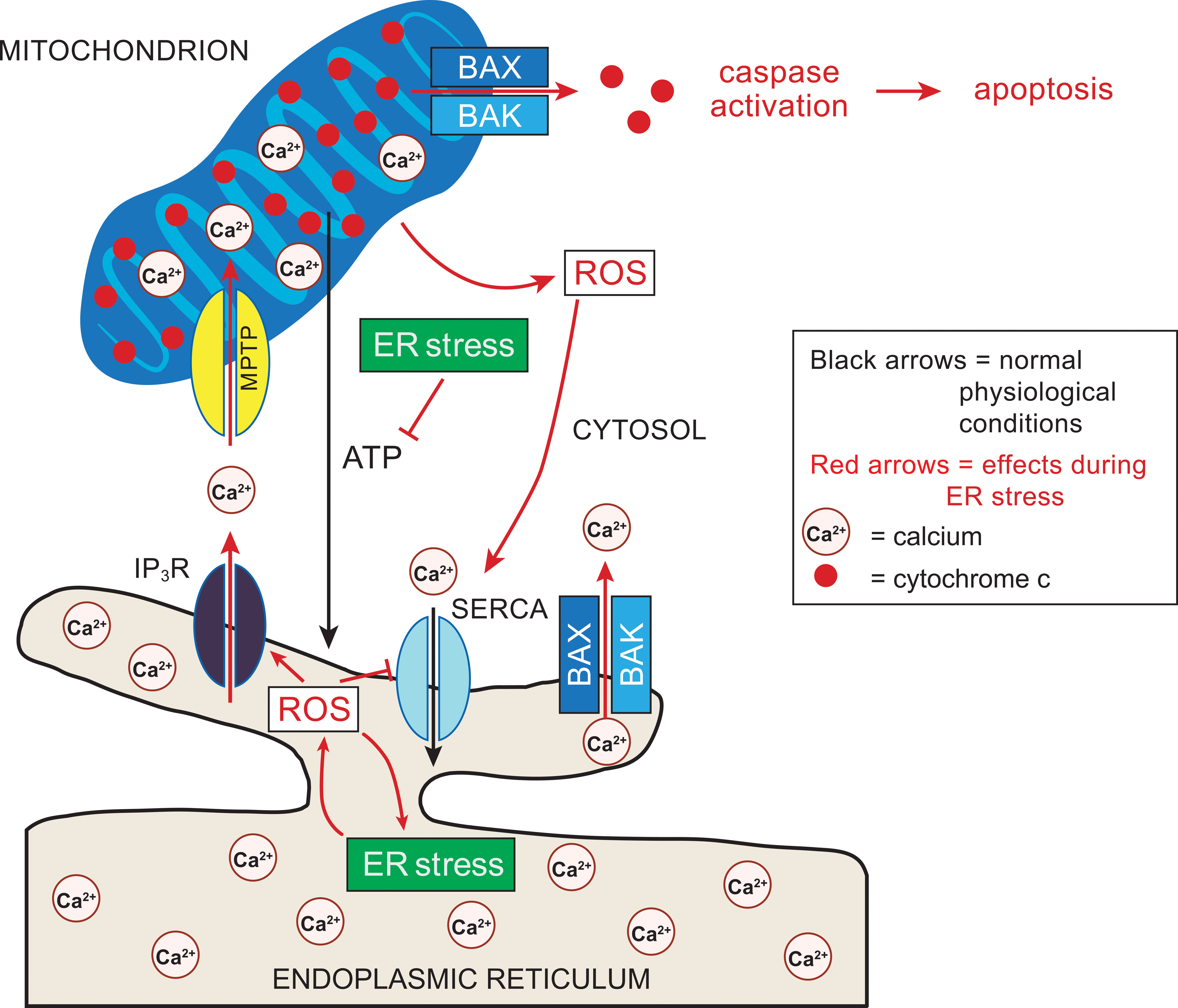
Interactions between ER stress, mitochondria, and oxidative stress. ROS can be produced by multiple mechanisms including mitochondrial oxidative phosphorylation or excessive ER oxidative protein folding which in turn affect ER localized Ca2+ channels/pumps and chaperones resulting in Ca2+ release into the cytosol. This excessive cytosolic Ca2+ can be taken up by mitochondria, causing opening of the mitochondrial permeability transition pore, depolarization of the inner mitochondrial membrane, generating further ROS exacerbating Ca2+ release from the ER and triggering oxidative stress, ER stress, cytochrome c release, and apoptosis. These interactions lead to a vicious cycle involving ROS production, depletion of ER Ca2+, and induction of ER stress. Mitochondrial cytochrome c release can also be mediated by BAK and BAX which oligomerize and form a pore in the outer mitochondrial membrane, given the appropriate pro-apoptotic signals. BAX and BAK can also insert into the ER membrane to mediate Ca2+ release into the cytosol activating calpains. Cytosolic cytochrome c activates downstream apoptotic pathways. Adapted from Zhang (2010). ER = endoplasmic reticulum; ROS = reactive oxygen species.
ER Stress Links with Apoptosis and Autophagy
Apoptosis
Apoptosis is a terminal, controlled cell death program characterized by cell rounding, nuclear condensation, DNA fragmentation, cytoskeleton disassembly, and plasma membrane blebbing. The cellular components are eventually encased in membrane-enclosed apoptotic bodies that are rapidly taken up and destroyed by phagocytes or neighboring cells. A prominent feature of apoptosis is the activation of a caspase cascade leading to proteolytic cleavage of intracellular components (Kurokawa and Kornbluth 2009). Two main apoptotic pathways regulate programmed cell death: the extrinsic pathway mediated by caspase-8 (or caspase-10) signal transduction downstream of death receptors (such as Fas) located on the plasma membrane, and the intrinsic pathway mediated by signal transduction associated with mitochondria (Salvesen and Dixit 1997). The intrinsic apoptotic pathway is the main apoptotic pathway induced by the UPR and is set in motion by mitochondrial release into the cytoplasm of certain mitochondrial proteins such as cytochrome c by BAK and BAX, which oligomerize and form a pore in the outer mitochondrial membrane. Cytosolic cytochrome c promotes oligomerization of the caspase recruitment domain-containing adaptor protein-1 (Apaf-1), which in turn recruits caspase-9 via apoptosome formation leading to activation of other executioner caspases such as caspase-3 (Kurokawa and Kornbluth 2009). When activated, BAX and BAK can also insert into the ER membrane to mediate Ca2+ release into the cytosol, which can trigger pro-apoptotic signaling mechanisms as described above (Rasola and Bernardi 2011). Other apoptosis regulating proteins include BH3-only proteins (e.g., BID [BH3 interacting-domain death agonist], BIM, and BAD [BCL2 associated death promoter]) which lead to the loss of mitochondrial membrane integrity disabling anti-apoptotic proteins (e.g. BCL-2, BCL-XL and Mcl-1) and triggering oligomerization of the pro-apoptotic proteins BAX and BAK that lead to permeabilization of the outer mitochondrial membrane (Giam, Huang, and Bouillet 2008). Conversely, BCL-2 can inhibit apoptosis through sequestration of BH3-only proteins such as BAD, BIM, Noxa, and Puma (Cheng et al. 2001). ER Ca2+ levels are also regulated by BI-1, an ER resident transmembrane cell death suppressor protein that interacts with BCL-2 and BCL-XL (Ishikawa et al. 2011). Overexpression of BI-1 decreases the ER Ca2+ concentration while BI-1 knockdown increases ER Ca2+ concentrations (Chae et al. 2004). Oligomerization of BI-1 during ER stress promotes Ca2+/H+ antiporter activity, thereby releasing ER-stored Ca2+, reducing acidification of the cytosol, and supporting cell survival (Robinson et al. 2011).
Several studies have indicated an association with the activation of the UPR and activation of the caspase-12-mediated intrinsic apoptotic pathway which activates downstream caspase-9 and caspase-3 (Nakagawa and Yuan 2000; Rao et al. 2001). Importantly, during caspase-12-mediated apoptosis, cytochrome c is not released from mitochondria (Morishima et al. 2002). Mice deficient in caspase-12 are resistant to ER stress-induced apoptosis but sensitive to apoptosis from other death stimuli (Nakagawa et al. 2000). Caspase-12 is associated with the ER and is specifically involved in apoptosis in response to ER stress at least in mouse cells. It is unclear whether this pathway is involved in humans since most humans encode an enzymatically inactive caspase-12 gene (Saleh et al. 2004), with only certain subpopulations expressing a functionally active enzyme (Yavari et al. 2012). Caspase-4 has been suggested to fulfill the role of caspase-12 in humans (Hitomi et al. 2004). Other studies have suggested caspase-12 is downstream of BAX/BAK-mediated mitochondrial permeabilization and that it is more involved in amplification of the apoptotic signal rather than initiation (Ruiz-Vela et al. 2005). Caspase-12 may be regulated by IRE1 signaling via recruitment by IRE1/TRAF2 (Yoneda et al. 2001), by calpain proteases activated by Ca2+ release from the ER to the cytosol mediated by BAK and BAX (Tan et al. 2006) or by caspase-7 (Rao et al. 2001). Activated caspase-12 can trigger caspase-9 activation which can lead to caspase-3 activation resulting in apoptosis. Given that apoptosis is an energy requiring process, cell death can result from necrosis if the stress is sufficiently severe to deplete ATP (Jaeschke et al. 2002).
Autophagy
Autophagy is a highly conserved pro-survival catabolic process used by a variety of cell types to maintain homeostasis by sequestering cytoplasmic cargo into double-membrane vesicles followed by fusion with lysosomes for degradation and recycling. Autophagy occurs at a low basal level in most if not all cell types to ensure homeostasis of protein and organelle turnover. However, the rate of autophagy can be enhanced such as during periods of increased nutrient and energy demands, growth factor withdrawal, developmental remodeling, oxidative stress, mitochondrial dysfunction, infections, and ER stress (Levine and Kroemer 2008). Because ER stress can induce autophagy, this could represent an alternate mechanism for removing unfolded proteins independently of the proteasome system and may be important for diseases associated with exhaustion of proteasome function and protein aggregation (Ogata et al. 2006; Bernales, McDonald, and Walter 2006). Although generally a survival mechanism, autophagy has been associated with induction of nonapoptotic cell death in several contexts (Levine and Kroemer 2008). Several reports have shown that autophagy can be directly activated by ER stress via the IRE1/JNK pathway and PERK/eIF2α/ATF4-dependent signaling (Rzymski et al. 2009; Ding et al. 2007; Kouroku et al. 2007). This may involve JNK-mediated phosphorylation of BCL-2, thereby affecting its interaction with the autophagy protein Beclin-1 (Pattingre et al. 2009). Conversely, inhibitors of ER stress as well as siRNA knockdown of IRE1 expression inhibited autophagy and cell death in cardiomyocytes (Younce and Kolattukudy 2010). Inhibition of autophagy can lead to the accumulation of misfolded proteins in neuronal cells and may be involved in chronic neurodegenerative diseases such as Huntington’s and Alzheimer’s disease (Zhang et al. 2007).
Normal Physiological Events Dependent on ER Stress and UPR Signaling
Healthy or unstressed cells can experience low levels of ER stress and UPR activation to maintain homeostasis and adjust ER folding capacity to meet normal fluctuations in protein demand. Although most cells may be dependent on the UPR at some point in their life cycle depending on environmental conditions or cellular differentiation, cells with a high secretory capacity such as hepatocytes, plasma cells, pancreatic β cells, exocrine pancreatic cells, osteoblasts, and gastric cells are particularly dependent on UPR signaling. Genetic disruption of the PERK or IRE1 pathway molecules causes severe abnormalities in these cells and affects their function (Lee et al. 2005; Iwawaki, Akai, and Kohno 2010; Huh et al. 2010; Harding et al. 2001). For example, the IRE1/XBP1 pathway is essential for B cell maturation into antibody producing plasma cells. As B cells differentiate into antibody producing plasma cells, the UPR is activated with increased expression of chaperones and the ER expands several fold to accommodate the large increase in immunoglobulin (Ig) production (Wiest et al. 1990). UPR activation, however, precedes significant Ig synthesis and B cells incapable of IgM secretion induced XBP1 activation normally upon differentiation (van Anken et al. 2003; Ma et al. 2010; Hu et al. 2009), suggesting increased Ig production is not the UPR activating signal but is dependent on a differentiation-dependent event. Studies in ATF6α-deficient B cells however showed that ATF6α is not required for the development of antibody-secreting cells (Aragon et al. 2012).
Other normal functions of the UPR include regulation of the production of hepcidin, a hepatic peptide hormone that regulates iron homeostasis (Vecchi et al. 2009). IRE1α and ATF6α are necessary to maintain hepatic lipid homeostasis during ER stress (Zhang et al. 2011; Yamamoto et al. 2010). ER stress can control gluconeogenesis via ATF6-mediated negative regulation of the CREB-regulated transcription coactivator 2 (CRTC2; Wang, Vera, et al. 2009). Brain-derived neurotrophic factor (BDNF) initiates XBP1 splicing to stimulate neurite outgrowth (Hayashi et al. 2007). XBP-1 regulates lipogenesis in the liver (Lee et al. 2008) and can help regulate glucose homeostasis by interactions with forkhead box O1 (FoxO1), directing it toward proteasomal degradation (Zhou et al. 2011). XBP1 has also been linked to DNA damage and repair pathways and targets Mist1, a regulator of differentiation (Acosta-Alvear et al. 2007). ER stress and UPR signaling is therefore critical for cellular homeostasis in addition to adapting to increased protein folding load.
ER Stress and Disease
ER stress is a central feature in the pathogenesis of many genetic or acquired diseases (Schroder and Kaufman 2005). From a tissue/organ standpoint, although cell survival and death are two contrasting outcomes from ER stress, it should be highlighted that cell survival resulting from adaptation may also be accompanied by cellular dysfunction which can lead to the pathogenesis of various diseases or conditions such as insulin resistance, impaired gluconeogenesis, inflammation, steatosis, and others (Table 1). It is not always clear, however, whether UPR deregulation causes/contributes to the disease, or whether UPR activation is a secondary response. Therapeutic intervention in modulating ER stress has mainly focused on assisting the pro-survival aspects of the UPR, such as with chemical chaperones that assist with protein folding thereby preventing ER stress-induced apoptosis. However, in other disease states, amplifying the apoptotic signaling pathways may be beneficial such as with cancer. ER stress is often a feature of cancer cells since the uncontrolled proliferation can lead to regions of hypoxia, hypoglycemia, and oxidative stress, all known activators of ER stress. Cancer cells can co-opt the protective mechanisms of the UPR while suppressing the pro-death signals or antiproliferative signals to gain a survival advantage in the hostile tumor environment. A comprehensive description of diseases linked with ER stress is beyond the scope of this review; however, this subject is covered in greater detail elsewhere (Kim, Xu, and Reed 2008; Schroder and Kaufman 2005; Chakrabarti, Chen, and Varner 2011; Ozcan and Tabas 2012). Table 1 lists some prominent examples of diseases or pathologic conditions associated with ER stress.
Genetic or acquired diseases and links to ER stress.
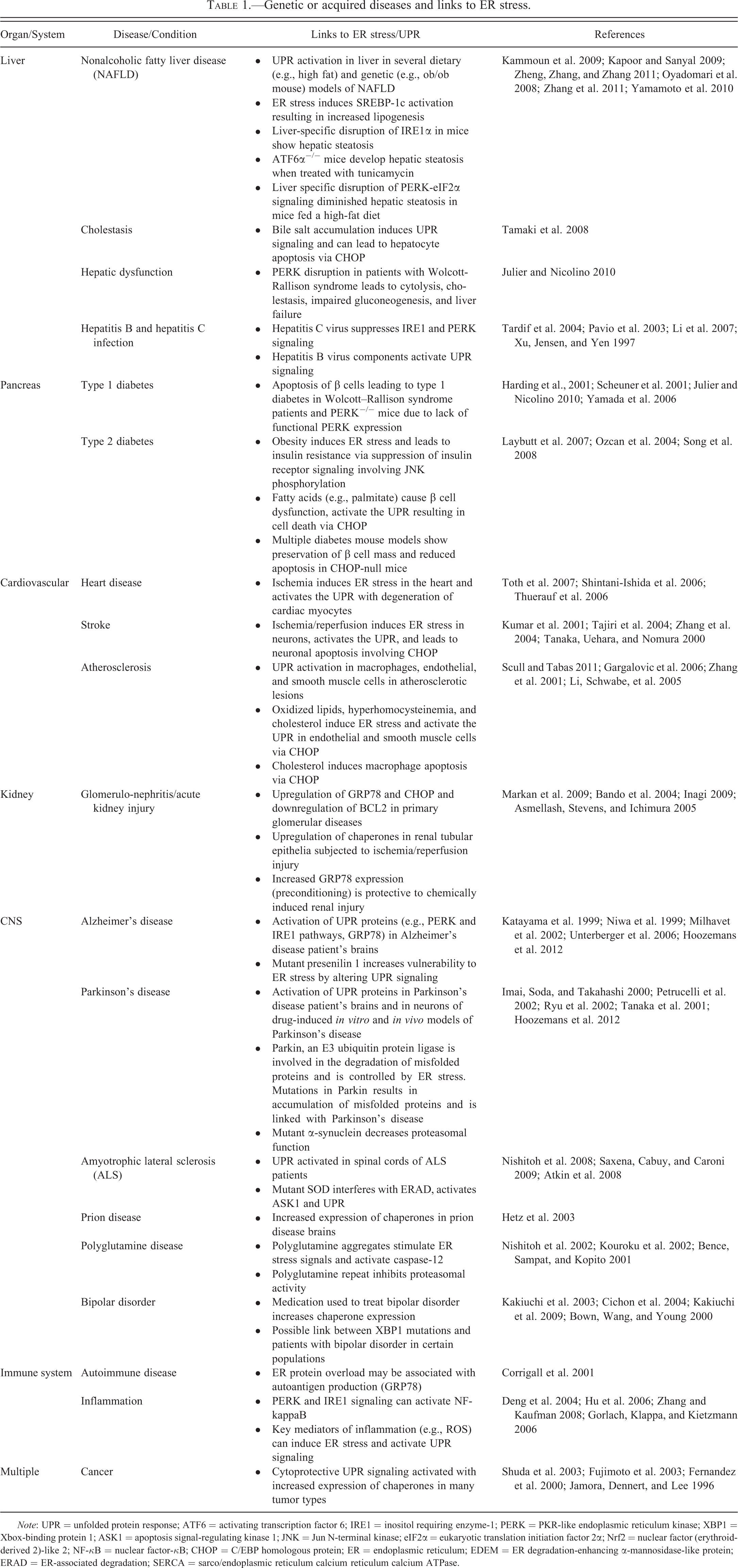
Note: UPR = unfolded protein response; ATF6 = activating transcription factor 6; IRE1 = inositol requiring enzyme-1; PERK = PKR-like endoplasmic reticulum kinase; XBP1 = Xbox-binding protein 1; ASK1 = apoptosis signal-regulating kinase 1; JNK = Jun N-terminal kinase; eIF2α = eukaryotic translation initiation factor 2α; Nrf2 = nuclear factor (erythroid-derived 2)-like 2; NF-κB = nuclear factor-κB; CHOP = C/EBP homologous protein; ER = endoplasmic reticulum; EDEM = ER degradation-enhancing α-mannosidase-like protein; ERAD = ER-associated degradation; SERCA = sarco/endoplasmic reticulum calcium reticulum calcium ATPase.
Xenobiotics with ER Stress-induced Cytotoxic Effects
A number of xenobiotics have been described that can cause ER stress or modulate the adaptive response resulting in apoptosis via a variety of different mechanisms (Table 2). These examples include perturbations in protein folding by interfering with N-linked glycosylation, disulfide bond formation, or inhibition of chaperone expression. Other xenobiotics can block protein transport from the ER to the Golgi, resulting in a backup of proteins in the ER, perturb protein folding by targeting factors that influence protein folding such as modulation of ER Ca2+ channels/pumps, or directly target specific UPR signaling molecules interfering with the adaptive response. Finally, inhibition of the unfolded protein degradation machinery such as ERAD is another way xenobiotics may cause enhancement of ER stress and reduced cell survival. These xenobiotics include a variety of classical tool compounds which robustly cause ER stress and activate the UPR in a variety of cell types such as tunicamycin, thapsigargin, brefeldin A, and DTT. Other xenobiotics with ER stress inducing activity are MG-132, Resveratrol, STF-083010, versipelostatin, and eeyerestatin I. Marketed pharmaceuticals developed for various diseases can also cause ER stress by either on- or off-target mechanisms for which they were developed including bortezomib, nelfinavir, atazanavir, ritonavir, lopinavir, sorafenib, indomethacin, and celecoxib.
Xenobiotics that modulate ER stress and/or have cytotoxic effects.
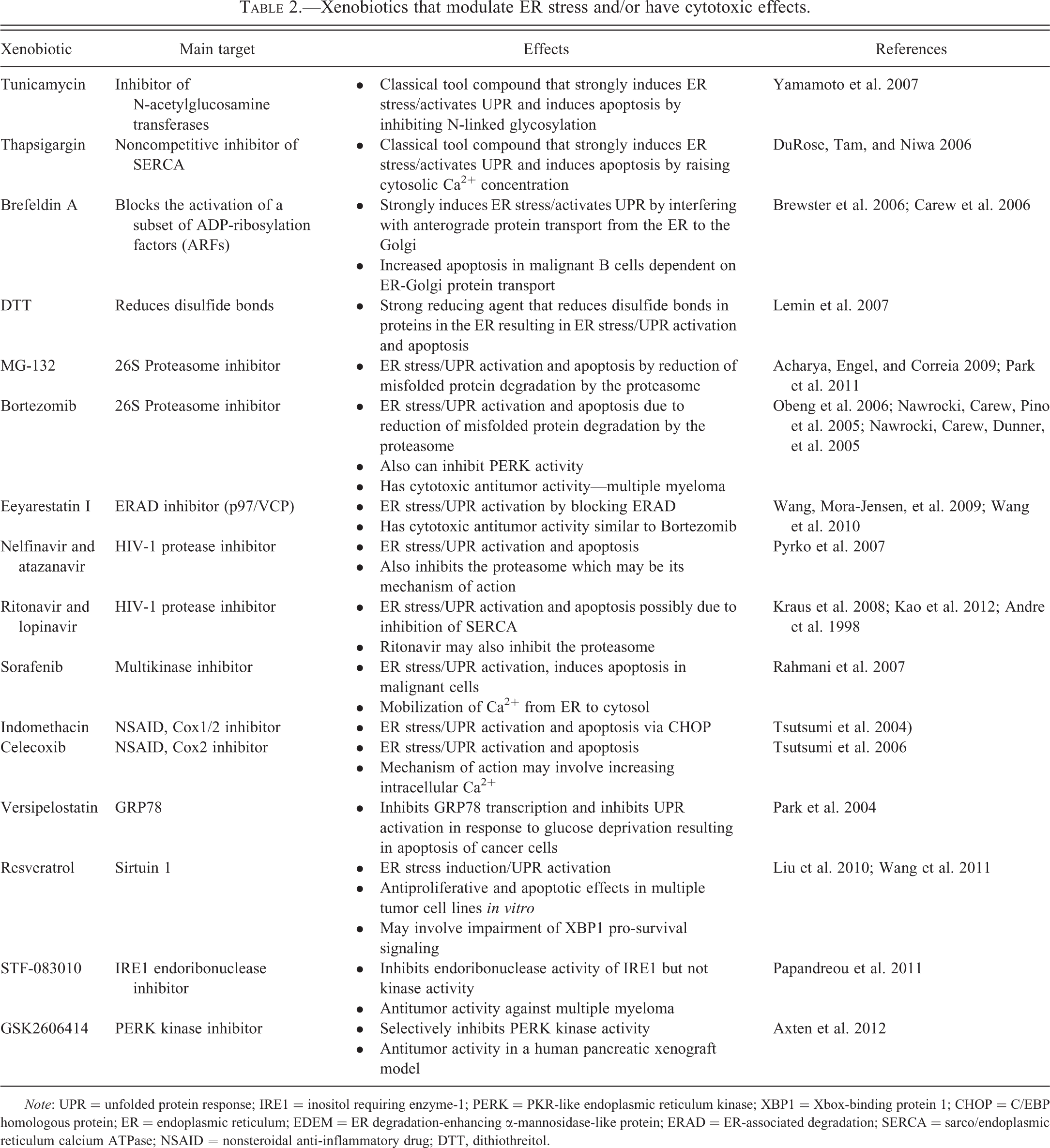
Note: UPR = unfolded protein response; IRE1 = inositol requiring enzyme-1; PERK = PKR-like endoplasmic reticulum kinase; XBP1 = Xbox-binding protein 1; CHOP = C/EBP homologous protein; ER = endoplasmic reticulum; EDEM = ER degradation-enhancing α-mannosidase-like protein; ERAD = ER-associated degradation; SERCA = sarco/endoplasmic reticulum calcium ATPase; NSAID = nonsteroidal anti-inflammatory drug; DTT, dithiothreitol.
An example of on-target ER stress modulation is seen with bortezomib (Velcade) developed as a proteasome inhibitor that induces ER stress in several cell types, particularly multiple myeloma cells which contribute to its anticancer cytotoxic activity (Lee et al. 2003; Frankland-Searby and Bhaumik 2012; Obeng et al. 2006). These malignant cells derived from plasma cells secrete extensive amounts of immunoglobulins and are thus particularly vulnerable to disruptions in ER homeostasis. ER stress induction by Bortezomib in these cells is partly due to their reduced ability to degrade misfolded proteins targeted to the proteasome (Nawrocki, Carew, Pino et al. 2005). Similar effects were also seen in pancreatic tumor models although bortezomib also appeared to inhibit PERK-mediated translation attenuation in addition to the proteasome, thereby exacerbating ER stress (Nawrocki, Carew, Dunner, et al. 2005).
Off-target effects causing ER stress can be seen for nelfinavir, atazanavir, ritonavir, and lopinavir, which although originally developed as HIV-1 protease inhibitors, were later found to cause ER stress and activate the UPR via other mechanisms possibly involving proteasome inhibition at high concentrations (Pyrko et al. 2007; Andre et al. 1998; Lum et al. 2007; Schmidtke et al. 1999). Ritonavir and lopinavir may also have effects on ER Ca2+ levels by inhibition of SERCA (Kao et al. 2012). A variety of cancer cell lines are sensitive to the action of HIV protease inhibitors in vitro (Gills et al. 2007) and induce cell death in malignant glioma xenografts in vivo involving ER stress induction and CHOP-mediated apoptosis (Pyrko et al. 2007). For some of the xenobiotics listed in Table 2, the exact mechanism leading to ER stress is not completely understood and may involve multiple mechanisms depending on the concentration as some xenobiotics only induce ER stress at very high concentrations. This may push the cells toward apoptosis due to a combination of cellular stresses (ER-UPR, mitochondria-UPR, cytosolic responses, oxidative stress, and others). Since the ER is the major site for metabolic biotransformation of xenobiotics, the observed effects on ER stress by xenobiotics may be due to the parent xenobiotic, a metabolite, or both.
Xenobiotics with Cytoprotective Effects
Table 3 shows examples of xenobiotics that have cytoprotective activities in a variety of in vitro and in vivo model systems. The mechanisms are varied and include increasing the ER protein-folding capacity with chemical chaperones or increased expression of protein chaperones as well as selectively enhancing the protective UPR signaling pathways. Antioxidants, Ca2+ channel blockers, and autophagy inducers can also show protective effects by ensuring proper ER homeostasis. A few examples of these protective effects are described in more detail below for sodium 4-phenylbutyrate (PBA), taurine-conjugated ursodeoxycholic acid (TUDCA), salubrinal/guanabenz, trans-4,5-dihydroxy-1,2-dithiane (DTTox), and valproate.
Xenobiotics that modulate ER stress and/or have cytoprotective activities.
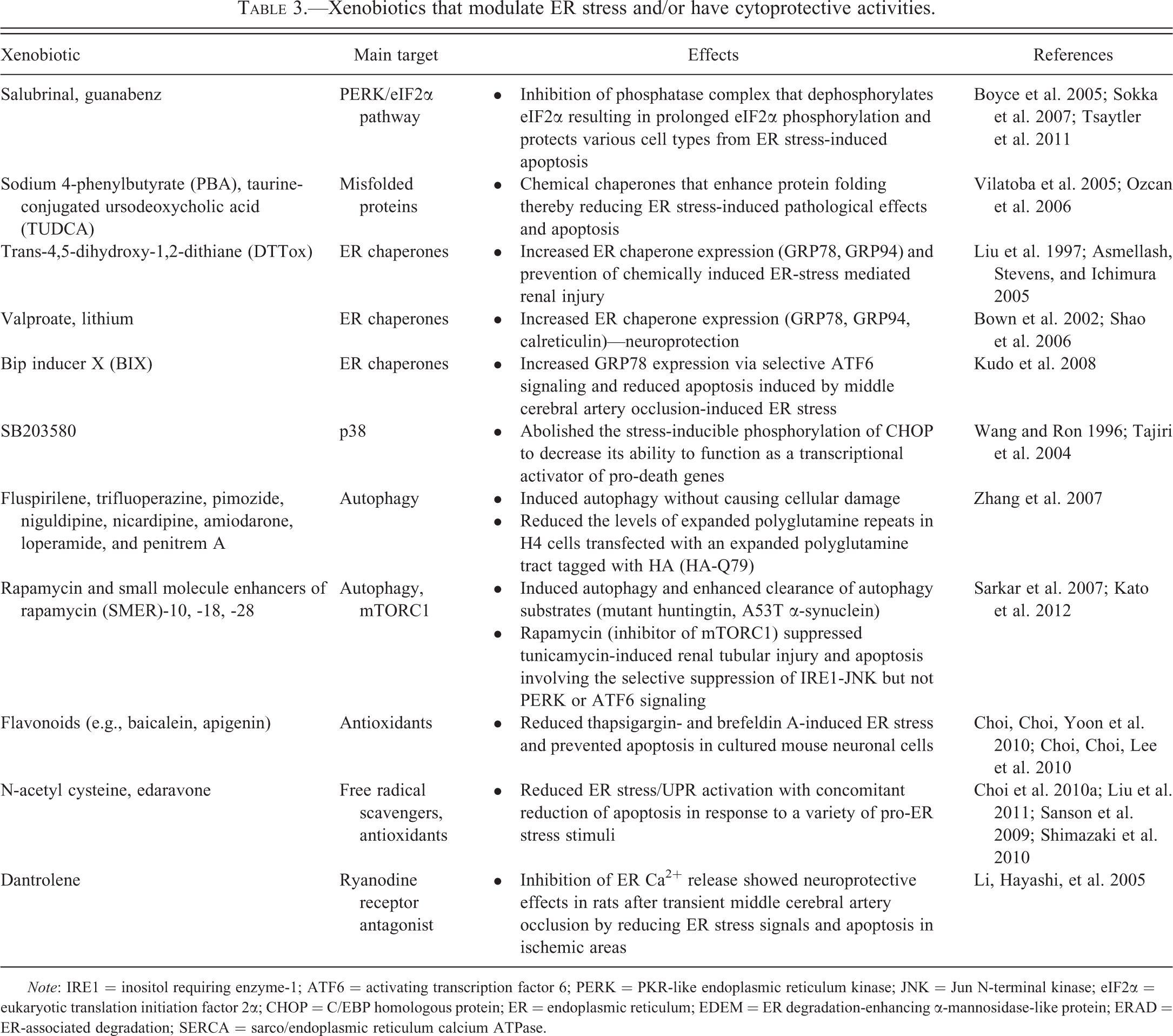
Note: IRE1 = inositol requiring enzyme-1; ATF6 = activating transcription factor 6; PERK = PKR-like endoplasmic reticulum kinase; JNK = Jun N-terminal kinase; eIF2α = eukaryotic translation initiation factor 2α; CHOP = C/EBP homologous protein; ER = endoplasmic reticulum; EDEM = ER degradation-enhancing α-mannosidase-like protein; ERAD = ER-associated degradation; SERCA = sarco/endoplasmic reticulum calcium ATPase.
Chemical Chaperones
In pathological conditions where ER stress is a major factor leading to injury, alleviating ER stress by enhancing the ER protein folding capacity with chemical chaperones may be of benefit. For example, obesity can induce ER stress which in turn plays a role in the development of insulin resistance and diabetes via activation of IRE1/JNK signaling which phosphorylates the insulin receptor substrate 1 (IRS1) at position Serine 307 resulting in decreased tyrosine phosphorylation by the insulin receptor (Ozcan et al. 2004). Obese and diabetic mice treated with the chemical chaperones PBA and TUDCA resulted in normalization of blood glucose levels, re-establishment of insulin sensitivity, and resolution of associated steatosis (Ozcan et al. 2006). In another injury model, PBA protected against liver ischemia–reperfusion injury by inhibition of ER-stress-mediated apoptosis (Vilatoba et al. 2005). Treatment of macrophages with PBA protected against palmitate-induced ER-stress-related apoptosis and atherosclerosis in ApoE−/− mice fed a Western diet (Erbay et al. 2009). Chemical chaperones may thus have potential for the treatment of a variety of pathological conditions where ER stress plays a role in mediating injury such as type 2 diabetes, ischemia–reperfusion injury, and atherosclerosis.
Salubrinal/guanabenz
Xenobiotics such as salubrinal or guanabenz (α2-adrenergic receptor agonist) have inhibitory activity toward the phosphatase complex that dephosphorylates eIF2α resulting in prolongation of eIF2α phosphorylation with extension of translation attenuation (Boyce et al. 2005; Tsaytler et al. 2011). This offers cytoprotection to certain cell types experiencing ER stress from a number of inducers. Salubrinal protected neuronal cells from apoptosis triggered by mutant proteins that promoted ER stress including α-synuclein mutants associated with Parkinson’s disease (Smith et al. 2005) and N-terminal Huntingtin proteins with expanded polyglutamine repeats associated with Huntington’s disease (Reijonen et al. 2008). Salubrinal also reduced brain damage and apoptosis after cerebral ischemia–reperfusion injury in rats (Nakka, Gusain, and Raghubir 2010). Elevated brain glutamate and activation of glutamate receptors accompanies neurological disorders, such as epilepsy and brain trauma. Salubrinal protected against cell death induced by the glutamate receptor agonist kainic acid both in vitro and in vivo (Sokka et al. 2007). Salubrinal also suppressed β-adrenergic receptor-stimulated ER stress and apoptosis in cardiac myocytes in mice (Dalal et al. 2012). These cytoprotective effects of salubrinal, however, are not observed in pancreatic β cells, as salubrinal alone induced apoptosis and exacerbated free fatty acid–induced ER stress and apoptosis in primary β cells (Cnop et al. 2007) and potentiated the lipotoxicity of oleate and palmitate in human islets (Ladriere et al. 2010). However, guanabenz protected β cells from apoptosis triggered by a mutant misfolding prone form of insulin (Tsaytler et al. 2011).
Acute and progressive renal diseases are major health concerns; recent reviews highlight a role for ER stress in the pathophysiology of both conditions (Inagi 2010; Quiros et al. 2011; Kitamura 2008; Cybulsky 2010). The glomerular and tubular components of the nephron are targets for nephrotoxic exposure or chronic nephropathies secondary to disease states (e.g., diabetes and hypertension), and ER stress in different cellular components of both structures is associated with disease progression (Inagi 2010). Numerous in vitro studies illustrate the importance of ER stress in nephrotoxicant-induced acute injury, but in vitro results are not necessarily replicated in vivo (Peyrou and Cribb 2007). Cyclosporine, a widely used immunosuppressant, prevents rejection of transplanted kidneys, however, longer-term treatment with cyclosporine is nephrotoxic and causes ER stress; salubrinal prevented cyclosporin-induced tubular injury in vivo and in vitro (Pallet et al. 2008; Bouvier et al. 2009). Cisplatin also induced renal ER stress in vivo, but administration of salubrinal exacerbated the injury and increased ATF4 and CHOP expression as well as cleavage of caspases-3, -9 and -12 (Wu et al. 2011). The authors suggested that longer-term eIF2α phosphorylation with translational attenuation exacerbated the oxidative stress associated with cisplatin toxicity. Thus, modulating the UPR or decoupling ER stress from downstream events including translational control can either prevent or exacerbate injury depending on the agent and mechanism of toxicity. Long-term cell survival requires protein synthesis, suggesting that a balance of protein synthesis would be required for optimal cell survival in cells treated with inhibitors that prolong translation attenuation.
DTTox
Pretreatment of rats with DTTox that increases the expression of GRP78 protected the proximal tubular epithelium against a subsequent challenge with the nephrotoxicant S-(1,1,2,2,-tetrafluoroethyl)-
Valproate
Valproate has been shown to be an effective epilepsy and bipolar treatment; however, its mechanism of action is not well understood (Bown et al. 2002). Valproate has been shown to induce expression of GRP78 in rat cerebral cortex and hippocampus tissues after chronic treatment (Chen, Wang, and Young 2000). These findings were confirmed in vitro with chronic valproate treatment of rat C6 glioma cells where valproate induced the expression of GRP78, GRP94, as well as other chaperone proteins (Bown, Wang, and Young 2000). This induction appeared to involve histone deacetylase inhibition (Shi et al. 2007). Valproate has also been shown to cause hepatic steatosis through inhibition of fatty acid oxidation (Silva et al. 2008; Aires et al. 2010) and has been shown to induce the expression of the chaperones GRP78, GRP94, PDI, and calreticulin in HepG2 cells (Kim et al. 2005). Pretreatment of cells with valproate protected HepG2 cells from tunicamycin treatment-related ER stress-induced lipid accumulation and apoptosis (Kim et al. 2005). This protection appeared to occur downstream of the ER stress response and may involve GSK3α/β (Kim et al. 2005). Treatment of rats with the chemical chaperone TUDCA resulted in decreased activation of IRE1 and PERK as well as protection from ischemia–reperfusion injury after partial hepatectomy (Ben Mosbah et al. 2010). Valproate is associated with hepatic steatosis and severe hepatotoxicity when administered at high doses to humans (Navarro and Senior 2006). Thus, the protective effects observed in vitro or in rat models of liver injury do not appear to translate to the clinical setting.
ER Stress Genes as Markers for ER Stress
From a morphological perspective using histopathology or electron microscopy, evidence of ER stress may include disturbances in cellular architecture such as disorganization or distension of the ER lumen (Scheuner et al. 2005). Other downstream effects of ER stress such as morphological features of apoptosis or hepatic steatosis may also be observed (Rutkowski et al. 2008). These, however, are not definitive markers, but rather effects sometimes associated with ER stress and may give an indication that ER stress might be involved. More definitive molecular markers of ER stress and UPR signaling are needed to confirm the presence of ER stress.
The ultimate UPR response induced by xenobiotics may vary depending on the nature of the ER stress inducing xenobiotic, the concentration (mild vs severe), and the duration of exposure (acute vs chronic). In addition, other external factors such as the cell type, nutrient levels, oxidative stress, state of differentiation, and others can all have an impact on the degree of UPR response to xenobiotics. Integration from translational attenuation in the PERK pathway, CHOP levels, mRNA degradation via IRE1, JNK signaling from IRE1, binding of IRE1 to BAX/BAK and BI1, as well as other effects come into play that determine the ultimate cellular fate. When assessing xenobiotics for effects on modulating ER stress and UPR signaling, which molecular markers are the most critical to measure? Several of the more prevalent UPR-associated end points with examples on how to measure them are listed in Table 4; however, this is not an exhaustive list with other end points possible, depending on the context of study. It is worth mentioning that while some of these end points are relatively specific for UPR activation, such as PERK and IRE1 phosphorylation and ATF6 cleavage, some of these end points can be activated by other cellular stresses, such as phosphorylation of eIF2α by the PERK family members GCN2 (general control of nitrogen protein kinase) activated by amino acid starvation (Kimball 2001), PKR (protein kinase R or interferon-induced double stranded RNA-activated protein kinase) activated by dsRNA viruses (Garcia, Meurs, and Esteban 2007), and HRI (heme-regulated eIF-2α kinase) activated by heme deficiency (Chen and London 1995). Therefore, care must be taken when interpreting results from a limited number of end points that are not completely UPR specific. Given that multiple types of activation mechanisms are in play during UPR signaling, such as phosphorylation events, protein cleavage, intron splicing, nuclear translocation, increased transcription, and so on, each UPR-related protein will have its own activation kinetics with some acting early (e.g., PERK and eIF2α phosphorylation), others with intermediate kinetics (e.g., CHOP), and others acting late (e.g., GADD34). Therefore, a time-based assessment may be required to fully understand the spectrum of UPR-related effects.
Major end points to monitor for modulation of ER stress/UPR signaling.
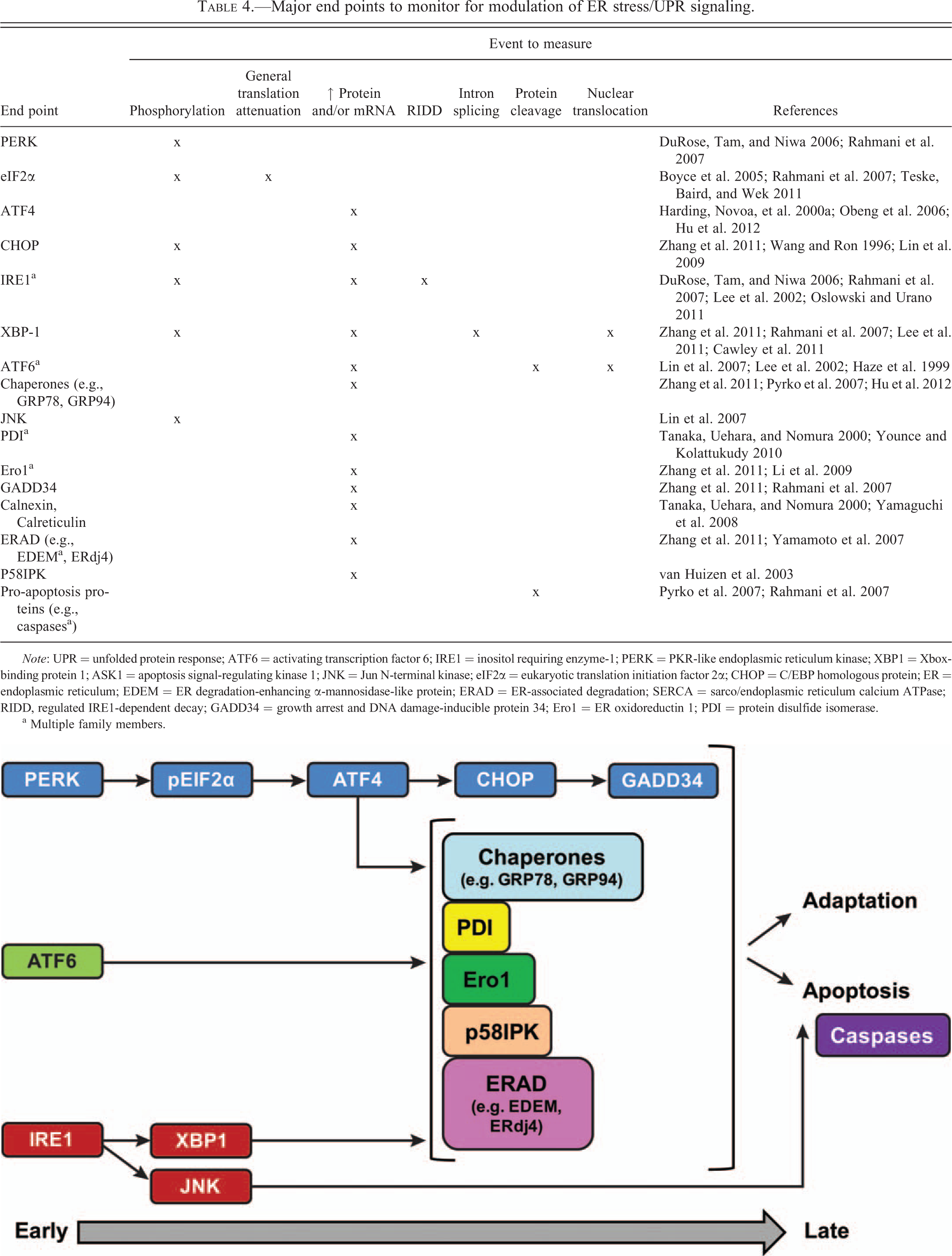
Note: UPR = unfolded protein response; ATF6 = activating transcription factor 6; IRE1 = inositol requiring enzyme-1; PERK = PKR-like endoplasmic reticulum kinase; XBP1 = Xbox-binding protein 1; ASK1 = apoptosis signal-regulating kinase 1; JNK = Jun N-terminal kinase; eIF2α = eukaryotic translation initiation factor 2α; CHOP = C/EBP homologous protein; ER = endoplasmic reticulum; EDEM = ER degradation-enhancing α-mannosidase-like protein; ERAD = ER-associated degradation; SERCA = sarco/endoplasmic reticulum calcium ATPase; RIDD, regulated IRE1-dependent decay; GADD34 = growth arrest and DNA damage-inducible protein 34; Ero1 = ER oxidoreductin 1; PDI = protein disulfide isomerase.
a Multiple family members.
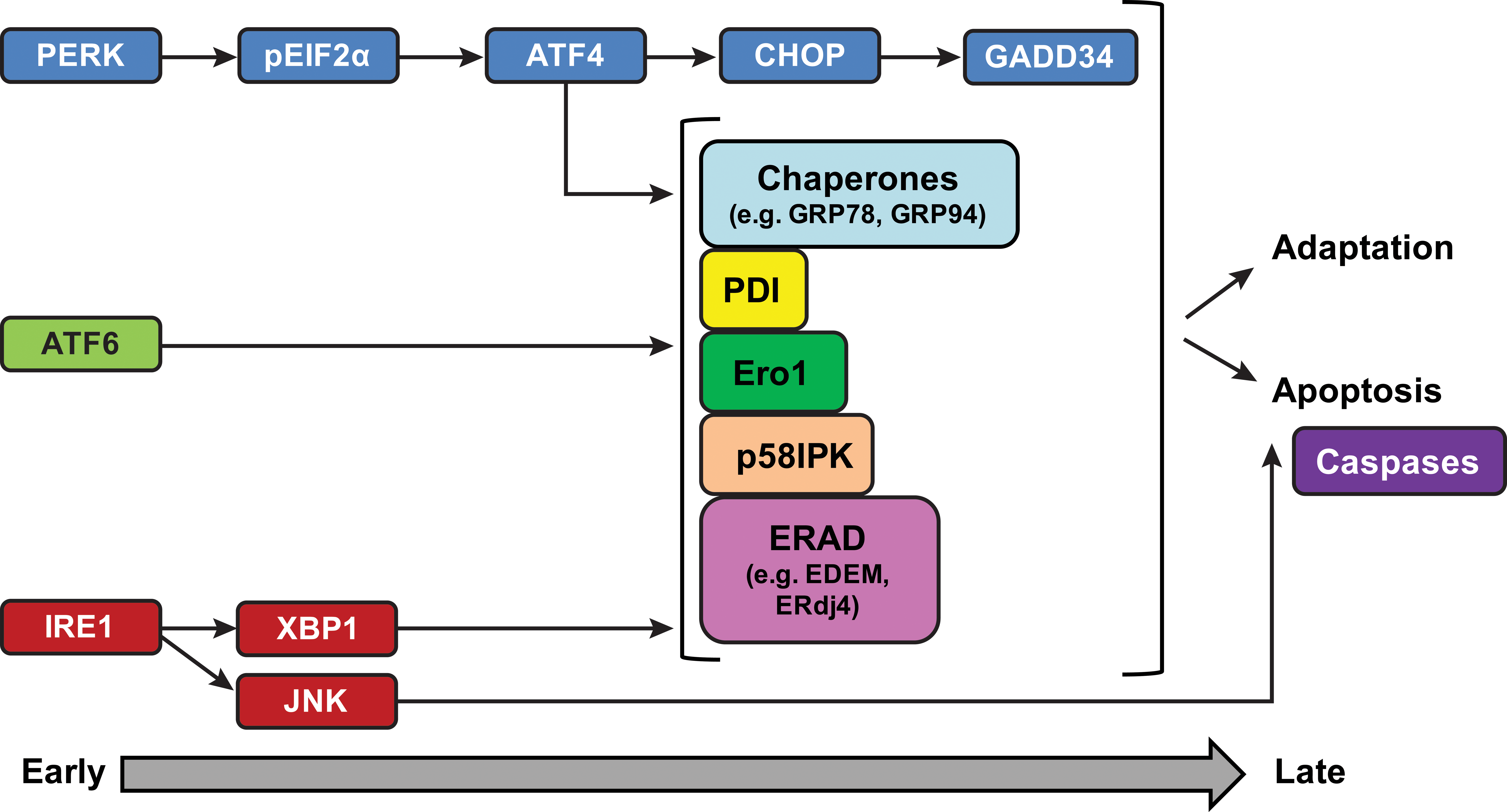
Most studies assessing UPR activation have not analyzed all UPR proteins listed in Table 4, with most groups choosing only a select few depending on the experimental context. Although not all UPR components necessarily need to be measured to assess UPR modulation, it is prudent to measure several of these components from the three different signaling arms to properly understand the spectrum of UPR signaling events as some cases of selective or differential modulation of the UPR arms have been reported (see below) which may affect cellular outcome.
Differential Activation of the UPR Pathways
Although each of the 3 UPR arms shows unique features in adaptation to ER stress, functional overlap is also observed in terms of upregulation of UPR genes to ensure cells have the best chance for survival when confronted with acute and chronic insults. Therefore, a robust induction of ER stress would likely lead to activation of all three arms of the UPR. However, some notable exceptions have been observed where only selective arms of the UPR have been activated. This is especially evident in different physiological events dependent on the UPR. Although B cells activate all three arms of the UPR in response to pharmacological ER stress inducers, plasma cell differentiation in response to LPS mounted only a partial response including IRE1 and ATF6 but not full activation of PERK (Gass et al. 2008). Pretreatment of B cells with LPS inhibited activation of PERK signaling by pharmacological ER stress inducers, suggesting that differentiation-induced signals specifically silence the PERK pathway (Ma et al. 2010). Another example is seen in differentiating thyroid cells where the synthesis and secretion of thyroglobulin (thyroid hormone precursor) was accompanied by increased ER chaperone expression (Sargsyan et al. 2002; Kim and Arvan 1993). Hormone-mediated thyroglobulin production in FRTL-5 rat thyroid cells showed modest activation of ATF6 and PERK pathways, but not IRE1-mediated XBP1 splicing in contrast to tunicamycin-treated cells which strongly activated all 3 arms (Sargsyan, Baryshev, and Mkrtchian 2004). Toll-like receptor (TLR) signaling may also initiate partial UPR signaling events necessary for optimal responses to invading pathogens (Martinon et al. 2010; Woo et al. 2009).
There are scattered examples of xenobiotic-induced differential UPR activation with sometimes opposing observations. In most cases, these include activation of all 3 UPR arms but with differential activation/attenuation kinetics or degree of activation intensity, with few examples of xenobiotics activating only 1 or 2 of the UPR arms. In terms of UPR activation kinetics, contrasting observations have been observed depending on the stimulus and cell type studied. In one report, ATF6 was found to be activated before the IRE1 arm in HeLa cells treated with tunicamycin and thapsigargin (Yoshida et al. 2001). In contrast, CHO cells treated with thapsigargin, showed delayed ATF6 activation compared to a more rapid response for PERK and IRE1. However, DTT-mediated ER stress induction resulted in rapid activation of all three UPR arms compared to thapsigargin or tunicamycin (DuRose, Tam, and Niwa 2006). Another study showed after rapid initial activation of all 3 UPR arms by tunicamycin and thapsigargin in HEK293 cells, the IRE1 and ATF6 activities were attenuated by persistent ER stress, however; PERK signaling and CHOP induction were maintained. Artificially sustained IRE1 activity resulted in enhanced cell survival, suggesting a causal link between the duration of IRE1 signaling and cell survival (Lin et al. 2007). Variability was observed, however, in terms of duration of UPR activation in response to a given stimulus in different cell lines (Lin et al. 2007). In another study using chemical-genetics to activate PERK and IRE1 individually independent of ER unfolded protein accumulation, sustained PERK signaling impaired cell proliferation and promoted apoptosis. However, IRE1 signaling enhanced cell proliferation without promoting apoptosis. Thus, prolonged PERK and IRE1 signaling showed opposite effects on cell viability (Lin et al. 2009). Given the spectrum of effects described above, the final outcome may vary depending on the cell type and ER stress inducing stimulus.
Adaptation to ER stress may be mediated by the differential stabilities of pro-survival and pro-death mRNAs and proteins rather than differential activation of the UPR sensors. By measuring the activation of UPR gene expression pathways with tunicamycin and thapsigargin, mild ER stress activated all three arms of the UPR. Cell survival was favored in these conditions as a result of the intrinsic instabilities of pro-apoptotic mRNAs and proteins (e.g., CHOP, ATF4) compared to those of the adaptive response (e.g., GRP78, GRP94) and not a result of selective activation of the ER stress sensors in these experiments (Rutkowski et al. 2006). The entirety of the UPR was activated in these experimental conditions by mild ER stress, but the downstream signaling molecules were differentially regulated depending on whether the final outcome was survival or death. The resulting improved ER protein folding capacity (via upregulation of chaperones) played a significant role in suppressing the UPR during adaptation (Rutkowski et al. 2006). The longer half-life of chaperones also allowed the cell to have a more sustained protein folding capacity to limit acute fluctuations in ER protein folding capacity. The half-lives of the various UPR components may however vary between cell types resulting in different sensitivities or responses to ER stress.
Differential Suppression of the UPR Pathways
There are not many xenobiotics that selectively suppress a particular arm of the UPR although some have been identified. Kato et al. reported that rapamycin-attenuated ER stress-induced apoptosis by tunicamycin or thapsigargin with selective suppression of IRE1-JNK signaling without affecting PERK and ATF6. In vivo administration of rapamycin suppressed renal tubular injury and apoptosis in tunicamycin-treated mice (Kato et al. 2012). The small molecule STF083010 selectively inhibits the IRE1 endoribonuclease function but not its kinase domain. This inhibitor has cytotoxic activity against multiple myeloma cells in vitro and in human multiple myeloma xenografts (Papandreou et al. 2011). A PERK selective small molecule inhibitor has also been recently reported that inhibits tumor growth in a human pancreatic xenograft model (Axten et al. 2012). Each of these xenobiotics show different effects depending on the target and the cell type studied. This is expected, given that although the three UPR arms show functional overlap, unique functions are apparent between the different arms, highlighted in the different phenotypes of UPR sensor knockout mice and genetic deficiencies described above.
How would one interpret xenobiotic-induced full or differential UPR pathway activation in terms of biological outcome in a particular cell or tissue when assessing toxicity of a xenobiotic? At present, the answer to this question is complex and not straightforward, given the variety of responses described above. A clear theme, however, of chronic or severe ER stress leading to cell death or cellular dysfunction and acute or mild ER stress leading to adaptation and survival is present in most studies. Depending on the mechanism of UPR activation by different UPR inducers, different UPR activation kinetics and responses may be seen. This will also likely vary depending on the cell type and its basal protein folding capacity as well as other conditions occurring inside the cell as described above. The response will also depend on whether the xenobiotic activates ER stress or suppresses signaling from a UPR protein. Therefore, a comprehensive approach measuring multiple UPR end points at different time points is required to fully understand the extent of the response to a xenobiotic. Further studies assessing these responses in multiple cell/tissue types for a number of UPR modulating xenobiotics will be useful as a means to link biomarkers of UPR activation with biological outcome.
Summary and Future Directions
Multiple mechanisms are in place to allow cells to respond and adapt to varying protein folding loads in different cellular compartments. Managing the expression of potentially malformed and improperly functioning extracellular proteins is important to protect an organism to cells that may inappropriately respond to their environment and thus may contribute to tissue dysfunction, uncontrolled growth, or differentiation. In general, acute or mild ER stress leads to cell survival while prolonged or severe ER stress leads to cellular dysfunction and/or death. Many xenobiotics have been shown to influence ER stress and UPR signaling with either pro-survival or pro-death features. The final outcome is dependent on many factors including the mechanism of action of the xenobiotic, concentration of xenobiotic, duration of exposure (acute vs. chronic), cell type affected, nutrient levels, oxidative stress, state of differentiation, and others. When characterizing the potential of a xenobiotic to affect ER stress and UPR signaling, a comprehensive assessment of effects on the three UPR arms is important to better understand the potential spectrum of outcomes associated with modulation of ER stress and UPR signaling.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Marc Lafleur and Jeff Lawrence are employees of Amgen. James Stevens is an employee of Eli Lilly and Company.