
Abstract
In the development of dental fluorosis, oxidative stress is considered as the key mechanism. Endoplasmic reticulum (ER) stress can induce oxidative stress and activate the important antioxidative factor nuclear factor erythroid 2-related factor 2 (Nrf2) in a PKR-like ER kinase (PERK)-dependent manner, but combining ER stress and oxidative stress, the role of PERK-Nrf2 signaling pathway involved in fluoride-regulated ameloblasts is not fully defined. Here, we studied the effect of fluoride on PERK-Nrf2 signaling pathway in mouse ameloblasts. We found that low-dose and continuous fluoride exposure increased binding immunoglobulin protein expression and activated PERK–activating transcription factor 4 signaling pathway. Meanwhile, the expression of Nrf2 and its target genes (glutamylcysteine synthetase and glutathione S-transferase-P1) enhanced following ER stress. Tunicamycin increased the expression of PERK, leading to Nrf2 nuclear import, and tauroursodeoxycholate suppressed Nrf2 activation through PERK during ER stress, indicating that PERK activation is required for Nrf2 nuclear entry. Furthermore, tert-butylhydroquinone triggered the overexpression of Nrf2 to reduce ER stress, but luteolin inhibited Nrf2 nuclear localization to elevate ER stress. In summary, this study proved that fluoride under certain dose can induce ER stress and promote Nrf2 nuclear import via PERK activation and suggested that antioxidation mechanism mediated by PERK-Nrf2 can alleviate fluoride-induced ER stress effectively.
Introduction
Fluoride is a valid caries prophylactic. However, higher levels of fluoride exposure result in dental and skeletal fluorosis, epithelial lung cell toxicity, kidney toxicity, and reproductive defect. 1 –3 Among these, attention has been focused on the role of fluoride in dental fluorosis because the most distinct effect of excess fluoride ingestion in susceptible individuals is white spots or dark stains on the teeth. Research data strongly suggest that fluoride can alter glutathione levels, 4 –6 often resulting in the excessive production of reactive oxygen species (ROS), leading to oxidative stress and the damage of cellular components. 7 –9
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor of the antioxidant defense system by directly binding to an antioxidant-responsive element (ARE) and modulating antioxidant genes. 10 Early experiments defined a role for Nrf2 in the expression of a subset of detoxifying enzymes including glutathione S-transferase-P1 (GST-P1) and glutamylcysteine synthetase (GCLC). 11,12 Therefore, the Nrf2-ARE pathway has been proved to be the key regulator of antioxidant and cytoprotective proteins. 13,14 Previous studies found that an excessive fluoride induced Nrf2 activation. 15 However, the upstream regulatory mechanism of the pathway involved in dental fluorosis remains elusive.
In eukaryotic cells, newly synthesized secretory and transmembrane proteins fold and assemble in the lumen of the endoplasmic reticulum (ER). The perturbation of ER functions causes accumulation of unfolded and misfolded proteins in the ER. Such alterations are referred as ER stress, and the response elicited is the unfolded protein response (UPR). 16 –18 The UPR pathway is orchestrated by three different branches, each of which is initialized by a proximal sensor anchored in the ER as transmembrane proteins, inositol-requiring enzyme 1 (IRE1) and PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6). 19 As unfolded proteins accumulate in the ER, ER chaperone binding immunoglobulin protein (BiP) is sequestered from these sensors allowing their activation, inducing UPR. 20 Known, the ER provides an environment that is highly optimized for oxidative protein folding. It is important to note that oxidative folding could contribute a significant source of ROS, suggesting that ROS accumulation during ER stress will facilitate oxidative stress. 21,22
Notably, the downstream signaling pathway for both ER stress and oxidative stress is somewhat overlapping. ER stress can induce Nrf2 nuclear translocation in a PERK-dependent manner, and this activation does not require the phosphorylation of α-subunit eukaryotic translation initiation factor 2 (eIF2α) or the accumulation of ROS. 21,23 In fact, PERK-Nrf2 signaling pathway is required for survival during the ER stress; the mechanism depends on the ability of Nrf2 target genes to promote redox homeostasis following ER stress induction. 24 Nevertheless, the ERS and Nrf2 interrelated mechanisms in the pathogenesis of dental fluorosis are rarely reported. Consequently, we hypothesized that ERS activates Nrf2 in a PERK-dependent manner and then initiates the antioxidation mechanism by the activation of Nrf2 signaling pathway after fluoride exposure. In this study, we demonstrate a functional cross talk between the ER stress and oxidative stress pathways in mouse ameloblasts cells under ER stress, and PERK-Nrf2 signaling pathway alleviated the ER stress response effectively.
Materials and methods
Cell culture
The ameloblasts, derived from mouse, were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum and penicillin/streptomycin mixture (200 U/mL penicillin G and 200 g/mL streptomycin), and incubated in a humidified atmosphere of 5% CO2 and 95% air. The experiment was designed to include the control group, sodium fluoride (NaF) alone group, ER stress inducer tunicamycin (TM) pretreatment group, ER stress inhibitor tauroursodeoxycholate (TUDCA) pretreatment group, Nrf2 inducer tert-butylhydroquinone (tBHQ) pretreatment group, and Nrf2 inhibitor luteolin pretreatment group. Cells in NaF alone groups were treated with DMEM containing 0.25, 0.50, 1.00, 2.00, and 4.00 mM of NaF for 24 and 48 h, respectively. Each pretreatment group was preincubated with 0.5 μg/mL TM (12 h), 0.5 mM TUDCA (12 h), 50 μM tBHQ (12 h), and 10 μM luteolin (24 h) and further incubated in DMED containing the varying doses of NaF for either 24 or 48 h. Cells in the control group were cultured with DMEM (NaF-free) for the same period.
Reverse transcription and semi-quantity polymerase chain reaction (PCR)
Mouse ameloblasts were seeded into 6-cm dishes at a density of 5 × 105 cells/mL with varying pretreatment and further grown in DMEM containing different doses of NaF for 24 and 48 h. Total ribonucleic acid (RNA) was extracted from mouse ameloblasts with TRIzol® reagent (Takara, Shiga, Japan). Reverse transcription was performed, followed by semi-quantity PCR using PrimeScript® RT-PCR Kit (Takara). Primer sequences for expression analyses and annealing temperatures are shown in Table 1. The reaction conditions were as follows: 94°C preheating for 3 min, followed by 32 cycles of 94°C for 30 s (denaturation), approximately 56–60°C for 30 s (annealing), and 72°C for 40 s (elongation). Glyceraldehyde-3-phosphate dehydrogenase was used as an internal control. PCR products were separated on 1.5% agarose gel, stained with ethidium bromide, scanned using the BIO-BEST140E transilluminator (SIM, LA, CA, USA), and quantified by Gel-Pro Analyzer-4 software.
PCR primers for gene expression analysis.

PCR: polymerase chain reaction; BiP: binding immunoglobulin protein; PERK: PKR-like ER kinase; ATF4: activating transcription factor 4; Nrf2: nuclear factor erythroid 2-related factor 2; GCLC: glutamylcysteine synthetase; GST-P1: glutathione S-transferase-P1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
Western blot analysis
Cells were plated in 10-cm dishes at a density of 5 × 105 cells/mL with varying pretreatment and further incubated in DMEM containing different concentrations of NaF for 24 and 48 h. Cytoplasm and nuclear proteins were extracted by Nucbuster™ Protein Extraction Kit (Merck, Darmstadt, Germany), and the protein concentration of extracts was measured by the BCA Protein Assay Kit (Dingguo, Beijing, China). A total of 50 µg protein per lane was run on approximately 4–10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Dingguo). After being blocked with 5% nonfat dried milk at room temperature for 2.5 h, the PVDF membranes were probed with primary antibodies anti-PERK (sc-13073; 1:600) or anti-Nrf2 (sc-13032; 1:600) and anti-β-actin (Bioss, Beijing, China; 1:1000) overnight at 4°C. Subsequently, the blots were washed with Tris-buffered saline containing 20% Tween-20 (TBST) and incubated with horseradish peroxidase-conjugated secondary antibody (Dingguo; 1:3000) at room temperature for 2 h. After washing with TBST, the protein bands were detected by ECL (Pierce, Woburn, MA, USA) and exposed to Kodak Medical X-ray Film (Kodak, Rochester, NY, USA). Gel-Pro Analyzer-4 software was used to quantify the expression of proteins.
Statistical analysis
All experiments were repeated three times. The data were described as mean ± standard deviation. Data were subjected to analysis of variance, followed by the Student–Newman–Keuls multiple comparisons test using SPSS Version 16.0. Significance was set at p < 0.05.
Results
NaF induces the ER stress
Given the multiple and essential roles in protein folding and activating ER stress sensors, BiP is considered as one of ER stress markers. As unfolded proteins accumulate in the ER, dissociation of BiP induces ER stress. The signal pathway mediated by PERK–activating transcription factor 4 (ATF4) has been proved to be involving in response to ER stress. 21 In an attempt to research the possible mechanism regulated by ER stress-dependent PERK activation, we investigate the effect of NaF exposure on the expression of BiP, PERK, and ATF4 in mouse ameloblasts. Cells were incubated with various doses of NaF for different periods.
The gene expression of BiP in mouse ameloblasts exposed to NaF is shown in Figure 1(a) and (b). It was substantially upregulated of BiP in 0.25∼1.00 mM groups for 24-h exposure than that in the control, but there was a gradual downregulation in 2- and 4-mM groups. The BiP messenger RNA (mRNA) significantly increased in 0.25- and 0.50-mM groups for 48-h exposure. However, there was a progressively decreased expression in approximatel 1.00–4.00-mM groups. At a different concentration, slightly increased expression of BiP was observed for 48-h treatment compared with the 24-h treatment. The results demonstrated that low-dose and continuous exposure to NaF induced varying degrees of ER stress in mouse ameloblasts.

NaF triggers ER stress. Cells were exposed to NaF at various concentrations, and the amounts of BiP (a), PERK (c), and ATF4 (e) transcripts were detected by semi-quantity PCR. Quantitative analyses of BiP (b), PERK (d), and ATF4 (f) were analyzed by Gel-Pro Analyzer-4. (g) Western blot of PERK. (h) Quantitative analysis of PERK protein levels. Values are expressed as mean ± SD. ap ≤ 0.05 versus control group. bp ≤ 0.05 versus 24 h. NaF: sodium fluoride; ER: endoplasmic reticulum; BiP: binding immunoglobulin protein; PERK: PKR-like ER kinase; ATF4: activating transcription factor 4; PCR: polymerase chain reaction; SD: standard deviation.
The gene expression of PERK slightly increased in 0.25-mM group, but markedly decreased in 4-mM group for 24-h exposure. The slightly upregulated expression of PERK gene was viewed in 0.25- and 0.50-mM groups for 48-h exposure (Figure 1(c) and (d)). It was significantly upregulated for PERK protein in 0.25∼1 mM groups for 24- and 48-h exposure (Figure 1(g) and (h)). ATF4 acted as the downstream signal pathways of PERK. It showed the significant upregulation of ATF4 mRNA in 0.25-mM group for 24-h exposure. The slightly increased expression of ATF4 was apparent for 48-h treatment compared with the 24-h treatment at a different concentration (Figure 1(e) and (f)). Their trends of expression were similar to the altered expression of BiP. These data suggested that PERK signal pathway was sensitive to the fluoride-induced ER stress.
NaF induces the Nrf2 and Nrf2 target genes
Nrf2 is a significant stress-responsive transcription factor of the “cap-and-collar” family, especially during oxidative stress. It mediates the expression of numerous Phase II enzymes (GCLC and GST-P1) to promote antioxidative stress. To identify whether NaF elevates the activation of Nrf2 and Nrf2-dependent gene expression, we assess the effect of NaF exposure on the expression of Nrf2, GCLC, and GST-P1 in mouse ameloblasts.
It was markedly upregulated of Nrf2 mRNA in 0.25∼2.00-mM groups for 48-h exposure than in the control, but there was a significant downregulation in 4-mM groups for 24-h exposure (Figure 2(a) and (b)). The slightly upregulated Nrf2 protein was viewed in 0.25∼2.00-mM groups for either 24- or 48-h exposure (Figure 2(g) and (h)). GCLC and GST-P1 acted as the downstream signal pathways of Nrf2. It showed the upregulation of GCLC and GST-P1 mRNA in low-dose group for 24 and 48 h (Figure 2(c) to (f)). It is indicated that Nrf2 is activated following ER stress response induced by fluoride.

NaF induces Nrf2 signal pathway. Cells were exposed to different doses of NaF as indicated, and the amounts of Nrf2 (a), GCLC (c), and GST-P1 (e) transcripts were detected by semi-quantity PCR. Quantitative analyses of Nrf2 (b), GCLC (d), and GST-P1 (f) were analyzed by Gel-Pro Analyzer-4. (g) Western blot of Nrf2. (h) Quantitative analysis of Nrf2 protein levels. Values are expressed as mean ± SD. ap ≤ 0.05 versus control group. bp ≤ 0.05 versus 24 h. NaF: sodium fluoride; Nrf2: nuclear factor erythroid 2-related factor 2; GCLC: glutamylcysteine synthetase; GST-P1: glutathione S-transferase-P1; PCR: polymerase chain reaction; SD: standard deviation.
Activation of PERK-Nrf2 signal pathway by fluoride-induced ER stress
A role for Nrf2 activation during the ER stress was established following the determination of Nrf2 as a PERK substrate. To evaluate whether ER stress-dependent induction of the Nrf2 required PERK activity after NaF exposure, we measured the expression of BiP, PERK, and Nrf2 in mouse ameloblasts pretreated with TM and TUDCA.
TM induces ER stress by inhibiting N-linked protein glycosylation and has been identified to induced ER stress sensors expression in other cell lines. Pretreatment of mouse ameloblasts was done with 0.5 μg/mL TM for 12 h and further incubated with different doses of significantly induced BiP mRNA, PERK mRNA, and protein overexpression than that in the corresponding dose of NaF group (Figure 3(a) to (d), (g), and (h)). Upregulation of them results in Nrf2 mRNA and protein overexpression via PERK activation (Figure 3(e), (f), (i), and (j)).
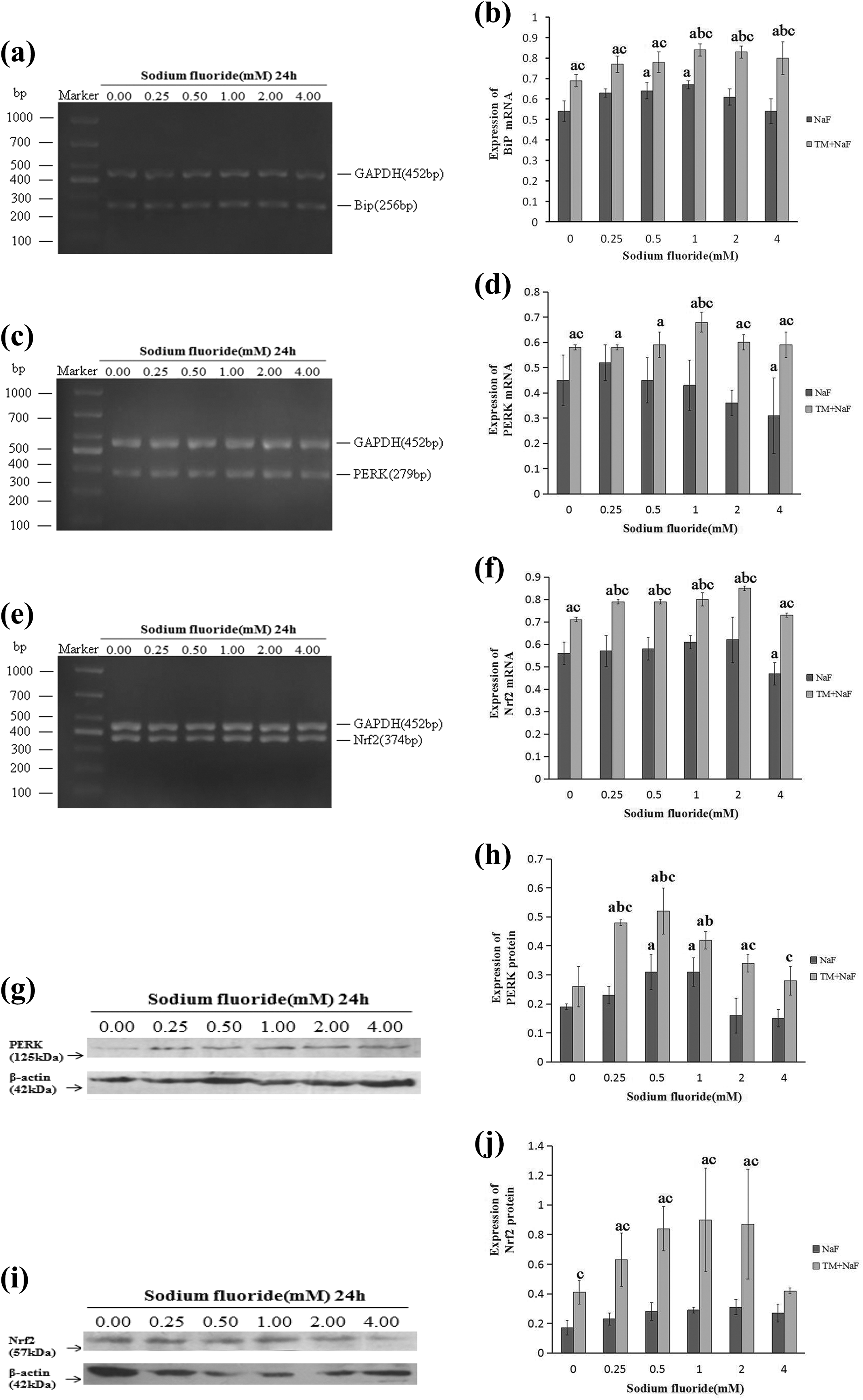
ER stress inducer TM activates PERK-Nrf2 signal pathway induced by NaF. Cells were pretreated with TM for 12 h and further incubated with NaF at various concentrations. BiP (a), PERK (c), and Nrf2 (e) transcripts were detected by semi-quantity PCR. Quantitative analyses of BiP (b), PERK (d), and Nrf2 (f) were analyzed by Gel-Pro Analyzer-4. Western blot of PERK (g) and Nrf2 (i). Quantitative analysis of PERK (h) and Nrf2 (j) protein levels. Values are expressed as mean ± SD. ap ≤ 0.05 versus control, bp ≤ 0.05 versus TM pretreatment group, cp ≤ 0.05 versus corresponding NaF group. ER: endoplasmic reticulum; TM: tunicamycin; PERK: PKR-like ER kinase; Nrf2: nuclear factor erythroid 2-related factor 2; NaF: sodium fluoride; BiP: binding immunoglobulin protein; PCR: polymerase chain reaction; SD: standard deviation.
To further confirm these results, we next assessed whether the inhibition of ER stress downregulates the expression of Nrf2. As a chemical chaperone, TUDCA effectively alleviate ER stress. TUDCA treatment resulted in a rapid decrease in BiP mRNA, PERK mRNA, and protein (Figure 4(a) to (d), (g), and (h)). Similar results were observed upon the downregulation of Nrf2 mRNA and protein (Figure 4(e), (f), (i), and (j)). These decreases likely reflect that the Nrf2 is triggered followed by ER stress via PERK-dependent regulation.

ER stress inhibitor TUDCA suppresses PERK-Nrf2 signal pathway induced by NaF. Cells were pretreated with TUDCA for 12 h and further incubated with NaF at various concentrations. BiP (a), PERK (c), and Nrf2 (e) transcripts were detected by semi-quantity PCR. Quantitative analyses of BiP (b), PERK (d), and Nrf2 (f) were analyzed by Gel-Pro Analyzer-4. Western blot of PERK (g) and Nrf2 (i). Quantitative analysis of PERK (h) and Nrf2 (j) protein levels. Values are expressed as mean ± SD. ap ≤ 0.05 versus control, bp ≤ 0.05 versus TUDCA pretreatment group, cp ≤ 0.05 versus corresponding NaF group. ER: endoplasmic reticulum; TUDCA: tauroursodeoxycholate; PERK: PKR-like ER kinase; Nrf2: nuclear factor erythroid 2-related factor 2; NaF: sodium fluoride; BiP: binding immunoglobulin protein; PCR: polymerase chain reaction; SD: standard deviation.
Nrf2 alleviates ER stress induced by fluoride
Studies suggested that Nrf2 promotes the expression of several genes implicated in protein folding, perhaps in an effort to reduce the amount of unfolded and misfolded proteins within the ER. To determine whether Nrf2 activation inhibited ER stress response induced by NaF, we measured the expression of BiP, PERK, and Nrf2 in mouse ameloblasts pretreated with tBHQ and luteolin.
Treatment with tBHQ, a known inducer of Nrf2 activity, increased Nrf2 gene and protein expression (Figure 5(e), (f), (i), and (j)). However, there was a downregulation of BiP mRNA in various groups for 24-h exposure. The expression PERK mRNA and protein also substantially decreased for 24-h exposure (Figure 5(a) to (d), (g), and (h)). To further investigate the role of Nrf2, we utilized luteolin-pretreated mouse ameloblasts. Cells were challenged with Nrf2 inhibitor, and downregulation of Nrf2 gene and protein is shown in Figure 6(e), (f), (i), and (j). On the other hand, it is markedly upregulation of BiP in different dose groups for 24-h exposure, but the expression of PERK mRNA significantly decreased in various groups for 24-h exposure (Figure 6(a) to (d), (g), and (h)). These data demonstrate that Nrf2 function is essential to antagonize ER stress.

Nrf2 inducer tBHQ alleviates ER stress induced by NaF. Cells were pretreatment with tBHQ for 12 h and further incubated with NaF at various concentrations. BiP (a), PERK (c), and Nrf2 (e) transcripts were detected by semi-quantity PCR. Quantitative analyses of BiP (b), PERK (d), and Nrf2 (f) were analyzed by Gel-Pro Analyzer-4. Western blot of PERK (g) and Nrf2 (i). Quantitative analysis of PERK (h) and Nrf2 (j) protein levels. Values were expressed as mean ± SD. ap ≤ 0.05 versus control, bp ≤ 0.05 versus tBHQ pretreatment group, cp ≤ 0.05 versus corresponding NaF group. Nrf2: nuclear factor erythroid 2-related factor 2; tBHQ: tert-butylhydroquinone; ER: endoplasmic reticulum; NaF: sodium fluoride; BiP: binding immunoglobulin protein; PERK: PKR-like ER kinase; PCR: polymerase chain reaction; SD: standard deviation.

Nrf2 inhibitor luteolin enhances ER stress induced by NaF. Cells were pretreated with luteolin for 24 h and further incubated with NaF at various concentrations. BiP (a), PERK (c), and Nrf2 (e) transcripts were detected by semi-quantity PCR. Quantitative analyses of BiP (b), PERK (d), and Nrf2 (f) were analyzed by Gel-Pro Analyzer-4. Western blot of PERK (g) and Nrf2 (i). Quantitative analysis of PERK (h) and Nrf2 (j) protein levels. Values were expressed as mean ± SD. ap ≤ 0.05 versus control, bp ≤ 0.05 versus luteolin pretreatment group, cp ≤ 0.05 versus corresponding NaF group. Nrf2: nuclear factor erythroid 2-related factor 2; ER: endoplasmic reticulum; NaF: sodium fluoride; BiP: binding immunoglobulin protein; PERK: PKR-like ER kinase; PCR: polymerase chain reaction; SD: standard deviation.
Discussion
In reviewing the literature, Cullinan and Diehl reported that PERK signal pathway, by activating Nrf2 transcription factors, coordinates the convergence of ER stress and oxidative stress signaling. 21 At present, little is known about the association between ER stress and oxidative stress involved in the development of dental fluorosis. Based on the document, the present research was designed to investigate the effect of fluoride on PERK-Nrf2 signal pathway in mouse ameloblasts. It showed that low-dose and continuous fluoride exposure enhanced BiP expression in mouse ameloblasts. As it is known, the primary sensor of ER stress is thought to be BiP, which interacts with IRE1, PERK, and ATF6. Research suggests that the binding of BiP to unfolded or misfolded protein results in the active release of IRE1, PERK, and ATF6 and then elicits signaling cascades resulting in the attenuation of protein synthesis and the induction of UPR genes to protect against various types of cell death induced by ER stress. 25,26 This result corroborated the previous reports in this field, which showed the upregulation of BiP is used as a sentinel marker for ER stress in ameloblast-like cell line treated with fluoride. 27 –29 Moreover, we found that a low concentration exposure to fluoride was related to activate the UPR-dependent PERK-ATF4 signal pathway. In response to ER stress, PERK phosphorylates eIF2α to attenuate the protein folding load at the ER, and phosphorylated eIF2α selectively enhances the translation of ATF4 that reduces the workload on the ER. 20,30,31 Consequently, PERK is activated during ER stress induced by fluoride.
In this study, our findings demonstrated that fluoride increased the expression of Nrf2 for either 24- or 48-h exposure and altogether resulted in enhanced GCLC and GST-P1 in mouse ameloblasts. The results may be closely related to earlier findings which indicated that oxidative stress results from ameloblasts exposed to fluoride. 9 Previous research studies have suggested that one of the significant cellular defense mechanisms that neutralize ROS is mediated by the activation of Nrf2, which binds to the ARE present in the promoter deoxyribonucleic acid of a number of detoxifying enzymes and antioxidant genes. 14,32 In addition, Nrf2 has been shown to be sequestered by cytoskeleton-anchored Keap1 protein in the cytosol as an inactive complex. 33 Keap1 has a cysteine-rich surface that is susceptible to extracellular stimuli. This obviously results in conformational changes to Keap1, leading to the dissociation of Nrf2. 34 –36 When mouse ameloblasts are under ER stress, the liberation of Nrf2 results in increased levels of Nrf2 and activation of its downstream antioxidant target genes, including GCLC and GST-P1. Preliminary work reported that ER stress-stimulated GCLC, NAD(P)H dehydrogenase quinone 1, and heme oxygenase 1 (HO-1) induction occurs through Nrf2 binding to ARE, consistent with our findings. 24,37
Of note, early evidence indicated that the transcription factor Nrf2 acts as a novel PERK substrate. PERK directly phosphorylates Nrf2, thereby evaluating Nrf2 as only the second known substrate for PERK. Unlike ATF4, Nrf2 is an immediate PERK substrate, and its activation is independent of eIF2α phosphorylation. 23 Consistent with this idea, our experiments revealed that ER stress activity increases PERK-Nrf2 signal pathway challenged with TM and decreases PERK-Nrf2 signal pathway challenged with TUDCA in each dose of fluoride and time, strongly suggesting that Nrf2 participates in ER stress signaling through PERK. Nrf2 activation has also been shown to be mediated through phosphorylation of Nrf2 by various kinases, such as protein kinase C, mitogen-activated protein kinases, and phosphoinositol 3 kinases. 38 –40 However, from the study reported here, fluoride can induce ER stress and trigger Nrf2 in a PERK-dependent manner in mouse ameloblasts. These current findings highlighted that, by dissolution of Nrf2/Keap1 complexes, this increases nuclear Nrf2 protein expression during ER stress (Figure 3). Early experiment showed that stress-dependent Nrf2 failed to nuclear import in PERK−/−cells; furthermore, dominant-negative PERK inhibited the activation of Nrf2, indicating that PERK phosphorylation of Nrf2 is sufficient for Nrf2/Keap1 dissociation and that ROS are not needed. 23 Similarly, long-term ER stress cascades PERK-Nrf2 signaling pathway in certain tumor cell. 41,42
In addition to genes encoding antioxidants, Nrf2 promotes the expression of genes implicated in cell growth, protein folding, protein degradation, and cell survival. 43,44 Interestingly, studies revealed that Nrf2 activity upon ER stress provides a negative feedback loop, preventing further folding of protein into the ER and thereby reducing the ER stress. 24 Our results indicated that the overexpression of Nrf2 will alleviate ER stress; however, the downexpression of Nrf2 will increase ER stress in mouse ameloblasts exposed to different doses and periods of fluoride. These observations have led to the recognition that Nrf2 plays an integral role in fluoride-induced ER stress responded to promote cell survival. It is known that ER stress has a dual effect on the cell fate: UPR-dependent cell survival and apoptosis following sustained ER stress. One of the possible protective effects of Nrf2 acts by promoting the transcription of some genes encoding proteasome subunits, thus facilitating proteasome assembly, suggests that PERK-Nrf2 signaling pathway may contribute to protein degradation during the ER stress. 43 A recent study has also reported that Nrf2 activation at the onset of ER stress provides a cytoprotective mechanism by which cells elevate the level of glutathione in an effort to weaken the deleterious effects that ER stress attacks on redox maintenance. 24 In addition, Nrf2-dependent protection from ER stress-induced cell death likely ascribes to its function as a regulator of genes encoding Phase II detoxifying enzymes. Stulnig et al. reported that HO-1 silencing resulted in increased levels of ROS and increased duration of ER stress and also augmented apoptosis as found by increased numbers of cleaved caspase-3-positive cells. 45 Consistent with these notions is current demonstration that PERK-dependent Nrf2 is an essential effector to alleviate ER stress response.
Taken together, data presented in this study suggested that fluoride can initiate ER stress and activate PERK-Nrf2 signaling pathway to reduce the fluoride-induced ER stress. Further exploration of Nrf2 downstream mechanisms responsible for ER stress involved in mouse ameloblasts treated with fluoride will be required to shed more light on this area.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Joint Natural Sciences Fund of the Department of Science and Technology and the First Affiliated Hospital of Guangdong Pharmaceutical University (Grant No. GYFYLH201301).