
Abstract
Particulate matter, with a mean aerodynamic diameter of ≤10 µm (PM10), exposure is considered as a risk factor for cardiovascular and respiratory diseases. The mechanism of cell damage induced by PM10 exposure is related to mitochondrial alterations. The aim of this work was to investigate the detailed alterations induced by PM10 on mitochondrial function. Since lung tissue is one of the most important targets of PM10 inhalation, isolated mitochondria from lung rat tissue were exposed to PM10 and structural alterations were analyzed by transmission electron microscopy. Mitochondrial function was evaluated by respiratory control index (RCI), membrane potential, adenosine triphosphate (ATP) synthesis, and activity of respiratory chain. Results showed that exposure to PM10 in isolated mitochondria from lung tissue caused enlarged intermembrane spaces and shape alterations, disruption of cristae, and the decrease in dense granules. Oxygraphic traces showed a concentration-dependent decrease in oxygen consumption and RCI. In addition, mitochondrial membrane potential, ATP synthesis, and activity of complexes II and IV showed an increase and decrease, respectively, after PM10 exposure. PM10 exposure induced disruption in structure and function in isolated mitochondria from lung rat tissue.
Introduction
Exposure to airborne particulate matter (PM) has been implicated as a risk factor for lung cancer and mortality (Pope et al. 2002, 2011; Brunekreef et al. 2002; Ferecatu et al. 2010), but the number of alterations detected in other tissues is increasing. In this regard, PM exposure causes alterations in the gastrointestinal tract (Mutlu et al. 2011), brown adipose tissue (Xu et al. 2011), and brain (Guo et al. 2012). These changes are associated with decreased lung function and growth among children (Roy et al. 2012). PM exposure with a mean aerodynamic diameter of ≤10 µm (PM10) is associated with an increase in emergency hospital admissions for respiratory and thromboembolism illness (Qiu et al. 2012; Martinelli et al. 2012), and with small size for gestational age at birth in humans (van den Hooven et al. 2012). In addition, it has been demonstrated that PM exposure increases the risk for several diseases including hypothalamus stimulation in obese rats predisposing them to higher cardiovascular risk (Balasubramanian et al. 2012). PM10 enhances cell damage induced by hydrogen peroxide in human alveolar epithelial cells (Chirino et al. 2010) and also aggravates apoptosis in cystic fibrosis in bronchial epithelial cells (Kamdar et al. 2008).
After exposure, PM can be internalized into cells by phagocytosis and localized in cytoplasm and mitochondria (Gualtieri et al. 2009), and it has been widely demonstrated that PM effects are strongly related to the higher production of reactive oxygen species (ROS; Chirino et al. 2010; Sánchez-Pérez et al. 2009). In this regard, inhibitors of mitochondrial electron transport chain blocked ROS generation induced by PM10 exposure (Kamdar et al. 2008). The association between PM10 exposure and mitochondrial alterations has also been found in workers exposed to high levels of PM10, which caused mitochondrial DNA damage in peripheral blood (Hou et al. 2010). Alterations in mitochondrial number and size in brown adipose tissue have been reported after concentrated fine ambient PM inhalation (Xu et al. 2011). Mitochondria have a central role not only for the cell metabolism but also as mediators in the PM-induced cell effects. For example, mitochondrion are required for the increase in DNA methyltransferase 1 expression and for p16 promoter methylation induced by PM2.5 exposure (Soberanes et al. 2012), and those cellular events are found in human lung cancer (Belinsky et al. 1998). Mitochondria are also required to induce apoptosis in alveolar epithelial cells exposed to PM2.5, through the signaling pathway that involves apoptosis signaling kinase 1 (ASK1) and c-Jun N-terminal kinase (JNK) enzymes, among others (Soberanes et al. 2009). Recently, it has been demonstrated that pulmonary exposure of PM10 in rats induced myocardial mitochondrial alterations (Golomb et al. 2012).
The above information suggests that PM exposure could execute their effects, at least in part, through mitochondrial alterations even in distant tissues; however, a detailed description about mitochondrial function after PM10 exposure has not been described. The aim of this work was to investigate whether direct interaction between PM10 and mitochondria induces respiratory dysfunction using isolated mitochondria from lung rat tissue to evaluate mitochondrial function parameters including oxygen consumption, ATP synthesis capability, and the activity of respiratory complexes. This work describes a characterization of the PM10 effects on mitochondrial structure and oxygen consumption, ATP synthesis, mitochondrial membrane potential (ΔΨm), and activity of respiratory complexes.
Materials and Methods
PM10 Sampling
PM10 was collected from an industrial zone of Mexico City using a high-volume particle collector with a flux of 1.13 m3/min (GMW model 1 200 VFC HVPM10; Sierra Andersen, Smyrna, GA). A 3-µm pore size membrane of cellulose nitrate (Sartorius AG, Goettingen, Germany) was used as a support to obtain particles with mean aerodynamic diameters equal to or smaller than 10 µm that were collected by the high-volume sampler. PM10 was stored at 4°C under darkness until particle removal. Particles were gently scraped off the membranes using an electric toothbrush (Oral-B, AdvancePower 400, Braun, Mexico) and maintained in endotoxin-free glass vials at 4°C until their use (Alfaro-Moreno et al. 2009).
Mitochondria Isolation from Lung Tissue
Experimental work followed the guidelines of Norma Oficial Mexicana for the use and care of laboratory animals (NOM-062-ZOO-1999) and for the disposal of biological residues (NOM-087-ECOL-1995). Mitochondria isolation from lung tissue was achieved by the conventional method with slight modifications (Drew and Leeuwenburgh 2003; Meng et al. 2007). Lung tissue was obtained from male Wistar rats weighing 220 to 250 g (10 weeks old). Before experiments, the animals were maintained under constant conditions of temperature (22°C) and lighting (12-hr light/dark cycle) and were fed with standard commercial rat chow diet from Harlan containing 18% protein (Harlan Teklad Global diet 2018S sterilized, Harland Teklad, Madison, WI) and water ad libitum. First, the lung tissues were rinsed with saline solution, weighed, and fixed in ice-cold isolation media containing 0.3 M mannitol, 2 mM ethylenediaminetetraacetic (EDTA), 1 mM dithiothreitol (DTT), 0.1% polyvinyl pyrrolidone (PVP) 40, 0.1% bovine serum albumin (BSA), pH 7.4. The tissue was diced, minced, and passed by a mesh sieve. Tissue was homogenized in a Potter–Elvehjem glass-glass homogenizer with Teflon pestle and then centrifuged at 2000g for 10 min at 4°C in a SS’34 rotor (Sorvall, Claremont, CA, USA). The supernatant fraction was decanted and centrifuged at 12,000g in an SS'34 rotor (Sorvall) for 10 min at 4°C. The mitochondrial pellet was centrifuged again with washing media containing 0.3 M mannitol, 1 mM EDTA, 0.1% BSA, pH 7.4 at 9600g. The mitochondrial protein concentration was determined by the Lowry method using BSA as a standard.
Mitochondria-enriched Preparations Exposed to PM10
PM10 was suspended in washing media and then 1 mg of isolated mitochondria was incubated with the following PM10 concentrations: 1, 5, 10, 30, and 50 µg for 1 hr. To test the effect of particle addition, 1 mg of mitochondria was exposed to 10 and 20 µg of titanium dioxide nanoparticles. TiO2 NPs (TiO2 NPs 99.7%) were purchased from Sigma-Aldrich (Sigma-Aldrich, China; Catalogue number 637254) as a dry material.
Transmission Electron Microscopy (TEM) for PM10-exposed Mitochondria
After treatments with PM10, mitochondrial preparations were fixed with glyceraldehyde 2% for 2 hr. Then, the samples were postfixed with the osmium tetroxide loaded and washed three times with a SORENSEN phosphates buffer supplemented with saccharose and 1% calcium chloride. Postfixed samples were dehydrated in a concentration gradient buffer-ethanol for 10 min. Samples were incubated overnight in 100% ethanol. Epoxy resin was used to embed samples and 70 µm sections were cut and analyzed under TEM (Jeol JEM-1010).
Mitochondrial Function
Samples were incubated under previous treatments and 0.1 mg of isolated mitochondria were added to 1 ml of assay buffer (KCl 125 mM, KH2PO4 125 mM, HEPES 20 mM, MgCl2 1 mM, BSA 0.4 mg/ml, EGTA 0.25 mM pH 7.4). Samples were mixed into a sealed chamber equipped with a magnetic stirrer, and glutamate (10 mM) and malate (5 mM) were used as mitochondrial substrates. Mitochondrial oxygen consumption was measured using a Clark-type electrode (Hansatech, United Kingdom). The respiratory control index (RCI) was calculated as the ratio of state III and state IV. State III was measured after the addition of ADP. The subsequent state IV was determined by measuring mitochondrial oxygen uptake upon complete ADP phosphorylation to ATP (Estabrook, 1967).
ATPase activity was measured with an enzyme-coupled assay following the reduction of NADP+ (Sigma-Aldrich, St. Louis, MO, USA) at 340 nm driven by the hexokinase and glucose-6-phosphate dehydrogenase (Sigma-Aldrich, St. Louis, MO, USA) activities (Cortés et al. 2000). Different ATP production activity within the mitochondrial fraction was subtracted from the total activity by performing the assay in the presence and absence of 50 µM oligomycin (Sigma-Aldrich, St. Louis, MO, USA).
Mitochondrial Membrane Potential (ΔΨm)
The fluorescent dye Rhodamine 123, which selectively accumulates in mitochondria, was used to measure the ΔΨm (Meng et al. 2007). Briefly, isolated mitochondria from lungs were incubated with previously mentioned PM10 concentrations for 1 hr and then with 2 µM rhodamine 123 for 60 min. After that, mitochondria were washed three times with phosphate-buffered saline. Each sample was mounted in a glass slide individually and analyzed using confocal microscopy Leica. ΔΨm was determined by three independent experiments, and the results were expressed as arbitrary fluorescence units (excitation: 488 nm; emission: 530 nm).
Activity of the Respiratory Chain
To measure the activity of mitochondrial complexes I to IV, the method described by Bénit et al. (2006) was used with some modifications. Diluted mitochondria (0.3 mg) in assay buffer (250 M sucrose, 50 mM phosphate pH 7.4, 0.01% Tween-20, and 0.2 mM EDTA) were incubated on ice for 30 min before the assay. First assay measured the redox change of cytochrome c (550 to 540 nm; extinction coefficient of 19.1 mM− 1cm− 1). To measure the activity of complex IV, 10 µM of reduced cytochrome c was added to assay buffer. The activity of complexes II + III was measured by the addition of 10 µM oxidized cytochrome c, 8 µM rotenone, 0.5 mM KCN, and 5 mM succinate. For complex III activity, 2 mM EDTA and 50 µM DBH2 were added, and 1 µM antimycin A was used to measure the antimycin-insensitive activity of complex III. Second assay was performed using dichloroindophenol sodium salt hydrate (DCPIP) after the activity of complexes I and II following redox changes (600 to 750 nm; extinction coefficient of 22 mM− 1cm− 1). Mitochondria in assay buffer with 1 mM NaN3 ere used to measure NADH dehydrogenase, adding 40 µM DCPIP, 1 mM KCN, and 50 µM NADH, and 8 µM rotenone. To measure thenoyl trifluoracetone (TTFA)-insensitive activity of complex II, 40 µM DCPIP, 5 mM Na succinate, 1 mM PMS, 4 mM TTFA, and 80 µM DQ were added. Measurements were done in an Aminco Dw2a™ UV-visible spectrophotometer with the OLIS DW2 conversion and OLIS software.
Statistical Analysis
Data are presented as mean ± standard error of mean (SEM). Different treatments were compared using analysis of variance followed by Bonferroni test for individual comparisons between group means. Differences at *p < .05 were considered as significant.
Results
PM10 Exposure Induced Structural Alterations
TEM analysis revealed normal outer and inner mitochondrial membrane in control samples. They had tightly packed cristae with parallel alignment, higher matrix density, and visible matrix dense granules. One milligram of mitochondria exposed to 1 µg and 5 µg of PM10 showed no structural alterations. However, 1 mg of mitochondria exposed to 10 µg of PM10 caused enlarged intermembrane spaces and shape alterations. The exposure to 30 µg and 50 µg of PM10 induced a higher disruption in cristae and the decrease in dense granules (Figure 1). At this concentration, titanium dioxide nanoparticles induced a decrease of 30% in RCI (Freyre-Fonseca et al. 2011). However, even if titanium dioxide nanoparticles and PM10 can induce a similar decrease in RCI, PM10 clearly induced higher number of alterations in mitochondria structure because of few enlarged intermembrane spaces (data not shown).
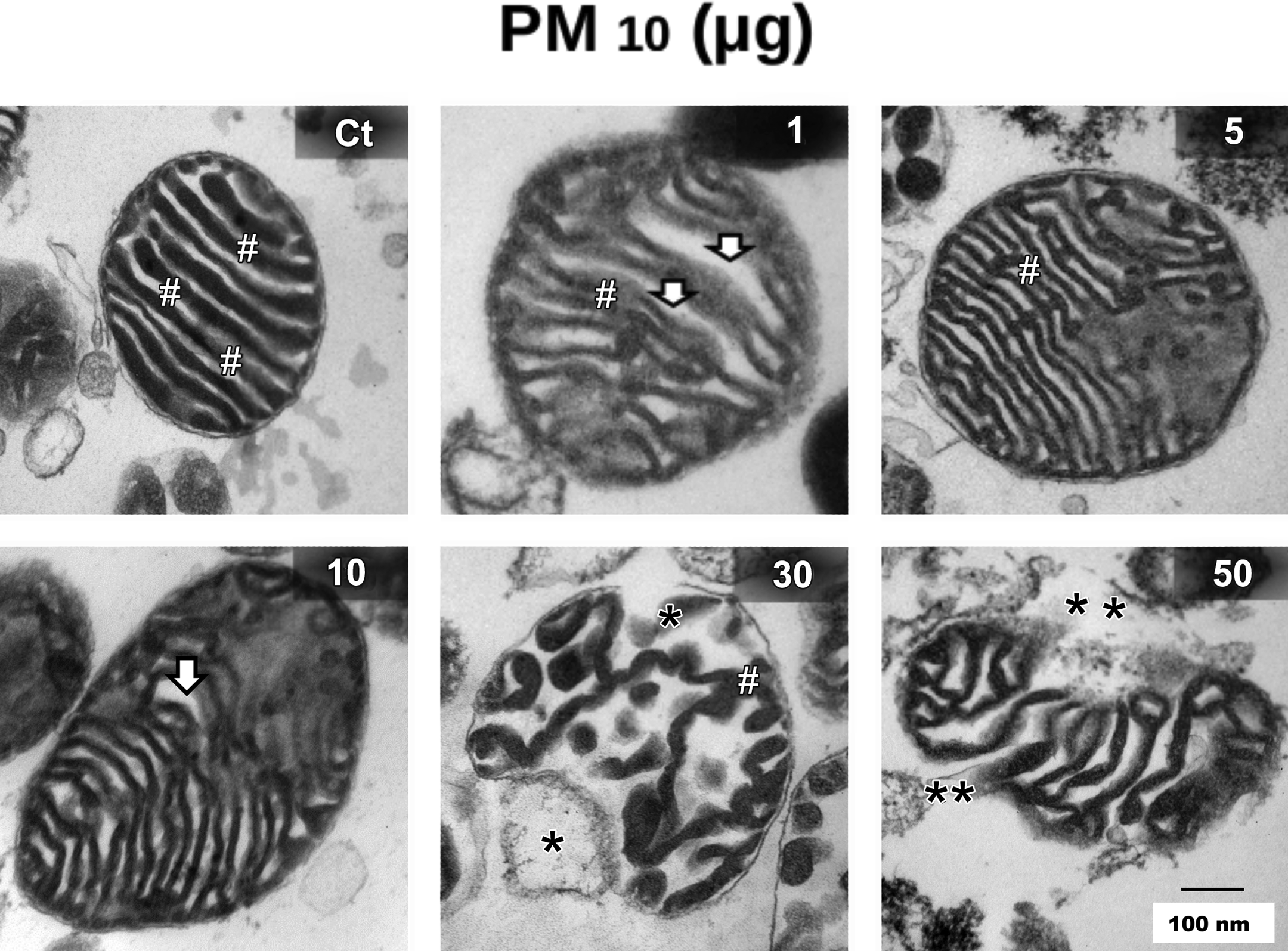
Mitochondrial transmission electron microscopy (TEM) from lung rat tissue. One mg of isolated mitochondria was exposed to the following PM10 amount: 0, 1, 5, 10, 30, and 50 µg for 1 hr. Mounted samples were cut in 70 µm sections and analyzed (Jeol JEM-1010). Normal outer and inner mitochondrial membrane in control (Ct) samples with tightly packed cristae with dense granules (#). White arrow shows enlarged intermembrane spaces and shape alterations caused by 10 µg of PM10 exposure. Asterisk shows a higher disruption of cristae caused by the exposure to 30 µg (*) and 50 µg (**) of PM10. Scale bar = 100 nm.
PM10 Induced a Decrease in Respiratory Function
PM10 induced a nonresponsive effect in the ADP-stimulated respiration in mitochondria exposed to an increased PM10 concentration and also a decrease in oxygen consumption. Oxygraphic traces under assay conditions showed an average consumption of 100 nmol O2 in control samples (Figure 2). The exposure to 1, 5, 10, 30, and 50 µg PM10 causes a consumption of 92.42, 72.83, 78.24, 45.3, and 31.4 nmol O2, respectively (Figure 2), which also is related to the decrease in RCI. Exposure of mitochondria to 1, 5, 10, 30, and 50 (µg/mg protein) of PM10 induced a decrease of 24.7%, 26.98%, 31.82%, 36.6%, and 51.35%, respectively, in state III, which measures ADP-stimulated respiration (*p < .05 vs. control; Figure 3A) without statistical changes in state IV (*p < .05 vs. control, Figure 3B), which represents oxygen consumption in the absence of ADP. As a result, the RCI, which is the ratio of state III to state IV respiration, decreased 15.97%, 11.99%, 32.21%, 54.29%, and 65.28% after 1, 5, 10, 30, and 50 µg of PM10 exposure (*p < .05 vs. control; Figure 3C). Lag phase, which is the time needed to consume the ADP added, increased to 1.99-fold and1.43-fold in samples exposed to 30 and 50 µg, respectively (*p < .05 vs. control; Figure 4A). Repolarization, which is the generated voltage during the ADP consumption, was increased 1.99- and 1.56-fold after exposure to 30 and 50 µg, respectively, in 1 mg of isolated mitochondria (*p < .05 vs. control; Figure 4B). However, we observed a nondependent concentration effect in the decrease of ATP synthesis (data not shown). A decrease in ΔΨm of 12.64%, 22.28%, and 31.31% was observed after 10, 30, and 50 µg of PM10, respectively (*p < .05 vs. control, Figure 5). We found that 10 µg of PM10 induced a decrease of 22% in RCI compared to control, which could be considered as a decrease between 5 and 10 µg/mg protein of PM10 exposure (Figure 3C). However, the ATP synthesis does not decrease after exposure to titanium dioxide nanoparticles (data not shown).
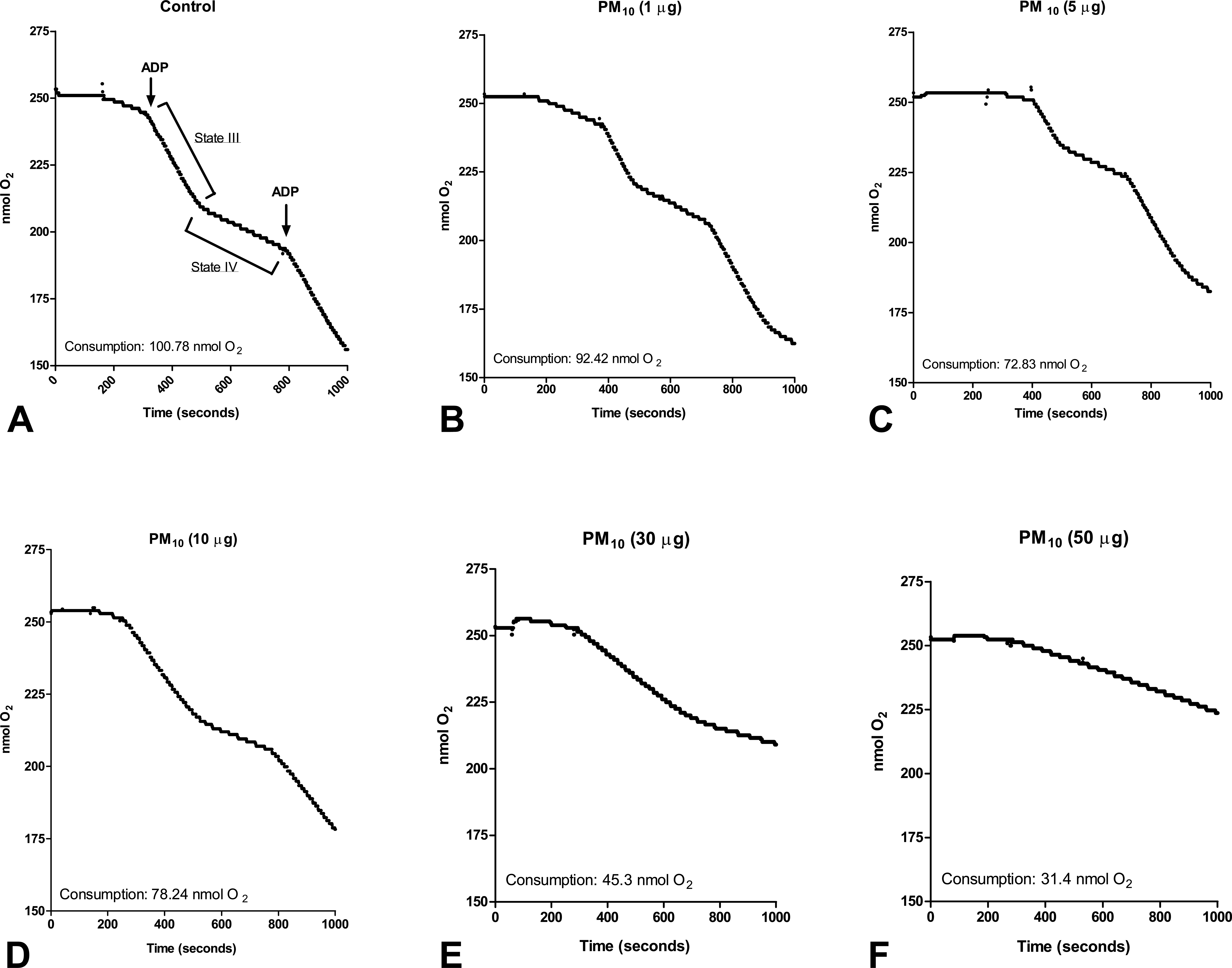
Representative graph of the particulate matter (PM10) effect on oxygen consumption in 1 mg of isolated mitochondria from lung rat tissue. Samples (1 mg of protein in each sample) were incubated with 0 (A), 1 µg (B), 10 µg (C), 15 µg (D), 30 µg (E), and 50 µg (F) for 1 hr. After incubations, the respiratory function was measured as indicated in methods. Malate/glutamate (10 mM/10 mM) was used as mitochondrial substrate and ADP 376 µM was added in the second 200 and 500.
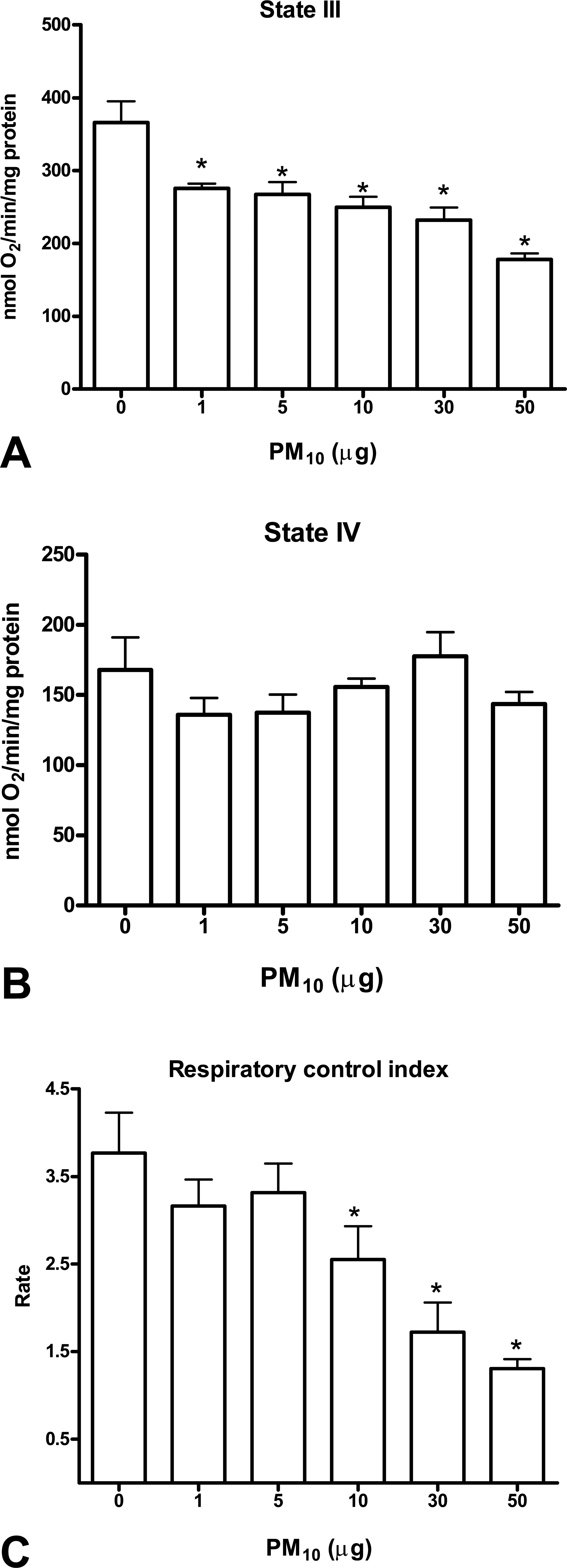
Effect of PM10 exposure on Respiratory Control Index (RCI). One milligram of mitochondria was incubated with 0, 1, 10, 15, 30, and 50 µg for 1 hr. RCI was calculated as the rate of stale III/state IV. Data are mean ± SEM of, at least, three independent experiments. *p < .05 versus control.
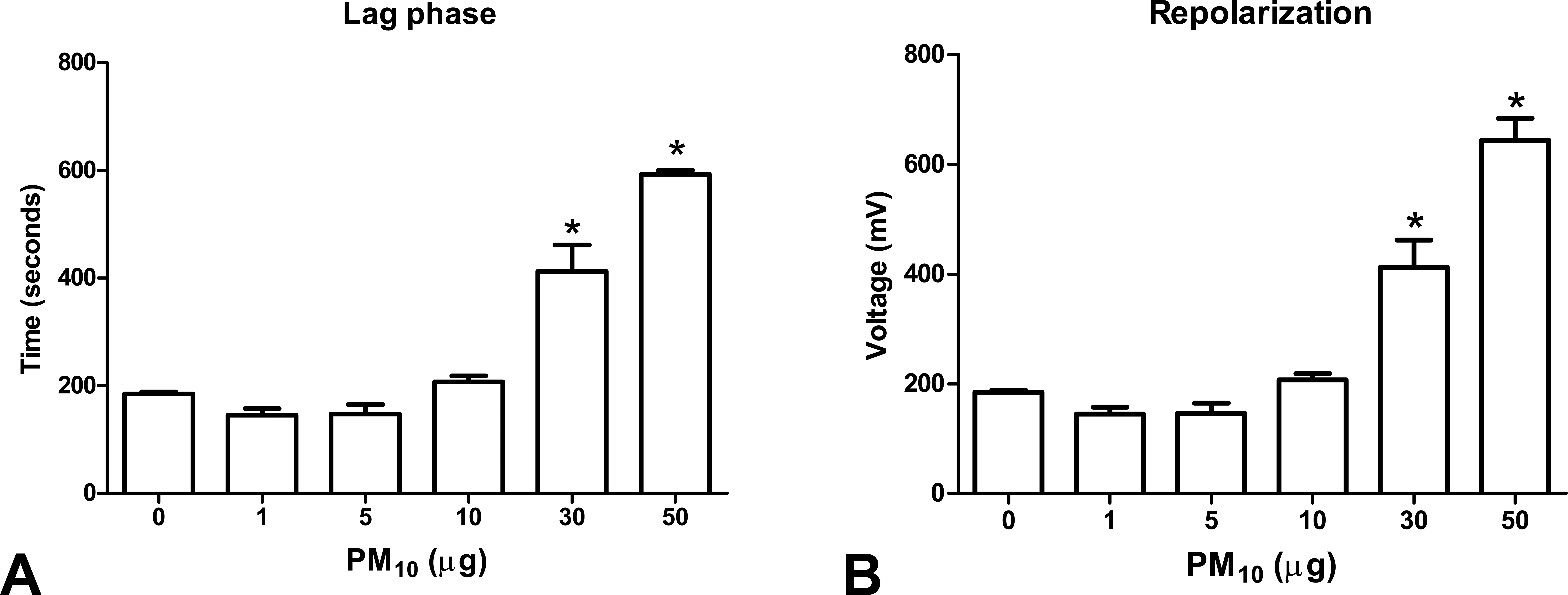
Effect of PM10 exposure on the lag phase and repolarization. One milligram of mitochondria was incubated with 0, 1, 10, 15, 30, and 50 µg during 1 hr. Data are mean ± SEM of, at least, three independent experiments. *p < .05 versus control.
PM10 Exposure Induced Increase in Succinate Deshydrogenase Activity and Decrease in Cytochrome c Oxidase Activity
On one hand, PM10 did not induce changes in activity of complexes I and III; however, the activity of complex II showed a 1.38-fold, 1.56-fold, and 1.66-fold increase in activity after 5, 10, and 30 µg of PM10 (p < .05 vs. control), respectively. On the other hand, the activity of complex IV showed a 40.38% and 35.92% decrease after 30 and 50 µg of PM10 (p < .05 vs. control), respectively (Figure 6). One milligram of mitochondria was also exposed to 10 µg titanium dioxide nanoparticles and the only change in respiratory chain was found in complex II, which showed 11% decrease (data not shown).
Continued
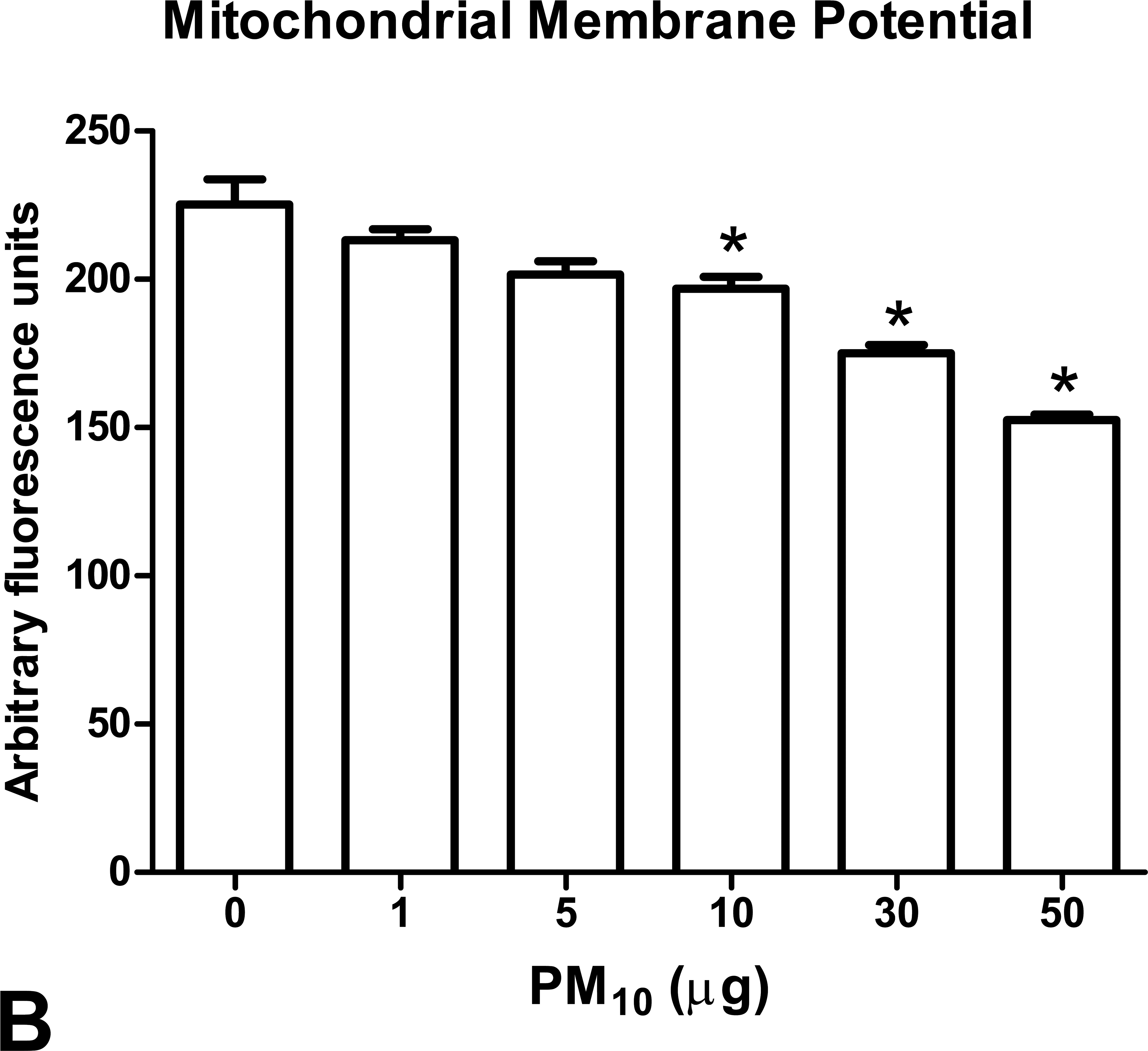
Effect of PM10 exposure on mitochondrial membrane potential. One milligram of mitochondria was incubated with 0, 1, 5, 10, 30, and 50 µg for 1 hr. After the PM10 exposure, samples were incubated with 2 µM Rodhamine 123 for 30 min at 37°C and (A) samples were mounted on slides and observed under confocal microscope. (B) Quantification of fluorescence intensity of mounted samples. Data are mean ± SEM of, at least, three independent experiments. *p < .05 versus control.
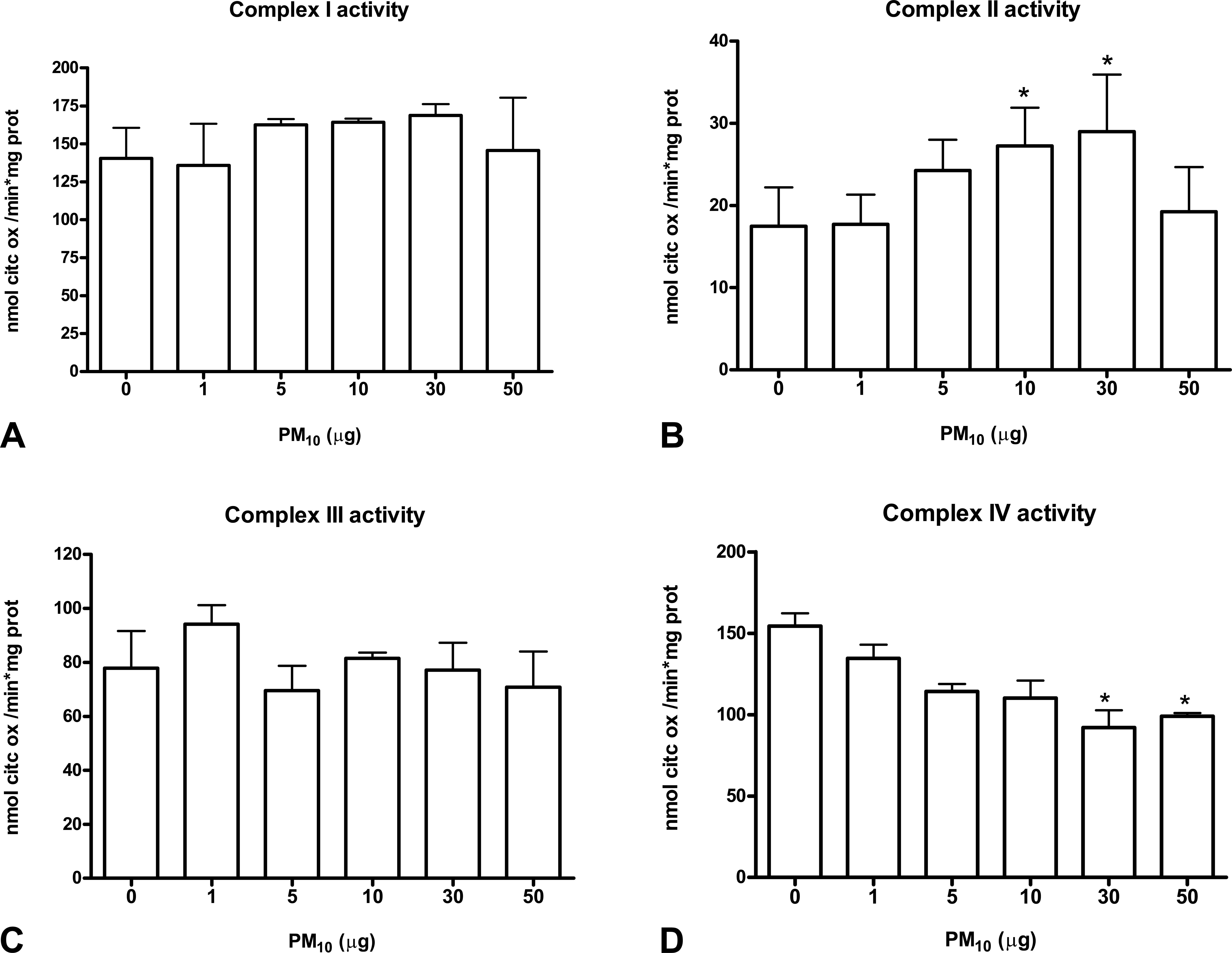
Effect of PM10 exposure on the activity of respiratory chain in mitochondria from lung tissue. One milligram of mitochondria was incubated with 0, 1, 5, 10, 30, and 50 µg for 1 h. After incubations, the activity of respiratory chain was measured as indicated in methods. (A) NADH dehydrogenase activity. (B) Succinate dehydrogenase activity. (C) cytochrome bc1 complex activity. (D) Cytochrome oxidase activity. Data are mean ± SEM of at least, 3 independent experiments. *p < .05 versus control.
Discussion
Epidemiological evidence sustains a positive association between PM10 exposure and the increase in cardiovascular diseases (Tonne et al. 2012; Pieters et al. 2012) and asthma (Delamater, Finley, and Banerjee 2012) on one hand and decrease in gestational age at birth on the other hand (van den Hooven et al. 2012). Also, PM10 exposure is associated with respiratory illness (Rodriguez-Villamizar, Castro-Ortiz, and Rey-Serrano 2012), decrease in lung function (Roy et al. 2012), higher blood pressure in children (Sughis et al. 2012), increase in hospital admission in aged people (Di Ciaula 2012), and increase in the risk of diabetes complications (Dales et al. 2012). The mechanism of cell damage induced by PM10 exposure has been investigated and the increase in ROS generation is one of the mechanisms involved in its cytotoxicity (Chirino et al. 2010). Mitochondria are one of the main sites of ROS generation, but also, mitochondria have been shown as a central target of PM-induced effects (Soberanes et al. 2009, 2012). Indeed, fine PM fraction can reach mitochondria, as has been observed in lung epithelial cell culture (Gualtieri et al. 2009). The mitochondrial alterations induced by PM are related to cellular effects such as apoptosis, for example, depletion of mitochondrial DNA protected cells from PM-induced apoptosis and these cells lack proteins involved in oxidative phosporylation (Mutlu et al. 2011), which could suggest that respiratory chain participates in the mechanism of cell damage under PM exposure. However, alterations in mitochondrial function have not been completely described.
This work describes alterations in mitochondrial structure and metabolism that include concentration-dependent changes in structural organization, decrease in oxygen consumption, RCI, and ΔΨm. Additionally, we performed some experiments using titanium dioxide nanoparticles (crystallite anatase size is in the sub-10 nm range) to investigate whether the mitochondrial exposure to particles induced the same response, but we found that titanium dioxide nanoparticles did not induce the same alterations in mitochondrial function even if the RCI decreases in the same magnitude.
First, we observed alterations in mitochondria after PM10 exposure, including unpacked and disrupted cristae with a decrease in parallel alignment and decrease in mitochondrial dense granules, which has been reported as calcium granules observed by TEM (Somlyo et al. 1974). Structural abnormalities are strongly associated with metabolic changes, as reported here, for example, disruption in membrane integrity affected RCI in isolated mitochondrial from endotoxin-induced liver damage (Crouser et al. 2002). Decrease in mitochondrial calcium granules are related to the dissipation of ΔΨm (Parihar, Parihar, and Ghafourifar 2008; Gáll et al. 2012). It has been reported that changes in cristae can be related to alterations in mitochondrial DNA and also to decrease in respiratory function and ATP synthesis (Gilkerson et al. 2000). In addition, it has been demonstrated that enlarged intermembrane spaces are related to release of cytochrome c and loss of ΔΨm (Sun et al. 2007), as we found in this work after PM10 exposure. It has been demonstrated that structural alterations in mitochondria are directly related to metabolic disturbances but also are associated into pathologies such as cancer, since there is evidence, for example, that reduction in mitochondrial area is observed in lung tissue from patients with lung cancer (Wu et al. 2009). Besides structural alterations, an increase in lag phase and repolarization was found after PM10 exposure. Lag phase represents the time needed to reestablish the ΔΨm after ADP phosporylation and repolarization and is the change in the voltage observed to complete ADP consumption. The increase in both parameters is related to decrease in oxygen consumption, which in turn, could be strongly related to the decrease in cytochrome c oxidase activity (complex IV), an enzyme that transfers electrons from cytochrome c turning oxygen into water. The decrease in cytochrome c oxidase activity induced by PM10 exposure could be related to mitochondrial lipid peroxidation, as it has been previously demonstrated in an oxidative stress model using mitochondrial membranes from lung tissue (Mokra et al. 2012). In this regard, we suggest that lipid peroxidation could be one of the first effects on mitochondrial membrane, since we previously reported that PM10 increases cellular levels of malondialdehyde, a final product and marker of lipid peroxidation (Chirino et al. 2010).
Some of the effects related to oxidative stress induced by PM10 exposure are related to the iron content (Chirino et al. 2010). Iron can catalyze the hydroxyl radical formation, and this radical is involved in the oxidation of lipids, proteins, and DNA. However, other metals contained in PM10, such as vanadium and nickel (Rosas-Pérez et al. 2007), could play a role as well, since it has been demonstrated that these metals can damage mitochondria (Di Pietro et al. 2011). However, mitochondrial damage induced by those metals can be actually transmitted to daughter cells in lung epithelial cells (Di Pietro et al. 2011). Endotoxin, which is another important component of PM10, could also contribute to mitochondrial damage. It has been previously demonstrated that endotoxin administration in animals caused an RCI decrease in isolated mitochondria from liver (Crouser et al. 2002), and PM10 endotoxin collected from Mexico City has high-endotoxin content (Osornio-Vargas et al. 2003; Alfaro-Moreno et al. 2007).
The above suggests that some other effects of PM10 exposure could be related to mitochondrial damage. For example, we previously reported that PM10 exposure induced senescence-like phenotype in lung epithelial cells (Sánchez-Pérez et al. 2009) and mitochondrial dysfunction is associated with senescence (Ljubicic, Menzies, and Hood 2010). We also thought that low but sustained exposure to PM10 could cause mitochondrial dysfunction, which may exacerbate the age-related mitochondrial dysfunction (Rooyackers et al. 1996; Dai, Rabinovitch, and Ungvari 2012).
In addition, we previously reported that PM10 induced senescence-like state in lung epithelial cells (Sánchez-Pérez et al. 2009), and this effect could be related partially to the mitochondrial dysfunction and it has been reported that decrease in respiratory function is associated with senescence (Ljubicic, Menzies, and Hood 2010). PM10 exposure increase in the last pregnancy month is associated with decrease in mtDNA content (Janssen et al. 2012), and as a result, a decrease in enzymes codified by this DNA may be absent, which may decrease the respiratory chain.
On the other hand, we recognize that an important number of studies showed that PM collected from different cities, including Ohio (Xu et al. 2011), Washington, D.C. (Mutlu et al. 2011), and Boston (Golomb et al. 2012) in United States, Paris, (Ferecatu et al. 2010), and Milan (Gualtieri et al. 2009), among others, shares some mitochondrial disturbances. However, we cannot discard the fact that some of the mitochondrial effects observed in this work could be attributed partially to the specific-PM10 composition collected from Mexico City.
This work concludes that PM10 exposure has the ability to induce structural alterations related to mitochondrial dysfunction, including a decrease in oxygen consumption, decrease in ΔΨm, and alterations in respiratory chain in isolated mitochondria from lung tissue. It could be possible that mitochondrial dysfunction induced by PM10 exposure can be associated with the increased risk for several illness including respiratory and cardiovascular diseases and diabetes.
Footnotes
Acknowledgments
The authors thank CONACYT 1298938, 102102 y PAPIIT IN201910, IN221611. V. Freyre-Fonseca has received fellowship from CONACyT (202805) and is a PhD student of Escuela Nacional de Ciencias Biológicas at Instituto Politécnico Nacional (Doctorado en Ciencias en Alimentos). The authors gratefully thank Dr. Darío Iker Tellez Medina from Escuela Nacional de Ciencias Biológicas for his kind help in TEM photograph process.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by CONACyT 118084, DGAPA IA202611-1, IB201112, PAPCA 2010–2011 (Project number 27), and PAPCA 2011-2012 (Project number 3).