
Abstract
Dilated cardiomyopathy (DCM) in A/J mice homozygous for the spontaneous thrombocytopenia and cardiomyopathy (trac) mutation results from a single base pair change in the Abcg5 gene. A similar mutation in humans causes sitosterolemia with high plant sterol levels, hypercholesterolemia, and early onset atherosclerosis. Analyses of CD3+ and Mac-3+ cells and stainable collagen in hearts showed inflammation and myocyte degeneration in A/J-trac/trac mice beginning postweaning and progressed to marked dilative and fibrosing cardiomyopathy by 140 days. Transmission electron microscopy (TEM) demonstrated myocyte vacuoles consistent with swollen endoplasmic reticulum (ER). Myocytes with cytoplasmic glycogen and irregular actinomyosin filament bundles formed mature intercalated disks with normal myocytes suggesting myocyte repair. A/J-trac/trac mice fed lifelong phytosterol-free diets did not develop cardiomyopathy. BALB/cByJ-trac/trac mice had lesser inflammatory infiltrates and later onset DCM. BALB/cByJ-trac/trac mice changed from normal to phytosterol-free diets had lesser T cell infiltrates but persistent monocyte infiltrates and equivalent fibrosis to mice on normal diets. B- and T-cell-deficient BALB/cBy-Rag1null trac/trac mice fed normal diets did not develop inflammatory infiltrates or DCM. We conclude that the trac/trac mouse has many features of inflammatory DCM and that the reversibility of myocardial T cell infiltration provides a novel model for investigating the progression of myocardial fibrosis.
Keywords
Introduction
Dilated cardiomyopathy (DCM) is the third most common form of heart failure with an incidence of 1:2,500. It is the most frequent indication for heart transplant. Approximately 30% of DCM appears to be familial with a number of genetic links identified (Maron et al. 2006). The clinical syndrome of dilative cardiomyopathy encompasses multiple causes with potentially overlapping pathogenic mechanisms. A variety of specific initiating causes include genetic alterations in myocardial contractile proteins, infection with viral bacterial or protozoal pathogens, and exposure to cardiotoxic exogenous or endogenous chemicals. Myocyte degeneration is often associated with inflammation. Inflammatory dilative cardiomyopathy (iDCM) is a serious complication of myocardial infections and may occur in relatively young patients infected with Coxsackie B3, adenovirus parvovirus B19, or Trypanosoma cruzi. Several recent studies suggest a final common pathway wherein autoimmune responses to myocyte proteins such as troponin-1 (Kaya et al. 2008; Kaya, Katus, and Rose 2010) and myosin (Caforio, Vinci, and Iliceto 2008; Zhang et al. 2009) drive myocardial inflammation. It has also been speculated that immune-mediated inflammation plays a role in ischemic heart disease and that the success of progenitor cell therapies may well rest on their ability to modulate the extent and progression of the myocardial inflammatory process (Gnecchi et al. 2008).
An axiom of cardiology is that the extent of fibrosis is more important than myocyte loss in restricting cardiac function in the progression to failure. Not only does fibrosis influence myocardial compliance (Krenning, Zeisberg, and Kalluri 2010), the deposition of collagen between myocytes interferes with the effect of overlapping action potentials in adjacent myocytes to dampen the propagation of any dysrhythmic depolarizations. Loss of this dampening effect greatly increases the chances of serious arrhythmias such as Torsades de Pointes (Xie et al. 2010). Recent experimental evidence separates the process of inflammation from the progression to fibrosis (Blyszczuk et al. 2009). The balance between matrix proteolysis and pro-fibrotic cytokine expression appears to determine whether some cases of myocarditis progress to severe fibrosis while others appear reversible (Krenning, Zeisberg, and Kalluri 2010). A variety of collagen secreting cell populations are postulated to contribute to myocardial fibrosis. These include resident fibroblast precursors, fibroblasts developing from endothelial cells via endothelial mesenchymal transition, and emigration of bone marrow derived precursors (Krenning, Zeisberg, and Kalluri 2010).
The most frequently studied mouse models of myocarditis are infection with coxsackievirus B3 and immunization with myosin fragments (Cihakova and Rose 2008). A variety of adoptive transfer experiments use myosin fragment transfected immune cells—particularly dendritic cells. Our laboratory has recently characterized a spontaneous mutation named thrombocytopenia and cardiomyopathy (trac) that occurred in A/J mice. The trac mutation disrupts the adenosine triphosphate (ATP)–binding cassette subfamily G, member 5 (Abcg5) gene. This gene encodes a member of the ABC transporter family that forms a heterodimer with the product of the Abcg8 gene and facilitates the exchange of cholesterol and other sterols across membranes (Chase et al. 2010). Mutations in the Abcg5 gene in humans affect cholesterol efflux through bile cannaliculus and phytosterol transport in intestinal epithelium. This human syndrome is called sitosterolemia and is characterized by the buildup of plant-derived phytosterols in plasma and tissues of affected individuals. Sitosterolemia patients also have increased levels of xanthoma formation, hypercholesterolemia, and early onset coronary atherosclerosis (Fitzgerald, Mujawar, and Tamehiro, 2010). A second human cardiovascular syndrome ascribed to mutation of this gene family affects the ABCA1 transporter and underlies the familial dyslipidemia known as Tangier disease. Mutation of ABCA1 inhibits cholesterol efflux from cells with consequent greatly diminished plasma high-density lipoprotein (HDL) and intracellular cholesterol accumulations that accentuate intimal foam cell formation (Fitzgerald, Mujawar, and Tamehiro, 2010).
The trac mutation is a G to A mutation in exon 10 of the Abcg5 gene. It alters a tryptophan codon (UGG) to a premature stop codon (UAG) that either alters function or prevents translation or posttranslational processing resulting in phytosterolemia (Chase et al. 2010). Mice homozygous for this mutation are slow growing, develop macrothrombocytopenia, megakaryocyte dysfunction, myocardial fibrosis, and are infertile. Platelets from affected mice show altered morphology characterized by enlargement, spherical shape, and changes in platelet microtubule coils. Megakaryocytes from these mice have reduced endomitosis and absence of a well-defined demarcation membrane system. Increased numbers of large megakayrocytes are found in spleen, bone marrow, and lungs, presumably in response to thrombocytopenia. Affected mice die prematurely with biventricular heart failure.
The objective of the present study was to characterize the progression of myocardial lesions caused by the trac mutation and determine whether the cardiomyopathy encompasses pathological changes of inflammatory DCM. Furthermore, we evaluated the role of phytosterolemia in the induction of myocardial lesions and analyzed whether restriction of dietary phytosterols prevented or reversed myocardial inflammation. Our findings demonstrate that myocyte degeneration elicits a T cell and monocyte-rich infiltrate associated with significant myocardial fibrosis. Lifetime dietary restriction of phytosterols prevents myocardial disease, while affected animals transferred to restricted diets after onset of cardiomyopathy have reduced inflammation but persistent myocardial fibrosis. Finally, B- and T-cell-deficient BALB/cBy-Rag1null trac/trac mice do not develop cardiomyopathy but remain thrombocytopenic throughout life.
Method
Mice
All mice in this study were raised in AALAC-accredited (Association for Assessment and Accreditation of Laboratory Animal Care) facilities at the Jackson Laboratory. The Abcg5trac mutation (hereafter referred to as trac) occurred on the A/J strain (Chase et al. 2010). A/J-trac/trac mice as well as heterozygotes (+/trac) and wild-type (+/+) controls were produced by intercrossing A/J +/trac heterozygotes. The trac mutation was backcrossed from A/J for 10 generations onto the BALB/cBJy strain background. CByJ.Cg-Abcg5trac /J homozygotes (hereafter abbreviated as BALB/cByJ-trac/trac mice) as well as +/trac and +/+ controls were produced by intercrossing BALB/cByJ +/trac heterozygotes. BALB/cBy +/trac mice were crossed with CByJ.129S7(Cg)-Rag1tm1Mom /Sz homozygotes (hereafter referred to as BALB/cByJ-Rag1null ) mice to generate a colony of BALB/cByJ-Rag1null mice segregating for the trac mutation. BALB/cByJ-Rag1null trac/trac mice as well as BALB/cByJ-Rag1null +/trac and BALB/cByJ-Rag1null +/+ controls were generated from intercrosses of BALB/cByJ-Rag1null +/trac mice.
Genotyping for alleles at the trac (Chase et al. 2010) and at the Rag1 (Shultz et al. 2000) locus was carried out on tail snip DNA as previously described (Chase et al. 2010). Mice were maintained on Purina 5K52 6% fat diet. A phytosterol-free diet (Purina 5TQM basal diet with no plant sterols) was used in experiments requiring the absence of dietary sterols. All mice received acidified water ad libitum.
Time Course of Myocarditis Development
Groups of A/J-trac/trac mice at 4 weeks (N = 4), 10 weeks (N = 4), and 20 weeks (N = 4) of age as well as an equal number of either A/J +/trac or +/+ age matched littermate control mice were evaluated. Mice were euthanized with CO2 and necropsies were performed. The heart, spleen, sections of liver, and kidney were fixed by immersion in 10% neutral buffered formalin. The lungs were fixed by intratracheal instillation of formalin at 20 cm H2O pressure. The heart was sagittally sectioned in a plane transecting the remaining right and left atria, left ventricle, and septum. Tissues were embedded in paraffin and 5-µm sections stained with hematoxylin and eosin (H&E) and Masson’s trichrome. Additional sections of heart were prepared for immunostaining of CD3 and Mac-3 as described below.
Echocardiography (ECHO)
Ultrasound studies were carried out on 7 A/J-trac/trac and +/trac animals in each genotype group (2 females, 5 males) using Vevo 770 High-Frequency Ultrasound Equipment (Visualsonics, Toronto, Ontario, Canada). Mice from a broad age range (9–20 weeks) were evaluated in order to have a sufficient sized group of animals from a limited breeding program for statistical comparisons of heart measurements. Anesthesia was induced in the mice with 5% isoflurane at 0.8 L/min. Anesthesia was maintained with 1–1.5% isoflurane at 0.8 L/min.
Effect of Diet Restriction on Cardiomyopathy
Groups of A/J-trac/trac mice were produced from A/J +/trac breeding pairs fed a normal diet. After weaning, these mice were maintained on a high-fat phytosterol-free diet (Purina 5TQM basal diet with no plant sterols) for 12 weeks (N = 3) and compared with A/J-trac/trac mice fed normal (phytosterol containing) diet (N = 3). To determine the long-term effect of phytosterol free diet, additional A/J-trac/trac mice were held for 85 weeks (N = 3) with two additional animals held for 115 weeks. Age matched pairs of heterozygous or homozygous normal mice served as controls in both studies. Trac/trac mice on normal diets were not included in the long-term study, as A/J-trac/trac mice on a normal chow diet containing phytosterols have a mean life span of only 20 weeks (Chase et al. 2010). Mice were euthanized with CO2 and necropsied as above. Tissues were fixed by immersion in Tellyesniczky/Fekete fixative. Hearts were sagittally sectioned in a plane bisecting all four chambers and embedded in paraffin. Five µm sections were stained with H&E as well as Masson’s trichrome stain.
Effect of Phytosterol Restriction on Lesion Progression
Groups of BALB/cBy-trac/trac as well as +/trac and +/+ control mice were raised from BALB/cBy+/trac breeding pairs fed normal diets. After weaning, these mice were fed normal diets until 10 (N = 3) or 14 (N = 2) weeks of age when they were switched to a phytosterol-free diet for an additional 8 weeks. An additional group of BALB/cBy-trac/trac mice were fed normal diets until euthanized at 16 weeks of age. Age-matched BALB/cBy +/trac or +/+ controls were included in each diet group. Mice were euthanized with CO2 and necropsied as above. Tissues were fixed by immersion in Tellyesniczky/Fekete fixative. Hearts were sectioned longitudinally to bisect all four chambers and embedded in paraffin. Five µm sections were stained with H&E as well as Masson’s trichrome stain. Additional sections of heart were prepared for immunostaining as described below.
Effect of Immunodeficiency in BALB/cBy-Rag1null trac/trac Mice
BALB/cBy-Rag1null trac/trac mice along with BALB/cBy-Rag1null +/trac and +/+ controls were maintained on normal diet for 37 to 64 weeks. A wide range of ages was necessary due to the complex breeding necessary to establish these crosses, but age ranges were matched between groups. Mice were euthanized and necropsied as outlined above. Hearts fixed in Tellyesniczky/Fekete fixative were embedded and stained with H&E, trichrome, as well as immunostained as described below.
Immunohistochemistry
Additional sections of paraffin-embedded heart and spleen were mounted on poly-L-lysine-coated slides for immunostaining. The following antibodies and dilutions were used for immunohistochemistry: CD3 (Abcam/Rabbit Polyclonal, 1:200) and Mac-3 (BD Biosciences/Rat Monoclonal, 1:200). Sections were stained with a Ventana Discovery XT automated IHC stainer and proprietary antigen retrieval and blocking solutions. Antigen retrieval was done for 20 min at 95°C followed by 4 min block at 37°C. Primary antibodies were incubated for 60 min at 37°C followed by buffer wash and incubation with OmniMap anti-Rabbit or Rat secondary antibodies. Reactions were developed with DAB/Hydrogen Peroxide for 8 min at room temperature and enhanced with Copper CM (Discovery XT Enhancer). Sections were counterstained with Mayer’s hematoxylin for 2 min.
Transmission Electron Microscopy (TEM)
A/J-trac/trac and littermate control mice were euthanized at 17 weeks age and fixed by intracardiac perfusion through the left ventricle with 2% glutaraldehyde, 2% paraformaldehyde, and 0.5% tannic acid in 0.1 M cacodylate buffer, pH 7.4. Tissue for TEM was harvested from the left ventricle, then submerged in EM fix, minced, fixed overnight, then postfixed with 1% osmium tetroxide (0.1 M cacodylate buffer, pH 7.4). The samples were then rinsed in the same buffer, dehydrated in a graded series of ethanols, and embedded in Epon Araldite resin (Electron Microscopy Sciences; Hatfield, PA). Ultrathin sections were poststained with uranyl acetate followed by lead citrate. Samples were imaged on a JEOL JEM-1230 electron microscope (Tokyo, Japan) and images were captured with AMT Advantage CCD 6 Mpix (Danvers, MA) and Image Capture Engine Software version 54.4.2.236.
Evaluation of Cardiac Lesions
H&E sections of heart, lung, liver, kidney, and spleen were evaluated by a veterinary pathologist without reference to genotype or treatment group assignment. Cardiac lesions including myocyte vacuole formation, collagen deposition, and interstitial inflammation, as detailed in the results, were recorded as present or absent. For studies using leukocyte markers, the number of cells positive for CD3 immunostain was counted in four 40× fields in each heart section. Each field was selected randomly at low magnification with one field each from the left ventricular apex, midsection, and heart base as well as the midsection of the interventricular septum. Only positive cell profiles that included a nucleus were counted. An average count per high-power field was calculated for each heart section. Replicate sections of myocardium were stained with Masson’s trichrome using standard techniques. Digital images were created from trichrome-stained slides using a whole slide scanner (Olympus VS110). The resulting images were analyzed with Visiopharm (Denmark) software (MicroImager and NewCAST) to calculate a V fibrosis/V left ventricle ratio. To accomplish this, the left ventricular free wall and interventricular septum were masked and systematically sampled using a random start with the MicroImager software. Microscopic fields from 35% of the masked region were collected at 20× magnification. Using a double lattice test grid of 9 × 81 points, the volume of fibrosis and cardiac tissue (reference volume) was estimated by point counting. Cardiac counts were normalized to the fibrosis counts so that a ratio of V fibrosis/V left ventricle was determined for each animal.
Results
A/J-trac/trac Mice Develop iDCM
Histopathology
A/J-trac/trac mice develop an iDCM characterized by mononuclear cell infiltration and vacuolar degeneration of cardiomyocytes. At 4 weeks (1–3 days postweaning), both wild-type and trac/trac mice had normal appearing cardiomyocytes. Overall, there was no difference in ventricular mass or atrial volume between wild-type and trac/trac mice at 4 weeks (Figure 1A and B). By 10 weeks, intramyocyte vacuolation was prominent and heterogeneous mononuclear infiltrates surrounded degenerative myocytes (Figure 1C and D). Trichrome staining demonstrated trac/trac mice had significant interstitial collagen deposition associated with moderate biventricular dilation and marked atrial dilation. Degenerative myocyte and inflammatory infiltrative changes persisted through 20 weeks but were increasingly accompanied by adjacent myocytes with pale staining cytoplasm with loosely arranged myofibrils (Figure 1E and F). Both right and left ventricular mass was greatly decreased with marked replacement of cardiomyocytes by trichrome positive material. Ventricular lumens were greatly enlarged (Figure 1F). Atrial dilation was marked with some animals developing atrial thrombi (data not shown).
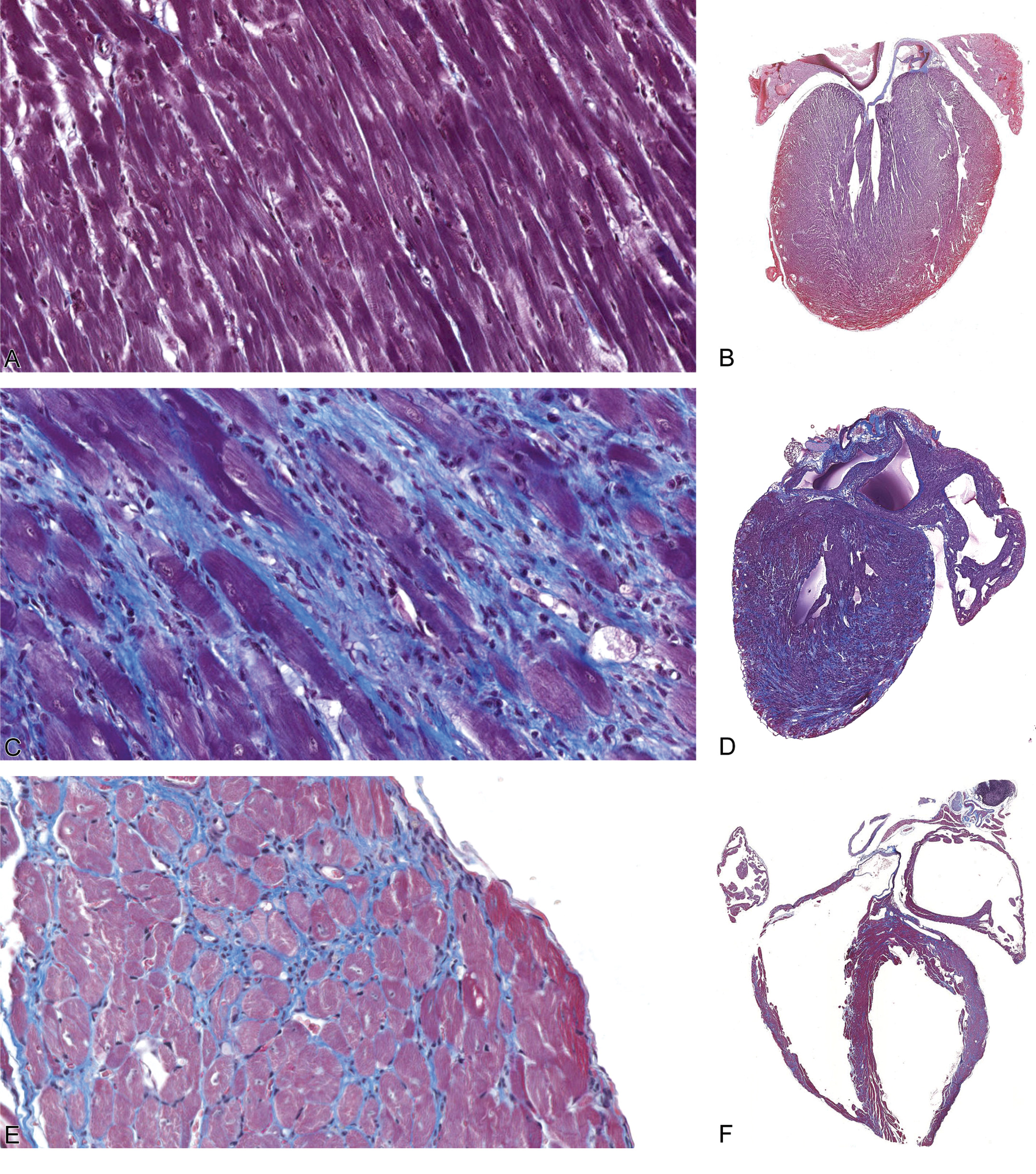
Progression of inflammatory cardiomyopathy in A/J-trac/trac mice postweaning: (A, B) At 4 weeks of age (2 days postweaning), ventricular walls are of normal thickness, myocytes are not affected, and there is no inflammatory infiltrate evident. (C, D) By 10 weeks, vacuolated myocytes are surrounded by inflammatory infiltrates and interstitial collagen. (E, F) At 14 weeks, both ventricular and atrial dilation is marked; fine strands of interstitial collagen are present between cardiomyocytes and interstitial inflammation persists. Masson’s trichrome, A, C, E ×400; B, D, E ×25.
Echocardiography
To evaluate cardiac function, A/J-trac/trac and littermate control mice were analyzed using echocardiography by an operator that was blinded to the genotypes of the mice tested. Average values for measurements from echocardiography are presented in Table 1. Mutant mice showed evidence of DCM, indicated by significant decreases in ejection fraction (p = .009) and fractional shortening (p = .008) when compared to heterozygotes. While the absolute values for left ventricular inner diameter (LVID) in diastole and systole did not differ significantly between genotype groups, there was a significant difference in the change in LVID from diastole to systole (p = .0004). Change in LVID in trac/trac mice was essentially half that of heterozygotes. Left ventricular mass differed significantly between groups, but this difference was not supported when LV mass was adjusted for body weight. Thickness of both left ventricular posterior wall and interventricular septum in diastole and systole was reduced in mutants compared to heterozygotes. Volume of the left ventricle did not differ significantly in diastole or systole between groups. Mice used for echocardiography ranged in age from 9 to 20 weeks. Phenotypes were consistent among groups regardless of age or sex. Representative echocardiogram traces for each genotype are presented in Figure 2.
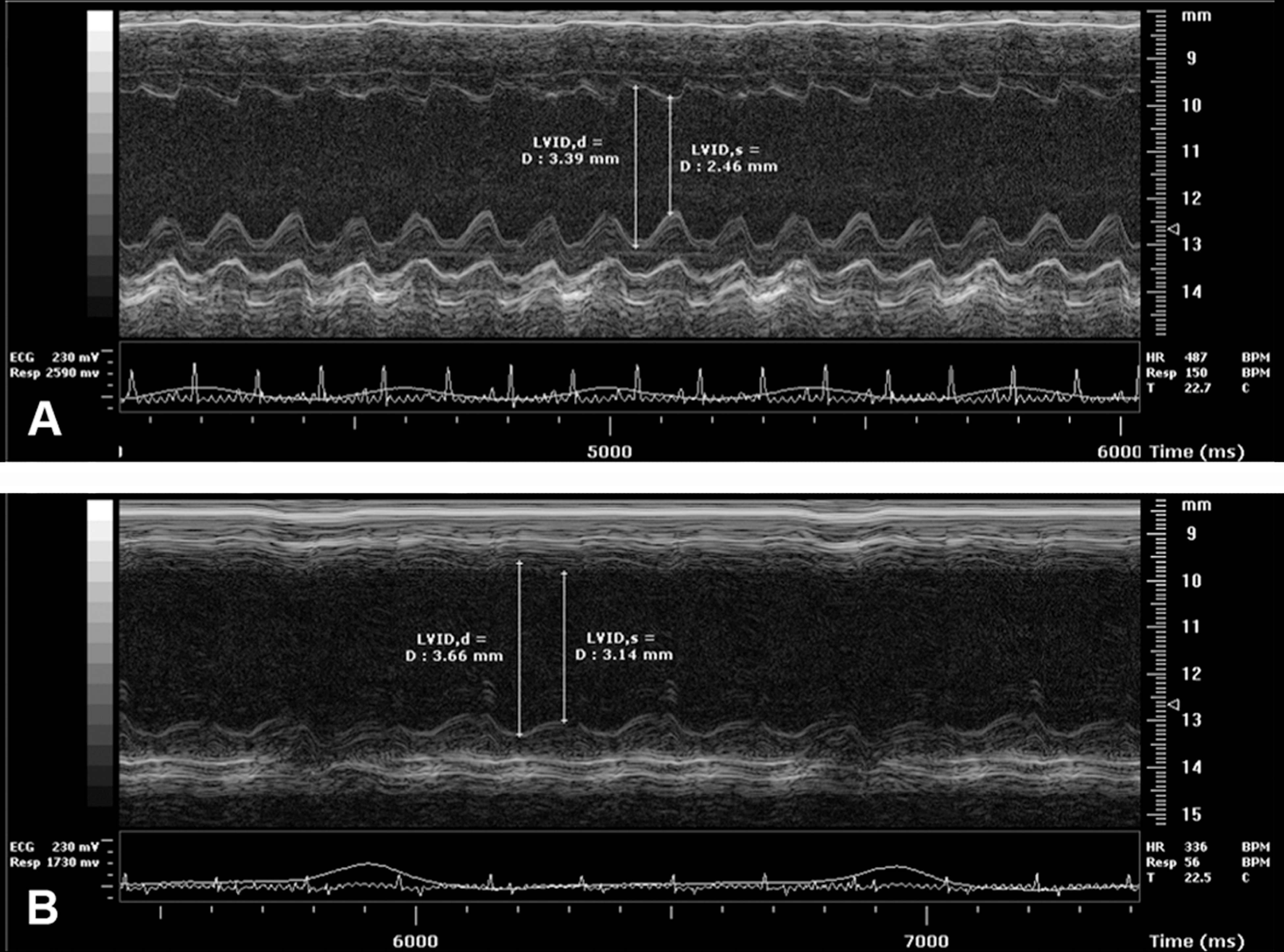
Representative M-mode echocardiogram traces of the left ventricle (A) A/J+/trac and (B) A/J-trac/trac mice. These traces compare males at 11 weeks of age. Mice were analyzed under isoflurane anesthesia to maintain heart rates between 350 and 500 beats per minute. Electrocardiogram traces appear below echographic images for each mouse. Vertical lines indicate measurement guides defining left ventricular inner diameter in diastole and systole.
Measurements from echocardiography.
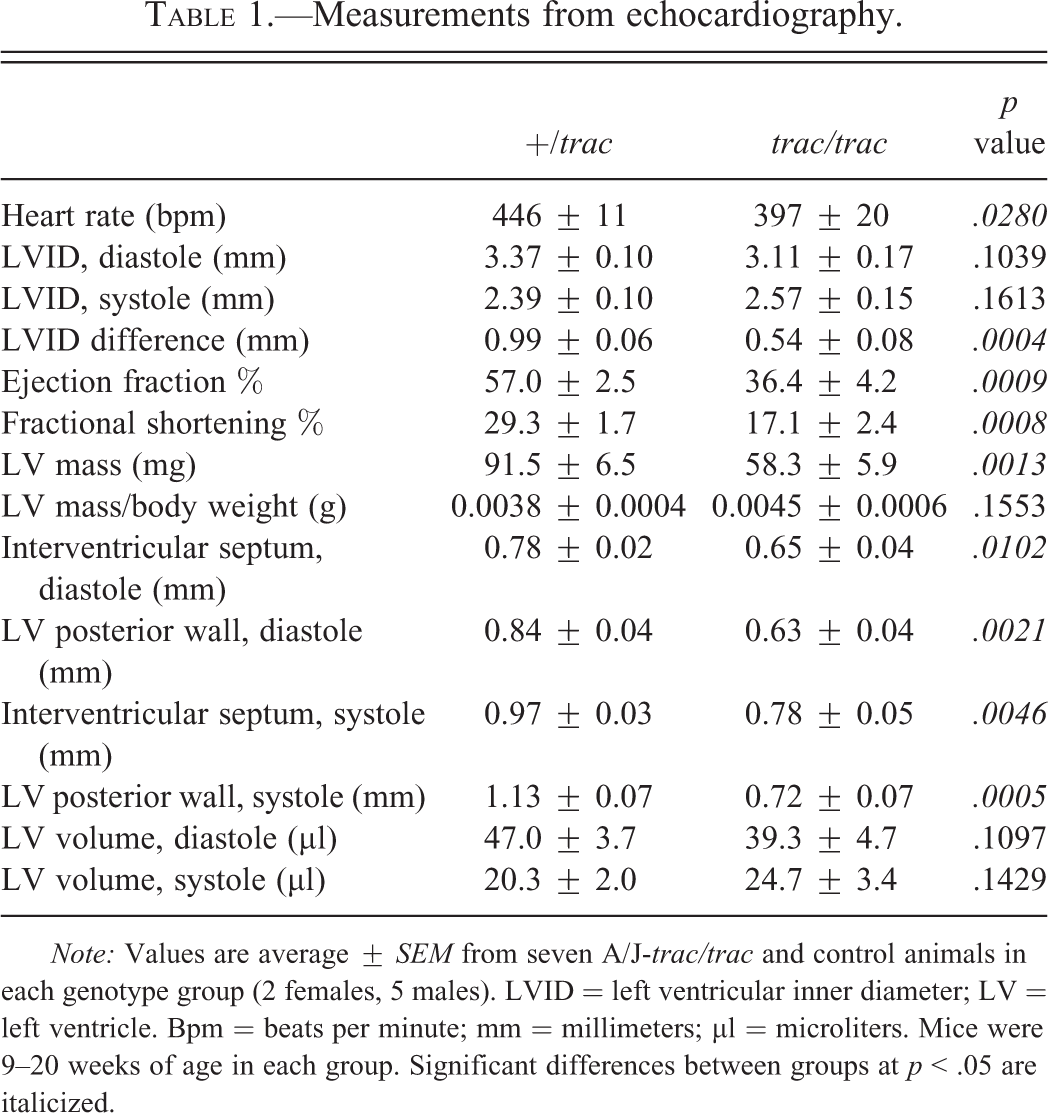
Note: Values are average ± SEM from seven A/J-trac/trac and control animals in each genotype group (2 females, 5 males). LVID = left ventricular inner diameter; LV = left ventricle. Bpm = beats per minute; mm = millimeters; µl = microliters. Mice were 9–20 weeks of age in each group. Significant differences between groups at p < .05 are italicized.
Myocardial Inflammation in A/J-trac/trac Mice Includes Significant Populations of T Cells and Monocytes
In order to better characterize the nature of infiltrating mononuclear cells in affected hearts, we performed immunostaining of the T cell marker CD3 and the monocyte/macrophage marker Mac-3. There were only rare CD3 positive cells in the myocardium of heterozygous mice early postweaning (4 weeks, Figure 3A) and no increase occurred at either 10 (Figure 3E) or 20 weeks (Figure 3I). A/J-trac/trac mice had focal interstitial infiltrates of CD3 positive cells at 4 weeks (Figure 3B). More cell dense coalescing infiltrates were evident in A/J-trac/trac mice by 10 weeks (Figure 3F) and similar infiltrates were present in 20-week-old trac/trac mice (Figure 3J). Modest numbers of MAC3 positive cells were present in the interstitium of heterozygote mice at all three ages evaluated (Figure 3C, F, K). Mac-3 positive cells in A/J-trac/trac mice were more frequent compared with littermate controls and had more extensive immunopositive cytoplasm. The increased cytoplasm was evident at 4 weeks (Figure 3D) with marked increases in cytoplasmic staining and cell numbers by 10 weeks (Figure 3H). Prominent Mac-3 positive cells were also evident in 20-week-old mice where they were admixed with interstitial deposits of extracellular matrix (Figure 3l). To more objectively evaluate CD3 and Mac-3 positive cell populations, counts were made of nuclei of positive cells in each of 40× fields (Figure 4A and B). At 4 weeks, there were no significant differences in CD3+ cell density between A/J-trac/trac mice versus +/trac control mice. By 10 weeks, CD3+ cell density in hearts of A/J-trac/trac mice increased 5-fold, and this increase persisted in hearts of affected animals at 20 weeks. Despite the appearance of increased amounts of Mac-3 positive cytoplasm in A/J-trac/trac mice, there was no increase in the numbers of cells compared with +/trac control mice as estimated by counts of nuclei of CD3+ cells at 4 weeks. The density of Mac-3 positive cells at 10 and 20 weeks of age paralleled that of CD3 positive cells (Figure 4B).
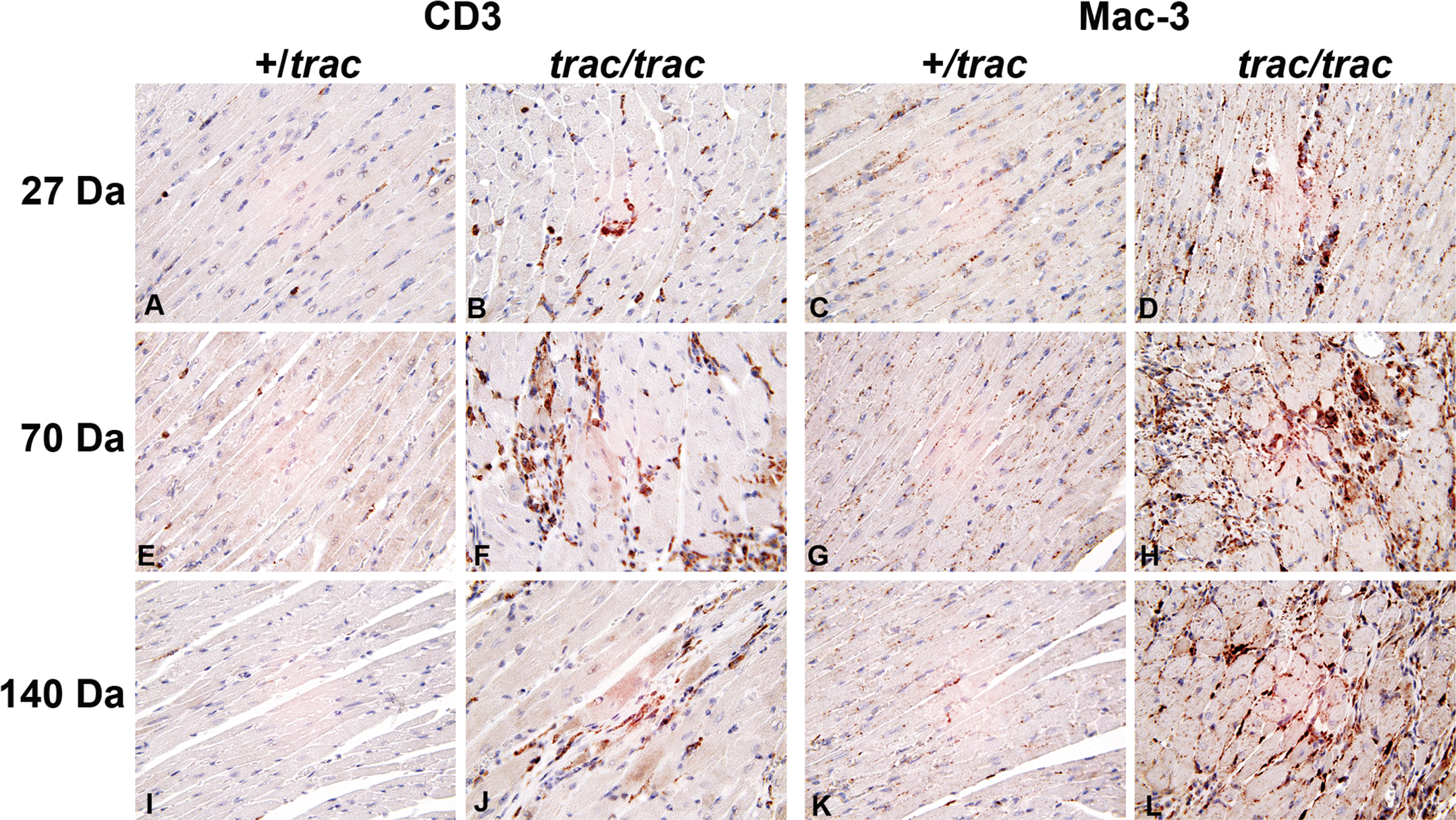
Immunophenotyping of mononuclear cells in myocardium of 4-week (A, B, C, D) 10-week (E, F, G, H), or 20-week (I, J, K, L) A/J-trac/trac and +/trac mice. Relative to age matched heterozygote mice (A, C) trac/trac mutants had modest increases in both CD3 (B) and MAC-3 (D) positive cells at 27 days. These infiltrates preceded histologically detectable degenerative myocyte changes. By 10 weeks, mononuclear cells infiltrating around degenerate or atrophic myocytes consisted on dense aggregates of CD3 (F) and MAC-3 (H) positive cells. Heterozygote mice remained unaffected at both 10 (E, G) and 20 (I, K) weeks. CD3 (J) and MAC-3 (L) positive infiltrates persisted as ventricular dilation proceeded through 20 weeks. All original magnifications 400×.
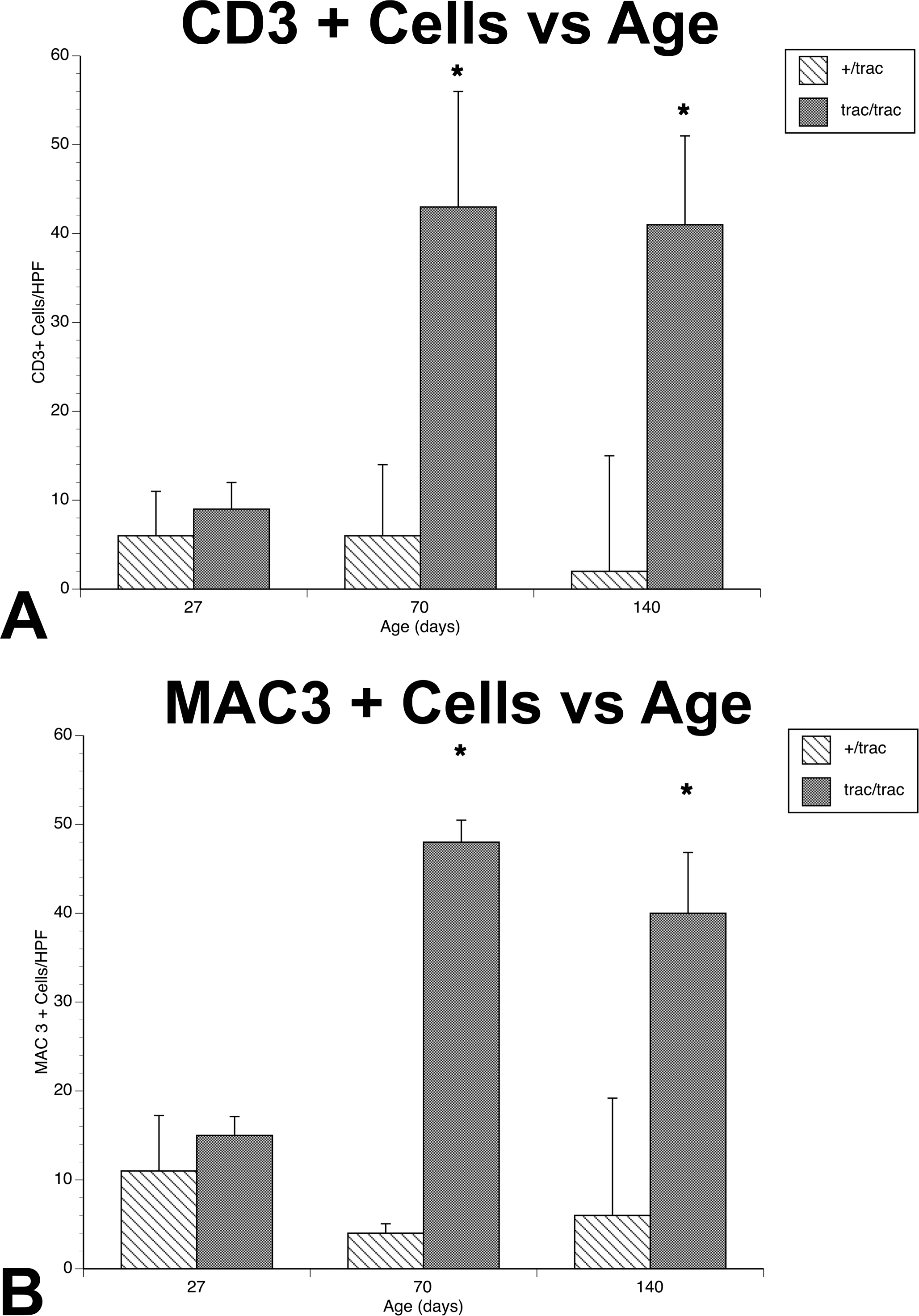
Counts of CD3 (A) and Mac-3 (B) positive cells in myocardium of A/J +/trac or trac/trac mice. Counts represent averages of nucleated positive cells in four 400× fields in left ventricle and septum of 4 mice from each age group. *Statistically different from wild type, p < .05.
Cardiac Myocyte Changes in A/J-trac/trac Mice Include Intramyocyte Membrane Bound Vacuoles and Evidence of Myofibril Genesis
The characteristic light microscopic changes in trac/trac mice were intracytoplasmic vacuoles and individual myocytes that had lightly staining cytoplasm. To further characterize these changes, hearts from 15-week-old A/J-trac/trac mice were evaluated by TEM. Regions with increased extracellular matrix contained myocytes with intracellular vacuoles surrounded by mononuclear cells and extensive deposition of parallel extracellular fibrils with periodicity characteristic of collagen (Figure 5A). Intramyocyte vacuoles were irregular but smoothly delimited from adjacent cytoplasm by intact membranes suggesting that they represent dilated endoplasmic reticulum. Adjacent sarcomere bundles were disorganized and lost parallel orientation to adjacent bundles. Mitochondria in vacuolated cells did not appear altered (Figure 5B). Additional myocytes in regions with inflammatory infiltrates were more electron lucent as a consequence of abundant amorphous cytoplasm containing slightly electron dense granules suggestive of glycogen. This cytoplasm was most prominent in the perinuclear region but also dissected between myofibrillar sarcomere bundles. Perinuclear regions also contained aggregates of parallel fibrils lacking Z bands (Figure 5C). Despite limited sarcomere formation, these electron lucent cells formed mature intercalated disks with adjacent cytologically normal myocytes (Figure 5D). The electron lucent myocytes were clearly evident on toluidine blue-stained thick sections and analogous cells could be identified in H&E-stained paraffin sections. These altered myocytes were not observed in H&E sections, EM thick sections, or by ultrastructural evaluation of hearts from A/J-+/trac littermate control animals (data not shown).
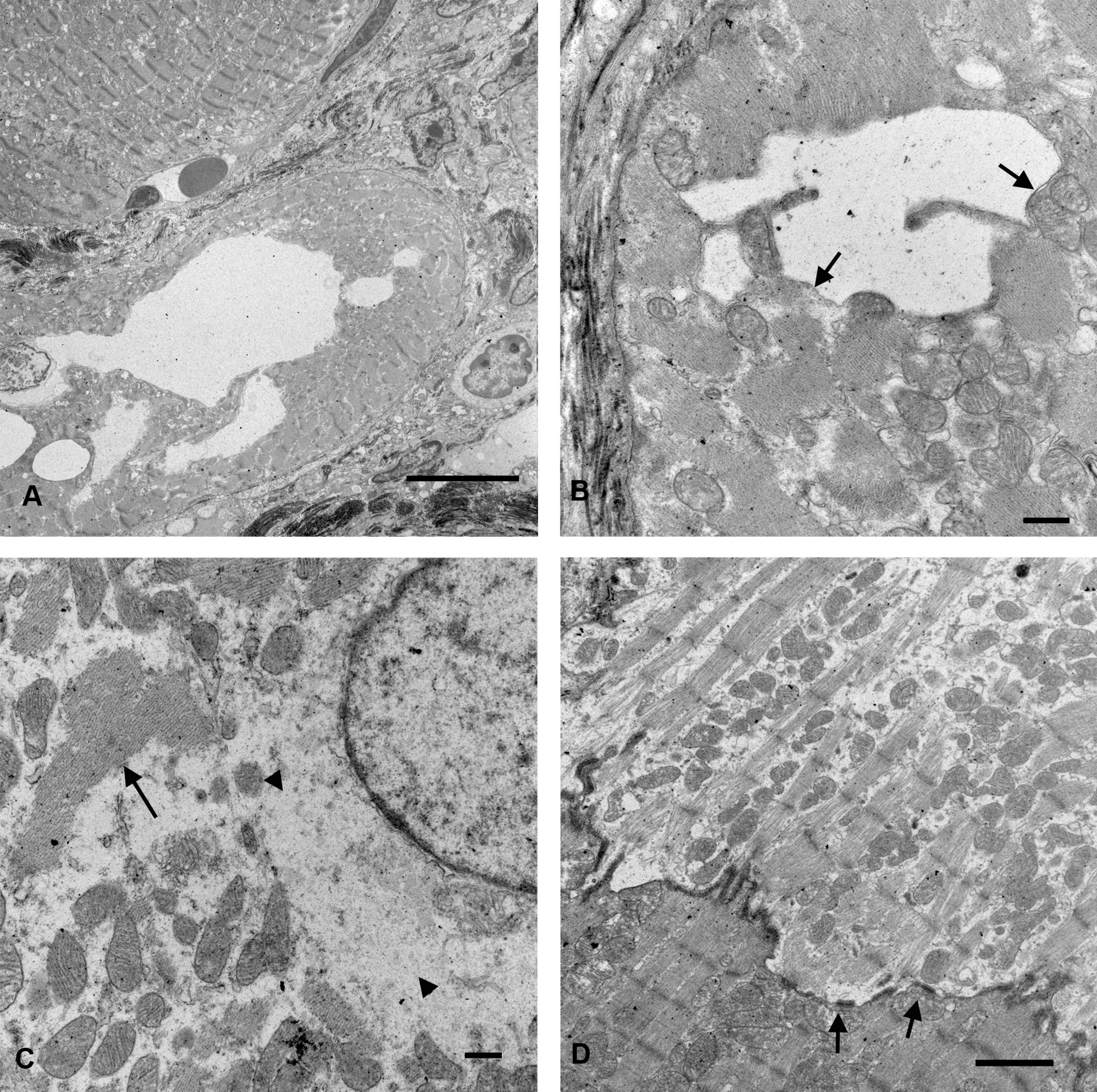
Ultrastructure of ventricle from A/J-trac/trac mouse at 17 weeks of age. (A) TEM intramyocyte membrane bound vacuoles displace myofibrils. Bar = 10 µm. (B) Myocyte vacuoles are membrane bound (arrows). Bar = 0.5 µm. (C) Perinuclear cytoplasm of electron lucent myocyte shows finely granular cytoplasm suggestive of glycogen accumulation (arrowheads) and aggregates of thick filaments without Z bands (arrow). Bar = 0.5 µm (D). Intercalated disk with discrete connexin-like junctions (arrows) between electron lucent and normal myocyte. Bar = 2 µm.
Inflammation and Cardiomyopathy in trac/trac Mice Is Dependent on Dietary Phytosterols
In humans with homozygous mutations at the ABCG5 or ABCG8 genes, hypercholesterolemia is evident before weaning, but sitosterolemia is only evident after beginning consumption of plant containing foods (Rios et al. 2010). While A/J-trac/trac mice do not have hypercholesterolemia (Chase et al. 2010), it is uncertain whether the myocardial lesions are a direct consequence of phytosterol accumulation or an alternative effect of the trac mutation. To determine the role of phytosterols in the development of cardiomyopathy, A/J-trac/trac and littermate control wild-type mice were nursed on dams given a normal diet but fed a phytosterol-free diet postweaning for 12 weeks. Histopathologic evaluation and trichrome staining showed significant inflammation and collagen deposition in all mice fed a normal diet (Figure 6A). Compared with A/J-trac/trac mice given normal chow diet (Figure 6B), none of the mice fed on a phytosterol-free diet had myocyte degeneration, inflammatory infiltration, or fibrosis, and there were no subjective differences in chamber volume or wall thickness from wild-type mice. In a separate experiment, A/J-trac/trac mice were kept on a phytosterol-free diet for 85 to 115 weeks, nearing the life span of this strain. These mice had minimal myocardial inflammation and collagen deposition (Figure 6C) and were not subjectively distinguished from heterozygote controls by histopathologic evaluation.
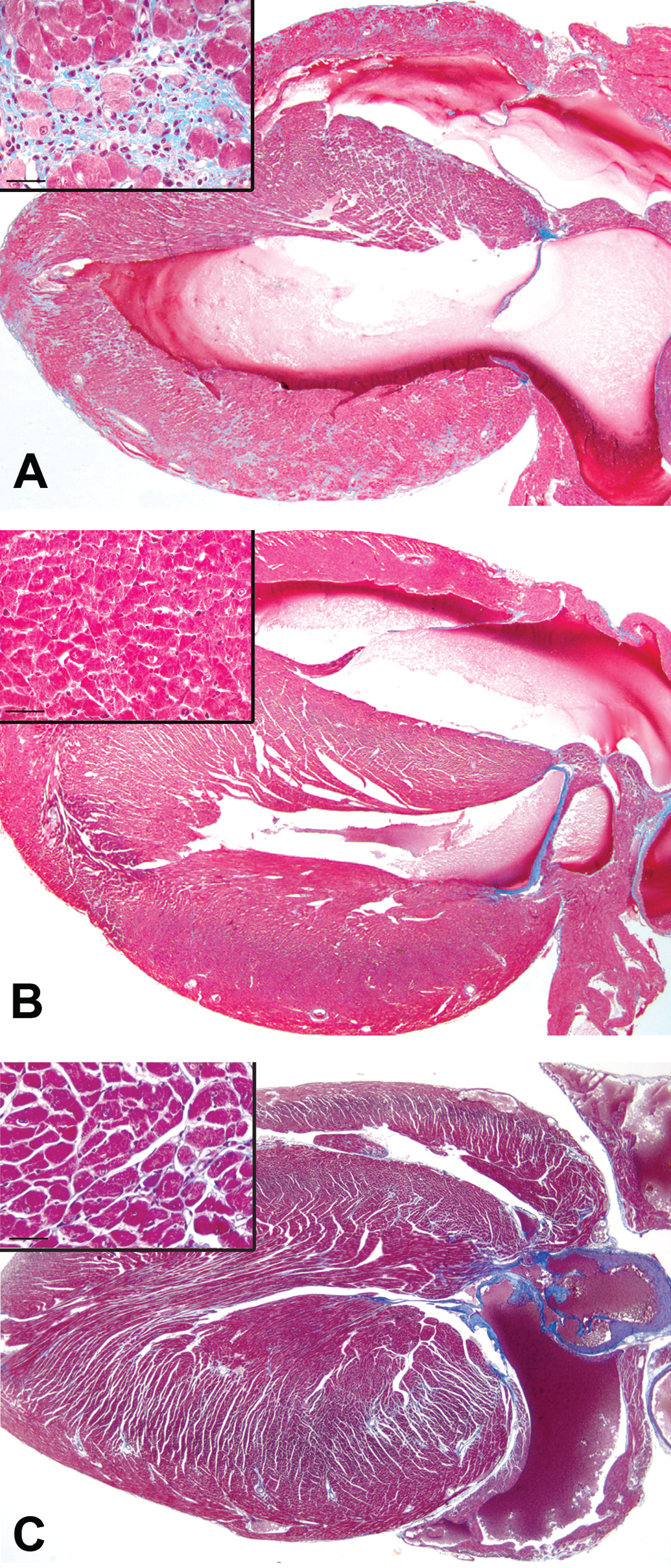
Effect of restriction of phytosterols in diet on development of cardiomyopathy in A/J-trac/trac mice. (A) A/J-trac/trac heart at 12 weeks of age maintained on a normal diet. (B) A/J-trac/trac heart at 12 weeks of age. This mouse was maintained on a phytosterol-free diet. (C) A/J-trac/trac heart from a mouse maintained on a phytosterol-free diet until 100 weeks of age. H&E 20×.
Removal of Phytosterols from Diet of trac/trac Mice Attenuates T Cell Infiltration but Monocytes Persist and the Extent of Fibrosis Is Not Altered
To determine the reversibility of the cardiac lesions, BALB/cBy-trac/trac mice were maintained on a normal diet (Purina 5K52) from weaning to 10 to 14 weeks of age and then switched to a phytosterol-deficient diet for an additional 8 weeks before necropsy. While a positive control group fed a normal diet throughout 18 weeks had ongoing vacuolar myocyte degenerative changes and mononuclear inflammatory infiltrate, mice switched to a phytosterol-free diet had markedly reduced inflammation and only limited myodegeneration. There was no difference in the extent of interstitial fibrosis between these two groups. On normal chow diet, BALB/cBy-trac/trac mice had fewer myocardial CD3 positive cells compared with A/J-trac/trac mice. Quantitative evaluation of CD3 and Mac-3 positive cells as well as volume density of fibrosis (Figure 7) confirmed these observations and demonstrated a reduction of CD3 positive cells in mice switched to a phytosterol-free diet. A modest numerical reduction in Mac-3 positive cells in the phytosterol-free diet group was not found to be statistically significant. Digital quantitation of the extent of trichrome positive extracellular matrix showed equivalent increases over heterozygote control mice for both normal and phytosterol-free diets.
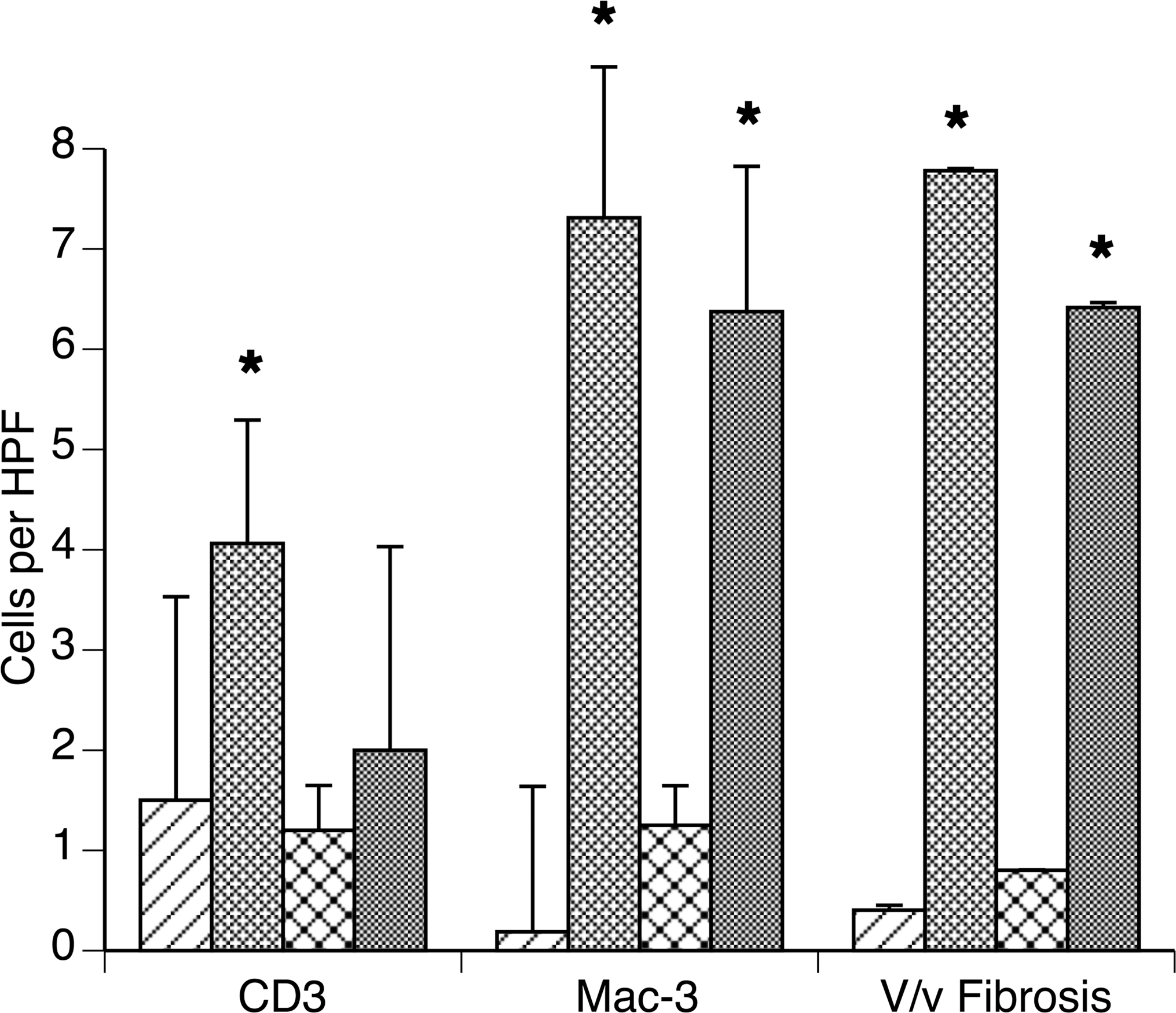
Effect of transfer to phytosterol diet on myocardial inflammation and fibrosis. BALB/cBy-trac/trac mice fed normal diet (ND) for 10 weeks postweaning followed by 8 weeks on normal or phytosterol-free diet (PFD). Counts of immunostained sections and stereologically determined volume percentage of trichrome positive matrix show phytosterol-free diet mice had fewer CD3 positive cells but equivalent MAC3 positive cells and equivalent volume percentage of ventricular myocardium with trichrome positive extracellular matrix.
Absence of Mature T and B Lymphocytes Prevents Cardiomyopathy in trac/trac Mice
To determine whether functional mature T and B lymphocytes play a critical role in development of cadiomyopathy in trac/trac mice, we carried out genetic crosses to develop T- and B-cell-deficient BALB/cBy-Rag1null trac/trac mice. Immunocompetent BALB/cBy-trac/trac mice evaluated at 37 to 42 weeks developed severe inflammatory cardiomyopathy with marked left ventricular and atrial dilation and interstitial fibrosis (Figure 8A). In contrast, BALB/cBy-Rag1null trac/trac mice did not develop cardiomyopathy (Figure 8B). Counts of CD3 (Figure 8C) and MAC-3 (Figure 8D) positive cells demonstrated significant infiltrates of both cell types in BALB/cBy-trac/trac mice. BALB/cBy-Rag1null trac/trac mice had only rare CD3 positive cells and no increase in MAC-3 cells in the myocardial interstitium. While BALB/cBy-trac/trac mice had longer life spans than AJ-trac/trac mice, both male and female BALB/cBy-Rag1null trac/trac mice had significantly increased life spans compared to immunocompetent BALB/cBy-trac/trac cohorts (Table 2). Although elimination of adaptive immunity in BALB/cBy-Rag1null trac/trac mice greatly reduces the severity of cardiomyopathy, nevertheless, these mice are severely thrombocytopenic throughout life (data not shown).
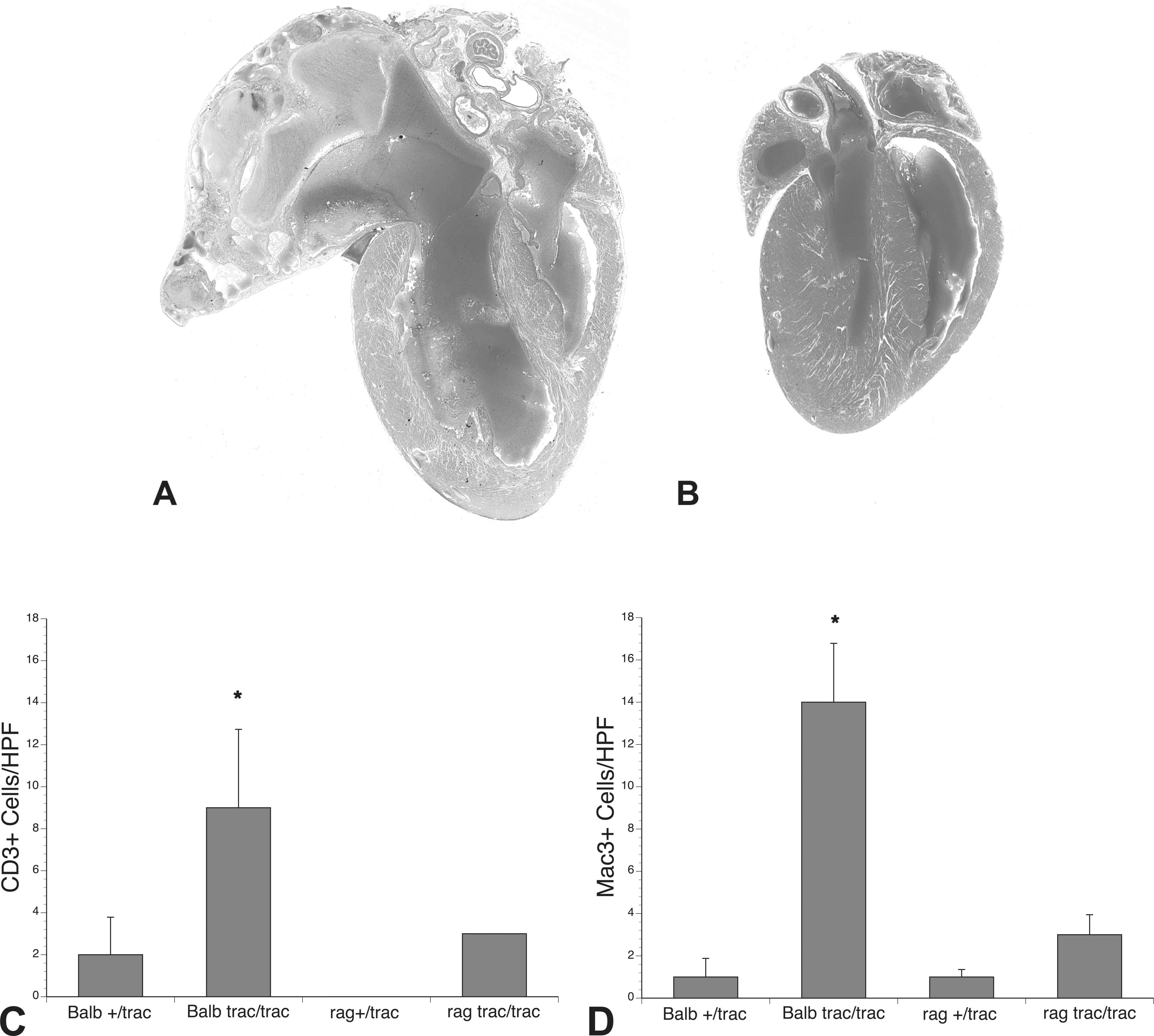
Effect of T and B cell deficiency on cardiomyopathy in BALB/cByJ-trac/trac mice. Mice with wild-type allele at the Rag1 locus gene develop profound dilative cardiomyopathy at 37 weeks (A); BALB/cBy-Rag1null trac/trac mice exhibit normal cardiac phenotype at 37 weeks (B); quantitation of inflammatory cell infiltrates show BALB/cBy-Rag1null trac/trac mice have only rare CD3 positive staining cells (C); and no increase in myocardial MAC3 positive cells (D).
Strain- and sex-specific survival of wild-type and trac/trac mice.

Note: Unmanipulated mice were kept in standard housing until death or euthanized when moribund except for BALB/cBy-Rag null trac/trac males which were euthanized between 600 and 900 days. A/J-trac/trac male versus BALB/cBy-trac/trac male: ***p < .0001. A/J-trac/trac female versus BALB/cBy-trac/trac female: ***p < .0001. BALB-trac/trac male versus BALB/cBy-Rag1null trac/trac male: **p < .0066.
BALB-trac/trac female versus BALB/cBy-Rag1null trac/trac female: ***p < .0001.
Discussion
While investigation of myocardial disease often focuses on consequences of infarction, generalized myocardial fibrosis associated with cardiomyopathy or systemic or pulmonary hypertension is becoming recognized as potentially more significant relative to risk for fatal arrhythmia. Recent research suggests that regulation of fibroblast stimulation, collagen synthesis, and matrix degradation in the heart is complex and mutifactorial with local responses to stretch, physiologic mediators, and even local angiotensin generating systems competing with systemic signals and inflammation (Kakkar and Lee 2010). Most models of iDCM use immunologic induction of inflammation that progress through an autoimmune manner. Our evaluation of trac/trac mice suggests a critical role for the immune system in myocardial degeneration, expansion of the monocyte population, and the induction of myocardial fibrosis. Dynamic evaluation by ultrasonography demonstrated marked interference with myocardial contractility, evidenced by markedly reduced systolic/diastolic differences in LVID in affected mice. This response is associated with serum phytosterol accumulation and prevented by maintenance of trac/trac mice on a phytosterol-free diet. This mutation thus represents an endogenous mouse model of iDCM that may have additional utility since the initiating T cell infiltrate can be diminished by dietary manipulation thus allowing experimental intervention that addresses the ongoing monocyte–fibroblast interface and the relative role of fibrosis in altered myocardial contractility.
Most human cases of inflammatory DCM are thought to have an autoimmune pathogenesis either subsequent to exposure of cardiac antigens through viral infection or familial predisposition based on genetic variations in MHC or other immune-associated proteins (Caforio, Vinci, and Iliceto 2008). Inflammatory infiltrates reflect immunogenic pathogenesis with a mixture of T cell and monocyte effector cells. While T-cell-mediated recruitment of monocytes contributes to the inflammatory phenotype, local factors also contribute, and experimental evidence demonstrates a complex interplay between monocytes, and TH-1 and TH-17 lymphocytes in regulation of inflammatory responses (Valaperti et al. 2008). Our experiments demonstrate that, similar to the human disease, T cell and monocytic infiltrates predominate in the progressive inflammation in trac/trac mice and that immune modulation prevents both inflammation and myocardial fibrosis.
Much interest has been focused on the potential for myocyte renewal in cardiac disease. The recognition that resident stem cells expressing contractile elements can participate in myocardial repair (Beltrami et al. 2003) suggests novel approaches for therapeutic intervention (Lyngbaek et al. 2007). Furthermore, infiltration of injured myocardium by bone marrow–derived stem cells with potential for differentiation into both vascular cells and myocytes suggests mechanisms that explain improved contractility and vascular supply in experimental models treated with stem cell transplants. Whether this improvement represents differentiation of progenitor cells into functional contractile cells or paracrine remodeling of existing cells remains controversial (Lyngbaek et al. 2007). Our ultrastructural studies demonstrate a population of cells in affected regions of heart that have organized actinomyosin structures yet have extensive regions of cytoplasm largely occupied by cytosol containing glycogen. Their appearance suggests either synthesis of contractile elements by reversibly injured myocytes or differentiation of precursors to functional contractile cells. Importantly, these cells demonstrate clear interface with adjacent morphologically normal cells through well-developed intercalated disks.
The induction of iDCM in this mouse model does not appear to be reflected in human sitosterolemia patients. Affected humans develop hypercholesterolemia and premature coronary atherosclerosis (Berge et al. 2000) presumably due to increases in intestinal absorption of cholesterol (Fitzgerald, Mujawar, and Tamehiro, 2010). Inflammatory myocardial disease has not been reported in human sitosterolemia patients. Significant differences in the extent and time of onset of iDCM were evident between the two mouse strains used in this study. The original spontaneous mutation was found in A/J strain mice and resulted in a rapid onset of severe myocardial disease with few individuals surviving beyond 22 weeks. Backcrossing of this mutation onto the BALB/cBy strain background resulted in a lesser inflammatory response and longer survival time but did eventually recapitulate a similar inflammatory and fibrotic ventricular failure. A/J strain mice have an inherent defect in another myocardial protein, dysferlin, that predisposes A/J mice to late onset myocardial fibrosis (Chase et al. 2009). Whether the combined dysferlin/Abcg5 defect explains these strain differences remains uncertain.
A variety of genetic alterations associated with DCM have been identified. Molecular mechanisms behind their pathogenesis include cytoskeletal proteins affecting interaction of the sarcomere with the Z disk, mutations in contractile proteins that interfere with Ca++ regulation of myosin function, and mutations driving myocyte apoptosis (Toko et al. 2010). Mutations identified in humans have been reproduced in a variety of mouse genetic models. Predictable hypertrophic and dilated cardiomyopathies occur (McCauley and Wehrens 2009), but the extent of myocardial fibrosis is generally significantly less that that observed in the sitosterolemia model (Toko et al. 2010) and the role of inflammation in the progression of these diseases has generally not been investigated.
Most models of inflammatory cardiomyopathy support the data from studies of the human condition implicating autoimmunity to myocardial contractile proteins in the development of iDCM. Among these, autoantibodies to the myosin heavy chain are often associated with myocardial inflammation. The recognition that T cell infiltrates are significant components of myocardial inflammation raises the question as to whether autoantibodies drive the inflammatory response or rather represent an epiphenomenon of T-cell-mediated damage induced by other immunogens. Of significance for the sitosterolemia model is the importance of adjuvant induction of toll-like receptor (TLR) responses in addition to autoimmune activation in the progression of experimental autoimmune iDCM (Blyszczuk et al. 2009). The extensive inflammation and fibrosis in the sitosterolemia model suggests this process occurs without exogenous activation of TLR.
The mechanism of initiation of the inflammatory response in this model remains uncertain. The protective effects of dietary restriction of phytosterols suggests a causative role but does not determine whether phytosterol accumulation drives myocardial degeneration with immune responses subsequent to release of myocyte proteins or alternatively that phytosterols act as proximate immunogens, perhaps by incorporation into myocyte cell membranes. The paucity of myocardial inflammation immediately postweaning in affected individuals is further evidence that the process is driven by dietary phytosterols. A recent case report demonstrated that a nursing infant later confirmed to have an ABCG5 mutation only developed sitosterolemia postweaning (Rios et al. 2010). This finding supports the concept that plant sterols rather than hypercholesterolemia initiate myocardial inflammation in this mouse model. Myocyte degeneration in this syndrome is characterized by dilated membrane-bound intracellular vacuoles that suggest osmotic dysregulation associated with dilated sarcoplasmic reticulum. Whether these represent chemical interference with myocyte homeostasis or T-cell-mediated myocyte cytotoxicity is uncertain.
This mouse model suggests several potential experiments that address aspects of inflammatory DCM. Only one-third of myocarditis cases progress to DCM through as yet incompletely understood mechanisms. Our experiments with immunodeficient BALB/cBy-Rag1null trac/trac mice document the necessity of a competent adaptive immune system in the development of myocardial inflammation in the sitosterolemia model. The rapid and quantifiable progression of inflammation and fibrosis observed with this model would potentiate experiments aimed at developing anti-inflammatory and immunosuppressive therapeutic interventions. While T-cell-mediated monocyte infiltration is thought to be a key aspect of iDCM (Cihakova and Rose 2008), recent studies suggest dissociation of T-cell regulation from the progression of myocardial fibrosis. In an adoptive immunity experiment, Myd88-deficient mice were protected from a myocardial fibrotic response despite similar inflammatory myocardial infiltrates. Chimeric mouse experiments further demonstrated Myd88 and IL-1β dependency occurred in bone marrow–derived fibroblast precursors in the myocardial infiltrate (Blyszczuk et al. 2009). Alternatively, a critical role for TH-17 cells has recently been described in the induction of myocardial IL1β expression, recruitment of monocytes, and progression to fibrosis (Baldeviano et al. 2010). The predictable and early onset of myocardial inflammation and fibrosis in this mouse model as well as its manipulation by diet provides a convenient model to examine these processes in an adjuvant free and endogenously induced myocardial inflammation.
Monocyte-macrophage lineage cells are key effectors in generating myocardial injury. For example, dysregulation of monocyte activation drives accentuated responses to both myosin and viral induced myocarditis in IL-13-deficient mice (Cihakova et al. 2008). Monocyte lineage cells are both effectors and modulators of myocarditis. Persistent IL-17-mediated monocyte inflammation drives accentuation of experimental allergic myocarditis in BALB/c interferon γ-deficient mice and a variety of interferon γ replacement interventions prevent this (Valaperti et al. 2008). Additional studies demonstrate prominin 1+ bone marrow precursors differentiate into myocardial fibroblasts through activation of transforming growth factor (TGFβ) signaling (Kania et al. 2009). Current concepts suggest an intricate interplay of paracrine and direct interactions between myocardial fibroblasts and myocyte development, hypertrophy, rhythmicity, and response to injury including matrix deposition (Kakkar and Lee 2010).
The reversibility of T cell infiltration by dietary manipulation in the Abcg5 mutant mouse model offers a simple approach to selective modulation of inflammatory cell populations and provides an opportunity to evaluate therapeutic intervention in the pro-fibrotic process. Future experiments should dissect lymphocyte subsets, cytokine networks, and stem cell/fibrocyte recruitment in this model.
Footnotes
Acknowledgment
This article is dedicated to the memory of Dr. Thomas H. Chase, talented pathologist, artist, and sailor, and great human being.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: HL 077642 (LDS) and NIH Cancer Core Grant CA34196 to The Jackson Laboratory.