
Abstract
The two-year rodent bioassay remains the mainstay for carcinogenicity testing, although numerous difficulties have been identified. Fundamentally, a chemical can increase the risk of cancer (1) by damaging DNA directly (DNA reactive) or (2) indirectly by increasing the number of DNA replications (non–DNA reactive). Mechanistic research has identified numerous precursor lesions in the sequence of key events necessary for neoplasia development. Based on these concepts, the author has proposed a short-term (thirteen-week) assay for screening for carcinogenic potential based on a mode of action analysis and on readily available, identifiable preneoplastic changes. A screening assay that detects all potential rodent hepatocarcinogens has been previously identified (Toxicol Pathol
Introduction
The current standard for evaluation of possible carcinogenic activity of a chemical in humans is the two-year bioassay in rodents. Two species are evaluated, usually rats and mice, but occasionally hamsters have been used. The details of this assay have been published extensively. Various aspects of the assay have been exhaustively evaluated and developed, primarily through the National Toxicology Program (NTP; see http://ntp.niehs.nih.gov), beginning with the standard assay developed in the 1960s by the United States National Cancer Institute. With some modifications, this is the standard for the evaluation of chemicals for possible carcinogenic activity for humans for all types of chemicals, whether pharmaceuticals, agrichemicals, consumer products, food additives, industrial chemicals, or chemicals found in the environment. Chemicals that occur naturally have also been evaluated by this protocol.
Numerous difficulties have been identified with this assay, including the enormous consumption of resources and time necessary to perform an adequate assay (Cohen 2004). The process begins with a shorter-term, usually thirteen-week, dose range finding study to determine the acceptable doses to be utilized in the two-year bioassay. Extensive clinical observations are performed during the course of the experiment, and extensive gross and histopathologic evaluations of all tissues are performed at autopsy. It usually takes approximately four years to complete the full range of studies and examinations necessary to come to a conclusion concerning the results of the two-year bioassay. The cost, depending on route of administration, number of doses to be examined, and chemical being evaluated can range from $2-4 million, or even more for certain routes of administration, such as inhalation. The highest dose given is a maximum tolerated dose (MTD) and usually includes some evidence of toxicity, such as decreased weight gain (Rhomberg et al. 2007). Extrapolation from the high doses used in these animal experiments to exposures in humans, particularly for nonpharmaceutical chemicals for which there frequently are several orders of magnitude difference in exposure between rodents in the bioassays and humans, has been the source of considerable controversy (Ames and Gold 1990; Rhomberg et al. 2007; Flamm and Winbush 1984; Slikker et al. 2004). Most importantly, during the past several years, several examples have been identified that raise the question of the relevance of the tumors induced in the rodent bioassay to assessing human risk (Cohen 2004; International Agency for Research on Cancer [IARC] 1999a; Meek et al. 2003; Cohen et al. 2004; Seed et al. 2005; Boobis et al. 2006, 2008).
Several hundred chemicals have been evaluated in the NTP bioassay (http://ntp.niehs.nih.gov), and several hundred if not thousands more have been evaluated by private industry in the evaluation of chemicals designed for commercial use. Based on the results of these studies and extensive mechanistic investigations on chemicals that produced tumors in these bioassays, considerable insight into interpretation of the results in these rodent bioassays has been gained. Based on these extensive evaluations, I have proposed that it is no longer necessary to perform the two-year bioassay to provide a rational basis for human cancer risk assessment for chemicals (Cohen 2004). Instead, a shorter-term evaluation, with an emphasis on mode of action and focus on interpretation of findings in rodents for their relevance to humans, is encouraged.
A critical part of this process is the assessment of mode of action (MOA) in rodents and its human relevance. Considerable guidance as to how this can be accomplished has been developed over the past decade in an evolving framework that began with the International Programme on Chemical Safety (IPCS) and has continued with activities of the International Life Sciences Institute (ILSI)/Risk Sciences Institute (RSI). The evolution of this framework has been published in a series of articles along with several case studies illustrating its application (Sonich-Mullin et al. 2001; Meek et al. 2003; Cohen et al. 2004; Seed et al. 2005; Boobis et al. 2006, 2008). It can be applied to chemicals producing cancer as well as other toxic endpoints. It was in the application of the framework to various cancer MOAs that it became apparent that there were always precursor lesions that could be identified that were critical for the development of the cancer (Meek et al. 2003; Boobis et al. 2006). For these interim key alterations, it is apparent that the changes are necessary, by definition, but are not sufficient, since not all animals that have these early changes ultimately develop tumors. This is most likely related to the stochastic processes involved in carcinogenesis as extensively described from our research (Greenfield, Ellwein, and Cohen 1984; Cohen and Ellwein 1990a, 1991) and from investigations by Moolgavkar and his associates (Moolgavkar and Knudson 1981; Luebeck and Moolgavkar 1996; Meza et al. 2008). Since these interim key events that lead to the development of cancer can be identified in rodents within thirteen weeks of administration of the carcinogen, it became apparent that a screening assay for carcinogenesis could be developed that involved thirteen weeks or less of administration of the compound. When necessary, the screening assay could be followed by more detailed investigations of the MOA in the animal to evaluate whether any findings were relevant to humans, both with respect to MOA and with respect to a detailed dose response (Cohen 2004).
In rodent bioassays the liver is the most common target organ (http://ntp.niehs.nih.gov; http://www.epa.gov; http://potency.berkeley.edu; Gold et al. 1991, 2005). In this article, I provide additional details for this shorter-term, mechanistic approach to cancer risk assessment based on a detailed examination of liver carcinogenesis. Previous discussions regarding MOAs for liver carcinogenesis were summarized in Holsapple et al. (2006).
It is important to keep in mind during any of these evaluations that the ultimate goal is an assessment of possible carcinogenic risk in humans, not in rodents. The rodent is serving merely as a model. As was stated by the mathematician George Box (1979) several decades ago, “Models: All are Wrong. Some are Useful.” It is incumbent on scientists to identify those aspects of the animal models we use that are relevant to humans (the “useful” aspects of the model).
Any time experiments are performed in animals with the intent of ultimately extrapolating to possible effects in humans, two fundamental assumptions are made: (1) the carcinogenic (or any other) effect at doses used in the bioassay will also occur at doses to which humans are exposed (dose extrapolation) and (2) chemicals that cause cancer (or other effects) in rodents will cause cancer in humans (species extrapolation). For most chemicals identified as carcinogens in rodents in the 1950s and 1960s, these assumptions appeared to be valid (Cohen and Ellwein 1991; Cohen 1998a, 1998b, 2004, 2008). The chemicals were carcinogenic in humans and in the animal models and appeared to be effective even at low dose exposures. Mechanistically this was thought to be reasonable since it was assumed that chemicals behaved much like radiation in producing cancer. Like radiation, these chemicals were shown to damage DNA, but in contrast to radiation, the chemicals usually required metabolic activation to the reactive electrophiles or free radicals before interacting with the DNA. Radiation appeared to produce free radicals that were the source of the damage to the DNA. This effect on DNA was the basis for the Ames' assay utilizing Salmonella mutagenicity assays along with a variety of other in vitro and in vivo assays that have been developed for evaluation of genotoxicity (Haseman, Hailey, and Morris 1998; Kirkland, Aardema, et al. 2005; Kirkland et al. 2006; Zeiger 2004). However, it became evident as more and more chemicals were tested that several of these were not positive in the genotoxicity assays and yet produced cancer in two-year bioassays in rodents (Weisburger and Williams 1981).
There are essentially two classes of chemical carcinogens, one that directly produces DNA damage and the other that does not (Weisburger and Williams 1981). These have been referred to as genotoxic versus nongenotoxic, although a more precise and restrictive classification is to classify them as DNA reactive and non–DNA reactive chemicals, respectively (Cohen and Ellwein 1991; Cohen 1998a, 2004; Weisburger and Williams 1981). Based on extensive analyses in animal models and in human epidemiologic investigations, it became apparent that, in addition to DNA reactivity, the other factor that needed to be taken into consideration was the number of chemically induced DNA replications (cell proliferation) (Greenfield, Ellwein, and Cohen 1984; Cohen and Ellwein 1991; Moolgavkar and Knudson 1981).
Evidence continues to accumulate demonstrating that cancer is due to a variety of genetic alterations, frequently occurring in somatic cells rather than being inherited in germ cells (Greenfield, Ellwein, and Cohen 1984; Moolgavkar and Knudson 1981; Knudson 1971). It is also evident that more than one genetic defect is required in a single cell for cancer to arise. It was also demonstrated soon after the structure of DNA was identified that DNA replication is extraordinarily precise, but not perfect. It has been estimated that the error rate is approximately 1 per 1010 nucleotides per DNA replication. Thus, for human cells with approximately 3 × 109 nucleotides in the genome, there is < 1 mistake per DNA replication. It is also critical that the mistake occurs in one of the genes in the DNA that is necessary for the development of cancer, rather than some other part of the DNA; and it is also critical that the damage that occurs in the DNA results in a defect in the function of the gene product. Numerous chemical modifications of the DNA appear to occur regularly, and yet mutations are relatively uncommon. This appears to be due to the extraordinary capacity for DNA repair that has evolved (Hanawalt 1995; Lindahl and Wood 1999; Dixon and Kopras 2004; Loeb and Harris 2008).
Screening for Short-Term Effects
Based on these assumptions and observations, it is clear that there are fundamentally only two ways that a chemical can alter the incidence of cancer: (1) directly damaging DNA (DNA reactive) or (2) increasing the number of DNA replications resulting in an increase in the spontaneous errors in DNA (Cohen and Ellwein 1991). It has become evident that most DNA-reactive carcinogens also produce an increase in DNA replications at high doses.
An increase in DNA replications can occur either by increasing cell births or decreasing cell deaths (Cohen 1998a, 1998b, 2004). Cell births can be increased either by direct mitogenesis (involving hormones and/or growth factors) or by cytotoxicity (cell death) with consequent regenerative proliferation. Cell deaths can be decreased (which leads to an accumulation of cells) either by inhibiting apoptosis or inhibiting cell differentiation (a cell death process). Increasing the number of cells increases the number of cells that can replicate. This amounts to an increase in the number of DNA replications even if the rate remains normal. The number of replications is the critical parameter, not the rate. All of the critical genetic events must occur in a single pluripotential (tissue stem) cell.
Based on these considerations, a shorter-term assessment of effects on various tissues can be made that can predict potential carcinogenic activity in humans (Cohen 2004). More importantly, the shorter-term assays can provide a more detailed dose response and mechanistic basis for the effect in the rodent, providing the basis for a more rational extrapolation to possible human effects rather than making the default assumptions of interspecies and dose extrapolations described above. This article focuses on the liver as an example of how such a series of short-term investigations could be utilized for cancer risk assessment rather than relying on the two-year bioassay. A similar process can be performed for all tissues. However, as I have described previously (Cohen 2004), many findings in the rodent involving some tissues (such as endocrine) are not predictive of carcinogenic activity in humans and thus should not be considered further in the assessment of human cancer risk.
To have such a short-term assessment, a screening process is necessary that will detect potential human carcinogens. Since we do not have a definitive indicator of the carcinogenic activity of all chemicals for the human, we rely on the results of two-year bioassays in rodents as a surrogate for such an evaluation. Fortunately, an extensive evaluation for possible markers in ninety-day screening assays used for development of dose selection for two-year bioassays provides for such a screening process. Allen et al. (2004) evaluated the results for 111 chemicals tested by the NTP over a ten-year period (1991–2001). They observed that if they utilized four indicators for screening purposes, all chemicals that produced liver tumors in mice or rats in two-year bioassays could be detected in this thirteen-week screen. The indicators that were utilized were hepatocellular necrosis, hepatocellular hypertrophy, hepatocellular cytomegaly, and increased liver weight. Increased liver weight seemed to be the most sensitive, but chemicals that produced liver tumors frequently induced multiple morphologic changes. There were no false negatives in this evaluation, but there were numerous false positives. That is, there were several chemicals that produced one or more of these four effects on liver that did not produce liver tumors in the full two-year bioassay. Since it is the intent of screening assays to be conservative, it is most important that there be no false negatives with respect to human carcinogenesis. The difficulty with false positives is that on a commercial basis, many useful compounds might be discarded unnecessarily based on these short-term screens. It is for that reason that the short-term screening requires a careful mechanistic evaluation to determine the basis of the positive result, whether an increase in liver weight or one of the other markers of liver alteration is used in the screen. For such analysis, we need to know the mode of action for which a chemical might produce one or more of these effects, and then we must evaluate whether it is a relevant finding for humans.
Several modes of action have been identified for liver carcinogenesis, both in humans and in rodent models. Those that are applicable to the rodent model are listed in Table 1 . Those that have evidence of human relevance are highlighted in bold letters. Liver carcinogens can be divided into those that are DNA reactive versus those that are non–DNA reactive and produce their carcinogenic effect by increasing cell proliferation. All of the known DNA-reactive liver carcinogens produce other alterations in the liver in a thirteen-week or shorter period when administered at high doses. This is most often evident as increased liver weight associated with cytotoxicity and regeneration (Allen et al. 2004; Peto et al. 1991), frequently accompanied by increased liver enzyme activities in the serum. As discussed above, Allen et al. (2004) showed that evaluating the liver for four features (liver weight, cytomegaly, hypertrophy, necrosis) after ninety days of treatment will detect all of the chemicals that will produce an increased incidence of liver cancer in the two-year bioassay. Although reliable in the liver, in some other tissues, morphologic or weight changes are not always sensitive enough to detect evidence of increased cell proliferation, and in those instances a labeling index for DNA replication is useful. The standard for such a determination at the present time is bromodeoxyuridine (BrdU), either by intraperitoneal (IP) injection or with the use of an osmotic mini-pump (Gratzner 1982; Eldridge et al. 1990). Evidence has been presented that for the liver an osmotic mini-pump produces the most reliable results (Eldridge et al. 1990). Other markers have also been developed to assess DNA replication that utilize endogenous substrates and do not require administration of an exogenous substance. These include Ki-67 (Scholzen and Gerdes 2000) and PCNA (Dietrich 1993). For each of these, careful attention needs to be paid to methodologic details to ensure reproducible and reliable results. Although based on the results of Allen et al. (2004) such a proliferation labeling index assay does not appear to be necessary for the liver, it would be worthwhile to consider its use for chemicals that are negative for the other parameters as a further assessment for screening.
Modes of action for hepatocellular carcinogenesis.
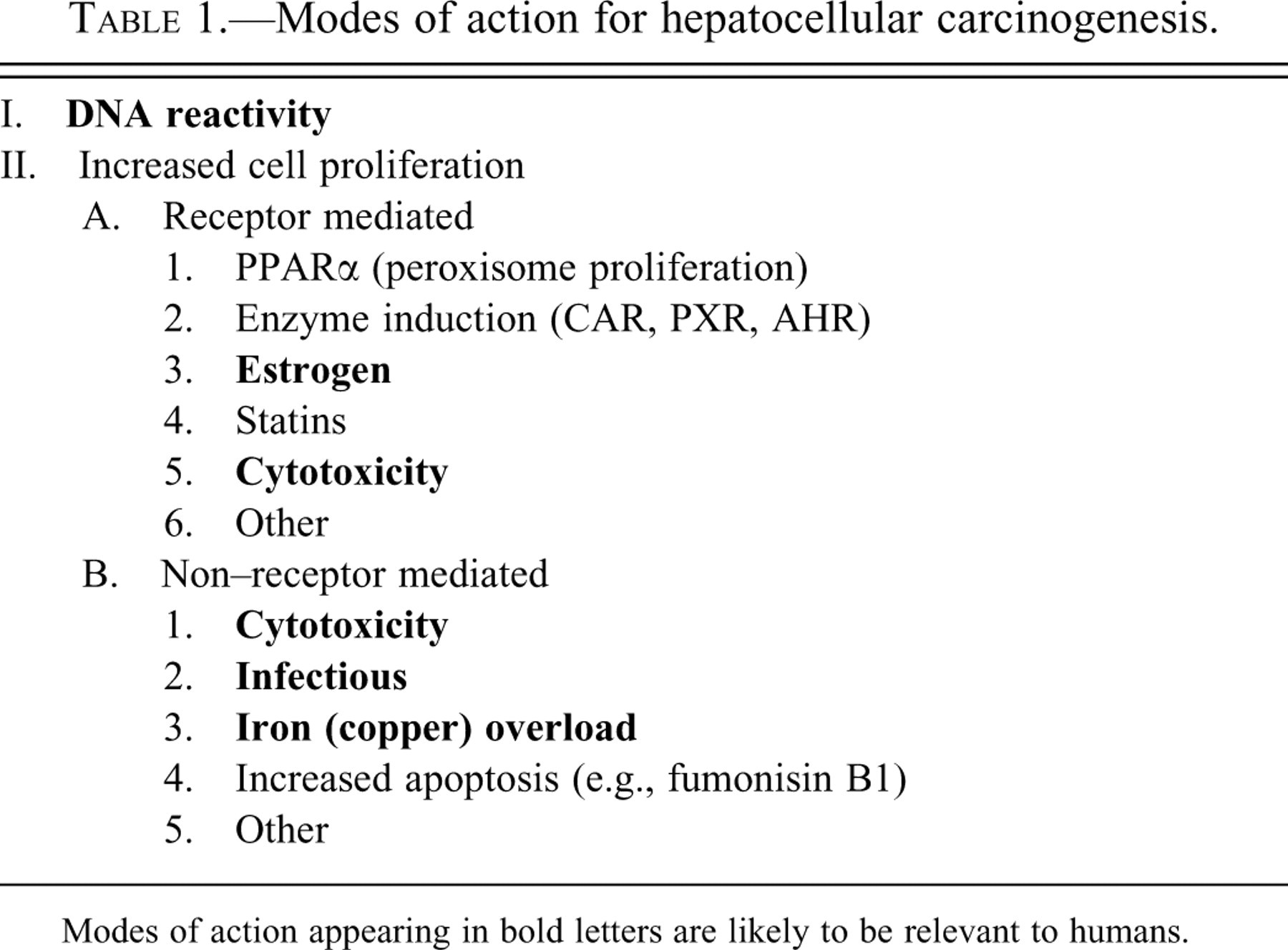
Modes of action appearing in bold letters are likely to be relevant to humans.
Chemicals that increase cell proliferation in the liver do so either through identifiable receptor-mediated mechanisms or by apparent non-receptor-mediated mechanisms. Importantly, once a positive result is identified in the initial screening, a small number of short-term follow-up studies can be performed that can quickly identify the possible MOA(s) through which the chemical might be acting. The decision model and the specific assays that can be evaluated are illustrated in Figure 1 . Many of the follow-up mechanistic studies require either short term evaluations (one to four weeks) or rely on other modalities, such as receptor binding assays or evaluation of effects on other tissues. The details of the various mechanisms and their evaluation for human relevance are presented below.

Proposed sequence of evaluation to screen for potential hepatocarcinogenesis. The initial screen involves the four criteria proposed by Allen et al. (2004). If one or more of these four signals are detected in the 13 wk. screening assay, follow-up mechanistic evaluations are performed to determine the MOA(s) and provide the basis for an assessment of human cancer risk.
DNA-Reactive Carcinogens
The primary distinction for chemicals with respect to MOA is between DNA-reactive carcinogens and those that act by increasing cell proliferation (Cohen and Ellwein 1990a, 1991, 1993; Weisburger and Williams 1981). Many of the hepatic carcinogens identified in animal models before the advent of the two-year bioassay were DNA-reactive carcinogens, such as aflatoxin, nitrosamines, alkoxybenzenes, aromatic amines, and azo dyes (Weisburger and Williams 1981). For pharmaceuticals, agrichemicals, and most consumer products, positivity in a screening assay for DNA reactivity will usually preclude further development (Snyder and Green 2001). Assessment of DNA reactivity can be made by in vitro and in vivo assays (Kirkland and Muller 2000; Thybaud et al. 2007; Kirkland, Aardema, et al. 2005; Kirkland, Henderson, et al. 2005; Kirkland et al. 2006; Kirkland, Pfuhler, et al. 2007; Kirkland, Aardema, et al. 2007). The easiest initial screening is for evaluation in the Ames' assay in vitro (Tennant et al. 1987). This assay is useful in detecting DNA reactive, mutagenic substances. However, this assay is not uniformly positive for hepatocarcinogens that ultimately are shown to be DNA reactive, such as tamoxifen (Tryndyak et al. 2006; Randerath et al. 1994). In vivo assays that can be utilized for an assessment of DNA reactivity include 32P-post-labeling and assays that directly evaluate DNA binding of the chemical or its metabolites (Phillips et al. 2000). Structure activity relationships (SAR) evaluated by computer programs have become increasingly effective in identifying the potential for DNA reactivity (Zeiger et al. 1996; McKinney et al. 2000).
Once DNA reactivity has been demonstrated for a given chemical, the dose response can be readily evaluated to determine if there are potential thresholds for metabolic activation or whether saturation of any of the enzymes involved occurs. More importantly, a comparison between metabolic activation and inactivation can be evaluated between species. This is particularly helpful if only the mouse or only the rat develops the liver changes. Utilization of a variety of “humanized” cell lines (Knasmuller et al. 2004) and animal models (Powley et al. 2009) that are now available for assessment of metabolic activation can be particularly useful in addressing species specificity. Toxicogenomics also have the potential of assessing DNA reactivity, although this has not been uniformly successful (Ellinger-Ziegelbauer et al. 2004, 2005, 2008; Nie et al. 2006; Thomas et al. 2007; Fielden, Brennan, and Gollub 2007; Fielden et al. 2008).
As indicated above, the DNA-reactive liver carcinogens also produce other effects in the liver, most commonly cytotoxicity, but are always associated with increased cell proliferation (Allen et al. 2004; Peto et al. 1991; Cohen and Ellwein 1991). This cell proliferative effect at higher exposure levels will significantly affect the dose response for tumorigenicity, as has been illustrated for aromatic amines (Cohen and Ellwein 1990b, 1993) and for nitrosamines (Peto et al. 1991). At low concentrations, when only DNA reactivity is occurring and increased cell proliferation is not evident based on computer models, there is estimated to be a low incidence of neoplasia in the animal models that usually is not detectable in experimental models with fifty to sixty animals per group (Cohen and Ellwein 1990b; Peto et al. 1991). Once the dose necessary for increased cell proliferation is achieved, the slope of the tumorigenic response significantly increases due to the synergistic interaction of DNA reactivity accompanied by an increase in the target cell population (cells that are undergoing replication) (Figure 2 ) (Cohen and Ellwein 1990b; Peto et al. 1991). Extrapolating from the animal models to humans based on the high dose response will give a misleadingly high estimate of risk to humans. Evaluation of a detailed dose response for the different factors involved with a DNA-reactive carcinogen can provide a more accurate assessment for extrapolation to humans, taking into account not only the dose response for DNA reactivity but also the dose response for the effect by which the chemical is increasing cell proliferation at the higher doses, whether through cytotoxicity or a mitogenic effect. The key events for DNA reactive carcinogens are listed in Table 2 .
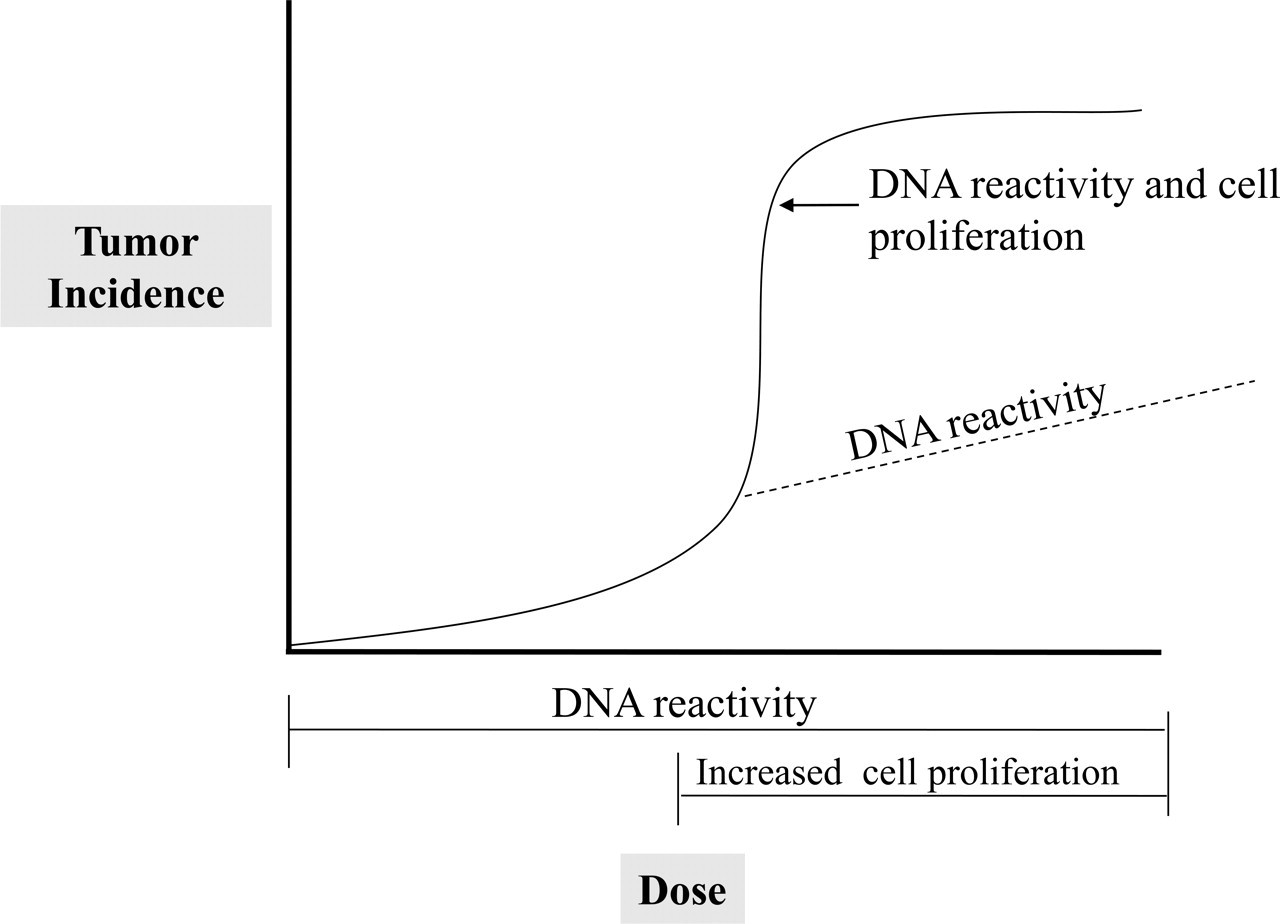
The synergistic effect of increased cell proliferation on the dose response for DNA reactive carcinogens. The solid line illustrates the usual dose response for DNA reactive carcinogens, with a sharp increase in the rate at doses where increased cell proliferation occurs in addition to DNA adduct formation. Based on computer models (Cohen and Ellwein, 1990a; 1990b), the dose response for the chemical can be estimated for the higher doses assuming the increased cell proliferation did not occur (the dotted line).
Mode of action for DNA-reactive liver carcinogens.

Receptor-Mediated, Non–DNA Reactive Carcinogens
Estrogen
Estrogen is a known carcinogen for humans, associated with tumors of the breast and endometrium, and possibly the ovary (Yu et al. 2003). In addition, there is evidence that increased estrogen exposure can induce a small increased incidence of hepatocellular carcinomas, particularly in young women who are exposed to high-estrogen-containing contraceptive pills (IARC 1999b). Although estrogens are more commonly associated with the induction of benign hepatocellular lesions, such as nodular hyperplasia and adenoma, there is evidence supporting an association with the induction of carcinomas. The mechanism by which this occurs remains controversial. There is no question that there is increased cell proliferation in response to increased estrogen levels, at least in the breast and endometrium, and it appears to also occur in the liver, presumably due to a direct mitogenic effect of estrogen resulting from binding to the estrogen receptor (Preston-Martin et al. 1990; IARC 1999b). However, there is also evidence that estrogen is metabolically activated to react with DNA, thereby forming purine adducts and leading predominately to apurinic sites (Gaikwad et al. 2008). Thus, although there is a specific receptor involved (estrogen receptor), the carcinogenic activity may be due to an interaction of DNA adduct formation with increased cell proliferation, similar to what is seen with classic liver carcinogens producing increased cell proliferation at higher exposures (see above). For this circumstance, there would actually be two concurrent modes of action that are operative, leading to a synergistic interaction. It is noteworthy that the hepatocellular carcinomas that occur in patients in response to estrogen exposure tend not to be related to situations involving chronic active inflammation or cirrhosis (IARC 1999b; Goodman and Terracciano 2007).
To detect estrogenic activity of a chemical in a ninety-day assay, it is more readily ascertained by a histologic assessment of the more typical estrogen-affected tissues such as the endometrium, cervix, or vagina (Moggs et al. 2004). For corroboration, binding of the chemical (or metabolite) to estrogen receptors in standard receptor assays can be accomplished (Gruber et al. 2002).
The key events of this MOA are listed in Table 3 . These include potentially two MOAs, one involving metabolic activation of estrogen with the formation of DNA adducts and, concurrently, an increase in cell proliferation secondary to binding to the estrogen receptor. Verification of increased proliferation activity can be accomplished by an assessment of DNA replication by BrdU, Ki-67, or PCNA labeling index. For estrogen-related chemicals, it is unclear whether DNA adduct formation is required for inducing hepatocellular carcinomas. The required pieces of data to support estrogenic activity as the MOA for a chemical that leads to positive changes in the ninety-day screening assay include histopathologic evidence of estrogenic activity in the endometrium, cervix, and/or vagina and evidence of increased cell proliferation of hepatocytes. An added feature would be reversibility of these findings upon discontinuance of administration of the compound, usually occurring within four weeks or less of discontinuation. Assessment of DNA reactivity will have occurred by standard procedures as described above.
Mode of action for estrogen and estrogenic chemicals inducing hepatocellular tumors.

It remains unclear whether DNA reactivity is necessary for estrogenic chemical-induced liver cancer.
CAR, PXR, and AHR
Induction of cytochrome P450 (CYP) enzyme activity is well known as a rodent hepatocarcinogenic process, with phenobarbital as the standard by which others have been compared (IARC 2001; Whysner, Ross, and Williams 1996; Lamminpaa et al. 2002). The sequence of key events are listed in Table 4 and include the recently identified observation of binding to the CAR receptor, producing CYP isozyme induction, leading to an increase in hepatocellular proliferation and ultimately to the induction of proliferative lesions in the liver, including foci, adenomas, and finally carcinomas. It remains unclear whether the CYP induction step is critical in the process or whether it is an indicator of activity of these chemicals that is secondary to binding to CAR (Wei et al. 2000; Ueda et al. 2002; Yamamoto et al. 2004). Metabolism of various endogenous substrates leading to products increasing cell proliferation might be modified by induction of various CYP isozymes. Clearly, activation of CAR in rodents leads to downstream activation of genes, which lead to hepatocellular proliferation, which is critical for development of these tumors (Yamamoto et al. 2004; Whysner, Ross, and Williams 1996). The exact mechanism by which CAR activation leads to increased cell proliferation remains unclear. In humans, binding to the CAR receptor and induction of CYP isozymes following phenobarbital administration occurs, although different enzymes appear to be induced in humans compared to rodents (Lambert et al. 2009). However, unlike in rodents, there is no evidence that this results in an increase in cell proliferation in the human liver. Furthermore, extensive epidemiologic investigations at exposure levels in humans that are comparable to those used in rodent bioassays are negative with respect to increased cancer risk (Whysner, Ross, and Williams 1996). Thus, although we do not know the specific molecular mechanisms by which CAR activation leads to an increase in cell proliferation in rodents, it is a critical key event in the carcinogenic process in rodents, and it does not occur in humans. Based on this MOA assessment, one can confidently conclude that phenobarbital will not be a hepatocarcinogen in humans, and this is corroborated by the epidemiologic evidence (IARC 2001).
Key events in the induction of liver tumors by CYP enzyme-inducing chemical acting through CAR activation.

The specific data requirements for assessment of this MOA are listed in Table 5 . These include demonstration that there is induction of CYP enzyme activity and, given the current state of our knowledge, a demonstration that there is binding to the CAR receptor. Furthermore, there needs to be demonstration that there is an increase in cell proliferation in the hepatocytes. For most of these compounds, including phenobarbital, the increase in labeling index actually appears very quickly, usually within one to two weeks, and the labeling index returns to normal by four weeks of administration (Yamada et al. 2009). Thus, a standard ninety-day study for assessment of labeling index will not be informative. The follow-up study to the ninety-day initial screening, which clearly shows evidence of enlargement of the liver, would be a much shorter study, indicating that there is an elevation of the labeling index. Although the labeling index returns to control levels within four weeks of administration, there is a significant increase in the number of hepatocytes associated with the enlargement of the liver, which continues for the length of time that the phenobarbital is administered. Since it is the number of DNA replications that is critical for carcinogenesis, not necessarily the rate of proliferation, there clearly is increased cell proliferation associated with phenobarbital (Cohen and Ellwein 1990a, 1991). Additional data that are helpful in identifying this MOA for a given chemical are the characteristic morphologic features of enzyme induction in rats and mice, including centrilobular hypertrophy by light microscopy and increased smooth endoplasmic reticulum by transmission electron microscopy (Yamada et al. 2009). Although helpful, the histopathologic changes that are readily evident in a ninety-day screening assay are not essential for demonstration of this MOA. More critical is demonstration of an increase in CYP enzyme activity and an increase in cell proliferation. These changes are readily reversible upon discontinuance of administration of the chemical. A similar sequence of events has been demonstrated for the pyrethrin metofluthrin (Yamada et al. 2009).
Data necessary to support CYP enzyme induction as a mode of action.

Similar to CAR activation, there are numerous chemicals that increase various CYP isozymes by binding to other receptors such as pregnane X receptor (PXR) (Moore et al. 2003) and aryl hydrocarbon receptor (AHR) (Perdew 2008; Schwarz and Appel 2005). These chemicals are also associated with increased cell proliferation in rodent liver, although the activation of specific downstream genes is not necessarily the same as those seen with CAR activation. For chemicals acting through the PXR and AHR receptors, evidence that there is induction of CYP isozymes and an increase in labeling index are critical pieces of data, as well as demonstration that the chemical (and/or a metabolite) binds to the respective receptor.
Similar to agents acting through CAR, most agents that act through activation of PXR or AHR receptor show reversibility of the early findings upon cessation of administration of the chemical. However, some of the chemicals acting through AHR activation are highly lipophilic (e.g., dioxin) and accumulate in the cells over time (Walker et al. 1998). Even upon cessation of administration of the chemical, exposure may continue for a long period, and biological effects will still be seen. Thus, reversibility may not be demonstrable for these agents.
PPARα Agonists
Another receptor-mediated MOA has been described for a wide variety of chemicals, including pharmaceuticals, plasticizers, and other agents, that act as PPARα agonists. These chemicals bind to PPARα and usually induce an increased incidence of hepatocellular tumors in rats and mice. Most also induce pancreatic acinar cell tumors and testicular Leydig cell tumors in rats. Details of the MOAs for this group of chemicals has been described by Klaunig et al. (2003).
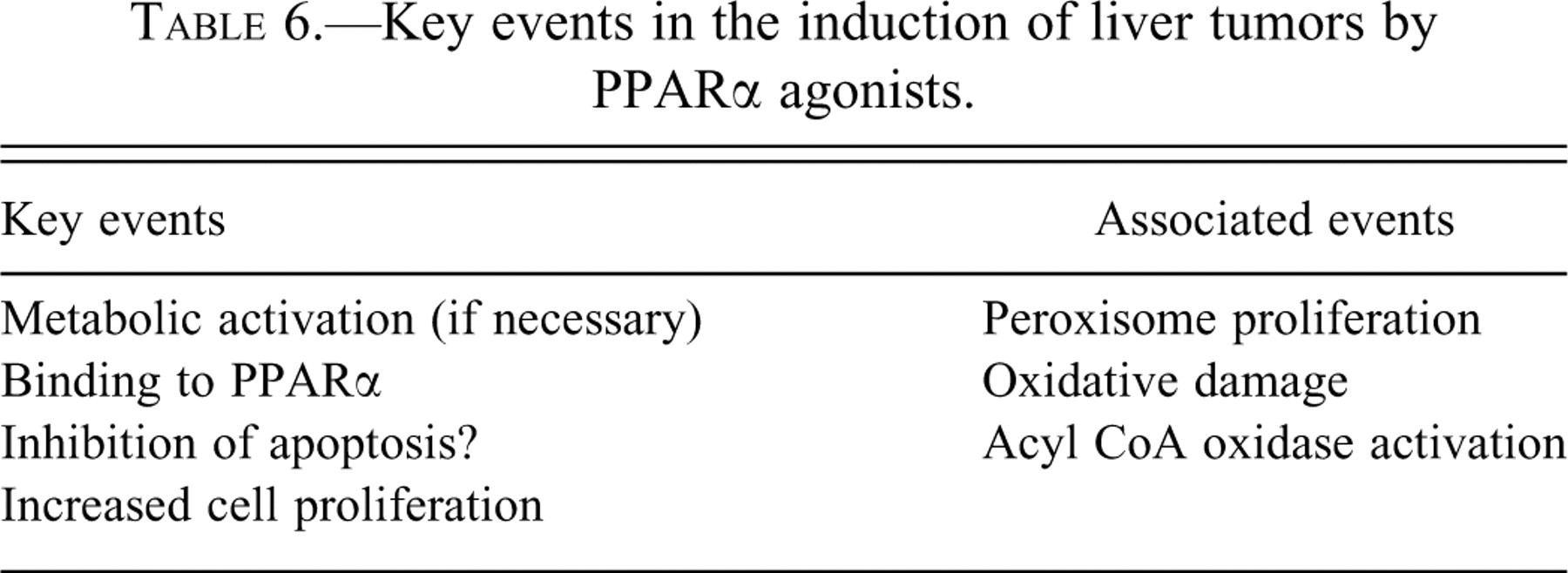
The key events involve binding to the receptor leading to peroxisome proliferation (the basis for the development of the gene name PPAR), which is associated with the production of several enzymes including acyl CoA-oxidase and other peroxisomal enzymes. Oxidative damage also occurs in association with the activation of peroxisomes. Ultimately, there is an increase in hepatocellular proliferation leading to the formation of lesions in the liver, including altered foci, adenomas, and carcinomas. There are effects on Kupffer cells, but it is unclear whether these are essential to the development of the tumor or an associated event. Also, there is considerable evidence that these chemicals inhibit apoptosis, which would lead to an accumulation of cells in combination with the increased DNA replication (increased labeling index). These key events are listed in Table 6 .
Key events in the induction of liver tumors by PPARα agonists.
There is also evidence that some of the PPAR agonists increase CAR activity, with some induction of CYP isozymes. This is most readily ascertained by a trancriptomics approach (Ross et al. 2009). It is unclear whether CAR activation in this setting is necessary for the carcinogenicity of these chemicals particularly since the level of activation is relatively low. There does not appear to be a correlation between the level of activation of CAR by these chemicals and their ultimate carcinogenic activity, and the spectrum of tumors is different than those seen with CYP enzyme inducers such as phenobarbital. Furthermore, some of the PPARα agonists shown cytotoxicity, which might contribute to the carcinogenic effect.
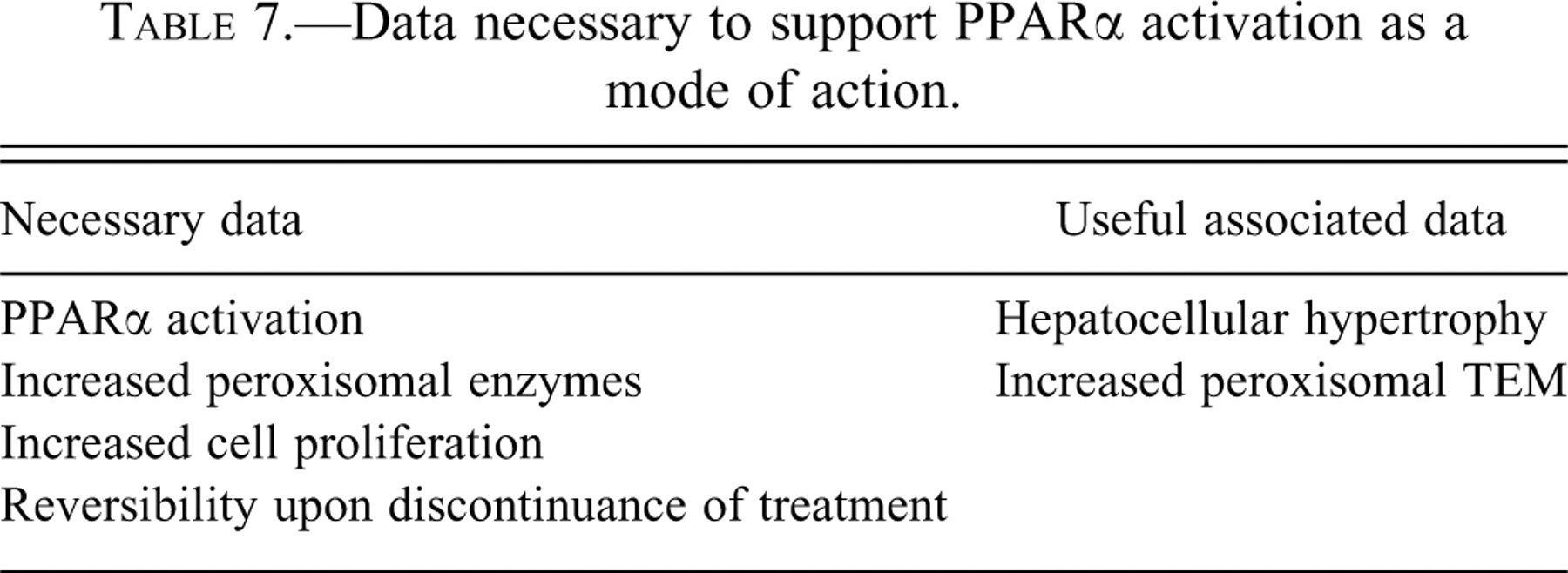
The essential pieces of data for determining if this MOA is involved for a given chemical are listed in Table 7 . PPARα activation will readily produce histologic hepatocellular hypertrophy and increased liver weight (Klaunig et al. 2003). Specific evidence indicating PPARα activation includes demonstrating binding to the PPAR receptor and confirming increased peroxisomal enzyme activity by demonstrating increased acyl CoA-oxidase activity. Increased numbers of peroxisomes can be detected by transmission electron microscopy, but this is not essential for demonstration of this MOA. An increase in the labeling index in the hepatocytes is essential, similar to what was discussed above for phenobarbital. Again, for most of these agents, reversibility of all of these findings occurs upon cessation of administration of the compound. Usually four weeks of discontinued administration is adequate for complete reversibility.
Data necessary to support PPARα activation as a mode of action.
In extrapolating the data regarding PPARα agonists from the rodent models to humans, significant qualitative and quantitative differences have been identified. To begin with, there are significantly fewer receptors present on human liver cells compared to mouse or rat, approximately by an order of magnitude (Klaunig et al. 2003). Furthermore, there is considerable evidence that the human does not respond to PPARα agonists with an increase in hepatocyte proliferation. Utilizing a wild-type mouse, Gonzalez and his colleagues have demonstrated that the hepatocytes respond by proliferating and by peroxisome proliferation when exposed to a PPARα agonist (Yang et al. 2008). However, when the mouse is “humanized” by adding the human receptor to the cells rather than the mouse receptor, the hepatocytes respond to PPARα agonists with appropriate changes indicative of peroxisome stimulation, but the cells do not respond with a mitogenic, proliferative response. These data strongly suggest that there are not only fewer receptors in humans cells, but they do not result in downstream activation of genes necessary for increased cell proliferation, in contrast to the effects in mice and rats. Thus, it appears that the carcinogenic response is unique to the rodent and does not occur in humans or in nonhuman primates.
Statins
Although not involving a receptor in the classic sense, statins act through binding to a specific enzyme, HMG-CoA-reductase, which in humans leads to marked reduction in cholesterol production (Slater and MacDonald 1988; Farwell et al. 2008). The statins have been widely used in human medicine to reduce serum cholesterol levels for more than twenty years. In rats and mice, they are known to produce a significant increase in hepatocellular tumors (MacDonald and Halleck 2004; MacDonald et al. 1988). In rodents, this is associated with increased cell proliferation that ultimately leads to the development of hepatocellular lesions and ultimately carcinomas.
The specific key events for these agents are not completely delineated but include demonstration of inhibition of HMG-CoA-reductase activity, increased cell proliferation, and ultimately development of tumors (MacDonald et al. 1988; MacDonald and Halleck 2004). Coadministration with mevalonic acid overcomes the enzyme inhibition and prevents the increased proliferative activity and the development of tumors. Thus, the critical pieces of data necessary to invoke this as the MOA are demonstration that there is HMG-CoA-reductase inhibition, increased proliferation of hepatocytes, and prevention of these activities by coadministration with mevalonic acid.
Humans respond to statins significantly different than do rodents. Rodents do not show a prolonged reduction in cholesterol following administration of statins (MacDonald and Halleck 2004). Most importantly, there is considerable epidemiologic evidence demonstrating that statins are not associated with either an increased or decreased incidence of liver cancer or any other type of cancer (Bonovas et al. 2006; Browning and Martin 2007; Dale et al. 2006; Farwell et al. 2008).
In a low number of patients, statins produce hepatocellular cytotoxicity, which results in the discontinuance of treatment (MacDonald and Halleck 2004). One could argue that if statins were continued to be administered to such individuals with continued cytotoxic effects, tumors could ensue. This is a theoretical consideration; for practical matters it is not relevant since individuals who develop hepatotoxicity secondary to statin treatment will have the treatment discontinued. Like other non–DNA reactive agents involving a variety of MOAs, the effects in the liver of these agents in rodents is completely reversible. In humans, the cytotoxicity also appears to be reversible upon discontinuance. This issue of prolonged exposure being required for the development of tumors in situations of cytotoxicity is described further below in the section on cytotoxicity. With respect to the statins, this MOA in rodents appears to be irrelevant to human carcinogenesis based on mechanistic understanding and extensive epidemiologic evidence involving evaluation of tens of thousands of individuals.
Non-Receptor-Mediated, Non–DNA Reactive Carcinogens
Cytotoxicity
Induction of cytotoxicity in the liver is a common toxicological phenomenon and is frequently associated with the development of hepatocellular tumors in rodents, including carcinomas. Cytotoxicity can be manifested by both DNA-reactive and non–DNA reactive carcinogens. As described above, even when a chemical is DNA reactive, cytotoxicity can greatly modify the dose response for tumorigenicity of these chemicals. If the chemical is non–DNA reactive, cytotoxicity is frequently not associated specifically with a known cellular receptor, although there frequently is metabolic activation by specific enzymes. Cytotoxicity is a threshold phenomenon, and since it is the key event in the development of the tumors, the carcinogenicity is also a threshold phenomenon (Cohen 2008; Andersen et al. 1998; Meek et al. 2003).
The key events of this MOA are the induction of cytotoxicity followed by regenerative cell proliferation and ultimately tumors (Table 8 ). The key events in the metabolic activation of specific chemicals can frequently be identified (see below). By identifying a detailed dose response, a specific threshold can be identified, and this can frequently be addressed in the human relevance evaluation (Andersen et al. 1998). The data necessary for demonstrating cytotoxicity and regenerative proliferation are listed in Table 9 .
Key events in the induction of liver tumors by cytotoxicity and regenerative proliferation.

Data necessary to support cytotoxicity and regenerative cell proliferation as a mode of action.

Cytotoxicity can be identified in nonclinical studies either histopathologically by evidence of necrosis or by an increase in serum enzyme activities that are known to be associated with liver toxicity, such as AST, ALT, LDH, and GGT (Holsapple et al. 2006; Meek et al. 2003). These enzyme changes are not specific individually, but if occurring together, they are strongly indicative of liver toxicity. Histopathologic verification is usually possible. Increased cell proliferation can be determined as described above. Like other MOAs involving non–DNA reactive chemicals, the process is reversible upon discontinuance of the chemical.
Cytotoxicity by chemicals can occur not only in rodents but in humans, frequently by the same chemicals. As indicated above, this is usually associated with a threshold dose response. Important for human risk assessment is that cytotoxicity be present for prolonged periods for carcinogenesis to occur. This rarely occurs with chemicals since discontinuance of exposure occurs if cytotoxicity is identified, as described above for the statins.
A classic example of cytotoxicity and liver carcinogenesis is chloroform (Andersen et al. 1998, 2000; Meek et al. 2003). The initial key event for chloroform is metabolic activation by CYP2E1 to phosgene, which reacts with cellular proteins leading to cell necrosis, cell regeneration, and ultimately tumors. These precursor key events can occur in humans, as was demonstrated with the toxicity associated with chloroform when it was widely used as an anesthetic in the first half of the past century (Meek et al. 2003; Andersen et al. 1998). However, there is no evidence of tumorgenicity in such instances, since the exposure was brief and liver and kidney toxicities were repairable. In detailed studies on chloroform, including physiologically based pharmacokinetic (PBPK) models of rodents and humans, the detailed dose response has been delineated, indicating a clear threshold (Andersen et al. 1998). The concept of thresholds in carcinogenesis has actually been upheld in the legal system utilizing the example of chloroform ( Chlorine Chemistry Council v. EPA 2000).
There are numerous other metabolic pathways and mechanisms of inducing cytotoxicity in the liver. Another example with detailed mechanistic information identified is thiamethoxam (Pastoor et al. 2005). It is metabolized to two different metabolites, one of which appears to be responsible for induction of cytotoxicity and the other to be involved more with enhancement of the cell proliferative response. Nevertheless, the ultimate effect of thiamethoxam treatment of mice is to induce cytotoxicity, which leads to regenerative proliferation and ultimately the development of tumors. The metabolic activation quantitatively is inadequate for cytotoxicity to occur in rats, and no tumors are produced in the rat with treatment. Likewise, human cells do not appear to react to thiamethoxam with generation of adequate amounts of these metabolites to produce cytotoxicity, and consequently it is highly unlikely that humans would have a carcinogenic response to exposure to thiamethoxam, regardless of the dose.
Hepatocellular toxicity can occur not only secondary to cell necrosis but also due to an increase in apoptosis. This was demonstrated for fumonisin B1, although the relationship of increased apoptosis to carcinogenesis was more specifically defined in the kidney (Dragan et al. 2001). In the liver, there was evidence of not only apoptosis but hepatocellular necrosis. Regardless of the cause of the liver cell death, the result was the presence of regenerative proliferation and ultimately the development of liver tumors.
Metal overload has also been identified as a potential MOA for liver carcinogenesis. However, the specifics of this mode of action are not entirely clear. There are several examples of accumulation of either iron (Kowdley 2004; Harrison and Bacon 2005) or copper (Powell 1994) in liver cells with the ultimate development of liver neoplasms, including carcinomas. The key events in this process presumably involve generation of oxidative damage as well as cytotoxicity associated with regeneration (Klaunig and Kamendulis 2004; Goodman and Terracciano 2007). However, there is evidence that abnormalities in porphyrin metabolism might be critical to the development of tumors in these circumstances (Greaves et al. 2005), although in humans porphyria cutanea tarda is frequently associated with hepatitis C infection (Goodman and Terracciano 2007). Liver carcinogenicity has occurred in animal models and in humans in association with iron and with copper overload. In humans, there are inherited disorders leading to accumulation of iron (hemochromatosis) and/or copper (Wilson’s disease), which lead to hepatocellular cytotoxicity, regenerative proliferation with chronic active inflammation, and cirrhosis (Goodman and Terracciano 2007). Many of these individuals, but not all, ultimately develop liver cancer. Again, the fact that not all of the individuals develop hepatocellular carcinomas, nor do all the animals that have toxicity develop carcinomas, is most likely related to a stochastic process related to increased cell proliferation. However, the exact details of this MOA are yet to be established.
Nevertheless, for metal overload there are several critical key events that can be established for this MOA. To begin with, there needs to be demonstration of increased iron or copper in the hepatocytes. For iron, this can be readily achieved by histologic evaluation, which may include the use of special stains such as Prussian blue for iron (Bancroft and Stevens 1982). For copper, the histologic identification is often not as straightforward, but if suspected, specific stains for copper can be performed as well as for the copper binding protein ceruloplasmin (Sheehan and Hrapchak 1980). Fundamentally, this mode of action is a specific form of cytotoxicity and regeneration.
In humans, the most common causes of liver cancer are hepatitis B and hepatitis C viruses (Goodman and Terracciano 2007). This is associated with chronic destruction of hepatocytes with associated chronic active hepatitis, ultimately leading to the formation of cirrhosis and occasionally liver cancer. Infectious organisms in the liver have also been identified as carcinogenic in rodents, specifically Helicobacter hepaticus in mice (Ward et al. 1994). This is a specific form of cytotoxicity, which can involve reactions of the immune system to the infectious organism or to the hepatocytes. In common for all of these non-receptor-mediated mechanisms is the presence of hepatocellular toxicity with associated regenerative proliferation. This is frequently associated with chronic active inflammation and, in humans, with cirrhosis.
In humans, ethanol is a common cause of hepatocellular carcinomas (Goodman and Terracciano 2007). Again, this is due to the chronic inflammatory process that is caused by high levels of consumption of ethanol leading to steatohepatitis, which can evolve into cirrhosis and occasionally the induction of liver tumors. This process has been difficult to replicate in animal models but appears to have been accomplished (Ramaiah, Rivera, and Arteel 2004). Again, it appears to be a threshold phenomenon and involves cytotoxicity and regeneration.
Short-Term Screening Assay for Hepatocellular Carcinogenesis
After a chemical has been identified as positive in the initial ninety-day screening as described utilizing the four criteria established by Allen et al. (2004), the detailed mechanistic studies described above and listed in Figure 1 will provide detailed information concerning the MOA of the chemical in producing the liver cell response. This will provide substantial information regarding potential relevance to humans. By looking at all of these parameters, more than one MOA could be identified, such as the combination of DNA reactivity and cytotoxicity seen with many DNA-reactive carcinogens, or the induction of CAR by many of the PPARα agonist. A detailed dose response can also be evaluated for the various key events. The dose response for the key events should run in parallel, with the most effective dose for subsequent key events being at the level of the previous key event or higher. Thus, even if an MOA is relevant to humans, a rational approach to the dose response can be evaluated, including the possibility of a threshold as is seen most commonly with cytotoxicity.
In this article, I have provided a detailed approach for a ninety-day screening method for the assessment of potential carcinogenic risk specifically for the liver. A similar approach can be identified for the other tissues (Cohen 2004). The approach involves a ninety-day screening assay in the whole animal, which would be part of a usual toxicity screening process. Liver weight and histopathology are routinely part of this process; also evaluated are blood for enzymes indicative of liver toxicity, such as AST, ALT, and GGT. Furthermore, routine histopathology that is currently performed on other tissues would include an examination of endometrium, cervix, and vagina, which would detect the possibility of an estrogenic effect. As I have described elsewhere, immunosuppression is another example of an effect that in humans corresponds to a markedly increased risk of certain types of cancers (Cohen 2004; Cohen, Purtilo, and Ellwein 1991). Although hepatocellular carcinomas are not usually present in such patients, such tumors are a possibility given the viral etiology of many hepatocellular carcinomas in humans. Nevertheless, routine histopathology in the ninety-day bioassay will readily detect immunosuppressive chemicals by their effects on the lymphoid system.
If none of the signals for the liver are detected in the ninety-day screening, one can be confident that the chemical will not be a hepatocarcinogen either for rodents or for humans. If one or more of the criteria for an effect on liver is detected in the ninety-day screening, the first mechanistic determination must be for a detailed assessment of DNA reactivity. In most instances, this will have been completed before a screening animal assay is performed and, if positive, would lead to discontinuance of development. However, for some chemicals already in commerce or naturally occurring, such information would be needed for an overall risk assessment. Screening for DNA reactivity should include the Ames' assay and structure-activity relationship assessment, with in vivo evaluations if indicated. I do not believe that other in vitro assessments of genotoxicity are appropriate, since these generally do not have adequate predictive value for an assessment of carcinogenicity (Zeiger 2004). If the chemical is DNA reactive, then details regarding metabolic activation and dose response are appropriate avenues to explore with respect to establishing the relationship of the findings in the rodents to the human situation.
Whether the chemical is DNA reactive or not, additional evaluation of other parameters for possible MOA analysis for the liver can be performed utilizing in vitro and/or in vivo methods, with most of the assays requiring less than four weeks of administration. Reversibility assays are also useful for many of the non–DNA reactive MOAs. The specific mechanistic evaluations involve assessment of binding to various receptors which are known to be related to liver carcinogenesis in rodents, such as PPARα, CAR, PXR, AHR, and the estrogen receptors. Histopathology and the ninety-day screening study will be helpful in directing additional investigations, such as centrilobular hypertrophy suggesting the possibility of CYP enzyme induction. Stains for iron or copper can also be performed on the tissue from the ninety-day study without having to perform them on tissues from additional studies. Evidence of cytotoxicity should also be apparent in the ninety-day screening study. In addition to the mechanistic studies, a detailed dose response evaluation is strongly recommended, which will be particularly useful for extrapolation to the human situation if the MOA appears to be relevant to humans.
In humans, hepatocellular tumors are usually the result of a chronic inflammatory process with continuous, long-term increased cell proliferation in the otherwise slowly proliferating hepatocytes and are rarely due to environmental chemicals (Goodman and Terracciano 2007). In contrast, in rodent bioassays the liver is the most common target organ, particularly in mice. In mice, there are several strains such as B6C3F1 and CD-1 that have extremely high background rates of hepatocellular tumors (Drinkwater, Hanigan, and Kemp 1990), suggesting that they might not be representative or indicative of potential risk to humans. In Europe, the mouse as a model system for carcinogenicity bioassays was not required in evaluation of pharmaceuticals because of the strong evidence that the tumorigenic response did not add predictive value of carcinogenicity to the rat bioassay (Van Osterhout et al. 1997). Whether a chemical produces liver tumors in both rats and mice or only in one species or the other does not appear to be predictive of whether the MOA is possibly relevant to humans. Thus, PPARα agonists and CAR-related chemicals frequently produce tumors in both rats and mice, and yet the MOA does not appear to be relevant to humans.
Toxicogenomics is increasingly useful for an evaluation of mechanism (Ellinger-Ziegelbauer et al. 2004, 2005, 2008; Nie et al. 2006; Thomas et al. 2007). It appears that as a screening tool it has limited usefulness, particularly as a way to predict carcinogenicity in general. For carcinogenesis screening, even if adequate patterns could be identified for such purposes, it would mean evaluating all tissues, with parameters for evaluation yet to be established. However, mechanistic information is increasingly becoming available by use of toxicogenomics. It can assist in the decision of DNA-reactive versus non–DNA reactive carcinogens, particularly with respect to activation of indirect forms of DNA damage such as oxidative damage, although a clear distinction between DNA-reactive and non–DNA reactive chemicals is not always possible. For liver carcinogenesis, it has the potential to assist in quickly identifying pathways that might be altered by a specific chemical, such as activation of PPARα, CAR, PXR, AHR, estrogen receptor, and so forth. It is not yet clear, however, whether such information obtained from toxicogenomic investigations will supplement information obtained from currently used methods such as those described above, or whether it will replace some or all of these. Additional investigation will be necessary to determine the role of toxicogenomics, as well as proteomics and metabolomics, in the evaluation of mechanism.
Once the above screen has been completed with the appropriate follow-up studies, an assessment of human relevance can be made (Table 1 and Figure 1). As described briefly above, and in greater detail in numerous publications, many of the MOAs described above do not appear to be relevant to humans. If such is the case, the finding of liver changes in the ninety-day screen followed by suitable mechanistic investigations provides ample evidence to reliably conclude that the chemical is not likely to be a human liver carcinogen.
After all of the mechanistic investigations that are listed are performed, there will be some chemicals for which no specific MOA is clearly identified. This will pose a challenge for regulatory approval of such a chemical, as it would for any toxicologic finding. Since the screening would identify DNA-reactive chemicals, one is left with a non–DNA reactive MOA. A margin of exposure could be identified based on the effect between the doses used in the animal studies compared to potential exposure in humans. Depending on the type of chemical and the agency regulating it, a margin of exposure approach to the acceptance of such a chemical may differ. It is my opinion that the company developing such a compound would be left with three alternatives: (1) rely entirely on margin of exposure estimates to predict safety for human exposures, (2) do the research necessary to identify the MOA by which the chemical produces the liver changes, or (3) perform a full two-year bioassay to address the issue of carcinogenicity. Obviously, as a scientist I prefer the second option, but I fully realize that this may be a significant hurdle to overcome, as it is particularly challenging to provide the necessary data for identifying the MOA when it is the first time that it is being established. Once the MOA is known, subsequent compounds with a similar MOA would require significantly less investigation to establish the MOA. The choice of which of these three options will be made will depend on the purpose for which the chemical is being developed and the economic consequences of the decision.
An additional benefit of this approach is that it provides a mechanistic basis for the liver changes that are observed in the ninety-day screen, which is essential for extrapolating potential liver toxicity in general to the human situation. The ninety-day assay is designed to pick up evidence of potential toxicity of any kind for humans, so that the appearance of any one of the four indicators identified by Allen et al. (2004) would need to be explained, and at the very least a dose response and margin of exposure with respect to humans would need to be identified.
Product Labeling
Since a large number of chemicals will give a positive signal in the ninety-day screen for the liver, how can we label such chemicals so that they are not all broadly branded as possible human carcinogens? I offer the following suggestion. If the chemical is DNA reactive, unless there is persuasive evidence that the metabolic activation cannot occur in humans, the chemical would have to be labeled as a possible human carcinogen. However, the possible quantitative risk assessment for humans needs to take into account any differences in metabolic activation and inactivation between species as well as possible differences in other aspects of toxicokinetics. Also to be taken into account are confounding factors such as cytotoxicity, direct mitogenesis, or saturation of metabolic pathways. For the non–DNA reactive chemicals, if an MOA can be identified from the mechanistic studies, then its relevance to humans can be directly addressed. Thus, if the compound is acting by an MOA that is unlikely to be relevant to humans, such as interaction with PPARα agonist or CAR binding, then the chemical would not be labeled as a possible carcinogen but merely indicated as to the specific toxicity issue that is identified (PPARα agonism, induction of cytochrome CYP isozymes, etc.). If the MOA that is identified is relevant to humans (e.g., estrogen, cytotoxicity), then the chemical can be indicated as being possibly carcinogenic to humans, but information regarding a dose response and possible threshold (as with cytotoxicity) can be added as a descriptor qualifying the carcinogenic response.
Conclusion
In conclusion, taking into account extensive experience with the two-year bioassay and the identification of key events in MOAs leading to the development of tumors, precursor changes can be readily identified in a ninety-day screening bioassay so that a full two-year bioassay for the ultimate development of liver tumors is unnecessary for an adequate, rational, and conservative approach to human cancer risk assessment. The follow-up detailed mechanistic and dose response studies can provide considerably more information regarding possible human cancer risk than the two-year bioassay is capable of achieving. The two-year bioassay merely tells you that the chemical produces tumors in rodents after two years. It gives you little information regarding a detailed dose response and no information with respect to MOA. The approach that I am suggesting is based on sound scientific principles, takes into account our knowledge of carcinogenesis, and provides a rational approach to human cancer risk assessment with utilization of considerably fewer animals, cost, and time.
Footnotes
Acknowledgments
This article is based on a presentation to the Third Workshop of the Standing Committee on Risk Analysis Issues and Reviews, National Academy of Sciences, Washington, D.C., November 8, 2007.